

Background: Mannose-binding lectin (MBL) activates complement by recognizing sugar arrays on pathogenic surfaces.
Results: Solution structures for the MBL monomer, dimer, trimer, and tetramer were determined by synchrotron x-ray scattering, analytical ultracentrifugation, and constrained modeling.
Conclusion: Near-planar solution structures were determined for the MBL dimer, trimer, and tetramer.
Significance: The structures clarify a likely molecular mechanism for MBL activation.
Keywords: Analytical Ultracentrifugation, Complement, Computer Modeling, Lectin, X-ray Scattering
Abstract
The complement system is a fundamental component of innate immunity that orchestrates complex immunological and inflammatory processes. Complement comprises over 30 proteins that eliminate invading microorganisms while maintaining host cell integrity. Protein-carbohydrate interactions play critical roles in both the activation and regulation of complement. Mannose-binding lectin (MBL) activates the lectin pathway of complement via the recognition of sugar arrays on pathogenic surfaces. To determine the solution structure of MBL, synchrotron x-ray scattering and analytical ultracentrifugation experiments showed that the carbohydrate-recognition domains in the MBL dimer, trimer, and tetramer are positioned close to each other in near-planar fan-like structures. These data were subjected to constrained modeling fits. A bent structure for the MBL monomer was identified starting from two crystal structures for its carbohydrate-recognition domain and its triple helical region. The MBL monomer structure was used to identify 10–12 near-planar solution structures for each of the MBL dimers, trimers, and tetramers starting from 900 to 6,859 randomized structures for each. These near-planar fan-like solution structures joined at an N-terminal hub clarified how the carbohydrate-recognition domain of MBL binds to pathogenic surfaces. They also provided insight on how MBL presents a structural template for the binding and auto-activation of the MBL-associated serine proteases to initiate the lectin pathway of complement activation.
Introduction
Mannose-binding lectin (MBL4; also called mannan-binding lectin or mannose-binding protein) is the primary recognition molecule of the lectin pathway of complement activation. Through its multiple carbohydrate-recognition domains (CRDs), MBL is able to bind to sugar arrays on microorganisms to activate the complement system. MBL circulates in the complex with MBL-associated serine proteases (MASPs-1, -2, and -3). Upon recognizing and binding appropriate sugars on the target surface, MBL and/or MASP undergo a conformational change that leads to activation of the MASPs. MASP-2 acts similarly to C1s of the classical pathway whereby it cleaves C4 and C2, resulting in the formation of the C3 convertase of the lectin pathway, C4b2a (1). Microorganisms are ultimately cleared following opsonization and phagocytosis or are lysed following activation of the membrane-attack pathway. Circulating human MBL consists of large oligomers assembled from structural monomers, each of which is formed from three identical polypeptide chains. The three polypeptide chains each consist of an N-terminal cysteine-rich domain, followed by a collagenous region containing 19 Gly-Xaa-Yaa triplet repeats, an α-helical neck region, and a C-terminal calcium-dependent CRD (Fig. 1A) (2, 3). In the rat, MBL forms oligomers, ranging only from monomers to tetramers, where the larger oligomers activate complement more effectively (4). The human MBL oligomers include dimers and trimers and extend up to hexamers and octamers; the major forms of human MBL are also trimers and tetramers, whereas pentamers and hexamers occur in much lower amounts. Three naturally occurring mutations in the collagenous region of MBL lead to reduced complement activation through the lectin pathway as a result of low levels of oligomerization of the structural units (5). MBL deficiency is the most common human immunodeficiency identified to date, and it increases the susceptibility to and the severity of infections or inflammatory diseases, including HIV (6), hepatitis (7), and cystic fibrosis (8). MBL replacement therapy may be an important application for the future to reduce the risk of specific diseases in children or immunosuppressed patients. For this to be effective, a better understanding of the MBL structure and oligomerization is required.
FIGURE 1.

