

Abstract
Recent findings from our group, obtained on experimental in vivo and ex vivo models of pancreatitis, reveal that this disease causes a profound dysfunction of key cellular organelles, lysosomes and mitochondria. We found that autophagy, the main cellular degradative, lysosome-driven process, is activated but also impaired in acute pancreatitis because of its’ inefficient progression/resolution (flux) resulting from defective function of lysosomes. One mechanism underlying the lysosomal dysfunction in pancreatitis is abnormal processing (maturation) and activation of cathepsins, major lysosomal hydrolases; another is a decrease in pancreatic levels of key lysosomal membrane proteins LAMP-1 and LAMP-2. Our data indicate that lysosomal dysfunction plays an important initiating role in pancreatitis pathobiology. The impaired autophagy mediates vacuole accumulation in acinar cells; furthermore, the abnormal maturation and activation of cathepsins leads to increase in intra-acinar trypsin, the hallmark of pancreatitis; and LAMP-2 deficiency causes inflammation and acinar cell necrosis. Thus, the autophagic and lysosomal dysfunctions mediate key pathologic responses of pancreatitis. On the other hand, we showed that pancreatitis causes acinar cell mitochondria depolarization, mediated by the permeability transition pore (PTP). Genetic (via deletion of cyclophilin D) inactivation of PTP prevents mitochondrial depolarization and greatly ameliorates the pathologic responses of pancreatitis. Further, our data suggest that mitochondrial damage, by stimulating autophagy, increases the demand for efficient lysosomal degradation and therefore aggravates the pathologic consequences of lysosomal dysfunction. Thus, the combined autophagic, lysosomal and mitochondrial dysfunctions are key to the pathogenesis of pancreatitis.
Keywords: lysosome, cathepsin, LAMP protein, permeability transition pore, pancreatic mitochondria
Introduction
Pancreatitis is an inflammatory disorder of the exocrine pancreas, with considerable morbidity and mortality and without specific treatment. Its pathogenesis remains obscure.1 Severe pancreatitis requires prolonged hospitalization and multiple interventions and thus places a heavy burden on the US healthcare system; from recent data,2 pancreatitis was the 5th most common nonmalignant cause of death from digestive diseases in the US. The disease is believed to initiate in acinar cells, the main cell type of the exocrine pancreas.1, 3 Hallmark responses of pancreatitis include the premature, intra-acinar conversion of digestive proteases from inactive zymogens into their active forms (e.g., trypsinogen to trypsin); accumulation of large vacuoles in acinar cells; inflammation; and parenchymal cell death through apoptosis and necrosis.1, 3–5
Alcohol abuse is a major etiologic factor for pancreatitis, accounting for 30–50% of cases of acute pancreatitis and 60–90% of chronic pancreatitis.1, 6 The pathogenic mechanism of alcoholic pancreatitis is poorly understood.6 Of note, clinically relevant disease develops in < 10% of heavy drinkers; also, ethanol feeding alone does not induce pancreatitis in animal (rodent) models. Thus, it is believed that alcohol alone is not sufficient to cause pancreatitis and that in addition to alcohol, other, yet unidentified, factors are important for the disease expression.
There are a number of experimental models of (nonalcoholic) acute pancreatitis that reproduce the responses of human disease – animal models, such as pancreatitis induced in rats or mice by administration of cerulein (CCK-8 analog), bile acid (e.g., taurocholate) or L-arginine, or by feeding mice a choline-deficient, ethionine supplemented (CDE) diet; and the ex vivo model of isolated pancreatic acinar cells hyperstimulated with supramaximal CCK-8 (CCK) or cerulein (CR).1, 3–7 As stated above, ethanol feeding by itself does not induce pancreatitis in rodents; therefore, models of alcoholic pancreatitis combine ethanol with another “hit”, for example, low dose of CCK or CR.6, 8–12 In both in vivo and ex vivo models, ethanol sensitizes pancreas to CCK/CR-induced pancreatitis and thus decreases the threshold for the initiation of pancreatic injury.8, 9, 12
The focus of research during the last 15 years has been primarily on signaling pathways involved in the pathologic responses of pancreatitis, leading to elucidation of the roles of a number of molecules mediating the inflammatory (e.g., NF-κB, cytokines/chemokines, adhesion molecules, and novel protein kinase C isoforms) and cell death responses (e.g., caspases).1, 3, 4, 13–15 By contrast, the role of intracellular organelles’ damage, such as the lysosomes and the mitochondria, in pancreatitis – and in particular, in disease initiation – has been under-appreciated and under-explored. This review summarizes our findings in experimental models of pancreatitis5, 6, 16–25 indicating critical roles for lysosomal/autophagic and mitochondrial dysfunctions in the pathogenesis of this disease.
