

Abstract
A series of dual inhibitors containing a 1,5-diarylpyrazole and a urea were designed, synthesized, and evaluated as novel COX-2/sEH dual inhibitors in vitro using recombinant enzyme assays and in vivo using a lipopolysaccharide (LPS) induced model of pain in rats. The best inhibition potencies and selectivity for sEH and COX-2 over COX-1 were obtained with compounds (21b, 21i and 21j) in which both the 1,5-diaryl-pyrazole group and the urea group are linked with a three-methylene group. Compound 21i showed the best pharmacokinetic profiles in both mice and rats (higher AUC and longer half-life). Following subcutaneous administration at 10 mg/kg, compound 21i exhibited anti-allodynic activity that is more effective than the same dose of either a COX-2 inhibitor (celecoxib) or a sEH inhibitor (t-AUCB) alone, as well as co-administration of both inhibitors. Thus, these novel dual inhibitors exhibited enhanced in vivo anti-allodynic activity in a nociceptive behavioral assay.
Introduction
The arachidonic acid (AA) cascade is the target of many pharmaceuticals therapies for various conditions such as cardiovascular, asthma and inflammatory diseases. For example, nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2 (COX-2) selective inhibitors (coxibs) block the conversion of AA to prostaglandins (PGs) to treat pain and inflammation.1 Lipoxygenase (LOX) inhibitors, in particular 5-LOX inhibitors, block the conversion of AA to leukotrienes (LTs) to reduce allergy.2 The concomitant inhibition of COX and LOX enzymes seems advantageous in various cardiovascular diseases and cancer therapy.3 Several dual inhibitors4 that inhibit cyclooxygenases (either COX-2 or both COX-1 and COX-2) and 5-LOX have been reported as potential agents for the treatment of arthritis. Licofelone (ML-3000) is an example of such an arthritis drug.5 And such dual inhibitors also have been prepared to treat inflammation,6 pain,7 and cancers.8
In addition to the COX and LOX pathways, there is a third major metabolic pathway in the AA cascade involving cytochrome P450 metabolism. This pathway leads to the formation of 20-hydroxyeicosatetranoic acid (20-HETE)9 and arachidonic acid monoepoxides known as epoxy-eicosatrienoic acids (EETs).10 The soluble epoxide hydrolase (sEH) enzyme catalyzes the conversion of these EETs into the corresponding diols, or dihydroxyeicosatrienoic acids (DHETs). EETs are known to exhibit vasodilatory,11 cardioprotective,12 anti-inflammatory,13 and anti-hyperalgesic14 properties, while the DHETs have greatly reduced activity in most assays.15
NSAIDs target cyclooxygenases which are key enzymes involved in prostaglandin (PG) biosynthesis from AA.16 However, morbidity and mortality due to NSAID-induced gastrointestinal (GI) toxicity are so significant and frequent worldwide to limit the therapeutic use of this drug class.17 To mitigate this side effect caused primarily by COX-1 inhibition, COX-2 selective inhibitors, or coxibs such as celecoxib and rofecoxib, were designed and developed. These coxibs were specialized to retain the beneficial anti-inflammatory and anti-hyperalgesic properties of NSAIDs but enhance GI tolerance.18 In spite of this design, COX-2 selective inhibitors retain some GI toxicity at higher doses and/or with long-term use. Moreover, COX-2 selective inhibitors may lose selectivity and inhibit COX-1 in vivo at higher doses, resulting in the undesirable side effects.19 High doses of COX-2 selective inhibitors also shift plasma thromboxane/prostacyclin ratio20,22 and increase the eicosanoid 20-HETE, which could potentially lead to thrombic events and hypertension.21 We have previously demonstrated that drug combinations with low doses of NSAIDs and soluble epoxide hydrolase inhibitors (sEHIs) produce synergistic effects when measuring anti-hyperalgesia and anti-inflammation outcomes. This observed sEHI synergy with NSAIDS reduces pain and inflammation while prospectively decreasing the side effects of coxibs such as cardiovascular toxicity.22
In general, there are safety concerns when administering combination therapy. Two drugs which are safe when used independently of each other cannot be assumed to be safe in combination, as drug-drug interaction warnings indicate. There are several tests that are necessary to find the optimal dose regiments including safety studies, a complex dosage ranging investigation, and drug-drug interaction analysis, all of which may significantly raise the practical cost and complexity of developing combination therapies.23 It is clear that this issue is also not exclusively due to metabolic shunting effects. For drug development, the prediction of pharmacodynamic and pharmacokinetic relationships is substantially less complex if polypharmacological action is derived from a single agent rather than from combination therapies (co-administration). Therefore, there has recently been a growing interest in designed multiple ligands (DMLs).24 The aim of DMLs is to enhance drug efficacy and improve drug safety by acting specifically on multiple targets (“targeted polypharmacology”), as opposed to drugs that address only a single target. DMLs have advantages over combination drugs or combination therapies because they circumvent the inherent problems associated with formulation of two or more drugs used for co-administration. In addition, the distinct differences in the pharmacodynamic and pharmacokinetic properties of individual drugs which may raise safety concerns, do not apply to DMLs.25 DMLs may also offer some advantage due to regulation of intellectual property. For all of these reasons dual inhibition of COX-2 and sEH through a single molecule is likely to be more advantageous than co-administration of the drugs using combination therapy.
The therapeutic targeting of the P450 branch of the AA cascade remains to be thoroughly explored and even less so using dual inhibitors. To date, there is only one current example of a dual inhibitor related to sEH in the literature, a sEH/11β-HSD1 dual inhibitor designed by GlaxoSmithKline.26 Herein, we report COX-2/sEH dual inhibitors as a new class of DMLs active in the AA cascade.
Chemistry
Diarylheterocycles have been extensively studied as COX-2 selective inhibitors (figure 1). Among them, celecoxib (Celebrex) which has a 1,5-diarylpyrazole skeleton, is the most successful compound. It is a potent COX-2 selective inhibitor with a good pharmacokinetic profile. A methyl sulfone group as in rofecoxib or a sulfonamide group as in celecoxib has been proven to be essential for the selective inhibition of COX-2 over COX-1,27 while N,N’-disubstituted ureas as well as their corresponding amides and carbamates have been thoroughly studied as sEH inhibitors.28 From this knowledge base, we sought to develop conjugates of pyrazole and urea having both COX-2 selective and sEH inhibitory activities as a new class of DML (Figure 1). Therefore we prepared the compounds 7, 11a–g, 15, and 21a that are linked with non-cleavable methylene chains between a pyrazole ring as a COX-2 fragment and a urea as a sEH fragment (Scheme 1) starting from compound 3a and 3b. These common intermediates were prepared by regioselective cyclization of either 4-methylsulfonylphenylhydrazine 1a or 4-amidosulfonylphenylhydrazine 1b with β-diketone 2.29 Compounds 21b,d–j were also prepared from the β-diketone 22 (Scheme 2). Compounds 11h and 21c were also prepared using similar methods (see Supporting Information). Compound 7 was prepared by the reaction with 3-(trifluoromethyl)phenyl isocyanate and an aminopyrazole 6 which was obtained by hydrolysis from corresponding ethyl carbamate 5. The Curtius reaction of carboxylic acid 4 in the presence of EtOH gave 5. Compounds 11a–g were prepared by the reaction of various isocyanates with amine 10a or 10b which was obtained by LAH reduction, mesylation, azide formation followed by Pd/C catalysed hydrogenation starting from the common intermediate 3a or 3b. Compound 15 was prepared by the reaction of 3-(trifluoromethyl)phenyl isocyanates with amine 14 which was obtained via Henry reaction, followed by LAH reduction from aldehyde 12b. Compound 21a was obtained by the reaction of 3-(trifluoromethyl)phenyl isocyanates with amine 20a which was obtained via sequential Horner-Wadsworth-Emmons reaction, Pd/C hydrogenation, LAH reduction, Mitsunobu reaction and hydrazine hydrolysis from aldehyde 12a. While compounds 21b,d–j were prepared using the reactions shown in Scheme 2. The desired 3C-linker in compound 18b was introduced by the reaction of p-amidosulfonylphenylhydrazine hydrochloride with 1-phenyl-1,3-dione-3-propanol 22 which was obtained by the reaction of acetophenone with γ-butyrolactone.30 The amine 20b was prepared by hydrogenation on Pd/C from an azide 23. A series of dual inhibitors 21b,d–j possessing three-methylene group as a linker were then obtained by reacting amine 20b with various isocyanates.
Figure 1.

Structures of reference COXs and sEH inhibitors and a general structure for COX-2/sEH dual inhibitors.
Scheme 1.

Synthesis of sEH/COX-2 Dual Inhibitors 7, 11a–h, 15, 21a.*
a Reagents and conditions: (a) EtOH, AcOH, reflux, 2h; (b) LiOH, THF, water, rt, 12h; (c) DPPA, Et3N, EtOH, 1,4-dioxane, reflux, 12h; (d) 10% NaOH, EtOH, reflux, 5h; (e) R3-NCO, Et3N, DMF, 0 °C to rt, 12 h; (f) LAH, THF, rt, 6h; (g) i) MsCl, Et3N, O °C, 2h, ii) NaN3, 1,4-dioxane, water, 80 °C, 2h; (h) Pd/C, H2, EtOAc, 2h; (i) R3-NCO, DMF, rt, 12 h; (j) PCC, DCM, rt, 6h; (k) i) MeNO2, AcONH4, reflux, 1h; (l) LAH, THF, rt, 6h; (m) R3-NCO, DMF, rt, 12 h; (n) triethyl phosphonoacetate, NaH, THF, 0 °C, 1 h; (o) Pd/C, H2, EtOAc, 2h; (p) LAH, THF, 6h; (q) PPh3, phthalimide, DIAD, THF, rt, 12 h; (r) 35% hydrazine, CH2Cl2, MeOH, rt, 1 d; (s) R3-NCO, DMF, rt, 12 h. * R2 = ph (see supporting information for the synthetic scheme of compounds having R2 = t-butyl or p-tolyl).
Scheme 2.

Synthesis of sEH/COX-2 Dual Inhibitors 21b,d–j.
a Reagents and conditions: (a) p-sulfoamidophenylhydrazine hydrochloride, EtOH, AcOH, reflux, 2h; (b) i) MsCl, Et3N, DCM, 0 °C, 2h, ii) NaN3, 1,4-dioxane, water, reflux, 6h; (c) Pd/C, H2, EtOAc, rt, 2h; (d) R3-NCO, DMF, rt, 12 h.
