

Abstract
AIM: To characterize the influence of diet-induced changes in body fat on colitis severity in SMAD3-/- mice.
METHODS: SMAD3-/- mice (6-8 wk of age) were randomly assigned to receive a calorie restricted (30% of control; CR), control (CON), or high fat (HF) diet for 20 wk and were gavaged with sterile broth or with Helicobacter hepaticus (H. hepaticus) to induce colitis. Four weeks after infection, mice were sacrificed and the cecum and colons were processed for histological evaluation.
RESULTS: Dietary treatment significantly influenced body composition prior to infection (P < 0.05), with CR mice having less (14% ± 2%) and HF-fed mice more body fat (32% ± 7%) compared to controls (22% ± 4%). Differences in body composition were associated with alterations in plasma levels of leptin (HF > CON > CR) and adiponectin (CON > HF ≥ CR) (P < 0.05). There were no significant differences in colitis scores between CON and HF-fed mice 4 wk post-infection. Consistent with this, differences in proliferation and inflammation markers (COX-2, iNOS), and infiltrating cell types (CD3+ T lymphocytes, macrophages) were not observed. Unexpectedly, only 40% of CR mice survived infection with H. hepaticus, with mortality observed as early as 1 wk following induction of colitis.
CONCLUSION: Increased adiposity does not influence colitis severity in SMAD3-/- mice. Importantly, caloric restriction negatively impacts survival following pathogen challenge, potentially due to an impaired immune response.
Keywords: SMAD3, Colitis, Adipokine, Obesity, Calorie restriction
INTRODUCTION
Adipose tissue (AT) is increasingly recognized as an active endocrine organ modulating a number of physiological processes. AT is a key regulator of insulin resistance[1,2] and contributes to systemic inflammation through production of a variety of proteins, hormones and cytokines collectively referred to as adipokines[3,4]. Many of these secretory products play important roles in energy homeostasis and the immune response[5]. Several pro-inflammatory cytokines, including interleukin (IL)-6, C-reactive protein (CRP) and leptin, are released from AT even in the absence of acute injury or inflammation, and their production is increased in proportion to AT mass[6-10]. Such altered production of these cytokines contributes to a number of pathophysiological processes including peripheral insulin resistance, inflammation, vascular disease, and immune dysfunction commonly observed in obesity[2,11].
Inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), are chronic conditions characterized by remittent inflammation resulting in extensive damage to the gastrointestinal tract[12-14]. CD can affect any part of the intestine[12], whereas UC is confined to the colon[14]. Although certain clinical features differ between these two conditions[13], both are thought to result from a dysregulated immune response in susceptible individuals[15].
Altered local and systemic levels of cytokines including tumor necrosis factor (TNF)-α, leptin, and adiponectin have been observed in individuals with IBD and are suggested to contribute to the disease pathogenesis[16]. Leptin is a 16-kDa product of the ob gene and is produced primarily by adipocytes[17]. Circulating levels of leptin are increased in obesity and show a positive correlation to body mass index[18,19]. Although leptin regulates energy metabolism by inhibiting food intake and increasing energy expenditure[20], it also has important immunomodulatory roles[21-23]. Increases in leptin levels in the serum[24], mesenteric AT[25], and in the colonic lumen[26] have been reported during the active stage of IBD. Leptin is also associated with susceptibility to experimental colitis in mice[27]. Colonocytes express the leptin receptor, and luminal administration of leptin induces epithelial wall damage and neutrophil infiltration, suggesting a local pro-inflammatory role for this protein[26].
Adiponectin is a high molecular weight protein secreted by AT that contributes to glucose homeostasis by increasing peripheral insulin sensitivity and reducing hepatic gluconeogenesis[28]. Pro-inflammatory mediators, including TNF-α and IL-6, suppress adiponectin secretion and serum levels are markedly reduced in obese individuals[29-31]. Adiponectin is generally considered anti-inflammatory due to antagonistic effects on cytokine signaling[32-34]. However, increased levels have been detected in serum and hypertrophied mesenteric AT in patients with active IBD[35,36]. A direct role for adiponectin during experimental colitis in animals has produced inconsistent results[37-39].
There is currently insufficient evidence to support a causal relationship between obesity and IBD; however, the conditions share similar inflammatory characteristics. Recent studies indicate that the constant low-grade inflammation associated with excess AT, including elevated serum levels of CRP, IL-6 and TNF-α, may contribute to the severity of IBD[40]. Additionally, overweight and/or obese individuals with CD were found to have more complications and more frequent disease relapses than normal weight individuals[41], providing a potential link between excessive AT and pathogenesis of IBD.
In the current study we evaluated the influence of adiposity on colitis severity in SMAD3-/- mice. SMAD3-/- mice have defective transforming growth factor (TGF)-β signaling and develop mild colitis within 4 wk following infection with Helicobacter spp. Dysfunctions in TGF-β signaling are commonly observed in human IBD and during colon cancer development. Maggio-Price et al[42] demonstrated that SMAD3-/-, but not SMAD3+/- mice develop chronic colitis and colon cancer in response to a bacterial infection. In the SMAD3-/- mouse model of colon cancer, initiation and progression is induced by a bacterial infection Helicobacter hepaticus (H. hepaticus). The bacterium colonizes the cecum and proximal colon persistently, low grade inflammation and immune cell infiltration observed eventually lead to mucinous adenocarcinoma formation at 15-30 wk post infection[42]. Importantly, these lesions are flatter, more aggressive and harder to diagnose in humans. It is widely hypothesized that chronic low levels of inflammation, whether induced by a pathogen or not, leads to cancer promotion and progression. Therefore, this model is highly relevant to the process of human colon carcinogenesis. Specifically, the SMAD model is very similar to the development of specific human cancers where a pathogen is necessary (but not sufficient) to cause dysplasia and tumor formation. Examples include hepatitis and liver cancer, Helicobacter pylori and stomach cancer, and human papillomavirus and cervical cancer. The contribution of this research was to understand how energy balance differentially modulates promotion/progression of inflammation and pathogen-induced cancers.
Mice were submitted to one of three dietary treatments (control, 30% caloric restriction, or high fat diet) to induce differing levels of adiposity after 20 wk, and were then infected with H. hepaticus to induce colitis. Plasma leptin and adiponectin were measured pre-infection and histological scoring was performed on cecum and colon tissue 4 wk post-infection.
MATERIALS AND METHODS
Animal husbandry
Mice (129-Smad3tm1Par/J, referred to hereafter as SMAD3-/-) were generously donated by Lillian Maggio-Price at the University of Washington. The mouse colony was developed by pairing SMAD3-/- males with SMAD3+/- females. Weaning and genotyping of subsequent litters was performed as described below at approximately 21 d after birth; only SMAD3-/- mice were used in this study. All mice were housed in 60 square inch plastic cages with micro-isolator lids and maintained in temperature and humidity controlled rooms with a 12-h light-dark cycle. Harlan Teklad 22/5 Rodent Diet 8640 (22% crude protein, 5% crude fat) was given ad libitum prior to the start of the study. All mouse procedures were approved by the Michigan State University Institutional Animal Care and Use Committee.