Lysosomal dysfunction and impaired autophagy in pancreatitis
The lysosome is the principal “digestive” organelle of eukaryotic cells serving both to eliminate obsolete components of the cell itself (autophagy) and to degrade material taken up from outside (endocytosis and phagocytosis).26, 27 The lysosome contains many types of hydrolytic enzymes (e.g., proteases, glycosidases, phospholipases) that usually exert their maximal enzymatic activity at low pH. The acidic milieu of lysosome (pH ~5) is maintained by a vacuolar ATPase (vATPase), which pumps protons from the cytosol into the lysosomal lumen.27 Cathepsins represent a major class of lysosomal proteases.28, 29 They are synthesized as inactive proforms that are activated through proteolytic processing (maturation) during cathepsins’ transit along the endolysosomal pathway. LAMPs (lysosome-associated membrane proteins) are integral membrane proteins that account for approximately half of the protein content in the lysosomal membrane. LAMPs maintain lysosomal structural integrity and function; they are highly glycosylated and abundant enough to form a carbohydrate “coat” on the inner surface of lysosomal membrane, thus protecting the membrane (and the cytoplasm) from the action of lysosomal hydrolases.26 LAMPs regulate the fusion of lysosomes with other organelles (e.g., endosomes), and are important for progression of autophagy.26
Macroautophagy (hereafter referred to as autophagy) is a multistep, lysosome driven, adaptive process whereby cells degrade cytoplasmic organelles and long-lived proteins.30, 31 At its initial stage (autophagy induction), an isolation membrane develops and wraps around material to be sequestered, e.g., mitochondria, forming a unique double-membrane vacuole, the autophagosome. Autophagy progression/resolution (flux) proceeds through fusion of autophagosomes with lysosomes, generating autolysosomes, in which sequestered material is degraded. Autophagy induction is controlled by a series of evolutionary conserved ATG genes.30 Autophagy progression and resolution critically depends on lysosomal function, i.e., the correct fusion of lysosomes with autophagosomes and efficient lysosomal proteolytic activity. Physiologically activated autophagy requires both the induction of autophagosome formation and efficient autophagic flux mediated by lysosomal degradation.30
Lysosomal and autophagic dysfunctions in pancreatitis
Using several experimental models, we showed that (nonalcoholic) pancreatitis has 2 effects on autophagy: autophagy is activated but its flux is impaired/retarded.5, 19, 20, 23, 25 Autophagy impairment in pancreatitis is caused by defective lysosomal degradation. Our data indicate that impaired lysosomal function is a common event in models of pancreatitis, a key manifestation of which is abnormal functioning of lysosomal hydrolases. In particular, a dramatic decrease in enzymatic activities of cathepsins in the lysosome-enriched pancreatic subcellular fraction (first shown by Steer and Saluja group 2 decades ago) is an early event in models of pancreatitis. We found5, 19, 20, 25 that cathepsin processing is impaired in experimental pancreatitis: there is a decrease in the fully mature (“double-chain”) cathepsin forms and accumulation of their immature forms. Our results further indicate that pancreatitis causes an imbalance between 2 major cathepsins, cathepsin B (CatB) and CatL, so that the ratio of their activities (CatB/CatL) increases in pancreatitis.