Monomer of MBL. A, schematic representation of the MBL polypeptide. Each polypeptide chain in rat MBL contains 221 residues and is composed of an N-terminal cysteine-rich region, a collagenous region with 19 Gly-Xaa-Yaa triplet repeats, an α-helical neck region, and a C-terminal carbohydrate recognition domain. B, MBL monomer is composed of three identical polypeptide chains. Potential sites of flexibility in the monomer include the swivel region, located between the collagenous and neck regions, the kink region where there is an interruption in the triplet repeats of the collagenous region, and the hub region between the N-terminal cysteine-rich region and the collagenous region.
The three-dimensional structures of the different forms of polymeric MBL are not well understood. Only the high resolution structures of the CRD and neck region of rat and human MBL have been determined by x-ray crystallography (9, 10). The size of MBL, together with its glycosylation, extended collagenous region, and anticipated flexibility, suggests that the crystal structure determination of intact MBL will be difficult to achieve. The overall structure of MBL oligomers has been observed by electron microscopy as either near-planar (termed “sertiform”) or three-dimensional bouquet-like structures, with rod-like collagen stems joined at a base and topped by globular CRDs in proximity to each other (11, 12). Three sites of potential flexibility have been identified within the MBL structure. Flexibility may arise in the swivel region between the collagenous domain and neck region of MBL (9), within the collagenous region at the site of an interruption in the Gly-Xaa-Yaa triplet repeat (13), and/or at the junction between the N-terminal cysteine-rich domain and collagenous domain (Fig. 1B) (14). Atomic force microscopy images suggest that the N-terminal cysteine-rich regions of the MBL oligomers form a small “hub,” and the structural monomers are splayed apart in near-planar structures from the start of the collagenous regions (14). An angle of around 40° between the monomers was shown to be preferred. These most recent images suggested that the monomers did not extend outward from the central hub of MBL in a symmetric fashion as proposed previously (15). However, the use of electron and atomic force microscopy may be affected by the experimental conditions, which may be harsh and may damage the sample or perturb the observed images. The use of solution structural determinations offers a powerful alternative approach.
The solution arrangement of the monomers in oligomeric MBL was determined by the combination of x-ray scattering and analytical ultracentrifugation data with their constrained modeling. This is a well established method for the overall solution structural determinations of large multidomain complement proteins and antibodies, where the structures are often verified by subsequent crystal structures. Examples include human complement factors H and I, complement receptor type 2, heparin and heparan sulfate, and the monomer, dimer, and secretory forms of human IgA (16, 17). Medium resolution molecular models are determined that are deposited in the Protein Data Bank. To yield novel insights into MBL function, we applied this approach here to determine the solution structure of monomeric rat MBL that was defined starting from known crystal structures for the CRD and collagen regions. Based on this monomer structure, we determined mostly sertiform-like near-planar structures for the dimeric, trimeric, and tetrameric forms of rat MBL. The resulting structures indicate that the CRD regions are positioned in proximity to each other and that oligomeric MBL in solution presents a multivalent surface to which the MASP proteins bind. These novel MBL structures are discussed in terms of clarifying the molecular mechanism for lectin pathway activation.