Genotyping
Ear tissue samples were obtained and DNA extracted with REDExtract-N-Amp™ Tissue PCR Kit (Sigma-Aldrich, St. Louis, MO) according to manufacturer’s recommendations. Four primers were used for polymerase chain reaction: 1271 (GGATGGTCGGCTGCAGGTGTCC) and 1272 (TGTTGAAGGCAAACTCACAGAGC) to recognize SMAD sequences and give a 130 bp product, and 506 (CGGCGAGGATCTCGTCGTGACCCA) and 507 (GCGATACCGTAAAGCACGAGGAAG) to recognize vector sequences. Thermal cycling of the samples was conducted with an initial denaturation at 94 °C for 3 min, 40 cycles of denaturation-annealing-extension (respectively 20 s at 94 °C, 30 s at 58 °C, and 1 min at 72 °C), and a final extension of 72 °C for 3 min. Polymerase chain reaction products were then evaluated on a 2% agarose gel and visualized under UV transillumination.
Helicobacter hepaticus culture
Isolates of H. hepaticus (strain 3B1, ATCC 51449) were kindly donated by Vince Young at University of Michigan. Bacteria were streaked onto sheep blood agar plates and incubated at 36 °C for 24-48 h under anaerobic conditions using GasPak™ pouch systems (BD, Franklin Lakes, NJ). After incubation, cultures were collected by the addition of Bacto™ Tryptic Soy Broth (BD, Franklin Lakes, NJ) and the optical density was assessed using a Bio-Tek Synergy HT multi-mode microplate reader (Bio-Tek, Winooski, VT) to ensure a constant bacterial population (≥ 1.8 at 600 nm wavelength).
Dietary treatments and experimental procedures
SMAD3-/- mice (6-8 wk of age) were randomly assigned to one of three Open Source diets (Research Diets Inc, New Brunswick, NJ): control (CON; formula D12450B: 20% protein, 70% carbohydrate, 10% fat), 30% calorie-restricted (CR; formula D03020702B: 27% protein, 54% carbohydrate, 6% fat) or high fat (HF; formula D12492: 20% protein, 20% carbohydrate, 60% fat) to induce differing levels of adiposity as previously described[43]. Mice were weighed weekly to assess body weight changes. Body composition was also assessed after 20 wk using an EchoMRI-100™ quantitative nuclear magnetic resonance machine (Echo Medical Systems, Houston, TX).
In a pilot study (data not shown; n = 73 mice), weight differences between dietary treatments were found to be maximal around 20 wk, therefore this time frame was chosen for induction of colitis. Mice were thus fed diets (n = 37 CON, 19 CR, and 36 HF) for 20 wk and then gavaged with 0.3 mL dosages of either bacteria-free control Tryptic Soy Broth or H. hepaticus, one dosage per day on two consecutive days. Continued weight monitoring was conducted on the gavaged mice and any animal that exhibited a weight loss of > 20% from one week to the next was euthanized. Four weeks after infection, mice were euthanized via carbon dioxide asphyxiation. Terminal bleeds were performed via cardiac puncture and blood was collected in a heparin-coated syringe. Blood samples were centrifuged at 12 000 × g for 15 min at 4 °C, and plasma was collected and frozen at -80 °C until further use.
Histopathology
The entire lower gastrointestinal tract was isolated and removed. Ceca were incised and cleared of fecal material with ice-cold phosphate buffered saline (PBS). Colons were similarly cleared, rinsed with PBS and sectioned. Cecum and colon samples were fixed for 24 h in a 10% formalin solution and then processed, stained, and scored by a board certified pathologist (Dr. Ingeborg Langohr) blinded to treatment for degree of colitis and dysplasia[44]. Grades were on a 1 to 4 scale both for inflammation (1, no inflammation; 2, mild inflammation; 3, moderate inflammation; 4, marked inflammation) and dysplasia (1, no dysplasia; 2, low grade dysplasia; 3, high grade dysplasia; 4, high grade dysplasia with invasion/adenocarcinoma). Briefly, low-grade dysplasia was characterized by thickened mucosa with elongated crypts with reduced numbers of goblet cells, but maintenance of cell polarity and nuclear morphology. High-grade dysplasia was characterized by thickened mucosa with elongated, irregularly branching glands, cytological and nuclear atypia including loss of differentiation and polarity, closely aggregated nuclei, and numerous mitotic figures. The two scores for colon and two scores for cecum tissue in each animal were combined such that a score of 4 indicated no inflammation or dysplasia and a score of 16 reflected maximal inflammation and neoplasia.
Quantification of serum adipokines by enzyme linked immunoabsorbance assay
Adiponectin and leptin were quantified by enzyme linked immunoabsorbance assay in plasma samples from mice prior to infection according to the manufacturer’s instructions (R and D Systems; Minneapolis, MN). Plasma (n = 5 per group) was diluted 1:10 for leptin and 1:10 000 for adiponectin in reagent diluent. Upon completion of the assay, the plate was read at 450 nm wavelength using a Synergy® HT plate reader (Bio-Tek; Winooski, VT).
Immunohistochemistry
Antibodies specific for cyclooxygenase (COX)-2, inducible nitric oxide synthase (iNOS), and F4/80 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), CD3 from AbCam (Cambridge, MA) and Ki67 from Novus Biologicals (Littleton, CO). A rat ABC detection kit (sc-2019) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and remaining secondary antibodies from DAKO (DAKO Co., Carpinteria, CA). All other reagents were obtained from Sigma Chemical Co. (St. Louis, MO) unless otherwise indicated.
Five-micron thick sections of formalin-fixed paraffin-embedded colon tissue were mounted on coated slides, and dietary differences in macrophage (F4/80) and T lymphocyte cell infiltration (CD3), proliferation (Ki67), and expression of COX-2 and iNOS were evaluated using peroxidase biotin-streptavidin immunohistochemistry. Epitope retrieval was carried out either by heating sections (92-95 °C) in 10 mmol/L citrate buffer (pH 6.0) for COX-2, iNOS, CD3, and Ki67 or with proteinase K digestion (Roche biochemicals) for F4/80. Slides were subsequently washed, treated with 3% H2O2 and incubated in 2.5% bovine serum albumin to reduce non-specific binding of antibody. Sections were incubated overnight at 4 °C with the primary antibody diluted in blocking buffer. After washing, sections were treated with appropriate biotinylated immunoglobulins followed by peroxidase-conjugated streptavidin at room temperature for 45 min each. Antigen-linked peroxidase was detected with the chromagen 3-3’-diaminobenzidine (DAB; 0.5 mg/mL) diluted in 10 mmol/L PBS (pH 7.2) containing 0.015% H2O2.