Another important (and early) lysosomal defect is a dramatic decrease in pancreatic levels of LAMP-1 and -2.23, 25, 32 We found this decrease in 4 dissimilar in vivo models of nonalcoholic pancreatitis induced in rats and mice by CR, L-arginine, and CDE diet, as well as in the ex vivo model. Furthermore, pancreatic LAMP-2 dramatically decreases in pancreatitis induced by ethanol feeding combined with low-dose CR.25 Marked decreases in LAMP-1 and -2 have been reported for human pancreatitis.32 Our data indicate that LAMP degradation in pancreatitis occurs in an acidic compartment and is mediated by cathepsins. Indeed, the decrease in LAMP levels was largely prevented by a broad-spectrum cysteine protease inhibitor E64d, the specific CatB inhibitor CA-074Me, and the inhibitor of vATPase, bafilomycin A. Of note, hydrolases are normally present in the lysosome as large multi-protein complexes which are spatially separated from LAMPs.27, 33 One may speculate that because of abnormal processing of cathepsins, their interactions and localization within the lysosome are altered in pancreatitis, making LAMPs accessible to proteolytic cleavage by cathepsins. In particular, our data indicate that pancreatitis primarily affects LAMPs C-terminus, which might result from the action of cathepsins at a site close to the transmembrane LAMP domain that becomes accessible to lysosomal hydrolases in pathologic conditions.34
Lysosomal dysfunction mediates autophagy impairment leading to pathologic responses of pancreatitis
Cathepsins’ impairment and LAMPs decrease both play an important role in pancreatitis (Figs. 1, 2). Our results5, 19, 20 indicate that defective cathepsin processing/activation leads to inefficient lysosomal degradation and retarded autophagic flux (Fig. 1). Autophagy impairment in pancreatitis19 is manifest, first of all, by accumulation of large autolysosomes containing partially degraded material (Fig. 2). In fact, accumulation of large vacuoles in acinar cells is a long-noted feature of both human and experimental pancreatitis;35–38 however, neither the identity of these vacuoles nor the mechanisms mediating their formation have been established.
Figure 1.
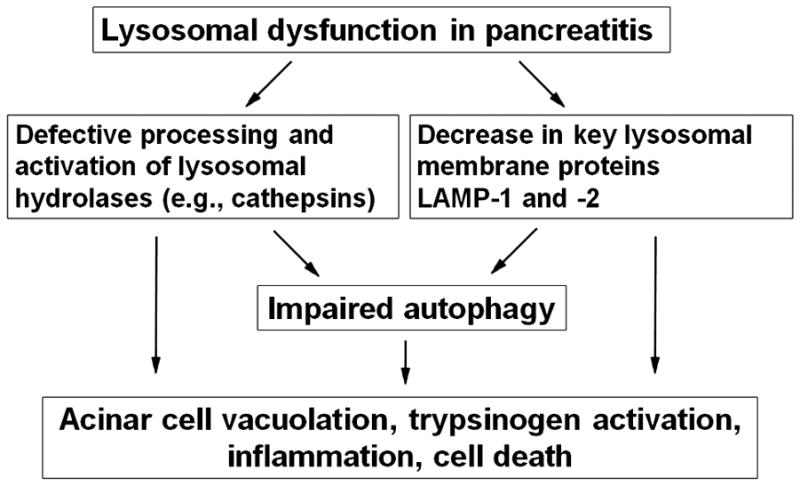
Lysosomal dysfunction leads to autophagy impairment and other pathologic responses of pancreatitis.
Figure 2.