Results and Discussion
Inhibition of COXs and sEH Activities
Known COX-2 selective inhibitors celecoxib and rofecoxib, the COX-1 preferred inhibitor indomethacin, and the sEH inhibitor t-AUCB31 (Figure 1) were evaluated for their cross activities against COX-2, COX-1, and sEH enzymes. Selected reference inhibitors showed expected selective inhibitory activity against each targeted enzyme (Table 1). To obtain dual COX-2/sEH inhibitors, we linked the 1,5-diarylpyrazole moiety of celecoxib to the urea pharmacophore of the sEH inhibitor. It has previously been shown that the 3-position of the pyrazolyl ring in celecoxib has very few steric restrictions in COX-2.32 Therefore, a series of compounds 11a–h were first synthesized by introducing urea groups at this position. Compound 11a was synthesized and tested because the methylsulfone group has been proven to give an excellent COX-2 selectivity over COX-1 as in rofecoxib. Furthermore, the adamantyl urea group has shown excellent inhibitory activity against recombinant human sEH and is detected at very low levels by LC/MS analysis.33 However, such a simple connection between two known pharmacophores (Figure 1) was not effective due to the lack of COX-2 inhibition and only weak sEH inhibition by compound 11a. Replacement of the adamantyl group of compound 11a by a cylcloheptyl group, which has been proven to exhibit excellent sEH inhibitory activity (11b), improved sEH inhibition by 10-fold, but still lacked COX-2 inhibition. Based on our previous SAR study for sEH inhibitors,34 replacement by an aryl group, especially when there is a substituent at para- or meta-position, showed excellent sEH inhibition and good pharmacokinetics. Therefore aryl groups were introduced as R3 groups such as compounds 11c–f. Surprisingly, introduction of a trifluoromethoxy group at the para-position (11d) gave slight COX-2 inhibition with moderate sEH inhibition. However, adding a trifluoromethyl group at the para-position not only decreased COX-2 inhibition but also unexpectedly decreased sEH inhibition approximately 20-fold. When the trifluoromethyl group was introduced on the meta-position on the phenyl ring, it regained COX-2 inhibition but poorly inhibited sEH. In general, sulfonamides have slightly better affinity than sulfones for the COX-2 active site,35 hence the methylsulfone group of compound 11f was replaced by an aminosulfone group 11g which became 2-fold more potent than the reference drug rofecoxib (COX-2 IC50 = 2 µM), but the potency for sEH inhibition did not improve from 11f. Replacement of a rigid phenyl group by a tert-butyl group in 11h increased sEH inhibition (sEH IC50 = 32±3 nM) but lost its COX-2 inhibitory activity.
Table 1.
Inhibitory Activities of Compounds 7, 11a–h, 15, and 21a–j.
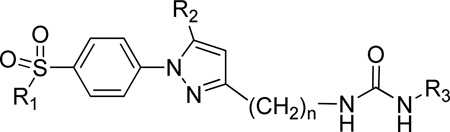 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | n | R3 | COX-1a inhibition (%)c |
COX-2a IC50 (µM)d |
sEHb IC50 (nM) |
| celecoxib | 41 | 0.01 | >10,000 | ||||
| rofecoxib | 17 | 2 | >10,000 | ||||
| Indomethacin | 84 | 17 | >10,000 | ||||
| t-AUCB | 25 | >100 | 0.5 ± 0.1 | ||||
| 11a | Me | phenyl | 1 | 1-adamantyl | 13 | >10 | 25 ± 1 |
| 11b | Me | phenyl | 1 | c-heptyl | 3 | >10 | 2.6 ± 0.3 |
| 11c | Me | phenyl | 1 | phenyl | 10 | >10 | 47 ± 4 |
| 11d | Me | phenyl | 1 | p-CF3O-phenyl | 10 | 7 | 6.0 ± 0.5 |
| 11e | Me | phenyl | 1 | p-CF3-phenyl | 11 | >10 | 110 ± 5 |
| 11f | Me | phenyl | 1 | m-CF3-phenyl | 8 | 2.5 | 72 ± 8 |
| 11g | NH2 | phenyl | 1 | m-CF3-phenyl | 17 | 1 | 84 ± 6 |
| 11h | Me | t-butyl | 1 | m-CF3-phenyl | 2 | >10 | 32 ± 3 |
| 7 | NH2 | phenyl | 0 | m-CF3-phenyl | 34 | 2 | 88 ± 5 |
| 15 | NH2 | phenyl | 2 | m-CF3-phenyl | 22 | 1 | 26 ± 3 |
| 21b | NH2 | phenyl | 3 | m-CF3-Phenyl | 27 | 0.7 | 4.1 ± 0.4 |
| 21a | Me | phenyl | 3 | m-CF3-phenyl | 6 | 3 | 3.4 ± 0.2 |
| 21c | NH2 | p-tolyl | 3 | m-CF3-phenyl | 12 | 2.8 | 10 ± 1 |
| 21d | NH2 | phenyl | 3 | 2,6-diiPr-phenyl | 6 | >10 | 1550 ± 70 |
| 21e | NH2 | phenyl | 3 | phenyl | 16 | >10 | 0.8 ± 0.1 |
| 21f | NH2 | phenyl | 3 | 1-adamantyl | 7 | 7 | 0.5 ± 0.1 |
| 21g | NH2 | phenyl | 3 | c-heptyl | 10 | 2 | 0.5 ± 0.1 |
| 21h | NH2 | phenyl | 3 | p-Cl-phenyl | 15 | 6 | 0.8 ± 0.1 |
| 21i | NH2 | phenyl | 3 | p-CF3-phenyl | 23 | 1.3 | 0.9 ± 0.1 |
| 21j | NH2 | phenyl | 3 | p-CF3O-phenyl | 14 | 0.9 | 0.5 ± 0.1 |
Values are the means ± SD of three independent experiments with COX Fluorescent Inhibitor Screening Assay Kit (Catalog No. 700100, Cayman Chemicals Inc., Ann Arbor, MI).
Determined via a kinetic fluorescent assay, results are means ± SD of three separate experiments.
% inhibition at 100 µM concentration.
Data points are triplicate average. We observed coefficient variation between 5 and 10%.
Comparison of dual inhibitors having variable-length spacer
These results suggested that 1,5-diarylpyrazole group is sterically hindered, which has been proven to decrease sEH inhibition.34 Subsequently, attempts to improve sEH inhibitory activity were initiated by increasing the length of the linker between the 1,5-diarylpyrazole group and the urea group to avoid steric repulsion. Compound 7 in which a biarylpyrazole ring and a urea group are directly connected, showed poor sEH inhibitory activity as with compound 11g. A two-methylene spacer between the two pharmacophores as in compound 15 improved the sEH inhibition only 3-fold compared to that of compound 11g. A further six-fold improvement was obtained in compound 21b by the addition of another methylene group (total of three methylene groups) between a COX-2 pharmacophore and a sEH pharmacophore, yielding an overall 20-fold improvement in potency against sEH compared to compound 11g, without changing the efficacy against COX-2.
SAR for the dual inhibitors having a three-methylene linker between two pharmacophores
The previous data suggested that a three-methylene spacer between two pharmacophores is critical for sEH inhibition in this series of compounds. A compound having an aminosulfonyl group as R1 in 21b was more potent as a COX-2 inhibitor than a methylsulfone group as R1 in 21a while sEH inhibition remained the same. Interestingly, compound 21c having a methyl group at para-position on a phenyl group at C-5 (R2) on the pyrazole ring similar to celecoxib diminished activities 3-fold for both COX-2 and sEH compared to compound 21b. Compound 21d that has two sterically hindered ortho-isopropyl groups next to a urea group (R3) was a poor sEH inhibitor in accordance of our previous SAR study for sEH inhibitors.34 Having developed compounds with reasonable COX-2 inhibitory, we began an effort to improve sEH inhibitory activity of the compounds. Again, we started introducing various groups at R3 based on our previous SAR study.34 This study showed that generally, a relatively bulky carbocyclic group and a meta- or para-substituted phenyl group increased the inhibitory activity of sEH inhibitors. In this case compounds followed our previous SAR, unlike compounds 11a–g. Compounds 21f became as potent as t-AUCB. Based on these results, we next further optimized their sEH inhibition activity by changing R3 group, while fixing a group for COX-2 inhibition (Scheme 2). Based on our previous SAR study for sEH inhibitors, several groups that increased sEH inhibition were introduced e.g., a bulky carbocyclic group as in 21f and 21g, and a para-substituted phenyl group as in compounds 21j, 21i, and 21h. Not surprisingly, compounds 21f–j in which R3 groups are carbocyclic or para-substituted phenyl groups inhibited sEH at picomolar concentrations. In addition, those compounds also showed good potency against COX-2 except compounds having a rigid ring as in 21f or a simple phenyl group 21e (or p-Cl phenyl group 21h). However compounds having a relatively flexible cycloheptyl group 21g and phenyl group with a substitution at the para-position such as a trifluoromethyl group in 21i or a trifluoromethoxy group in 21j kept their COX-2 inhibitory activity while retaining their sEH inhibitory activity. Among them, compounds having a CF3 group as in 21b, 21i and 21j gave better COX-2 inhibition. They showed IC50 values better than rofecoxib for COX-2, good to excellent sEH inhibition and showed reasonable COX-1/COX-2 selectivity. In addition, it has been reported that celecoxib possesses inhibitory activity against 5-LOX, an enzyme that produces proinflammatory leukotrienes (LTs).36 Our compounds showed similar potency with celecoxib against 5-LOX (i.e. % inhibition of at 10 µM: celecoxib inhibits 80% of human 5-LOX while 21i yields 83% inhibition; Table S1).
Docking compound 21i with COX-2 and sEH enzymes
To understand the observation that the dual inhibitors inhibit both sEH and COX-2, we manually docked inhibitor 21i into the active site of sEH and COX-2 (Figure 2) . For this, we used the published X-ray crystal structure of human sEH complexed with a urea-based ligand N-cyclohexyl-N’-(4-iodophenyl)urea, CIU (PDB accession number 1VJ5).37 Between two plausible binding modes for 21i, the orientation given in Figure 2A was favorable. The other binding mode resulted in steric clashes between the two aryl groups of the inhibitor and the residues of the binding site, such as Leu395, Leu406, Leu427, Pro266 and Phe265. These results might explain why we obtained unexpectedly poor sEH inhibition with compounds having a single methylene linker such as 11a vs 21f, 11e vs 21i, and 11g vs 21b, suggesting that these compounds, and, presumably, other compounds in the same series (Table 1), could not orient themselves to avoid an unfavorable interactions with the residues at the active site. As seen in Figure 2A, compound 21i was bound primarily through interactions with Tyr381, Tyr465 and Asp333 with the urea pharmacophore. We further manually docked inhibitor 21i into the active site of COX-2 (Figure 2B) using the published X-ray crystal structure (PDB accession number 1CX2) of murine COX-2 complexed with a biarylpyrazole-based ligand 1-phenylsulfonamide-3-trifluoromethyl-5-parabromophenylpyrazole 24 (SC-558).38 One of the oxygen atoms in the sulfonamide group of compound 21i forms a hydrogen bond to His90 in a similar fashion to celecoxib. In addition, we found that one of the NH groups in the urea of compound 21i could form the other H-bonding with Tyr365. Presumably, the presence of extra H-bonding in 21i might explain the slightly increased activity of compounds having a three-methylene linker compared to others in that the linker could provide enough space the urea group to interact with this residue.
Figure 2.

Compound 21i docked into (A) the co-crystal structure of human soluble epoxide hydrolase and (B) the co-crystal structure of murine COX-2. Black lines indicate possible hydrogen bonds.