For quantification of Ki67+ cells, a researcher blinded to treatments evaluated 10-20 full-length crypts/animal. The total number of nuclei (Ki67+ and Ki67-) lining one side of the crypt and extending from the base of the crypt to the lumen was recorded. Proliferative index (number of Ki67+ cells/total cells) was then calculated and analyzed. Colonic staining of COX-2, iNOS, CD3, and F4/80 were quantified using Nikon software and an inverted light microscope (Nikon; Kanagawa, Japan) equipped with a color camera (DS-U2, Nikon; Kanagawa, Japan). Using a 20 × objective, areas surrounding full length crypts in the proximal colon were traced and the positive stained area (total number of pixels) was quantified. Data are expressed as a percentage of positive stained area in relation to the total surface area. For each stain, at least 10 measurements/animal were taken.
Statistical analysis
Data for body weight and composition, colitis scores, and plasma adiponectin and leptin levels were analyzed with analysis of variance using Prism software (Graph Pad; San Diego, CA). Prior to analysis, normal distribution of the data was tested and when appropriate, data were transformed prior to statistical analysis. When statistical differences were detected, individual comparisons were made using Bonferroni’s multiple comparison test.
RESULTS
Effect of dietary treatment on body fat composition and plasma adipokines
Dietary treatments significantly influenced AT stores in mice prior to initiation of H. hepaticus infection. HF-fed mice weighed significantly more and had a higher percent body fat than CON or CR mice (Table 1). Conversely, CR mice weighed less than both HF and CON mice primarily due to lower amount of AT.
Table 1.
Pre-infection body weights and body composition of SMAD3-/- mice after 20 wk on dietary treatment
| Diet | Weight change (%) | Lean tissue (%) | Adipose (%) |
| CON | 35 ± 17.8 | 68 ± 5.0 | 22 ± 4.0 |
| CR | 32 ± 11.8 | 73 ± 3.0a | 14 ± 2.0a |
| HF | 45 ± 18.4 | 59 ± 4.0d | 32 ± 7.0d |
P < 0.05 vs control animals;
P < 0.01 vs control and calorie restricted animals. CON: Control; CR: Calorie restricted; HF: High fat.
Dietary treatment also affected plasma levels of metabolic hormones (Figure 1). Adiponectin was significantly lower in CR and HF mice compared to CON mice (Figure 1B, P < 0.05) whereas plasma concentrations of leptin were lowest in CR and highest in HF mice (Figure 1A, P < 0.05).
Figure 1.
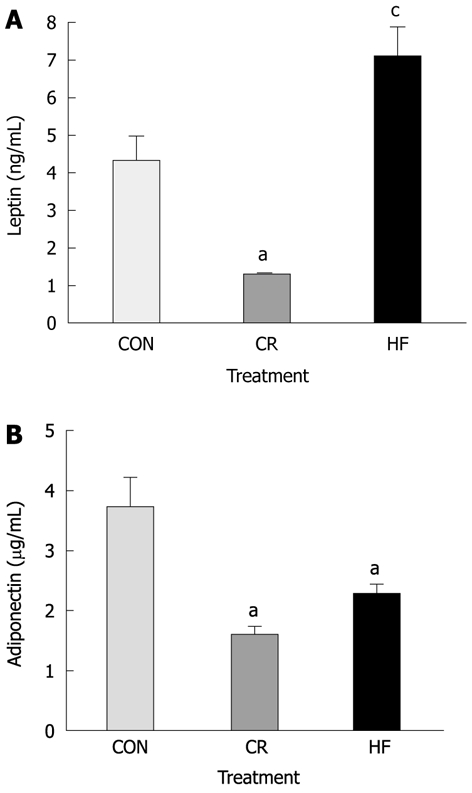
Plasma adipokines in SMAD3-/- mice after 20 wk on dietary treatment. A: Average plasma concentrations of leptin between diet groups prior to infection. Calorie restricted (CR) mice had significantly lower concentrations, whereas high fat (HF) mice had significantly higher concentrations of leptin compared to control (CON) mice (P < 0.01); B: Average plasma concentrations of adiponectin between diets prior to infection. Both CR and HF diet mice had significantly lower concentrations of adiponectin than mice on CON diet (P < 0.05). aP < 0.05 vs control; cP < 0.05 vs all other groups.
Effect of diet treatment on colitis and dysplasia scores 4 wk post-infection
After 20 wk on dietary treatment, mice were infected with H. hepaticus to determine if pre-infection adiposity would influence colitis scores 4 wk following infection. Unexpectedly, we found that CR mice had higher mortality rates beginning at 1 wk post-infection (Figure 2). Because only 40% of CR mice (n = 2) survived the infection period, we were unable to obtain enough tissue to appropriately evaluate colitis in this group.
Figure 2.
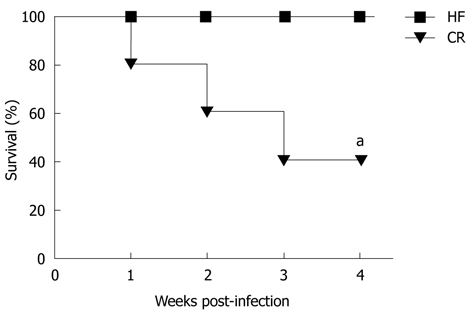
Survival curve of high fat, control and calorie restricted mice after infection with Helicobacter hepaticus. Calorie restricted (CR) mice experienced increased mortality after infection, with only 40% of CR mice surviving 4 wk post-infection. aP < 0.05 vs baseline. HF: High fat.
HF and CON mice were sacrificed 4 wk following infection and dietary differences in colitis and dysplasia were scored in the cecum and colon. Compared to the uninfected SMAD3-/- mice, CON and HF-fed mice exhibited significant infiltration of immune cell populations into the lamina propria following infection (Figure 3A). We found no significant differences in combined scores between CON (7.9 ± 1.9) and HF (7.8 ± 2.1) mice following infection (Figure 3B). In order to examine the potential effect of time, we allowed some mice to remain infected for up to 6 wk, and compared scores at 4, 5 and 6 wk post-infection. However, there were no further significant changes between or within dietary treatments across time (Figure 3C).
Figure 3.