In pancreatitis, defective cathepsin processing/activation, resulting in impaired autophagy, causes accumulation of large autophagic vacuoles and active trypsin in acinar cells. (A). Electron micrograph showing abnormally large autophagic vacuoles in pancreas of a rat with cerulein pancreatitis. Boxed area demonstrates various types of autophagic vacuoles containing intact or partially degraded cargo (e.g., zymogen granules). (B). Schematic illustrating the hypothesis that in pancreatitis, the pathologic, intra-acinar trypsin accumulation results from an imbalance between the activities of CatB, which converts trypsinogen to trypsin, and CatL, which degrades both trypsin and trypsinogen. The stimulatory and inhibitory effects of pancreatitis on these cathepsins are shown by (+) and (−) symbols, respectively.
Among lysosomal hydrolases, cathepsins B and L are particularly relevant to pancreatitis because both play a role in the pathologic, intra-acinar cell accumulation of active trypsin, a hallmark of human and experimental pancreatitis.1, 3 Importantly, these cathepsins have opposite roles in trypsin accumulation: whereas CatB converts trypsinogen to trypsin,3, 19, 39 CatL does not activate trypsinogen but, on the contrary, degrades both trypsin and trypsinogen.19, 40 As stated above, our data19, 20 indicate that lysosomal dysfunction in pancreatitis results in an imbalance between CatB and CatL, such that CatL activity is decreased relative to CatB and is not sufficient to degrade trypsin, resulting in its accumulation in autophagic vacuoles (Fig. 2).
To determine the significance of LAMP decrease in pancreas damage, we assessed the effects of LAMP-2 genetic ablation41 on exocrine pancreas. Accumulation in pancreatic acinar cells of abnormally large autophagic vacuoles containing partially degraded material was already prominent in LAMP-2 null mice at the age of 1 month, as seen with both light and electron microscopy. This was associated with progressive increase in the inflammatory cell infiltration, particularly macrophages, and in acinar necrosis. Thus, pancreatitis-like injury spontaneously develops in LAMP-2 null mice. The mechanisms by which LAMP deficiency causes impaired autophagy in pancreas remain to be determined. In other cells and organs, LAMP deficiency was shown to impair the ability of lysosomes to fuse with endosomes and autophagosomes.41
Thus, our data indicate that lysosomal dysfunction is a critical initiating event of pancreatitis. The 2 features of lysosomal dysfunction in pancreatitis are defective processing/activation of cathepsins and the decreases in LAMP-1 and LAMP-2. These defects lead to impaired autophagy, trypsinogen activation, inflammation, and acinar cell vacuolation and death – the key pathologic responses of pancreatitis (Figs. 1, 2).
Mitochondrial dysfunction in pancreatitis
Mitochondria play a central role in regulating both cell life and death.42, 43 These organelles generate most of the cell’s energy in the form of ATP supply. On the other hand, mitochondrial membrane permeabilization is a universal trigger of both apoptosis and necrosis and is often considered “the point of no return” in the chain of events leading to cell death. However, the molecular mechanisms mediating mitochondrial permeabilization are not fully understood.42, 44 Key manifestations of mitochondrial permeabilization triggering apoptotic and necrotic pathways are, respectively, the release of the mitochondria resident protein cytochrome c (as well as other apoptosis-inducing factors) into the cytosol and mitochondrial depolarization. Once in the cytosol, cytochrome c stimulates activation of specific cysteine proteases, the caspases, which mediate the downstream apoptotic events. On the other hand, loss of the mitochondrial membrane potential, ΔΨm, ultimately leads to ATP depletion and necrosis. Thus, mitochondrial permeabilization is a central event in both apoptotic and necrotic cell death.
Permeability transition pore
The permeability transition pore (PTP) is a key mediator of mitochondrial permeabilization.42, 44 PTP is a high conductance channel permeating both the outer and inner mitochondrial membranes, a multi-protein complex (Fig. 3A) a critical component of which is cyclophilin D (CypD). A major regulator of PTP opening is its sensitivity to Ca2+, which is affected by various factors, including pyridine nucleotides, lipid composition of mitochondrial membrane, etc.44 Transient PTP opening likely servesphysiologic purposes by regulating mitochondrialCa2+ level. In contrast, sustained, pathologic PTP opening, caused primarily by Ca2+ overload, results in mitochondria permeabilization to small (<1.5 kDa) solutes and the collapse of ΔΨm. As a result of PTP opening, water may penetrate the matrix, causing rupture of the outer mitochondrial membrane. Mice with genetic ablation of CypD45 provide a specific tool to inactivate PTP and test its physiologic and pathologic roles.