EXPERIMENTAL PROCEDURES
Purification and Composition of MBL Oligomers
Recombinant rat MBL-A was produced in a Chinese hamster ovary cell expression system and purified by affinity chromatography on mannose-Sepharose columns (4, 18). Oligomers were initially resolved by ion-exchange chromatography on a Mono Q HR 5/5 column (GE Healthcare) equilibrated in 50 mm Tris, pH 8.2, containing 10 mm EDTA at a flow rate of 1 ml/min at 25 °C. Oligomers were eluted from the column using a 0–500 mm NaCl gradient in the same buffer over 45 min. The absorbance was monitored at 280 nm, and 0.5-ml fractions were collected. MBL dimers, trimers, and tetramers were eluted as overlapping peaks from the column and were identified based on their migration pattern on SDS-polyacrylamide gels, under nonreducing conditions. The larger oligomers (dimers, trimers, and tetramers) were further purified by gel filtration on a Superdex 200 column (16–60; GE Healthcare) equilibrated in phosphate-buffered saline, pH 7.2 (without Ca2+ or magnesium; Invitrogen), at a flow rate of 1 ml/min. SDS-PAGE was used to ensure the MBL oligomers were suitably pure for structural experiments (4, 19), and the samples were stored at 4 °C. The MBL amino acid composition was computed from its sequence (Swiss-Prot accession code P19999). The glycosylation of MBL was represented by four O-linked glucosylgalactosyl disaccharides per MBL polypeptide chain. The molecular mass of the MBL monomer was calculated to be 74.3 kDa (24.8 kDa per polypeptide chain), including these known post-translational modifications (4). The unhydrated volume of the MBL monomer was determined to be 94.9 nm3. Its hydrated volume was calculated to be 125.2 nm3, based on a hydration of 0.3 g of H2O/g of glycoprotein and an electrostricted volume of 0.0245 nm3 per bound water molecule (20). A partial specific volume v̄ of 0.723 ml/g was obtained. The absorbance coefficient at 280 nm (1%, 1 cm path length) was calculated to be 6.16. X-ray scattering and analytical ultracentrifugation experiments were performed in 50 mm NaH2PO4, 150 mm NaCl, pH 7.3. The buffer density at 20 °C was measured using an Anton-Paar DMA5000 density meter to be 1.011062 g/ml.
X-ray Scattering Data Collection and Analyses
X-ray scattering data were obtained on the Beamline ID02 at the European Synchrotron Radiation Facility (Grenoble, France), operating with a ring energy of 6.0 GeV (21). Data were collected for MBL oligomers in four-bunch mode using storage ring currents of 22–41 mA. The sample-to-detector distance was set to 2.0 m, which yielded a Q range from 0.04 to 2.4 nm−1 (where Q = 4 π sin θ/λ; 2θ = scattering angle; λ = wavelength). Concentration series for MBL oligomers were prepared in 50 mm NaH2PO4, 150 mm NaCl, pH 7.3, with concentrations between 0.31 and 0.46 mg/ml for MBL monomers, 0.29 and 1.15 mg/ml for MBL dimers, 0.23 and 0.92 mg/ml for MBL trimers, and 0.43 and 0.70 mg/ml for MBL tetramers. 100 μl of sample was loaded into a capillary flow cell, with a diameter of 1.7 mm. The sample was moved continuously during beam exposure to minimize radiation damage. Experimental data were collected at 20 °C. Four sets of 10 time frames, each of duration of 0.25 or 0.5 s, were acquired for each sample. Buffers were measured in alternation with the sample to minimize background subtraction errors. Following on-line checks to confirm the absence of radiation damage at low Q, the 10 frames were averaged for data analysis. Other details including the data reduction procedure have been described elsewhere (22).
In a given solute-solvent contrast, the radius of gyration Rg is a measure of structural elongation if the internal inhomogeneity of scattering densities within the protein has no effect. Guinier analysis at low Q gives the Rg, and the forward scattering at zero angle I(0) (23) as shown in Equation 1,
This expression is generally valid in a Q·Rg range up to 1.5. If the structure is elongated, the mean cross-sectional radius of gyration Rxs and the cross-sectional intensity at zero angle (I(Q)Q)Q→0 is obtained from the curve analyzed in a Q range that is larger than and not overlapping with the Q used for the Rg determination as shown in Equation 2,
The Rg and Rxs analyses lead to the triaxial dimensions of the macromolecule if the structure can be represented by an elliptical cylinder, where L = √12(Rg2 − Rxs2) and L is its length (23). The Rg and Rxs analyses were performed using an interactive PERL script program SCTPL75 on Silicon Graphics OCTANE workstations. Indirect transformation of scattering data I(Q) in reciprocal space into real space to give the distance distribution function P(r) was carried out using the GNOM program (24) as shown in Equation 3,
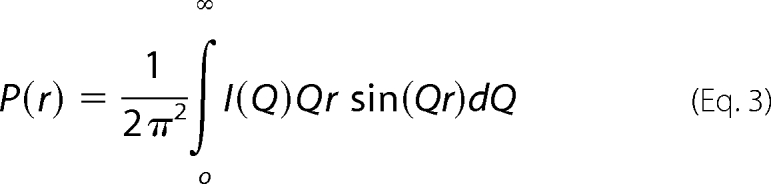 |
The P(r) curve corresponds to the distribution of distances r between volume elements. For the MBL monomer, the x-ray I(Q) curves utilized 167 points in the Q range between 0.1 and 1.2 nm−1. For the MBL dimer, 174 points were used in the Q range between 0.1 and 2.0 nm−1. For the MBL trimer, 213 points were used in the Q range between 0.0 and 1.5 nm−1. For the MBL tetramer, 143 points were used in the Q range between 0.1 and 1.1 nm−1.