Pharmacokinetic Screening in mice and rats
Having developed compounds with the desired inhibitor effects, we then investigated in vivo properties of selected inhibitors 21b,e–j. Pharmacokinetic screening was performed by oral cassette dosing in mice. As can be seen in Table 2, compounds 21i and 21j showed good PK parameters by demonstrating higher Cmax and AUC among selected inhibitors 21b,e–j. Even though the compound 21b exhibited similar inhibitory activities compared to 21i and 21j, it showed 15-fold lower Cmax and 8-fold decrease in AUC, respectively (Table 2). Based on these murine PK data, we further investigated two compounds, 21i and 21j in rats (Table 2). Comparing these compounds, 21i displayed better PK parameters in rats with a 2-fold higher Cmax and greater AUC. Therefore compound 21i was selected for in vivo study to determine its anti-nociceptive activity in a model of pain in rats.
Table 2.
PK Parameters of the Compounds 21b,e–j in Mice by Oral Cassette Dosing at 5 mg/kg (n =1) and in Rats by Oral Administration at 1 mg/kg (n=4).
| species | Inhibitors | Tmax (h) | Cmax (nM) | AUC (nM*h) | t1/2 (h) |
|---|---|---|---|---|---|
| Mouse | 21h | 0.8 | 190 | 375 | 2.4 |
| 21i | 0.8 | 260 | 980 | 2.6 | |
| 21j | 0.8 | 290 | 1335 | 2.4 | |
| 21e | 0.5 | 100 | 125 | 3.1 | |
| 21b | 0.5 | 19 | 35 | 1.0 | |
| 21g | 0.5 | 42 | 100 | 3.5 | |
| 21f | 0.5 | 20 | 22 | 1.7 | |
| Rat | 21i | 1.5 ± 0.6 | 415 ± 139 | 2764 ± 804 | 13 ± 4 |
| 21j | 1.0 ± 0.7 | 180 ± 71 | 1230 ± 521 | 12 ± 6 | |
In vivo anti-allodynic effects of COX-2/sEH dual inhibitors
A LPS induced model of inflammatory pain39 was used to compare the pain reducing activity of the COX-2/sEH dual inhibitors to individual and combination therapies. Specifically, a von Frey mechanical nociceptive assay was used to measure allodynia induced by LPS and the attenuation of this pain (Figure 3). Here we compared the effects of the dual inhibitor 21i, to the individual and co-administration of celecoxib and the t-AUCB, from different rat groups. Male Sprague-Dawley rats (n = 6) injected with LPS (10 µg per rat; intraplantar) developed allodynia indicated by a decrease in paw withdrawal latency to a non-noxious mechanical stimulus. Post LPS challenge, the average latency for hindpaw withdrawal dropped to 54 ± 4% (SEM) within 60 minutes and remained at or below this score compared to vehicle controls, which did not vary significantly from 100% for a six hour time-course. Prophylactic subcutaneous administration of the dual inhibitor 21i (10 mg/kg), a combination of celecoxib and t-AUCB (10 mg/kg, respectively), celecoxib alone (10mg/kg), and t-AUCB alone (10mg/kg) all showed anti-allodynic effects (Figure 3). While t-AUCB or celecoxib exerted a slight anti-allodynic effect, both the dual inhibitor 21i and co-administration of t-AUCB with celecoxib synergized effects in reducing allodynia. Remarkably, the compound 21i at 10 mg/kg exerted a stronger effect than the combination treatment at 10 mg/kg of each drug (celecoxib + t-AUCB), suggesting that the dual inhibitor could compensate for different pharmacokinetic properties of the co-administered individual compound. This high efficacy also is interesting because 21i is a stronger COX-2 inhibitor than rofecoxib but a far weaker inhibitor than celecoxib (Table 1.). Additional quantitative measurements of changes in animal behavior were not made beyond the von Frey mechanical assay. However, the animals were observed and handed over nine hours on the test day and there were no overt signs of any behavioral changes or acute observable symptoms of toxicity such as lacrimation, exuding porphyrin, frozen or hunched postures, gasping, or chattering from the rats treated by the compound 21i. In addition, we did not observe any acute toxicity on U937 cell line.
Figure 3.

Effect of sEH, COX-2 and dual inhibitor (21i) in a model of inflammatory pain. Male Sprague Dawley rats (n = 6 per group) were pretreated with inhibitors (Inh.) 60 minutes before LPS injection (10 µg/rat hindpaw intraplantar injection). A. Time course of allodynia using Von Frey Mechanical assay. Results are expressed as percentage of baseline (Bl.) that was measured prior to inhibitor treatment. LPS: ○; t-AUCB (10 mg/kg s.c.):  ; celecoxib (10 mg/kg s.c): ■; celecoxib + t-AUCB (10 mg/kg each injected separately s.c.):
; celecoxib (10 mg/kg s.c): ■; celecoxib + t-AUCB (10 mg/kg each injected separately s.c.):  ; 21i (10 mg/kg s.c.):▼. B. Overall response estimated by calculating area under the curve (AUC) from panel A.
; 21i (10 mg/kg s.c.):▼. B. Overall response estimated by calculating area under the curve (AUC) from panel A.
Conclusion
A series of novel COX-2/sEH dual inhibitors were rationally designed and synthesized to provide the basis for a structure-activity relationship (SAR) study. The compounds synthesized as DMLs demonstrate significant inhibition to both COX-2 selectively and sEH. This study has demonstrated that a grouping of diarylpyrazole (COX-2 pharmacophore) and urea (sEH pharmacophore) can improve the inhibitory activity of the resulting molecule on both COX-2 and sEH in vitro and in vivo. This study has demonstrated that the COX-2/sEH dual inhibitor 21i displays improved in vivo efficacy as compared to both the individual and combination therapies of celecoxib and t-AUCB, as observed using a pain model in rats. Furthermore, the dual inhibitor is also expected to show a better safety profile than that of the combination therapy.
Experimental Section
General
Celecoxib was a gift from Pfizer Corporation, Indomethacin was purchased from TCI America Fine Chemicals (Portland, USA), rofecoxib was purchased from Fisher Scientific (Chicago, USA). All other reagents and solvents were obtained from commercial suppliers and were used without further purification. All reactions, unless otherwise described, were performed under an inert atmosphere of dry nitrogen. Melting points were determined on an OptiMelt melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded at 300 and 75 MHz, respectively. Elemental analyses were determined at Midwest Microlab, Indianapolis, IN. Mass spectra were measured by LC-MS equipped with a Waters 2790 and a Waters PDA 996 using electrospray (+) ionization. Flash chromatography was performed on silica gel. All compounds were characterized by NMR and LC/MS. All final compounds, 7, 15, 11a–h, and 21a–j, had a purity of ≥95%. The purity of final compounds was determined by elemental analysis (within ± 0.4% of the calculated value).
5-Phenyl-1-(4-sulfamoyl-phenyl)-1H-pyrazole-3-carboxylic acid ethyl ester (3b)
To a solution of potassium bis(trimethylsilyl)amide (8.88 g, 44.5 mmol) in 400 mL of THF was added dropwise acetophenone (4.85 mL, 41.6 mmol) at −78 °C. After 1h, diethyl oxalate (6.2 mL, 45.8 mmol) was added dropwise at the same temperature. The reaction mixture was allowed to warm up to room temperature and stirred overnight. The solvent was removed in vacuo. The resulting yellowish solid was filtered and washed with Et2O to give the title compound 2 (10.5 g, 98%) as a potssium salt of ethyl 2-hydroxy-4-oxo-4-phenyl-2-butenoate (2).40 To a solution of potassium salt of 2 in EtOH (300 mL) was added 4-amidosulfonylphenylhydrazine 1b (10 g, 44.7 mmol) at room temperature. The reaction mixture was stirred for 1h and then 2.4 mL of AcOH was added. The reaction mixture was refluxed for 2h. After cooling, the solvent was removed in vacuo. The resulting solid was purified by recrystallization with MeOH to afford the titled compound (13.5 g, 89% yield) as a white solid. mp 191.5–194.6 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.87 (d, J = 8 Hz, 2H), 7.58-7.48 (m, 4H), 7.44-7.38 (m, 3H), 7.34-7.27 (m, 2H), 7.16 (s, 1H), 4.35 (q, J = 7 Hz, 2H), 1.32 (t, J = 7 Hz, 3H). MS (ESI) m/z: 372.10 (M + H+).
The preparation of the compound 7
5-Phenyl-1-(4-sulfamoyl-phenyl)-1H-pyrazole-3-carboxylic acid (4)
To a solution of compound 3b (0.37g, 1 mmol) in THF (10 mL) was added LiOH (35 mg, 1.5 mmol) followed by 2 mL of water at room temperature. The reaction mixture was stirred overnight. The reaction was quenched by adding 1N HCl. The solvent was evaporated in vacuo. The resulting white solids were collected by suction filtration and washed with water. The crude product was recrystallized from MeOH to give the titled compound (0.33 g, 96% yield) as a white solid. mp 188.5–190.9 °C. 1H NMR (300 MHz, DMSO-d6): δ 13.10 (s, 1H), 7.87 (d, J = 8 Hz, 2H), 7.55-7.48 (m, 4H), 7.44-7.37 (m, 3H), 7.34-7.26 (m, 2H), 7.10 (s, 1H). MS (ESI) m/z: 344.07 (M + H+).
[5-Phenyl-1-(4-sulfamoyl-phenyl)-1H-pyrazol-3-yl]-carbamic acid ethyl ester (5)
To a solution of acid 4 (0.74 g, 2 mmol) in 10 mL of EtOH and 10 mL of 1,4-dioxane was added diphenylphosphonylazide (0.52 mL, 2.4 mmol) followed by Et3N (0.34 mL, 2.4 mmol). The reaction mixture was refluxed overnight. After cooling the reaction mixture to room temperature, EtOAc and water were added. The organic layer was separated, washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography to give the titled compound (0.48 g, 62 %) as a white solid. mp 120.9–127.5 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.30 (s, 1H), 7.79 (d, J = 8 Hz, 2H), 7.50-7.34 (m, 7H), 7.33-7.25 (m, 2H), 6.74 (s, 1H), 4.14 (q, J = 7 Hz, 2H), 1.24 (t, J = 7 Hz, 3H). MS (ESI) m/z: 387.11 (M + H+).
4-(3-Amino-5-phenyl-pyrazol-1-yl)-benzenesulfonamide (6)
To a solution of an ethyl carbamate 5 (0.39 g, 1 mmol) in EtOH (10 mL) was added 10% NaOH solution (3.2 mL) at room temperature. The reaction mixture was refluxed for 5h. The reaction mixture was cooled to room temperature and water was added. The resulting white precipitate was filtered and washed with water thoroughly to give the titled compound (0.22 g, 70%) as a white solid. mp 286.3–290.2 °C. 1H NMR (DMSO-d6): δ 7.70 (d, J = 9 Hz, 2H), 7.43-7.20 (m, 9H), 5.89 (s, 1H), 5.17 (br s, 2H). MS (ESI) m/z: 315.09 (M + H+).