Figure 3.
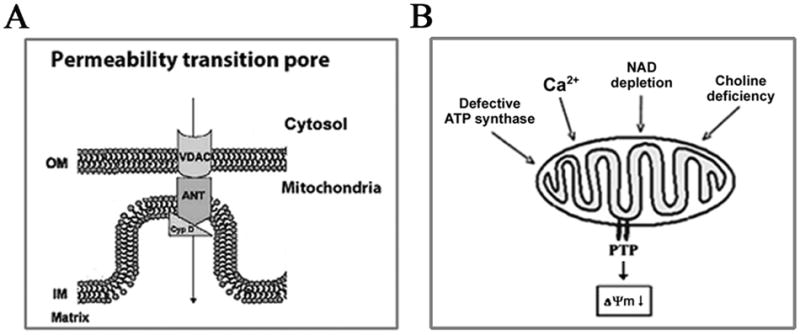
Permeability transition pore (PTP) opening, leading to loss of the mitochondrial membrane potential ΔΨm, is a common event in models of pancreatitis. (A). Schematic of PTP. OM, the outer mitochondrial membrane; IM, the inner mitochondrial membrane; VDAC, voltage-dependent anion channel; ANT, adenine nucleotide translocase: CypD, cyclophilin D. (B). PTP opening in experimental pancreatitis is mediated by different mechanisms: through an increase in cytosolic Ca2+ in the cerulein and bile acid models (Ref. 21); a decrease in ATP-synthase activity in arginine-induced pancreatitis (Ref. 46); and a decrease in NAD level in alcoholic pancreatitis (Ref. 24).
Mitochondrial depolarization in pancreatitis
We and other groups showed that loss of pancreatic mitochondria membrane potential is an early common event in models of nonalcoholic acute pancreatitis, including those induced by CR, L-arginine or taurocholate administration, and by CDE diet.5, 14, 16–18, 21, 46–48 Moreover, we showed24 that incubation of acinar cells with 50–100 mM ethanol causes mitochondrial depolarization, which is further potentiated by low-dose CCK. Mitochondrial depolarization in both nonalcoholic and alcoholic pancreatitis is mediated through PTP opening as it is largely prevented in CypD null mice (Mareninova OA et al, submitted). Thus, PTP-mediated mitochondrial depolarization emerges as a key early pathologic event of acute pancreatitis.
Interestingly, the data indicate that the mechanisms leading to PTP opening and ΔΨm loss differ between models of pancreatitis (Fig. 3B). In particular, PTP opening in the CCK/CR- and bile acid-induced models of pancreatitis is mediated by Ca2+ increase.14, 16, 17, 21, 47, 48 Experiments on isolated pancreatic mitochondria showed that Ca2+ directly depolarizes the mitochondria.5, 16, 17, 21, 24 Moreover, it appears that the pancreatic mitochondria PTP is highly sensitive to Ca2+ – as compared, for example, to the “classical” liver mitochondria.5, 16, 17, 21 Differently, the PTP opening in L-arginine pancreatitis is caused by arginine-induced impairment of ATP synthase.46 Further, our recent data24 show that ethanol depolarizes pancreatic mitochondria by decreasing the mitochondrial NAD level necessary to maintain PTP in the closed state.