Analytical Ultracentrifugation Data Collection and Analyses
Sedimentation velocity experiments were performed at 20 °C using two Beckman XL-I analytical ultracentrifuges (Beckman Coulter Inc., Palo, Alto, CA) at rotor speeds of 40,000 and 50,000 rpm. The instrument was equipped with an eight-hole AnTi50 rotor with standard double-sector cells with column heights of 12 mm. Sedimentation was monitored using absorbance optics at 280 nm and interference optics. The MBL dimer and trimer samples were prepared in 50 mm NaH2PO4, 150 mm NaCl, pH 7.3, at concentrations of 0.39 and 0.15 mg/ml, respectively. The sedimentation boundaries were analyzed using direct boundary Lamm fits of up to 300 scans using SEDFIT software (version 11.7) (25, 26). The continuous c(s) size distribution analysis algorithm assumes that all species have the same frictional ratio f/f0 in each fit. The final SEDFIT analyses used a fixed resolution of 200 and optimized the c(s) fit by floating the meniscus, bottom of the cell, the base line, and the frictional ratio f/f0 (initially 1.2) until the overall root mean square deviation and visual appearance of the fits were deemed satisfactory. The percentage of each MBL oligomer in the total loading concentration was derived using the c(s) integration function.
Generation of the MBL Monomer Model
The starting model for the rat MBL monomer was created using the unmodified crystal structure of the rat CRD/neck region (PDB code 1RTM) (10) and the crystal structure of a synthetic collagen triple helix peptide ((Pro-Hyp-Gly)4-Pro-Gly-(Pro-Hyp-Gly)5)3 (PDB code 1EI8) (27). The collagen triple helix was manipulated using INSIGHT II 98.0 molecular graphics software (Accelrys, San Diego) on Silicon Graphics OCTANE workstations. Collagen fragments were created that were superimposed upon one another to obtain a structure with the sequence (Gly-Xaa-Yaa)5-Gly-Xaa-(Gly-Xaa-Yaa)12 to correspond to that of rat MBL collagen. The residues of this synthetic collagen triple helix were then substituted with those of rat MBL using the Biopolymer module in INSIGHT II. To join the CRD and collagen structures, 5,000 conformationally randomized linkers 88SRAI91 were created for superimposition onto the C-terminal Ser residue on the collagen domain and the N-terminal Ile residue on the CRD region. This linker peptide was initially created in an extended β-strand conformation. The randomized conformations were generated from this starting β-strand model by molecular dynamics simulation using DISCOVER3 at a temperature of 773 K (28, 29). First, the peptide structure was subjected to energy minimization for 300 iterations. After a temperature equilibrium step of 5,000 fs, the simulation was run for 500,000 fs, saving this every 100 fs to produce the 5,000 models. A linear model of monomeric MBL was created by defining one vector along the long axis of the collagen triple helix region (Cα atoms of Gly-36 and Gly-86 on chains B and C) and another on that of the CRD region (Cα atoms of Val-93 and Thr-131 on chains B and C) and then measuring the angle between the two vectors. Models with vector angles close to 180° were visually inspected to identify the most satisfactory linear structure. The cysteine-rich N-terminal tails were constructed in extended β-strand conformations using Biopolymer and were superimposed onto the MBL collagen N-terminal peptides. Four glycosylgalactosyl disaccharides were added to residues Lys-44, Lys-47, Lys-79, and Lys-82 in each of the three collagen polypeptide chains to create the starting model for the rat MBL monomer.