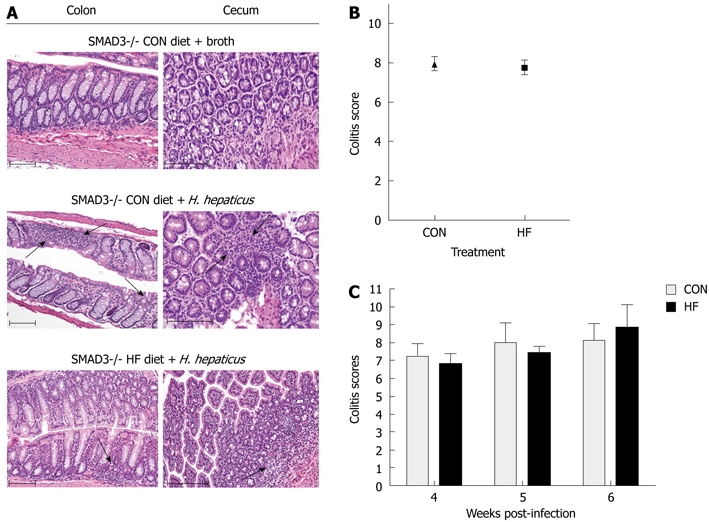
Effect of dietary treatment on colitis severity in SMAD3-/- mice. A: HE stained sections from the colon and cecum of SMAD3-/- control (CON)-fed mice treated with broth and SMAD3-/- CON- or high fat (HF)-fed mice 4 wk post-infection. Four weeks following infection, the number of inflammatory cells in the lamina propria is increased in the colon and cecum of both CON and HF diet animals, consistent with mild inflammation (arrows denote). Scale bars represent 100 μm; B: Average combined colitis and dysplasia scores between diet groups at 4 wk post-infection. There was no significant difference in theses scores between the CON and HF diet treatment groups; C: Average combined colitis and dysplasia scores between CON and HF mice at 4 wk, 5 wk and 6 wk post-infection. There was no difference between CON and HF diet groups at any point, and there was no effect of time on the scores between diets. H. hepaticus: Helicobacter hepaticus; HE: Hematoxylin and eosin.
Differences in pre-infection body weight on colonic proliferation and inflammatory markers
Immunohistochemistry analysis on proliferation and inflammatory markers in colon sections from CON-fed uninfected mice and CON and HF-fed mice 4 wk post-infection are presented in Figure 4. There was no significant difference in percentages of colon epithelia positively stained for Ki67, a marker of cellular proliferation, or F4/80 macrophages between CON or HF diet mice at 4 wk post-infection as compared to uninfected mice. A higher percentage of iNOS and COX-2 immunoreactivity (P > 0.05) and significantly higher levels of CD3+ lymphocytes (P < 0.05) was observed after infection in CON and HF fed mice compared to broth-treated mice, consistent with the increased colitis scores; however, there was no difference between infected animals on either diet (Figure 4).
Figure 4.
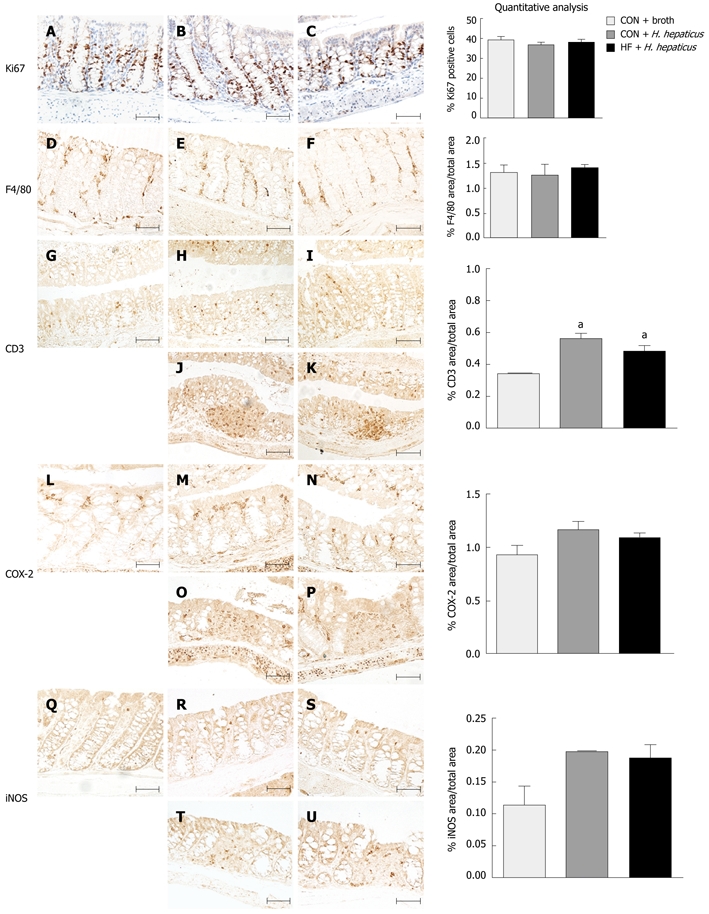
Effect of diet on proliferation and inflammatory markers following infection with Helicobacter hepaticus in SMAD3-/- mice. Immunohistochemical staining for Ki-67 (A-C), F4/80 (D-F), CD3 (G-K), COX-2 (L-P) and iNOS (Q-U) in proximal colon sections of SMAD3-/- mice fed control (CON) diet and treated with broth (A, D, G, L, Q), CON-fed mice treated with Helicobacter hepaticus (H. hepaticus) (B, E, H, J, M, O, R, T), or fed a high fat (HF) diet and treated with H. hepaticus (C, F, I, K, N, P, S, U) 4 wk post-infection. Normal appearing proximal colon segments (A-I, L-N, Q-S) and inflamed colon segments with lymphoid infiltrate (J, K, O, P, T, U). Scale bars represent 100 μm. There were no significant differences in proliferation indices or in macrophage infiltration. CON and HF diet mice had slightly increased staining for CD3+ T lymphocytes, cyclooxygenase (COX)-2, and inducible nitric oxide synthase (iNOS) post-infection compared to broth-treated controls, but averages were not statistically significant between diets (P > 0.05). aP < 0.05 vs CON animals.
DISCUSSION
Local and systemic alterations in adipokines are implicated in the pathogenesis of IBD[16]. In the current study, we investigated whether diet-induced changes in adiposity prior to induction of colitis would influence inflammatory changes in the colon of SMAD3-/- mice. After 20 wk on dietary treatment, significant changes in body composition were observed, with CR mice having the least and high fat-fed mice the most body fat compared to controls. Consistent with differences in adiposity, plasma concentrations of adipokines were significantly altered. Leptin is secreted in proportion to white AT mass whereas plasma adiponectin concentrations are markedly reduced in obese individuals[45]. In the current study, leptin levels were 1.5-fold higher in HF mice and 4.5 lower in CR mice, whereas low plasma adiponectin concentrations were observed in both HF and CR mice. Significantly lower levels of adiponectin were reported in individuals with anorexia and bulimia prior to treatment, which was restored following refeeding[46], suggesting a critical fat mass may be necessary for secretion[46].