Mitochondrial and lysosomal dysfunctions act in concert to promote impaired autophagy leading to pancreatitis
As discussed above, we showed5, 19, 20, 22, 25 that (nonalcoholic) pancreatitis has 2 effects on autophagy: it stimulates autophagy induction and at the same time impairs late stages of autophagy, resulting in retarded, inefficient autophagic flux. The autophagic flux impairment in pancreatitis is caused by lysosomal dysfunction (i.e., defective cathepsins and LAMPs). The mechanisms regulating autophagy induction in pancreatitis are yet to be determined. Our results indicate that the pathologic consequences of impaired autophagic flux are most pronounced when the level of autophagy induction is high. Therefore, inhibition of autophagy induction could be a promising approach to alleviate pancreatitis. Indeed, inhibiting autophagy induction by genetic or pharmacologic means markedly improved CR pancreatitis.19, 49 Of note, mitochondrial depolarization prominently activates autophagy, in particular mitophagy (autophagic removal of mitochondria).50 It is tempting to speculate that mitochondrial depolarization in pancreatitis triggers autophagy induction. Conversely, the impaired autophagy caused by lysosomal dysfunction in pancreatitis results in accumulation of dysfunctional mitochondria that fail to synthesize ATP, and therefore promotes both necrosis and further autophagy induction. Thus, the combined lysosomal and mitochondrial dysfunctions create a “vicious cycle” resulting in persistent autophagy impairment and mitochondrial depolarization, mediating the pathobiology of pancreatitis (Fig. 4).
Figure 4.
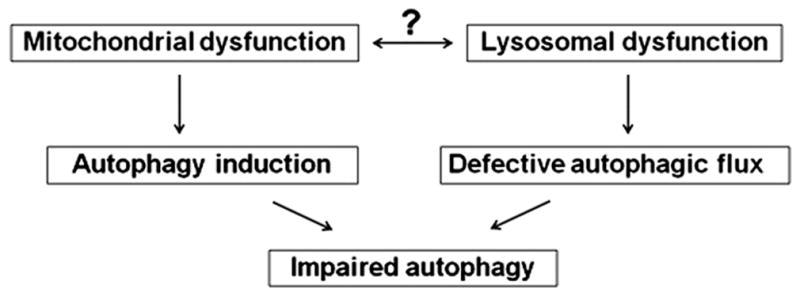
Schematic illustrating the hypothesis that mitochondrial and lysosomal dysfunctions in pancreatitis potentiate each other, promoting impaired autophagy.
Acknowledgments
Southern California Research Center for Alcoholic Liver and Pancreatic Diseases and Cirrhosis (NIH P50AA11999); Department of Veterans Affairs; NIH R01 grants DK59936 and AA19730.
Footnotes
Conflict of Interest
No conflict of interest has been declared by the authors.
References
- 1.Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;132:1127–51. doi: 10.1053/j.gastro.2007.01.055. erratum: ibid, 133: 1056. [DOI] [PubMed] [Google Scholar]
- 2.Everhart JE, Ruhl CE. Burden of digestive diseases in the United States Part III: Liver, biliary tract, and pancreas. Gastroenterology. 2009;136:1134–44. doi: 10.1053/j.gastro.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 3.Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–69. doi: 10.1146/annurev.physiol.69.031905.161253. [DOI] [PubMed] [Google Scholar]
- 4.Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology. 2004;4:567–86. doi: 10.1159/000082182. [DOI] [PubMed] [Google Scholar]
- 5.Gukovsky I, Pandol SJ, Gukovskaya AS. Organellar dysfunction in the pathogenesis of pancreatitis. Antioxid Redox Signal. 2011;15:2699–710. doi: 10.1089/ars.2011.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandol SJ, Lugea A, Mareninova OA, et al. Investigating the pathobiology of alcoholic pancreatitis. Alcohol Clin Exp Res. 2011;35:830–7. doi: 10.1111/j.1530-0277.2010.01408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lerch MM, Adler G. Experimental animal models of acute pancreatitis. Int J Pancreatol. 1994;15:159–70. [PubMed] [Google Scholar]
- 8.Pandol SJ, Periskic S, Gukovsky I, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;117:706–16. doi: 10.1016/s0016-5085(99)70465-8. [DOI] [PubMed] [Google Scholar]
- 9.Gukovskaya AS, Hosseini S, Satoh A, et al. Ethanol differentially regulates NF-kappaB activation in pancreatic acinar cells through calcium and protein kinase C pathways. Am J Physiol Gastrointest Liver Physiol. 2004;286:G204–13. doi: 10.1152/ajpgi.00088.2003. [DOI] [PubMed] [Google Scholar]