4-{5-Phenyl-3-[3-(3-trifluoromethyl-phenyl)-ureido]-pyrazol-1-yl}-benzenesulfonamide (7)
To a solution of amine 6 (0.1 g, 0.32 mmol) in 5 mL of DMF was added 3-(trifluoromethyl)phenyl isocyanate (0.12 g, 0.62 mmol) followed by triethylamine (0.05 mL, 0.32 mmol) at 0 °C. The reaction mixture was warmed up to room temperature and stirred overnight. The reaction mixture was poured into water, and the resulting precipitates were collected and washed with water. The crude product was purified by column chromatography by dry loading method to give the titled compound (0.13 g, 81% yield). mp 195.1–197.0 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.48 (s, 1H), 9.21 (s, 1H), 8.07 (s, 1H), 7.80 (d, J = 9 Hz, 2H), 7.61-7.26 (m, 12H), 6.81 (s, 1H). MS (ESI) m/z: 502.12 (M + H+). Anal. (C23H18F3N5O3S˙0.36CH4O) C, H, N.
The preparation of the compounds 11a–g
1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazole-3-carboxylic acid ethyl ester (3a)
Compound 3a was synthesized in a manner similar to the synthesis of 3b using 4-(methylsulfonyl)phenylhydrazine hydrochloride (10 g, 44.7 mmol) to give 3a (13.6 g, 82% yield) as a white solid. mp 199.4–202.9 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.99 (d, J = 9 Hz, 2H), 7.58 (d, J = 9 Hz, 2H), 7.43-7.38 (m, 3H), 7.34-7.27 (m, 2H), 7.17 (s, 1H), 4.34 (q, J = 7, 2H), 3.27 (s, 3H), 1.31 (t, J = 7, 3H). MS (ESI) m/z: 371.11 (M + H+).
General Procedure for the Synthesis of Hydroxymethylpyrazoles 8a and 8b
To a solution of compound 3a or 3b in THF was added LiAlH4 (3.2 eq) at room temperature. The reaction mixture was stirred for 6h. EtOAC was added and excess LiAlH4 was quenched by adding minimum amounts of water. The solution was dried with MgSO4 and white precipitates were filtered off. The filtrate was concentrated in vacuo. The crude product was purified by column chromatography (EtOAc/Hexanes in aequate proportions) to give the compound 8a or 8b.
[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-yl]-methanol (8a)
was synthesized as a white solid (3.25 g, 76% yield) from the ester 3a (5 g, 17.1 mmol) by the general procedure for the Synthesis of Hydroxymethylpyrazoles 8a and 8b and purified by column chromatography with Hexanes/EtOAc (1:1). mp 126.5–128.5 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.88 (d, J = 9 Hz, 2H), 7.49 (d, J = 9 Hz, 2H), 7.41-7.34 (m, 3H), 7.26-7.21 (m, 2H), 6.56 (s, 1H), 4.81 (d, J = 6 Hz, 2H), 3.05 (s, 3H), 2.13 (t, J = 6 Hz, 1H). MS (ESI) m/z: 329.10 (M + H+).
4-(3-Hydroxymethyl-5-phenyl-pyrazol-1-yl)-benzenesulfonamide (8b)
was synthesized as a white solid (2.8 g, 75% yield) from ester 3b (4.18 g, 11.3 mmol) by the general procedure for the Synthesis of Hydroxymethylpyrazoles 8a and 8b and purified by column chromatography with Hexanes/EtOAc (4:6). mp 173.8–180.5 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.81 (d, J = 8 Hz, 2H), 7.50-7.35 (m, 7H), 7.30-7.23 (m, 2H), 6.64 (s, 1H), 5.28 (t, J = 6 Hz, 1H), 4.53 (d, J = 6 Hz, 2H). MS (ESI) m/z: 330.09 (M + H+).
General Procedure for the Synthesis of Azidomethylpyrazoles 9a and 9b
To a solution of alcohol 8a or 8b in THF was added methanesulfonyl chloride (1.3 eq) followed by Et3N (1.6 eq) at 0 °C. The reaction mixture was stirred for 2h. After adding water, the product was extracted with EtOAc. The organic layer was dried with MgSO4 and the solvent was removed in vacuo. To a solution of a mesylate obtained, in 1,4-dioxane/water (1:1) was added NaN3 (2.5 eq). The reaction mixture was heated at 80 °C for 3h. After cooling, the solvent were removed in vacuo. The residue was purified by column chromatography (EtOAc/Hexanes in adequate proportions) to give the compound 9a or 9b as a viscous liquid.
3-Azidomethyl-1-(4-methanesulfonyl-phenyl)-5-phenyl-1H-pyrazole (9a)
was synthesized as a viscous liquid (0.7 g, 99% yield) from alcohol 8a (0.5 g, 2 mmol) by the general procedure for the Synthesis of Azidomethylpyrazoles 9a and 9b and purified by column chromatography with Hexanes/EtOAc (6:4). 1H NMR (300 MHz, DMSO-d6): δ 7.95 (d, J = 9 Hz, 2H), 7.51 (d, J = 9 Hz, 2H), 7.45-7.39 (m, 3H), 7.33-7.26 (m, 2H), 6.76 (s, 1H), 4.53 (s, 2H), 3.26 (s, 3H). MS (ESI) m/z: 354.10 (M + H+).
4-(3-Azidomethyl-5-phenyl-pyrazol-1-yl)-benzenesulfonamide (9b)
was synthesized as a viscous liquid (0.48 g, 90% yield) from ester 8b (0.5 g, 1.52 mmol) by the general procedure for the Synthesis of Azidomethylpyrazoles 9a and 9b and purified by column chromatography with Hexanes/EtOAc (1:1). 1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, J = 9 Hz, 2H), 7.51-7.37 (m, 7H), 7.31-7.24 (m, 2H), 6.75 (s, 1H), 4.53 (s, 2H). MS (ESI) m/z: 355.10 (M + H+).
General Procedure for the Synthesis of Aminomethylpyrazoles 10a and 10b
To a solution of 9a or 9b in EtOAc was added 10% palladium on carbon. The solution was filled with H2 and the reaction mixture was stirred for 2h. After the solution was filtered through Celite, the filtrate was concentrated in vacuo. The crude product was used for the next step without further purification.
C-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-yl]-methylamine (10a)
91% yield. 1H NMR (300 MHz, CDCl3): δ 7.87 (d, J = 9 Hz, 2H), 7.48 (d, J = 9 Hz, 2H), 7.40-7.32 (m, 3H), 7.26-7.19 (m, 2H), 6.48 (s, 1H), 3.97 (s, 2H), 3.04 (s, 3H), 1.65 (s, 2H).
4-(3-Aminomethyl-5-phenyl-pyrazol-1-yl)-benzenesulfonamide (10b)
90% yield. 1H NMR (300 MHz, CDCl3): δ 7.87 (d, J = 9 Hz, 2H), 7.48 (d, J = 9 Hz, 2H), 7.39-7.31 (m, 3H), 7.27-7.19 (m, 2H), 6.48 (s, 1H), 3.98 (s, 2H), 3.05-3.02 (m, 4H).
1-Adamantan-1-yl-3-[1-(4-methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-ylmethyl]-urea (11a)
The titled compound was prepared in 68% yield from 1-adamantyl isocyanate using the procedure detailed for compound 7. mp 150.0–155.3 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.93 (d, J = 9 Hz, 2H), 7.48 (d, J = 9 Hz, 2H), 7.44-7.38 (m, 3H), 7.30-7.23 (m, 2H), 6.54 (s, 1H), 6.13 (t, J = 6 Hz, 1H), 5.71 (s, 1H), 4.22 (d, J = 6 Hz, 2H), 3.25 (s, 3H), 2.04-1.96 (m, 3H), 1.90-1.85 (m, 6H), 1.63-1.57 (m, 6H). MS (ESI) m/z: 505.22 (M + H+). Anal. (C28H32N4O3S) C, H,N.
1-Cycloheptyl-3-[1-(4-methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-ylmethyl]-urea (11b)
The titled compound was prepared in 68% yield from cycloheptyl isocyanate using the procedure detailed for compound 7. mp 103.9–109.9 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.93 (d, J = 9 Hz, 2H), 7.48 (d, J = 9 Hz, 2H), 7.44-7.38 (m, 3H), 7.29-7.24 (m, 2H), 6.54 (s, 1H), 6.18 (t, J = 6 Hz, 1H), 5.94 (d, J = 8 Hz, 1H), 4.26 (d, J = 6 Hz, 2H), 3.66-3.54 (m, 1H), 3.25 (s, 3H), 1.82-1.70 (m, 2H), 1.61-1.29 (m, 10H). MS (ESI) m/z: 467.21 (M + H+). Anal. (C25H30N4O3S˙0.25CH4O) C, H, N.
1-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-ylmethyl]-3-phenyl-urea (11c)
The titled compound was prepared in 68% yield from phenyl isocyanate using the procedure detailed for compound 7. mp 95.7–104.2 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.62 (s, 1H), 7.94 (d, J = 9 Hz, 2H), 7.50 (d, J = 9 Hz, 2H), 7.43-7.37 (m, 5H), 7.31-7.18 (m, 4H), 6.89 (t, J = 7 Hz, 1H), 6.67-6.61 (m, 1H), 6.62 (s, 1H), 4.38 (d, J = 6 Hz, 2H), 3.25 (s, 3H). MS (ESI) m/z: 447.15 (M + H+). Anal. (C24H22N4O3S) C, H, N.
1-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-ylmethyl]-3-(4-trifluoromethoxy-phenyl)-urea (11d)
The titled compound was prepared in 68% yield from 4-(trifluoromethoxy)phenyl isocyanate using the procedure detailed for compound 7. mp 152.9–156.3 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.87 (s, 1H), 7.94 (d, J = 9 Hz, 2H), 7.55-7.46 (m, 4H), 7.43-7.37 (m, 3H), 7.31-7.19 (m, 4H), 6.73 (t, J = 6 Hz, 1H), 6.62 (s, 1H), 4.39 (d, J = 6 Hz, 2H), 3.25 (s, 3H). MS (ESI) m/z: 531.13 (M + H+). Anal. (C25H21F3N4O4S) C, H, N.
1-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-ylmethyl]-3-(4-trifluoromethyl-phenyl)-urea (11e)
The titled compound was prepared in 68% yield from 4-(trifluoromethyl)phenyl isocyanate using the procedure detailed for compound 7. mp 129.3–131.7 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.09 (s, 1H), 7.94 (d, J = 9 Hz, 2H), 7.65-7.54 (m, 4H), 7.50 (d, J = 9 Hz, 2H), 7.43-7.38 (m, 3H), 7.31-7.25 (m, 2H), 6.83 (t, J = 6 Hz, 1H), 6.64 (s, 1H), 4.41 (d, J = 6 Hz, 2H), 3.25 (s, 3H). MS (ESI) m/z: 515.14 (M + H+). Anal. (C25H21F3N4O3S˙0.375CH4O) C, H, N.