We next evaluated whether differences in adiposity and adipokine levels would influence severity of colitis. We found that restricting caloric intake to 70% that of control animals significantly impacted survival of mice following H. hepaticus infection, with only 40% of mice surviving the full infection period. These results were somewhat unexpected as moderate calorie restriction delays or reduces severity of autoimmune disorders[47,48], prolongs life span[49], as well as inhibiting tumorigenesis at several different sites[50,51]. Additionally, Shibolet et al[52] found that calorie restricted mice were protected from chemically-induced colitis, which was associated with a decrease in pro-inflammatory cytokine release and an increase in NK1.1 + T lymphocytes. However, in experimental models of infection, CR increases susceptibility to bacterial[53] and parasitic[54] infections, as well as viral infections[55-57], consistent with our findings.
Leptin is recognized to play a pivotal role in both innate and adaptive immune responses by stimulating T cell proliferation[58], chemotaxis of neutrophils[59], NK cell maturity and activation[60], differentiation of dendritic cells[61], eicosanoid synthesis and cytokine release by monocytes and macrophages[62-64], as well as in preventing thymocyte apoptosis[65]. The increased mortality observed in this study was not investigated and is the aim for future projects. However, in a parallel study, we found baseline NK cell populations are reduced in the SMAD3-/- compared to SMAD3+/- mice (Fenton, JI, unpublished observations). Therefore, it is possible that reduced immune cell populations combined with lower circulating leptin in CR mice contributed to immune suppression and reduced capability to mount a response to H. hepaticus. In support of this, Clinthorne et al[56] recently reported that short term refeeding restored leptin in CR mice, improved survival, and attenuated the decline in NK cell function following influenza infection. Low circulating leptin in tuberculosis patients was also associated with increased disease severity[66], suggesting a causal relationship between adiposity, leptin, and immune response.
The development of IBD results from a complex interaction between genetic, immune and environmental factors. Diet is an important environmental factor in IBD pathogenesis; diets high in dairy products, refined sugar and fast food are associated with an increased risk of developing IBD[67,68]. However, there is little conclusive epidemiological evidence for a causal relationship between dietary intake and onset of IBD[68]. Importantly, after the onset of IBD, malnutrition (resulting from decreased food intake, malabsorption and increases in both nutrient loss and energy requirements) is common[69]. These changes in intake and energy expenditure may result from circulating inflammatory mediators associated with the pathophysiology of IBD, such as TNF-α, IL-1 and IL-6. These cytokines can increase catabolism and lead to anorexia[68,70]. Malnutrition is also associated with adverse outcomes in IBD progression, exacerbating immunodeficiency, perpetuating malabsorption and increasing risk of infections, particularly via bacterial translocation. This differs somewhat from what is modeled in this study. The caloric restriction diet met 100% of all nutrient needs and was only deficient by 30% of energy. The model caloric restriction of experimental-colitis is consistent with increased mortality in IBD related to energy deficit but not malnutrition[70-73]. Given the importance of leptin and immune function, our data do imply that reduced AT and leptin production (directly related to fat cell size and number) may further impair innate immune response to a pathogen. Similar mortality effects were observed in caloric restriction and influenza infection in mice discussed above.
Although low body fat stores and reduced circulating levels of leptin may impair immune responses to infectious stimuli, elevated leptin and peripheral leptin resistance is commonly observed in obese individuals. Previous studies suggest that obesity exacerbates colonic inflammation[40,74-77]. Increased mesenteric fat and fat creeping were also observed in inflamed intestinal regions in patients with CD[78]. Additionally, overweight and/or obese individuals with CD have more complications from and more frequent disease relapses than normal weight individuals[41]. In the current study, we did not observe any overall changes in colitis severity between control and HF mice, despite differences in body fat and serum adipokines. To determine whether general inflammatory markers were altered, we stained for COX-2 and iNOS, which are induced by pro-inflammatory cytokines in a variety of pathological conditions including UC in humans[79,80]. Further, we examined colons for T lymphocyte and macrophage infiltration to determine whether higher body fat would influence specific subsets of inflammatory cells. Both control and high fat mice treated with H. hepaticus had moderately elevated levels of CD3+ T lymphocytes, as well as COX-2 and iNOS immunoreactivity in epithelia-associated myofibroblasts and macrophages, compared to broth-treated mice. However no further changes were observed between dietary treatments. We also did not observe any differences in the proliferative index in the proximal colon segments between treatments, demonstrating that higher body fat does not influence disease severity in our model.
Results from the current study suggest that moderately increased adiposity induced by high fat feeding does not influence colitis severity in SMAD3-/- mice despite changes in plasma adipokines. Although we were able to induce a body fat percentage of 32% in the SMAD3-/- mice, this percentage body fat may be insufficient to induce chronic inflammation associated with obesity observed in other mouse strains that approach 50%-60% AT. More importantly, we found that calorie restricted mice had a higher mortality in response to infection with H. hepaticus. Future studies examining the association between percent body fat, leptin, and immune responses to infectious stimuli leading to IBD are warranted.
COMMENTS
Background
Recent studies indicate that the constant low-grade inflammation associated with obesity or excess adipose tissue (AT), including elevated serum levels of C-reactive protein, interleukin-6 and tumor necrosis factor-α, may contribute to the severity of inflammatory bowel diseases (IBD). Additionally, overweight and/or obese individuals with Crohn’s disease were found to have more complications and more frequent disease relapses than normal weight individuals, providing a potential link between excessive AT and pathogenesis of IBD.
Research frontiers
There is currently insufficient evidence to support a causal relationship between obesity and IBD; however, the conditions share similar inflammatory characteristics. In this study, the authors demonstrate that moderately increased adiposity with associated changes in adipokines does not influence colitis severity in SMAD3-/- mice.
Innovations and breakthroughs
Previous studies suggest that obesity exacerbates colonic inflammation. Malnutrition is associated with adverse outcomes in IBD progression, exacerbating immunodeficiency, perpetuating malabsorption and increasing risk of infections. We report that moderate obesity did not influence the severity of colon inflammation in experimentally-induced colitis. Importantly, caloric restriction negatively influences survival following pathogen challenge, potentially due to an impaired immune response.
Applications
Further studies should examine the role of energy balance, both positive and negative, on the pathogenesis of IBD. Understanding how caloric restriction increases mortality in this preclinical model of experimentally-induced colitis may lead to therapeutic strategies for bacterially-driven IBD in humans.