- 10.Gukovskaya AS, Mareninova OA, Odinokova IV, et al. Cell death in pancreatitis: effects of alcohol. J Gastroenterol Hepat. 2006;21 (Suppl 3):S10–13. doi: 10.1111/j.1440-1746.2006.04571.x. [DOI] [PubMed] [Google Scholar]
- 11.Perides G, Tao X, West N, Sharma A, Steer ML. A mouse model of ethanol dependent pancreatic fibrosis. Gut. 2005;54:1461–7. doi: 10.1136/gut.2004.062919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gukovsky I, Lugea A, Shahsahebi M, et al. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G68–79. doi: 10.1152/ajpgi.00006.2007. [DOI] [PubMed] [Google Scholar]
- 13.Rakonczay Z, Jr, Hegyi P, Takacs T, McCarroll J, Saluja AK. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57:259–67. doi: 10.1136/gut.2007.124115. [DOI] [PubMed] [Google Scholar]
- 14.Gukovskaya AS, Gukovsky I, Jung Y, Mouria M, Pandol SJ. Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells. Roles in cell injury processes of pancreatitis. J Biol Chem. 2002;277:22595–604. doi: 10.1074/jbc.M202929200. [DOI] [PubMed] [Google Scholar]
- 15.Mareninova OA, Sung KF, Hong P, et al. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem. 2006;281:3370–81. doi: 10.1074/jbc.M511276200. [DOI] [PubMed] [Google Scholar]
- 16.Odinokova IV, Sung KF, Mareninova OA, Hermann K, Gukovsky I, Gukovskaya AS. Mitochondrial mechanisms of death responses in pancreatitis. J Gastroenterol Hepatol. 2008;23 (Suppl 1):S25–30. doi: 10.1111/j.1440-1746.2007.05271.x. [DOI] [PubMed] [Google Scholar]
- 17.Odinokova IV, Sung KF, Mareninova OA, et al. Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut. 2009;58:431–42. doi: 10.1136/gut.2007.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sung KF, Odinokova IV, Mareninova OA, et al. Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp Cell Res. 2009;315:1975–89. doi: 10.1016/j.yexcr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mareninova OA, Hermann K, French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340–55. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gukovsky I, Gukovskaya AS. Impaired autophagy underlies key pathological responses of acute pancreatitis. Autophagy. 2010;6:428–9. doi: 10.4161/auto.6.3.11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gukovskaya AS, Gukovsky I. Which way to die: the regulation of acinar cell death in pancreatitis by mitochondria, calcium, and reactive oxygen species. Gastroenterology. 2011;140:1876–80. doi: 10.1053/j.gastro.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mareninova OA, Yakubov I, French SW, et al. Ethanol feeding causes lysosomal dysfunction in exocrine pancreas similar to pancreatitis; but in contrast to pancreatitis, ethanol down-regulates autophagy (Abstract) Gastroenterology. 2010;138(Suppl 1):S148. [Google Scholar]
- 23.Mareninova OA, Yakubov I, Jia W, Gukovsky I, Gukovskaya AS. Experimental acute pancreatitis causes pathologic changes in lysosomal membrane protein and lipid composition (Abstract) Gastroenterology. 2010;138(Suppl 1):S125–S6. [Google Scholar]
- 24.Shalbueva N, Mareninova OA, Pandol SJ, Gukovskaya AS. Alcohol promotes pancreatic mitochondria depolarization by sensitizing the permeability transition pore to Ca2+ (Abstract) Pancreas. 2010;39:1347. [Google Scholar]
- 25.Mareninova OA, Yakubov I, Pandol SJ, Gorelick FS, Gukovskaya AS, Gukovsky I. Disordering of lysosomal and autophagic pathways in nonalcoholic and alcoholic experimental pancreatitis (Abstract) Pancreas. 2010;39:1332. [Google Scholar]
- 26.Saftig P. Lysosomal Membrane Proteins. In: Saftig P, editor. Lysosome. New York: Springer Science and Business Media; 2005. pp. 37–49. [Google Scholar]