1-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-ylmethyl]-3-(3-trifluoromethyl-phenyl)-urea (11f)
The titled compound was prepared in 68% yield from 3-(trifluoromethyl)phenyl isocyanate using the procedure detailed for compound 7. mp 107.5–113.0 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.05 (s, 1H), 8.00 (s, 1H), 7.94 (d, J = 9 Hz, 2H), 7.56-7.36 (m, 7H), 7.32-7.21 (m, 3H), 6.82 (t, J = 6 Hz, 1H), 6.63 (s, 1H), 4.40 (d, J = 6 Hz, 2H), 3.25 (s, 3H). MS (ESI) m/z: 515.13 (M + H+).
4-{5-Phenyl-3-[3-(3-trifluoromethyl-phenyl)-ureidomethyl]-pyrazol-1-yl}-benzenesulfonamide (11g)
The titled compound was prepared in 68% yield from 3-(trifluoromethyl)phenyl isocyanate using the procedure detailed for compound 7. mp 184.6–189.6 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.04 (s, 1H), 8.00 (s, 1H), 7.81 (d, J = 9 Hz, 2H), 7.56-7.35 (m, 9H), 7.30-7.21 (m, 3H), 6.81 (t, J = 6 Hz, 1H), 6.61 (s, 1H), 4.39 (d, J = 6 Hz, 2H). MS (ESI) m/z: 516.13 (M + H+).
For the preparation of compound 11h see Supporting Information.
The preparation of the compound 15
4-(3-Formyl-5-phenyl-pyrazol-1-yl)-benzenesulfonamide (12b)
To a solution of alcohol 8b (3 g, 9.1 mmol) and 3 g of powdered 4 Å molecular sieves in 150 mL of DCM, was added PCC (2.94 g, 13.7 mmol) at room temperature. After 6h, the reaction mixture was filtered through a 0.75-in. pad of Celite. The filtrate was concentrated in vacuo then the residue was purified by column chromatography to afford the titled compound (2.1 g, 70%) as a white solid. mp 177.3–179.8 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.03 (s, 1H), 7.88 (d, J = 8 Hz, 2H), 7.61-7.49 (m, 4H), 7.48-7.37 (m, 3H), 7.35-7.26 (m, 2H), 7.20 (s, 1H). MS (ESI) m/z: 328.08 (M + H+).
4-[3-((E)-2-Nitro-vinyl)-5-phenyl-pyrazol-1-yl]-benzenesulfonamide (13)
To a solution of aldehyde 10 (0.96 g, 2.9 mmol) in MeNO2 (5.9 mL, 109 mmol) was added AcONH4 (0.34 g, 4.4 mmol). The reaction mixture was refluxed for 1h. After cooling, MeNO2 was evaporated in vacuo. The residue was purified by column chromatography with Hexanes/EtOAc (7: 3) to give the corresponding nitroethylene compound (0.5g, 46%) as a yellow solid. mp 189.2–191.8 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.24 (d, J = 14 Hz, 1H), 8.01 (d, J = 14 Hz, 1H), 7.85 (d, J = 9 Hz, 2H), 7.54-7.47 (m, 4H), 7.46-7.40 (m, 3H), 7.37 (s, 1H), 7.32-7.26 (m, 2H). MS (ESI) m/z: 371.08 (M + H+).
4-[3-(2-Amino-ethyl)-5-phenyl-pyrazol-1-yl]-benzenesulfonamide (14)
To a solution of the compound above (0.42 g, 1.1 mmol) in 10 ml of THF was added LiAlH4 (0.21 g, 5.7 mmol) at room temperature. The reaction mixture was stirred for 6 h and quenched by adding water (10 mL). The mixture was extracted with EtOAc and the combined organic layers were dried with MgSO4. The solution was evaporated in vacuo. The crude product was used next step without further purification (0.3 g, 77% yield). mp 189.2–191.8 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.79 (d, J = 9 Hz, 2H), 7.43-7.36 (m, 5H), 7.28-7.23 (m, 2H), 6.55 (s, 1H), 5.23-3.88 (m, 4H), 2.88 (t, J = 7 Hz, 2H), 2.71 (t, J = 7 Hz, 2H).
4-(5-Phenyl-3-{2-[3-(3-trifluoromethyl-phenyl)-ureido]-ethyl}-pyrazol-1-yl)-benzenesulfonamide (15)
The titled compound was prepared in 84% yield from 3-trifluoromethylphenyl isocyanate using the procedure detailed for compound 7. mp 121.9–124.7 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.97 (s, 1H), 8.00 (s, 1H), 7.80 (d, J = 8 Hz, 2H), 7.52-7.36 (m, 8H), 7.29-7.20 (m, 4 H), 6.61 (s, 1H), 6.41 (t, J = 6 Hz, 1H), 3.49 (dd, J = 13 and 7 Hz, 2H), 2.85 (t, J = 7 Hz, 2H). MS (ESI) m/z: 530.15 (M + H+).
The preparation of the compound 21a
1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazole-3-carbaldehyde (12a)
The titled compound was prepared in 65% yield from the compound 3a using the procedure detailed for compound 12b. mp 133.0–135.5 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.03 (s, 1H), 8.02 (d, J = 9 Hz, 2H), 7.63 (d, J = 9 Hz, 2H), 7.47-7.39 (m, 3H), 7.37-7.29 (m, 2H), 7.22 (s, 1H), 3.29 (s, 3H). MS (ESI) m/z: 327.08 (M + H+).
(E)-3-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-yl]-acrylic acid ethyl ester (16)
To a solution of aldehyde 12a (1.4 g, 4.3 mmol) and triethyl phosphonoacetate (0.94 mL, 4.73 mmol) in 50 mL of THF was added 60% sodium hydride in oil (0.19 g, 4.73 mmol) at 0 °C. The reaction mixture was stirred for 1h. The reaction was quenched by adding 5 mL of water, and then mixture was extracted with EtOAc. After drying with MgSO4, the solvent was removed in vacuo. The residue was purified by column chromatography with 7:3 (hexanes:EtOAc) to give the titled compound (1.32 g, 77% yield) as a white solid. mp 163.5–168.4 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.96 (d, J = 9 Hz, 2H), 7.58 (d, J = 16 Hz, 1H), 7.55 (d, J = 9 Hz, 2H), 7.47-7.40 (m, 3H), 7.34-7.27 (m, 2H), 7.28 (s, 1H), 6.72 (d, J = 16 Hz, 1H), 4.21 (q, J = 7 Hz, 2H), 3.27 (s, 3H), 1.27 (t, J = 7 Hz, 3H). MS (ESI) m/z: 397.12 (M + H+).
3-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-yl]-propionic acid ethyl ester (17)
To a solution of 16 (0.17 g, 0.42 mmol) in 5 mL of EtOAc was added 10% palladium on carbon. The solution was filled with H2 and the reaction mixture was stirred for 2h. After the solution was filtered through Celite, the filtrate was concentrated in vacuo. Purification by column chromatography (3:7 EtOAc-haxanes) gave the title compound, 0.15 g (90 %) as a white solid. mp 96.5–99.1 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.91 (d, J = 9 Hz, 2H), 7.45 (d, J = 9 Hz, 2H), 7.44-7.37 (m, 3H), 7.29-7.23 (m, 2H), 6.57 (s, 1H), 4.09 (q, J = 7 Hz, 2H), 3.24 (s, 3H), 2.93 (t, J = 7 Hz, 2H), 2.74 (t, J = 7 Hz, 2H), 1.18 (t, J = 7 Hz, 3H). MS (ESI) m/z: 399.14 (M + H+).
3-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-yl]-propan-1-ol (18a)
To a solution of ester (1.27 g, 4 mmol) in 40 mL of THF was added LiAlH4 (0.24 g, 6.4 mmol). The reaction mixture was stirred for 6h at room temperature. EtOAC (5 mL) and water (10 mL) were added successively. The solvent was removed in vacuo and the resulting solid was filtered. The crude product was purified by column chromatography (EtOAc/Hexanes =7:3) to give the titled compound (0.97 g, 85%) as a white solid. mp 119.5–123.4 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.91 (d, J = 9 Hz, 2H), 7.46 (d, J = 9 Hz, 2H), 7.43-7.37 (m, 3H), 7.31-7.25 (m, 2H), 6.56 (s, 1H), 4.52 (t, J = 5 Hz, 1H), 3.54-3.46 (m, 2H), 3.24 (s, 3H), 2.79-2.60 (m, 2H), 1.88-1.77 (m, 2H). MS (ESI) m/z: 357.12 (M + H+).
2-{3-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-yl]-propyl}-isoindole-1,3-dione (19)
To a solution of alcohol 18 (0.3g, 0.84 mmol), PPh3 (0.22g, 0.84 mmol), and phthalimide (0.12g, 0.84 mmol) in 10 mL of THF was added dropwise DIAD (0.17g, 0.84 mmol), at room temperature. The reaction mixture was stirred overnight. The solvent was evaporated, and the residue was purified by column chromatography with hexanes/EtOAc (1:1) to afford 0.37g (90%) of the title compound as a white solid. mp 62.4–72.2 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.88 (d, J = 8 Hz, 2H), 7.83-7.74 (m, 5H), 7.43-7.29 (m, 4H), 7.17-7.09 (m, 2H), 6.51 (s, 1H), 3.72 (t, J = 7 Hz, 2H), 3.24 (s, 3H), 2.71 (t, J = 7 Hz, 2H), 2.15-2.02 (m, 2H). MS (ESI) m/z: 486.15 (M + H+).
1-{3-[1-(4-Methanesulfonyl-phenyl)-5-phenyl-1H-pyrazol-3-yl]-propyl}-3-(3-trifluoromethyl-phenyl)-urea (21a)
35 wt% Hydrazine hydrate (0.16 g, 1.73 mmol) was added to a solution of compound 19 (0.42 g, 0.86 mmol) in DCM (15 mL) followed by MeOH (15 mL) at room temperature. The reaction mixture was stirred for 1d. The white precipitates were filtered off and the solvent was removed in vacuo. The residue was dissolved in aqueous 1N HCl solution and washed with DCM. Aqueous layer was basified with excess 1N NaOH solution and then extracted with DCM. After drying with MgSO4, the solvent was evaporated affording the amine 20a. To a solution of amine obtained was an added 3-trifluoromethylphenyl isocyanate (0.16 g, 0.86 mmol). The reaction mixture was stirred overnight. After adding water, precipitates were collected by suction filter. The white solid was purified by column chromatography with hexanes/EtOAc (1:1) to give the titled compound (0.37 g, 80 mmol) as a white solid. mp 150.1–151.8 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.86 (s, 1H), 7.99 (s, 1H), 7.91 (d, J = 9 Hz, 2H), 7.53-7.36 (m, 7H), 7.31-7.25 (m, 2H), 7.24-7.19 (m, 1H), 6.59 (s, 1H), 6.39 (t, J = 6 Hz, 1H), 3.26-3.17 (m, 2H), 3.24 (s, 3H), 2.70 (t, J = 8 Hz, 2H), 1.94-1.80 (m, 2H). MS (ESI) m/z: 543.17 (M + H+). Anal. (C27H25F3N4O3S) C, H, N.