Terminology
In the current study,the authors evaluated the influence of adiposity on colitis severity in SMAD3-/- mice. SMAD3 is a transcription factor for transforming growth factor (TGF)-β. TGF-β is a protein that controls proliferation and cellular differentiation, both in normal cells and in the early stages of carcinogenesis. SMAD3-/- mice have defective TGF-β signaling and develop mild colitis within 4 wk following infection with Helicobacter spp. Dysfunctions in TGF-β signaling are commonly observed in human IBD and during colon cancer development.
Peer review
It is a relevant paper dealing with a demanding issue too much neglected in the biologic-era treatment of IBD. The suggestion is implementing discussion with some speculation on the potential negative influence of malnutrition in IBD.
Footnotes
Peer reviewer: Giuseppe Chiarioni, Dr., Gastroenterological Rehabilitation Division of the University of Verona, Valeggio sul Mincio Hospital, Azienda Ospedale di Valeggio s/M, Valeggio s/M 37067, Italy
S- Editor Tian L L- Editor Rutherford A E- Editor Zheng XM
References
- 1.Lyon CJ, Law RE, Hsueh WA. Minireview: adiposity, inflammation, and atherogenesis. Endocrinology. 2003;144:2195–2200. doi: 10.1210/en.2003-0285. [DOI] [PubMed] [Google Scholar]
- 2.Vettor R, Milan G, Rossato M, Federspil G. Review article: adipocytokines and insulin resistance. Aliment Pharmacol Ther. 2005;22 Suppl 2:3–10. doi: 10.1111/j.1365-2036.2005.02587.x. [DOI] [PubMed] [Google Scholar]
- 3.Balistreri CR, Caruso C, Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. 2010;2010:802078. doi: 10.1155/2010/802078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lago F, Gómez R, Gómez-Reino JJ, Dieguez C, Gualillo O. Adipokines as novel modulators of lipid metabolism. Trends Biochem Sci. 2009;34:500–510. doi: 10.1016/j.tibs.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Juge-Aubry CE, Henrichot E, Meier CA. Adipose tissue: a regulator of inflammation. Best Pract Res Clin Endocrinol Metab. 2005;19:547–566. doi: 10.1016/j.beem.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92:347–355. doi: 10.1079/bjn20041213. [DOI] [PubMed] [Google Scholar]
- 7.Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, Robert JJ, Capeau J, Hainque B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab. 2002;87:2084–2089. doi: 10.1210/jcem.87.5.8450. [DOI] [PubMed] [Google Scholar]
- 8.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131–2135. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 9.Park HS, Park JY, Yu R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res Clin Pract. 2005;69:29–35. doi: 10.1016/j.diabres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Valle M, Martos R, Gascón F, Cañete R, Zafra MA, Morales R. Low-grade systemic inflammation, hypoadiponectinemia and a high concentration of leptin are present in very young obese children, and correlate with metabolic syndrome. Diabetes Metab. 2005;31:55–62. doi: 10.1016/s1262-3636(07)70167-2. [DOI] [PubMed] [Google Scholar]
- 11.Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: an update. Clin Endocrinol (Oxf) 2006;64:355–365. doi: 10.1111/j.1365-2265.2006.02474.x. [DOI] [PubMed] [Google Scholar]
- 12.Shanahan F. Crohn’s disease. Lancet. 2002;359:62–69. doi: 10.1016/S0140-6736(02)07284-7. [DOI] [PubMed] [Google Scholar]
- 13.Sanders DS. The differential diagnosis of Crohn’s disease and ulcerative colitis. Baillieres Clin Gastroenterol. 1998;12:19–33. doi: 10.1016/s0950-3528(98)90084-7. [DOI] [PubMed] [Google Scholar]
- 14.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 15.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 16.Karmiris K, Koutroubakis IE, Kouroumalis EA. The emerging role of adipocytokines as inflammatory mediators in inflammatory bowel disease. Inflamm Bowel Dis. 2005;11:847–855. doi: 10.1097/01.mib.0000178915.54264.8f. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu H, Shimomura Y, Hayashi R, Ohtani K, Sato N, Futawatari T, Mori M. Serum leptin concentration is associated with total body fat mass, but not abdominal fat distribution. Int J Obes Relat Metab Disord. 1997;21:536–541. doi: 10.1038/sj.ijo.0800437. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz MW, Prigeon RL, Kahn SE, Nicolson M, Moore J, Morawiecki A, Boyko EJ, Porte D. Evidence that plasma leptin and insulin levels are associated with body adiposity via different mechanisms. Diabetes Care. 1997;20:1476–1481. doi: 10.2337/diacare.20.9.1476. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F, Chen Y, Heiman M, Dimarchi R. Leptin: structure, function and biology. Vitam Horm. 2005;71:345–372. doi: 10.1016/S0083-6729(05)71012-8. [DOI] [PubMed] [Google Scholar]
- 21.Faggioni R, Feingold KR, Grunfeld C. Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB J. 2001;15:2565–2571. doi: 10.1096/fj.01-0431rev. [DOI] [PubMed] [Google Scholar]
- 22.Fernández-Riejos P, Najib S, Santos-Alvarez J, Martín-Romero C, Pérez-Pérez A, González-Yanes C, Sánchez-Margalet V. Role of leptin in the activation of immune cells. Mediators Inflamm. 2010;2010:568343. doi: 10.1155/2010/568343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conde J, Scotece M, Gómez R, Gómez-Reino JJ, Lago F, Gualillo O. At the crossroad between immunity and metabolism: focus on leptin. Expert Rev Clin Immunol. 2010;6:801–808. doi: 10.1586/eci.10.48. [DOI] [PubMed] [Google Scholar]
- 24.Tuzun A, Uygun A, Yesilova Z, Ozel AM, Erdil A, Yaman H, Bagci S, Gulsen M, Karaeren N, Dagalp K. Leptin levels in the acute stage of ulcerative colitis. J Gastroenterol Hepatol. 2004;19:429–432. doi: 10.1111/j.1440-1746.2003.03300.x. [DOI] [PubMed] [Google Scholar]
- 25.Barbier M, Vidal H, Desreumaux P, Dubuquoy L, Bourreille A, Colombel JF, Cherbut C, Galmiche JP. Overexpression of leptin mRNA in mesenteric adipose tissue in inflammatory bowel diseases. Gastroenterol Clin Biol. 2003;27:987–991. [PubMed] [Google Scholar]
- 26.Sitaraman S, Liu X, Charrier L, Gu LH, Ziegler TR, Gewirtz A, Merlin D. Colonic leptin: source of a novel proinflammatory cytokine involved in IBD. FASEB J. 2004;18:696–698. doi: 10.1096/fj.03-0422fje. [DOI] [PubMed] [Google Scholar]