- 27.Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–32. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 28.Erickson AH. Biosynthesis of lysosomal endopeptidases. J Cell Biochem. 1989;40:31–41. doi: 10.1002/jcb.240400104. [DOI] [PubMed] [Google Scholar]
- 29.Ishidoh K, Kominami E. Processing and activation of lysosomal proteinases. Biol Chem. 2002;383:1827–31. doi: 10.1515/BC.2002.206. [DOI] [PubMed] [Google Scholar]
- 30.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fortunato F, Burgers H, Bergmann F, et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137:350–60. doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Kreutzer R, Kreutzer M, Sewell AC, Techangamsuwan S, Leeb T, Baumgartner W. Impact of beta-galactosidase mutations on the expression of the canine lysosomal multienzyme complex. Biochim Biophys Acta. 2009;1792:982–7. doi: 10.1016/j.bbadis.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 34.Cuervo AM, Mann L, Bonten EJ, d’Azzo A, Dice JF. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003;22:47–59. doi: 10.1093/emboj/cdg002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helin H, Mero M, Markkula H, Helin M. Pancreatic acinar ultrastructure in human acute pancreatitis. Virchows Arch A, Pathol Anat Histol. 1980;387:259–70. doi: 10.1007/BF00454829. [DOI] [PubMed] [Google Scholar]
- 36.Aho HJ, Nevalainen TJ, Havia VT, Heinonen RJ, Aho AJ. Human acute pancreatitis: a light and electron microscopic study. Acta Pathol Microbiol Immunol Scand A, Pathology. 1982;90:367–73. [PubMed] [Google Scholar]
- 37.Watanabe O, Baccino FM, Steer ML, Meldolesi J. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. Am J Physiol. 1984;246:G457–67. doi: 10.1152/ajpgi.1984.246.4.G457. [DOI] [PubMed] [Google Scholar]
- 38.Niederau C, Grendell JH. Intracellular vacuoles in experimental acute pancreatitis in rats and mice are an acidified compartment. J Clin Invest. 1988;81:229–36. doi: 10.1172/JCI113300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halangk W, Lerch MM, Brandt-Nedelev B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773–81. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wartmann T, Mayerle J, Kahne T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology. 2010;138:726–37. doi: 10.1053/j.gastro.2009.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eskelinen EL, Illert AL, Tanaka Y, et al. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol Biol Cell. 2002;13:3355–68. doi: 10.1091/mbc.E02-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 43.Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med. 2004;25:365–451. doi: 10.1016/j.mam.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Ricchelli F, Sileikyte J, Bernardi P. Shedding light on the mitochondrial permeability transition. Biochim Biophys Acta. 2011;1807:482–90. doi: 10.1016/j.bbabio.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 45.Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 46.Biczo G, Hegyi P, Dosa S, et al. The crucial role of early mitochondrial injury in L-lysine-induced acute pancreatitis. Antioxid Redox Signal. 2011;15:2669–81. doi: 10.1089/ars.2011.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voronina SG, Barrow SL, Gerasimenko OV, Petersen OH, Tepikin AV. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem. 2004;279:27327–38. doi: 10.1074/jbc.M311698200. [DOI] [PubMed] [Google Scholar]
- 48.Booth DM, Murphy JA, Mukherjee R, et al. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140:2116–25. doi: 10.1053/j.gastro.2011.02.054. [DOI] [PubMed] [Google Scholar]
- 49.Hashimoto D, Ohmuraya M, Hirota M, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol. 2008;181:1065–72. doi: 10.1083/jcb.200712156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]