The preparation of the compound 21b and 21d–j
4-[3-(3-Hydroxy-propyl)-5-phenyl-pyrazol-1-yl]-benzenesulfonamide (18b).40
60% dispersion of sodium hydride in mineral oil (4.0 g, 100 mmol) was washed twice with hexane (100 mL each) and dried under a stream of nitrogen. Ether (300 mL) was added followed by dropwise addition of ethanol (0.25 mL) and γ-butyrolactone (4.0 mL, 52 mmol). The mixture was cooled to 10 °C and acetophenone (5.8 mL, 50 mmol) in ether (40 mL) was added dropwise over 1 hour. The mixture was warmed to 25 °C and stirred overnight. The mixture was cooled to 0 °C and quenched with ethanol (5 mL) followed by 10% aqueous ammonium sulfate (100 mL). The organic solution was separated, dried over Mg2SO4 and concentrated. The residue was chromatographed on silica gel with 1:1 hexane/ethyl acetate to give the desired diketone 22 (3.4 g, 33%) as oil. Pyridine (0.34 mL, 4.2 mmol) and the diketone 22 (700 mg, 3.4 mmol) in methanol (3 mL) were added to slurry of 4-sulfonamidophenylhydrazine-HCl (750 mg, 3.4 mmol) in methanol (8 mL). The mixture was stirred at 25 °C overnight and concentrated in vacuo. The residue was dissolved in DCM and the solution washed with 1N HCl. The organic solution was separated, dried and concentrated. The residue was chromatographed on silica gel using EtOAc to give the desired alcohol 18b (1 g, 90%) as a white solid. mp 203.5–205.4 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.79 (d, J = 9 Hz, 2H), 7.45-7.35 (m, 7H), 7.28-7.23 (m, 2H), 6.53 (s, 1H), 4.51 (t, J = 5 Hz, 1H), 3.50 (dd, J = 12 and 6 Hz, 2H), 2.84-2.56 (m, 2H), 1.88-1.77 (m, 2H). MS (ESI) m/z: 358.12 (M + H+).
4-[3-(3-Azido-propyl)-5-phenyl-pyrazol-1-yl]-benzenesulfonamide (23)
The titled compound was prepared in 88% yield using the compound above using the procedure detailed for compound 9b. mp 114.0–115.6 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.80 (d, J = 9 Hz, 2H), 7.48-7.36 (m, 7H), 7.28-7.23 (m, 2H), 6.58 (s, 1H), 3.46 (t, J = 7 Hz, 2H), 2.72 (t, J = 8 Hz, 2H), 2.00-1.89 (m, 2H). MS (ESI) m/z: 383.13 (M + H+).
3-[5-Phenyl-1-(4-sulfamoyl-phenyl)-1H-pyrazol-3-yl]-propyl-ammonium chloride (20b)
was synthesized as a white solid (68% yield) from compound 23 using the General Procedure for the Synthesis of Aminomethylpyrazoles 10a and 10b, except the titled compound was prepared as an HCl salt with anhydrous HCl (g). mp 152.3–270.0 °C dec. 1H NMR (300 MHz, TFA-d): δ 11.81-11.52 (m, 3H), 8.07 (d, J = 8 Hz, 2H), 7.57 (d, J = 8.18 Hz, 2H), 7.50-7.43 (m, 1H), 7.40-7.32 (m, 2H), 7.26-7.21 (m, 2H), 7.05 (br s, 2H), 6.88 (s, 1H), 3.48-3.36 (m, 2H), 3.26-3.06 (m, 2H), 2.51-2.35 (m, 2H). MS (ESI) m/z: 357.14 (M + H+).
4-(5-Phenyl-3-{3-[3-(3-trifluoromethyl-phenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide (21b)
The titled compound was prepared in 84% yield from 3-(trifluoromethyl)phenyl isocyanate using the procedure detailed for compound 7. mp 175.5–176.4 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.85 (s, 1H), 7.98 (s, 1H), 7.79 (d, J = 9 Hz, 2H), 7.53-7.35 (m, 9H), 7.29-7.18 (m, 3H), 6.57 (s, 1H), 6.38 (t, J = 6 Hz, 1H), 3.25-3.17 (m, 2H), 2.69 (t, J = 8 Hz, 2H), 1.94-1.80 (m, 2H). MS (ESI) m/z: 544.16 (M + H+). Anal. (C26H24F3N5O3S) C, H, N.
4-(3-{3-[3-(2,6-Diisopropyl-phenyl)-ureido]-propyl}-5-phenyl-pyrazol-1-yl)-benzenesulfonamide (21d)
The titled compound was prepared in 86% yield from 2,6-diisopropylphenyl isocyanate using the procedure detailed for compound 7. mp 137.9–140.7 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.79 (d, J = 9 Hz, 2H), 7.46-7.34 (m, 9H), 7.29-7.16 (m, 3H), 7.14-7.07 (m, 2H), 6.55 (s, 1H), 3.22-3.10 (m, 4H), 2.75-2.61 (m, 2H), 1.90-1.73 (m, 2H), 1.12 (d, J = 7 Hz, 12H). MS (ESI) m/z: 560.27 (M + H+). Anal. (C31H37N5O3S) C, H, N.
4-{5-Phenyl-3-[3-(3-phenyl-ureido)-propyl]-pyrazol-1-yl}-benzenesulfonamide (21e)
The titled compound was prepared in 90% yield from phenyl isocyanate using the procedure detailed for compound 7. mp 171.0–172.1 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.43 (s, 1H), 7.79 (d, J = 9 Hz, 2H), 7.47-7.36 (m, 9H), 7.29-7.16 (m, 4H), 6.87 (t, J = 7 Hz, 1H), 6.57 (s, 1H), 6.23 (t, J = 6 Hz, 1H), 3.25-3.14 (m, 2H), 2.69 (t, J = 8 Hz, 2H), 1.92-1.78 (m, 2H). MS (ESI) m/z: 476.18 (M + H+). Anal. (C25H25N5O3S˙0.3CH4O) C, H, N.
4-{3-[3-(3-Adamantan-1-yl-ureido)-propyl]-5-phenyl-pyrazol-1-yl}-benzenesulfonamide (21f)
The titled compound was prepared in 84% yield from 1-adamantyl isocyanate using the procedure detailed for compound 7. mp 139.4–143.5 °C. 1H NMR (300 MHz, CDCl3): δ 7.75 (d, J = 9 Hz, 2H), 7.37-7.28 (m, 5H), 7.21-7.15 (m, 2H), 6.36 (s, 1H), 5.70-5.64 (m, 2H), 4.68-4.60 (m, 1H), 4.29 (s, 1H), 3.23 (q, J = 7 Hz, 2H), 2.78 (t, J = 7, 2H), 2.08-1.87 (m, 9H), 1.67-1.59 (m, 6H), 1.29-1.22 (m, 2H). MS (ESI) m/z: 534.25 (M + H+).
4-{3-[3-(3-Cycloheptyl-ureido)-propyl]-5-phenyl-pyrazol-1-yl}-benzenesulfonamide (21g)
The titled compound was prepared in 91% yield from cycloheptyl isocyanate using the procedure detailed for compound 7. mp 164.2–169.8 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.79 (d, J = 9 Hz, 2H), 7.47-7.36 (m, 7H), 7.29-7.22 (m, 2H), 6.55 (s, 1H), 5.82-5.70 (m, 2H), 3.63-3.49 (m, 1H), 3.13-3.02 (m, 2H), 2.63 (t, J = 8 Hz, 2H), 1.84-1.66 (m, 4H), 1.60-1.27 (m, 10H). MS (ESI) m/z: 496.24 (M + H+).
4-(3-{3-[3-(4-Chloro-phenyl)-ureido]-propyl}-5-phenyl-pyrazol-1-yl)-benzenesulfonamide (21h)
The titled compound was prepared in 92% yield from 4-chlorophenyl isocyanate using the procedure detailed for compound 7. mp 103.2–110.4 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.60 (s, 1H), 7.79 (d, J = 9 Hz, 2H), 7.47-7.35 (m, 9H), 7.29-7.21 (m, 4H), 6.57 (s, 1H), 6.29 (t, J = 6 Hz, 1H), 3.26-3.13 (m, 2H), 2.68 (t, J = 8 Hz, 2H), 1.92-1.77 (m, 2H). MS (ESI) m/z: 510.14 (M + H+). Anal. (C25H24ClN5O3S˙CH4O) C, H, N.
4-(5-Phenyl-3-{3-[3-(4-trifluoromethyl-phenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide (21i)
The titled compound was prepared in 85% yield from 3-trifluoromethylphenyl isocyanate using the procedure detailed for compound 7. mp 184.8–185.6 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.90 (s, 1H), 7.79 (d, J = 9 Hz, 2H), 7.63-7.53 (m, 4H), 7.46-7.36 (m, 7H), 7.28-7.22 (m, 2H), 6.57 (s, 1H), 6.41 (t, J = 6 Hz, 1H), 3.27-3.15 (m, 2H), 2.69 (t, J = 8 Hz, 2H), 1.93-1.78 (m, 2H). MS (ESI) m/z: 544.16 (M + H+). Anal. (C26H24F3N5O3S) C, H, N.
4-(5-Phenyl-3-{3-[3-(4-trifluoromethoxy-phenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide (21j)
The titled compound was prepared in 84% yield from 3-trifluoromethylphenyl isocyanate using the procedure detailed for compound 7. mp 205.7–206.6 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.67 (s, 1H), 7.79 (d, J = 9 Hz, 2H), 7.49 (d, J = 9 Hz, 2H), 7.45-7.36 (m, 7H), 7.29-7.18 (m, 4H), 6.57 (s, 1H), 6.30 (t, J = 5 Hz, 1H), 3.27-3.15 (m, 2H), 2.68 (t, J = 8 Hz, 2H), 1.92-1.79 (m, 2H). MS (ESI) m/z: 560.16 (M + H+). Anal. (C26H24F3N5O4S) C, H, N.
Molecular Modeling
Molecular modeling was performed using “Scigress Explorer Standard ver. 7.7.0.49” software (Fujitsu Computer Systems Corporation). The atomic coordinates of the crystal structures of human sEH complex with CIU (N-cyclohexyl-N′-(4-iodophenyl)urea) and of murine COX-2 complexed with 2438 were retrieved from Protein Data Bank (entry 1VJ5 and 1CX2, respectively). 21i was docked into the ligand-binding pocket manually by superposition with the parent molecule (CIU or 2438) and minimized on MM geometry (MM3). The image was produced using freewares VMD 1.8.6 (www.ks.uiuc.edu/Research/vmd) and POV-Ray 3.6 (www.povray.org).
sEH IC50 assay
For the recombinant affinity purified sEHs (human, mouse and rat), we used a fluorescent-based assay to determine IC50s.5 Enzymes (~1 nM human sEH) were incubated with inhibitors for 5 min in 25 mM Bis-Tris/HCl buffer (200 µL; pH 7.0) at 30°C before substrate (cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyl-oxyran-2-yl)methyl carbonate, CMNPC) was added ([S]final = 5 µM). Activity was assessed by measuring the appearance of the fluorescent 6-methoxynaphthaldehyde product (λem.= 330 nm, λex = 465 nm) at 30 °C during a 10 min incubation (Spectramax M2; Molecular Device, Inc., Sunnyvale, CA). The IC50s were calculated from at least three separate runs, each in triplicate, to obtain the standard deviation given in the results section. The IC50 was determined from at least 4 points in the linear region of the inhibition curve with at least one point above and one below the IC50.