- 27.Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology. 2002;122:2011–2025. doi: 10.1053/gast.2002.33631. [DOI] [PubMed] [Google Scholar]
- 28.Lara-Castro C, Fu Y, Chung BH, Garvey WT. Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease. Curr Opin Lipidol. 2007;18:263–270. doi: 10.1097/MOL.0b013e32814a645f. [DOI] [PubMed] [Google Scholar]
- 29.Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, Paschke R. Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2003;301:1045–1050. doi: 10.1016/s0006-291x(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 30.Kappes A, Löffler G. Influences of ionomycin, dibutyryl-cycloAMP and tumour necrosis factor-alpha on intracellular amount and secretion of apM1 in differentiating primary human preadipocytes. Horm Metab Res. 2000;32:548–554. doi: 10.1055/s-2007-978684. [DOI] [PubMed] [Google Scholar]
- 31.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 32.Kobashi C, Urakaze M, Kishida M, Kibayashi E, Kobayashi H, Kihara S, Funahashi T, Takata M, Temaru R, Sato A, et al. Adiponectin inhibits endothelial synthesis of interleukin-8. Circ Res. 2005;97:1245–1252. doi: 10.1161/01.RES.0000194328.57164.36. [DOI] [PubMed] [Google Scholar]
- 33.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 34.Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380:24–30. doi: 10.1016/j.cca.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto K, Kiyohara T, Murayama Y, Kihara S, Okamoto Y, Funahashi T, Ito T, Nezu R, Tsutsui S, Miyagawa JI, et al. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn’s disease. Gut. 2005;54:789–796. doi: 10.1136/gut.2004.046516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karmiris K, Koutroubakis IE, Xidakis C, Polychronaki M, Voudouri T, Kouroumalis EA. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:100–105. doi: 10.1097/01.MIB.0000200345.38837.46. [DOI] [PubMed] [Google Scholar]
- 37.Pini M, Gove ME, Fayad R, Cabay RJ, Fantuzzi G. Adiponectin deficiency does not affect development and progression of spontaneous colitis in IL-10 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G382–G387. doi: 10.1152/ajpgi.90593.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fayad R, Pini M, Sennello JA, Cabay RJ, Chan L, Xu A, Fantuzzi G. Adiponectin deficiency protects mice from chemically induced colonic inflammation. Gastroenterology. 2007;132:601–614. doi: 10.1053/j.gastro.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 39.Nishihara T, Matsuda M, Araki H, Oshima K, Kihara S, Funahashi T, Shimomura I. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology. 2006;131:853–861. doi: 10.1053/j.gastro.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 40.Florin TH, Paterson EW, Fowler EV, Radford-Smith GL. Clinically active Crohn’s disease in the presence of a low C-reactive protein. Scand J Gastroenterol. 2006;41:306–311. doi: 10.1080/00365520500217118. [DOI] [PubMed] [Google Scholar]
- 41.Blain A, Cattan S, Beaugerie L, Carbonnel F, Gendre JP, Cosnes J. Crohn’s disease clinical course and severity in obese patients. Clin Nutr. 2002;21:51–57. doi: 10.1054/clnu.2001.0503. [DOI] [PubMed] [Google Scholar]
- 42.Maggio-Price L, Treuting P, Zeng W, Tsang M, Bielefeldt-Ohmann H, Iritani BM. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 2006;66:828–838. doi: 10.1158/0008-5472.CAN-05-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yakar S, Nunez NP, Pennisi P, Brodt P, Sun H, Fallavollita L, Zhao H, Scavo L, Novosyadlyy R, Kurshan N, et al. Increased tumor growth in mice with diet-induced obesity: impact of ovarian hormones. Endocrinology. 2006;147:5826–5834. doi: 10.1210/en.2006-0311. [DOI] [PubMed] [Google Scholar]
- 44.Maggio-Price L, Bielefeldt-Ohmann H, Treuting P, Iritani BM, Zeng W, Nicks A, Tsang M, Shows D, Morrissey P, Viney JL. Dual infection with Helicobacter bilis and Helicobacter hepaticus in p-glycoprotein-deficient mdr1a-/- mice results in colitis that progresses to dysplasia. Am J Pathol. 2005;166:1793–1806. doi: 10.1016/S0002-9440(10)62489-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 46.Tagami T, Satoh N, Usui T, Yamada K, Shimatsu A, Kuzuya H. Adiponectin in anorexia nervosa and bulimia nervosa. J Clin Endocrinol Metab. 2004;89:1833–1837. doi: 10.1210/jc.2003-031260. [DOI] [PubMed] [Google Scholar]
- 47.Fernandes G, Good RA. Inhibition by restricted-calorie diet of lymphoproliferative disease and renal damage in MRL/lpr mice. Proc Natl Acad Sci USA. 1984;81:6144–6148. doi: 10.1073/pnas.81.19.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandrasekar B, McGuff HS, Aufdermorte TB, Troyer DA, Talal N, Fernandes G. Effects of calorie restriction on transforming growth factor beta 1 and proinflammatory cytokines in murine Sjogren’s syndrome. Clin Immunol Immunopathol. 1995;76:291–296. doi: 10.1006/clin.1995.1128. [DOI] [PubMed] [Google Scholar]
- 49.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003;54:131–152. doi: 10.1146/annurev.med.54.101601.152156. [DOI] [PubMed] [Google Scholar]
- 51.Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis. 2010;31:83–89. doi: 10.1093/carcin/bgp280. [DOI] [PubMed] [Google Scholar]
- 52.Shibolet O, Alper R, Avraham Y, Berry EM, Ilan Y. Immunomodulation of experimental colitis via caloric restriction: role of Nk1.1+ T cells. Clin Immunol. 2002;105:48–56. doi: 10.1006/clim.2002.5260. [DOI] [PubMed] [Google Scholar]
- 53.Sun D, Muthukumar AR, Lawrence RA, Fernandes G. Effects of calorie restriction on polymicrobial peritonitis induced by cecum ligation and puncture in young C57BL/6 mice. Clin Diagn Lab Immunol. 2001;8:1003–1011. doi: 10.1128/CDLI.8.5.1003-1011.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kristan DM. Chronic calorie restriction increases susceptibility of laboratory mice (Mus musculus) to a primary intestinal parasite infection. Aging Cell. 2007;6:817–825. doi: 10.1111/j.1474-9726.2007.00345.x. [DOI] [PubMed] [Google Scholar]
- 55.Ritz BW, Aktan I, Nogusa S, Gardner EM. Energy restriction impairs natural killer cell function and increases the severity of influenza infection in young adult male C57BL/6 mice. J Nutr. 2008;138:2269–2275. doi: 10.3945/jn.108.093633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clinthorne JF, Adams DJ, Fenton JI, Ritz BW, Gardner EM. Short-term re-feeding of previously energy-restricted C57BL/6 male mice restores body weight and body fat and attenuates the decline in natural killer cell function after primary influenza infection. J Nutr. 2010;140:1495–1501. doi: 10.3945/jn.110.122408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritz BW, Gardner EM. Malnutrition and energy restriction differentially affect viral immunity. J Nutr. 2006;136:1141–1144. doi: 10.1093/jn/136.5.1141. [DOI] [PubMed] [Google Scholar]
- 58.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 59.Caldefie-Chezet F, Poulin A, Vasson MP. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic Res. 2003;37:809–814. doi: 10.1080/1071576031000097526. [DOI] [PubMed] [Google Scholar]
- 60.Tian Z, Sun R, Wei H, Gao B. Impaired natural killer (NK) cell activity in leptin receptor deficient mice: leptin as a critical regulator in NK cell development and activation. Biochem Biophys Res Commun. 2002;298:297–302. doi: 10.1016/s0006-291x(02)02462-2. [DOI] [PubMed] [Google Scholar]
- 61.Mattioli B, Straface E, Quaranta MG, Giordani L, Viora M. Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J Immunol. 2005;174:6820–6828. doi: 10.4049/jimmunol.174.11.6820. [DOI] [PubMed] [Google Scholar]
- 62.Mancuso P, Canetti C, Gottschalk A, Tithof PK, Peters-Golden M. Leptin augments alveolar macrophage leukotriene synthesis by increasing phospholipase activity and enhancing group IVC iPLA2 (cPLA2gamma) protein expression. Am J Physiol Lung Cell Mol Physiol. 2004;287:L497–L502. doi: 10.1152/ajplung.00010.2004. [DOI] [PubMed] [Google Scholar]
- 63.Raso GM, Pacilio M, Esposito E, Coppola A, Di Carlo R, Meli R. Leptin potentiates IFN-gamma-induced expression of nitric oxide synthase and cyclo-oxygenase-2 in murine macrophage J774A.1. Br J Pharmacol. 2002;137:799–804. doi: 10.1038/sj.bjp.0704903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zarkesh-Esfahani H, Pockley G, Metcalfe RA, Bidlingmaier M, Wu Z, Ajami A, Weetman AP, Strasburger CJ, Ross RJ. High-dose leptin activates human leukocytes via receptor expression on monocytes. J Immunol. 2001;167:4593–4599. doi: 10.4049/jimmunol.167.8.4593. [DOI] [PubMed] [Google Scholar]
- 65.Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, Lechler RI, Bloom SR. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J Clin Invest. 1999;104:1051–1059. doi: 10.1172/JCI6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Crevel R, Karyadi E, Netea MG, Verhoef H, Nelwan RH, West CE, van der Meer JW. Decreased plasma leptin concentrations in tuberculosis patients are associated with wasting and inflammation. J Clin Endocrinol Metab. 2002;87:758–763. doi: 10.1210/jcem.87.2.8228. [DOI] [PubMed] [Google Scholar]
- 67.O’Sullivan M, O’Morain C. Nutrition in inflammatory bowel disease. Best Pract Res Clin Gastroenterol. 2006;20:561–573. doi: 10.1016/j.bpg.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 68.Lomer MC. Dietary and nutritional considerations for inflammatory bowel disease. Proc Nutr Soc. 2011;70:329–335. doi: 10.1017/S0029665111000097. [DOI] [PubMed] [Google Scholar]
- 69.Shamir R. Nutritional aspects in inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2009;48 Suppl 2:S86–S88. doi: 10.1097/MPG.0b013e3181a15ca0. [DOI] [PubMed] [Google Scholar]
- 70.Lucendo AJ, De Rezende LC. Importance of nutrition in inflammatory bowel disease. World J Gastroenterol. 2009;15:2081–2088. doi: 10.3748/wjg.15.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mijac DD, Janković GL, Jorga J, Krstić MN. Nutritional status in patients with active inflammatory bowel disease: prevalence of malnutrition and methods for routine nutritional assessment. Eur J Intern Med. 2010;21:315–319. doi: 10.1016/j.ejim.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 72.Gassull MA. Nutrition and inflammatory bowel disease: its relation to pathophysiology, outcome and therapy. Dig Dis. 2003;21:220–227. doi: 10.1159/000073339. [DOI] [PubMed] [Google Scholar]
- 73.Pirlich M, Schütz T, Kemps M, Luhman N, Burmester GR, Baumann G, Plauth M, Lübke HJ, Lochs H. Prevalence of malnutrition in hospitalized medical patients: impact of underlying disease. Dig Dis. 2003;21:245–251. doi: 10.1159/000073342. [DOI] [PubMed] [Google Scholar]
- 74.Li H, Lelliott C, Håkansson P, Ploj K, Tuneld A, Verolin-Johansson M, Benthem L, Carlsson B, Storlien L, Michaëlsson E. Intestinal, adipose, and liver inflammation in diet-induced obese mice. Metabolism. 2008;57:1704–1710. doi: 10.1016/j.metabol.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 75.Miyamoto S, Tanaka T, Murakami A. Increased visceral fat mass and insulin signaling in colitis-related colon carcinogenesis model mice. Chem Biol Interact. 2010;183:271–275. doi: 10.1016/j.cbi.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 76.Bassaganya-Riera J, Ferrer G, Casagran O, Sanchez S, de Horna A, Duran E, Orpi M, Guri AJ, Hontecillas R. F4/80hiCCR2hi macrophage infiltration into the intra-abdominal fat worsens the severity of experimental IBD in obese mice with DSS colitis. e-SPEN J. 2009;4:e90–e97. [Google Scholar]
- 77.Chapman-Kiddell CA, Davies PS, Gillen L, Radford-Smith GL. Role of diet in the development of inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:137–151. doi: 10.1002/ibd.20968. [DOI] [PubMed] [Google Scholar]
- 78.Sheehan AL, Warren BF, Gear MW, Shepherd NA. Fat-wrapping in Crohn’s disease: pathological basis and relevance to surgical practice. Br J Surg. 1992;79:955–958. doi: 10.1002/bjs.1800790934. [DOI] [PubMed] [Google Scholar]
- 79.Kong W, Yen JH, Vassiliou E, Adhikary S, Toscano MG, Ganea D. Docosahexaenoic acid prevents dendritic cell maturation and in vitro and in vivo expression of the IL-12 cytokine family. Lipids Health Dis. 2010;9:12. doi: 10.1186/1476-511X-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vedin I, Cederholm T, Freund-Levi Y, Basun H, Hjorth E, Irving GF, Eriksdotter-Jönhagen M, Schultzberg M, Wahlund LO, Palmblad J. Reduced prostaglandin F2 alpha release from blood mononuclear leukocytes after oral supplementation of omega3 fatty acids: the OmegAD study. J Lipid Res. 2010;51:1179–1185. doi: 10.1194/jlr.M002667. [DOI] [PMC free article] [PubMed] [Google Scholar]