Cyclooxygenase Inhibition Assay
The ability of the test compounds 7, 11a–h, 15, and 21a–j to inhibit ovine COX-1 and human recombinant COX-2 (% inhibition at 100 µM and IC50 values (µM), respectively) was determined using an COX Fluorescent Inhibitor Screening Assay Kit (catalog number 700100, Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s instructions. Stock solutions of test compounds were dissolved in a minimum volume of DMSO. Briefly, to a series of supplied reaction buffer solutions (150 µl, 100 mM Tris-HCl, pH 8.0) with either COX-1 or COX-2 (10 µl) enzyme in the presence of Heme (10 µl) and fluorometric substrate (10 µl) were added 10 µl of various concentrations of the test compound solutions ([I]final between 0.01 and 100 µM). The reactions were initiated by quickly adding 10 µl of arachidonic acid solution and then incubated for two minutes at room temperature. Fluorescence of resorufin that is produced by the reaction between PGG2 and the fluorometric substrate, ADHP (10-acetyl-3,7-dihydroxyphenoxazine) were analyzed with an excitation wavelength of 535 nm and an emission wavelength of 590 nm. The intensity of this fluorescence is proportional to the amount of resorufin, which is proportional to the amount of PGG2 present in the well during the incubation. Percent inhibition was calculated by comparison from the 100% initial activity sample value (no inhibitor). The concentration of the test compound causing 50% inhibition of COX-2 (IC50, µM) was calculated from the concentration-inhibition response curve (triplicate determinations).
Pharmacokinetic protocol in Mice
All the animal experiments were performed according to protocols approved by the Animal Use and Care Committee of University of California, Davis. The PK study of compounds 21b,e–j in a murine model was conducted according to the protocol previously reported by Liu et al.42 Male CFW mice (8 week old, 24–30 g) were purchased from Charles River Laboratories. Compounds 21b,e–j (1 mg each) were dissolved in 1 mL of oleic ester-rich triglyceride containing 20% polyethylene glycol (average molecular weight: 400) to give a clear solution for oral administration. Cassette 1 contained compounds 21e, 21b, 21g, and rofecoxib, cassette 2, compounds 21i, 21j, 21f and celecoxib, cassette 3 compounds 21h, and t-AUCB. Each cassette was orally administered to 4 different mice at a dose of 5 mg/kg in 120–150µl of vehicle depending on animal weight. Blood (10 µL) was collected from the tail vein using a pipette tip rinsed with 7.5% EDTA(K3) at 0, 0.5, 1, 1.5, 2, 4, 6, 8, 24 hours after oral administration. The extraction of compounds from blood was performed by following our previous method except that ethyl acetate (200 µL) was added after the addition of internal solution I (10 µL, 500 nM solution of 1-(4-chloro-3-trifluoromethanylphenyl-3-(1-cyclopropanecarboxylpiperidin-4-yl)urea in methanol) to each thawed blood sample instead of prior to the addition of internal solution I. Blood samples were analyzed using an Agilent 1200 Series HPLC equipped with a 4.6 mm X 150 mm Inertsil ODS-4 3 µm column (GL Science Inc., Japan) held at 40 °C and coupled with an Applied Biosystems 4000 QTRAP hybrid, triple-quadrupole mass spectrometer. The instrument was equipped with a linear ion trap and a Turbo V ion source and was operated in negative ion MRM mode. The solvent system consisted of water/acetic acid (999/1 v/v, solvent A) and acetonitrile/acetic acid (999/1 v/v; solvent B). The gradient was begun at 30% solvent B and was linearly increased to 100% solvent B in 5 min. This was maintained for 3 min, then returned to 30% solvent B in 2 min. The flow rate was 0.4 mL/min. The injection volume was 10 µL and the samples were kept at 4 °C in the auto sampler. The PK parameters of individual mice were calculated by fitting the time dependent curve of blood inhibitor concentration to a non-compartmental analysis with the WinNonlin software (Pharsight, Mountain View, CA). Parameters determined include the time of maximum concentration (Tmax), maximum concentration (Cmax), half-life (t1/2), and area under the concentration–time curve to terminal time (AUCt).
Pharmacokinetic protocol in rats
Four male Sprague-Dawley rats (8 week old, 250–300 g) were used for pharmacokinetic study for dual inhibitors. Compounds 21i and 21j were given by oral administration at the dose of 1mg/kg. Inhibitors were dissolved in oleic ester rich triglyceride containing 10% PEG 400 to form a clear solution. Blood (10 µL) was collected from the tail vein by using a pipette tip rinsed with 7.5% EDTA(K3) at 0, 0.5, 1, 1.5, 2, 4, 6, 8, and 24 hour after oral dosing with the inhibitor. Each blood sample was immediately transferred to a tube containing 50 µL of water and mixed by Vortex for 1 min, all samples were stored at −80°C until analysis. Blood sample preparation and drug level quantification by LC/MS/MS were the same as previous study.42 The pharmacokinetic parameters of individual rat were calculated by fitting the data from blood concentration - time dependent curve to a non-compartmental analysis with the WinNonlin software (Pharsight, Mountain View, CA). Cmax, Tmax, t 1/2, and AUC were analyzed to characterize the pharmacokinetic profile of Compounds 21i and 21j.
Von Frey mechanical nociceptive assay
Male Sprague-Dawley rats weighing approximately 250–300 grams were obtained from Charles River Laboratories. On the day of the test rats were brought to the testing apparatus and allowed to acclimate. After 30 minutes the rats were tested with a von Frey aesthesiometer for their non-treated baseline withdrawal latency to mechanical stimulation. The von Frey apparatus is a raised metal mesh platform that has clear acrylic chambers to enclose rats but allow them to move freely. A rigid tip von Frey probe is used to probe the plantar surface of the rat hind paw though the mesh floor to elicit a withdrawal response. The measurements recorded are grams of force applied to the hind paw required to elicit a withdrawal. The pretreatment baseline score was considered the pain-free baseline (BL) and is assigned 100% for further response calculations. For the pain assay one of three compounds sEHI, celecoxib, compound 21i or a combination of sEHI + celecoxib formulated in 100% PEG400 was injected subcutaneously 60 minutes prior to the intraplantar LPS injection. After the 10ug LPS injection, rats were tested at 15, 30, 60, 120, 180, 240, 300, and 360 minutes with the von Frey aesthesiometer for their withdrawal latency. Reported scores are an average of 6 rats (3 trials per rat) with SEM for the group. The measures were then calculated as a percent of the pretreatment baseline score.
Supplementary Material
Acknowledgment
This work was supported in part by NIEHS grant ES02710, NIEHS Superfund grant P42 ES04699, and NIHLB grant HL059699. Karen M. Wagner was supported by Award Number T32ES007059 from the National Institute of Environmental Health Sciences. Aaron T. Wecksler was support by Award Number T32CA108459 from the National Institutes of Health. Bruce D. Hammock is a George and Judy Senior Fellow of the American Asthma Society. We gratefully acknowledge T.R. Holman for generously providing the recombinant Human 5-LOX enzyme.
Abbreviations
- t-AUCB
trans-4-[4-(3-Adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid
- sEH
soluble epoxide hydrolase
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- 5-LOX
5-lipoxygenase
- LPS
lipopolysaccharide
- AUC
area under the concentration-time curve
- AA
arachidonic acid
- PGs
prostaglandins
- EETs
epoxyeicosatrienoic acids
- DHETs
dihydroxyeicosatrienoic acids
- 20-HETE
20-hydroxyeicosatetranoic acid
- DML
designed multiple ligand
Footnotes
Supporting Information Available: Elemental analysis data and HRMS data, Synthesis of compounds 11h and 21c, Human 5-LOX activity data, PK analysis in mice and rats and data for LPS pain model. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Green GA. Understanding NSAIDs: From aspirin to COX-2. Clinical Cornerstone. 2001;3:50–59. doi: 10.1016/s1098-3597(01)90069-9. [DOI] [PubMed] [Google Scholar]
- 2.Poff CD, Balazy M. Drugs that target lipoxygenases and leukotrienes as emerging therapies for asthma and cancer. Curr. Drug Targets Inflamm. Allergy. 2004;3:19–33. doi: 10.2174/1568010043483917. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hyde CAC, Missailidis S. Inhibition of arachidonic acid metabolism and its implication on cell proliferation and tumor-angiogenesis. Int. Immunopharmaclology. 2009;9:701–715. doi: 10.1016/j.intimp.2009.02.003. [DOI] [PubMed] [Google Scholar]; (b) De Gaetano G, Donati MB, Cerletti C. Prevention of thrombosis and vascular inflammation: benefits and limitations of selective or combined COX-1, COX-2 and 5-LOX inhibitors. Trends Pharmacol. Sci. 2003;24:245–252. doi: 10.1016/S0165-6147(03)00077-4. [DOI] [PubMed] [Google Scholar]; (c) Koontongkaew S, Monthanapisut P, Saensuk T. Inhibition of arachidonic acid metabolism decreases tumor cell invasion and matrix metalloproteinase expression. Prostag. Oth. Lipid M. 2010;93:100–108. doi: 10.1016/j.prostaglandins.2010.07.002. [DOI] [PubMed] [Google Scholar]; (d) Hyde CAC, Missailidis S. Inhibition of arachidonic acid metabolism and its implication on cell proliferation and tumor-angiogenesis. Int. Immunopharmacol. 2009;9:701–715. doi: 10.1016/j.intimp.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 4.(a) Julémont F, Dogné JM, Pirotte B, de Leval X. Recent development in the field of dual COX/5-LOX inhibitors. Mini Rev. Med. Chem. 2004;4:633–638. doi: 10.2174/1389557043403747. [DOI] [PubMed] [Google Scholar]; (b) Charlier C, Michaux C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur. J. Med. Chem. 2003;38:645–659. doi: 10.1016/s0223-5234(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 5. Laufer SA, Augustin J, Dannhardt G, Kiefer W. J. Med. Chem. 1994;37:1894–1897. doi: 10.1021/jm00038a021.. Licofelone (ML3000) has been develop as a dual inhibitor of COX and LOX pathways. However Koeberle, A. et al. reported it is a dual inhibitor of 5-LOX activating protein and microsomal PGE2 syntase-1 in the following article; Koeberle A, Siemoneit U, Bühring U, Northoff H, Laufer S, Albrecht W, Werz O. Licofelone Suppresses Prostaglandin E2 Formation by Interference with the Inducible Microsomal Prostaglandin E2Synthase-1. J. Pharmacol. Exp. Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444..
- 6.(a) Janusz JM, Young PA, Ridgeway JM, Scherz MW, Enzweiler K, Wu L. New Cyclooxygenase-2/5-Lipoxygenase Inhibitors. Part 1. 7-tert-Butyl-2,3-dihydro-3,3-dimethylbenzofuran Derivatives as Gastrointestinal Safe Antiinflammatory and Analgesic Agents: Discovery and Variation of the 5-Keto Substituent. J. Med. Chem. 1998;41:1112–1123. doi: 10.1021/jm970679q. [DOI] [PubMed] [Google Scholar]; (b) Kirchner T, Argentieri DC, Barbone AG, Singer M, Steber M. Evaluation of the Antiinflammatory Activity of a Dual Cyclooxygenase-2 Selective/5-Lipoxygenase Inhibitor, RWJ 63556, in a Canine Model of Inflammation. J. Pharmacol. Exp. Ther. 1997;282:1094–1101. [PubMed] [Google Scholar]; (c) Barbey S, Goossens L, Taverne T, Cornet J, Choesmel V, Rouaud C, Gimeno G, Yannic-Arnoult S, Michaux C, Charlier C, Houssin R, Hénichart JP. Synthesis and Activity of a New Methoxytetrahydropyran Derivative as Dual Cyclooxygenase-2/5-Lipoxygenase Inhibitor. Bioorg. Med. Chem. Lett. 2002;12:779–782. doi: 10.1016/s0960-894x(02)00013-6. [DOI] [PubMed] [Google Scholar]
- 7.Praveen Rao PN, Chen Q-H, Knaus EE. Synthesis and Structure-Activity Relationship Studies of 1,3-Diarylprop-2-yn-1-ones: Dual Inhibitors of Cyclooxygenases and Lipoxygenases. J. Med. Chem. 2006;49:1668–1683. doi: 10.1021/jm0510474. [DOI] [PubMed] [Google Scholar]
- 8.Pommery N, Taverne T, Telliex A, Goossens L, Charlier C, Pommery J, Goossens J�F, Houssin R, Durant F, Hénichart J-P. New COX-2/5-LOX Inhibitors: Apoptosis-Inducing Agents Potentially Useful in Prostate Cancer Chemotherapy. J. Med. Chem. 2004;47:6195–6206. doi: 10.1021/jm0407761. [DOI] [PubMed] [Google Scholar]
- 9.Liu J�Y, Li N, Yang J, Li N, Qiu H, Ai D, Chiamvimonvat N, Zhu Y, Hammock BD. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc. Natl. Acad. Sci. USA. 2010;107:17017–17022. doi: 10.1073/pnas.1011278107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation: molecular and functional properties of the arachidonate monooxygenase. J. Lipid Res. 2000;41:163–181. [PubMed] [Google Scholar]
- 11.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 2007;292:C996–C1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 12.(a) Chaudhary KR, Pharm B, Abukhashim M, Hwang SH, Hammock BD. Inhibition of Soluble Epoxide Hydrolase by trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid Is Protective Against Ischemia-Reperfusion Injury. J. Cardiovasc. Pharmacol. 2010;55:67–73. doi: 10.1097/FJC.0b013e3181c37d69. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li N, Liu J�Y, Timofeyev V, Qiu H, Hwang SH, Tuteja D, Lu L, Yang J, Mochida H, Low R, Hammock BD, Chiamvimonvat N. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J. Mol. Cell. Cardiol. 2009;47:835–845. doi: 10.1016/j.yjmcc.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory Properties of Cytochrome P450 Epoxygenase-Derived Eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol. Sci. 2000;21:125–127. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- 14.Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18901–18906. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxy-eicosatrienoic acids (EETs): metabolism and biochemical function. Prog. Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 16.(a) Dannhard G, Kiefer W. Cyclooxygenase inhibitors-current status and future prospects. Eur. J. Med. Chem. 2001;36:109–126. doi: 10.1016/s0223-5234(01)01197-7. [DOI] [PubMed] [Google Scholar]; (b) Carter JS. Inhibitors of cyclooxygenase-2: November 1999 – April 2000. Exp. Opin. Ther. Pat. 2000;10:1011–1020. [Google Scholar]; (c) Talley JJ. Selective Inhibitors of Cyclooxygenase-2 (COX-2) Prog. Med. Chem. 1999;36:201–234. doi: 10.1016/s0079-6468(08)70048-1. [DOI] [PubMed] [Google Scholar]; (d) Rao PNP, Knaus EE. Evolution of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): Cyclooxygenase (COX) Inhibition and Beyond. J. Pharm. Pharmaceut. Sci. 2008;11:81s–110s. doi: 10.18433/j3t886. [DOI] [PubMed] [Google Scholar]; (e) Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J. Lipid Res. 2009;50:S29–S34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fries JF. NSAID Gastropathy: Epidemiology. J. Musculoskeletal Med. 1991;8:21–28. [Google Scholar]
- 18.For a review see: Reitz DB, Seibert K. Selective Cyclooxygenase Inhibitors. Annu. Rep. Med. Chem. 1995;30:179–188..
- 19.Meyer-Kirchrath J, Schror K. Cyclooxygenase-2 Inhibition and Side-effects of Nonsteroidal Antiinflammatory Drugs in Gastrointestinal Tract. Curr. Med. Chem. 2000;7:1121–1129. doi: 10.2174/0929867003374219. [DOI] [PubMed] [Google Scholar]
- 20.FitzGerald GA. Coxibs and Cardiovascular Disease. N. Engl. J. Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 21.Williams JM, Murphy S, Burke M, Roman RJ. 20-hydroxyeicosatetraeonic acid: a new target for the treatment of hypertension. J. Cardiovasc. Pharmacol. 2010;56:336–344. doi: 10.1097/FJC.0b013e3181f04b1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmelzer KR, Inceoglu B, Kubala L, Kim I-H, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal antiinflammatory drugs and soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. USA. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frantz S. The trouble with making combination drugs. Nature Reveiws. 2006;5:881–882. doi: 10.1038/nrd2188. [DOI] [PubMed] [Google Scholar]
- 24.Morphy R, Kay C, Rankovic Z. From magic bullets to designed multiple ligands. Drug Discovery Today. 2004;9:641–651. doi: 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]; (b) Morphy JR, Rankovic Z. Designed multiple ligands, an emerging drug discovery paradigm. J. Med. Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]; (c) Morphy R, Rankovic Z. The physicochemical challenges of designing multiple ligands. J. Med. Chem. 2006;49:4961–4970. doi: 10.1021/jm0603015. [DOI] [PubMed] [Google Scholar]
- 25.(a) Morphy R, Rankovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]; (b) Sreedhar D, Subramanian G, Udupa N. Combination drugs: Are they rational? Curr. Sci. India. 2006;91:406. [Google Scholar]
- 26.Marino JP, Brooks CA, Eidam P, Mcatee JJ. sEH and 11β-HSD1 Dual Inhibitors. PCT/US2009/051678, July 24, 2009. [Google Scholar]
- 27.Copeland rA, William JM, Glannaras J, Nurnberg S, Covington M, Pinto d, Pick S, Trzaskos JM. Proc. Natl. Acad. Sci. USA. 1994;91:11202–11206. doi: 10.1073/pnas.91.23.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marino JP. Soluble Epoxide Hydrolase, a Target with Multiple Opportunities for Cardiovascular Drug Discovery. Curr. Top. Med. Chem. 2009;9:452–463. doi: 10.2174/156802609788340805. [DOI] [PubMed] [Google Scholar]
- 29.Pommery N, Taverne T, Telliez A, Goossens L, Charlier C, Pommery J, Goossens J-F, Houssin R, Durant F, Hénichart J-P. New COX-2/5-LOX Inhibitors: Apoptosis-Inducing Agents Potentially Useful in Prostate Cancer Chemotherapy. J. Med. Chem. 2004;47:6195–6206. doi: 10.1021/jm0407761. [DOI] [PubMed] [Google Scholar]
- 30.Talley JJ, Penning TD, Collins PW, Rogier DJ, Jr, Malecha JW, Miyashiro JM, Bertenshaw SR, Khanna IK, Graneto MJ, Rogers RS, Carter JS, Docter SH, Yu SS. Preparation of pyrazolylbenzenesulfonamides as cyclooxygenase inhibitors for treatment of inflammation. U.S. (2002), 55 pp. Cont.-in-part of U.S. 6,413,960. CODEN: USXXAM US 6492411 B1 20021210. [Google Scholar]
- 31.Hwang SH, Tsai H-J, Liu J-Y, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase (sEH) inhibitors. J. Med. Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib) J. Med. Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe T, Schulz D, Morisseau C, Hammock BD. High-throughput pharmacokinetic method: Cassette dosing in mice associated with minuscule serial bleedings and LC/MS/MS analysis. Anal. Chim. Acta. 2006;559:37–44. doi: 10.1016/j.aca.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang SH, Morisseau C, Do Z, Hammock BD. Solid-phase combinatorial approach for the optimization of soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2006;16:5773–5777. doi: 10.1016/j.bmcl.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prasit P, Wang Z, Brideau C, Chan C-C, Charleson S, Cromlish W, Ethier D, Evans JF, Ford-Hutchinson AW, Gauthier JY, Gordon R, Guay J, Gresser M, Kargman S, Kennedy B, Leblanc Y, Leger S, Mancini J, O’Neill GP, Ouellet M, Percival MD, Perrier H, Riendeau D, Rodger I, Tagari P, Therien M, Vikers P, Wong E, Xu L-J, Young RN, Zamboni R, Boyce S, Rupniak N, Forrest M, Visco D, Patrick D. The discovery of rofecoxib, [MK 966, Vioxx, 4-(4′-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone], an orally active cyclooxygenase-2 inhibitor. Bioorg. Med. Chem. Lett. 1999;9:1773–1778. doi: 10.1016/s0960-894x(99)00288-7. [DOI] [PubMed] [Google Scholar]
- 36.Maier TJ, Tausch L, Hoernig M, Coste O, Schmidt R, Angioni C, Metzner J, Groesch S, Pergola C, Steinhilber D, Werz O, Geisslinger G. Celecoxib inhibits 5-lipoxygenase. Biochem. Pharmacol. 2008;76:862–872. doi: 10.1016/j.bcp.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 37.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 2004;43:4716–4723. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 38.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 39.Kanaan SA, Saade NE, Haddad JJ, Abdelnoor AM, Atweh SF, Jabbur SJ, Safieh-Garabedian B. Endotoxin-induced local inflammation and hyperalgesia in rats and mice: a new model for inflammatory pain. Pain. 1996;66:373–379. doi: 10.1016/0304-3959(96)03068-0. [DOI] [PubMed] [Google Scholar]
- 40.Brecker L, Pogorevc H, Griengl H, Steiner W, Kappe T, Ribbons DW. Synthesis of 2,4-diketo acids and their aqueous solution structures. New J. Chem. 1999;23:437–446. [Google Scholar]
- 41.Talley JJ, Penning TD, Collins PW, Rogier DJ, Jr, Malecha JW, Miyashiro JM, Bertenshaw SR, Khanna IK, Graneto MJ, Rogers RS, Carter JS, Docter SH, Yu SS. Preparation of pyrazolylbenzenesulfonamides as cyclooxygenase inhibitors for treatment of inflammation. U.S. (2002), 55 pp. Cont.-in-part of U.S. 6,413,960. US 6492411 B1 20021210. [Google Scholar]
- 42.Liu JY, Tsai H-J, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Brit. J. Pharmacol. 2009;156:284–296. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.