Constrained Modeling of Monomeric MBL
The starting model for the MBL monomer was optimized using four fit searches to assess the interdomain linker structures. The first search fit optimized the linker between the collagen triple helix and the CRD of chain A of MBL. To enable the search of wider conformational orientations, a longer six-residue linker peptide 87DSRAIE92 was initially created in an extended β-strand conformation and then 5,000 random conformations were generated following the above procedure. Each of the 5,000 MBL models for the MBL monomer was created by superimposition of the N- and C-terminal residues of each linker conformation onto the C- and N-terminal residues of the collagen and CRD regions, respectively. After this, the duplicated superimposed residues were deleted from the MBL monomer model prior to carrying out the scattering calculations. The second, third, and fourth search fits for the MBL monomer optimized the connection between the collagen triple helix and each of the three cysteine-rich N-terminal peptides using the best-fit MBL model from the first cycle of fits. A linker library containing 5,000 different conformations of the arbitrarily selected sequence 35CGRDGRD41 was generated as before. These linkers were used to connect the extended N-terminal peptides to the collagen triple helix in chains A, B, and C. In each search fit, the best-fit model for one N-terminal peptide was identified and retained for the next search fit until all three N-terminal peptides had been positioned.
To calculate the modeled x-ray scattering curves for comparison with the experimental curves, each MBL monomer model was converted into Debye spheres. A cube side length of 0.538 nm with a cutoff of four atoms consistently gave models within 3% of their total unhydrated volume of 94.9 nm3 (see above). The hydration shell surrounding glycoproteins is detected by x-ray scattering, and thus the sphere models were accordingly adapted by adding spheres to the surface of the unhydrated models using HYPRO based on a hydrated volume of 125.2 nm3 (see above) (30, 31). The optimal total of hydrated spheres for the MBL monomer was 828. The scattering curve I(Q) was calculated using the Debye equation adapted to spheres. No instrumental corrections were applied because these are negligible for the pinhole optics of Beamline ID02. Potential steric overlap between the CRD and collagen regions was assessed using the number of spheres N in the models following grid transformation, where models showing less than ∼97% of the required total of 828 spheres were discarded. In all four fit searches, less than half the models met this “absence of steric overlap” criterion. The models were then assessed by calculation of their Rg and Rxs−1 values in the same Q ranges as were used for the experimental Guinier fits. Models that passed the N, Rg, and Rxs−1 filters were then ranked using a goodness-of-fit (R-factor) defined by analogy with protein crystallography using the experimental curves in a Q range extending up to 1.2 nm−1. Sedimentation coefficients s20,w for the best-fit scattering models were calculated directly from the atomic coordinates in the HYDROPRO shell modeling program using the default value of 0.31 nm for the atomic element radius for all atoms to represent the hydration shell (32).
Constrained Modeling of Dimeric MBL
The best-fit MBL monomer model was used to generate MBL dimer models using a procedure employed for dimeric IgA1 (16). Initially, a planar starting model for the dimer was formed by arranging two monomers end-to-end in which the cysteine-rich peptides were adjacent to each other. One MBL monomer was then rotated about a dummy atom created between the cysteine-rich peptides of the two monomers, whereas the other monomer was held stationary. All orientations were tested using rotations of 10° between 0 and 180° on each of the x, y, and z axes, in which the two starting monomers shared a common y axis, resulting in 193 or 6,859 models. The first search did not employ a dummy atom, and the two monomers were directly superimposed upon one another. In the subsequent searches 2–4, the monomer separations were set at 3.3, 4.8, and 6.6 nm, respectively, between the Cα atoms of Glu-46 at the start of the two collagen triple helix regions. The scattering curves from each search were calculated and compared with the experimental data. The unhydrated volume of dimeric MBL was 189.8 nm3, whereas the hydrated volume was 250.5 nm3. Using the same cube side length and atom cut-off for the monomer, the dimer coordinates were converted into Debye sphere models, with 1,219 spheres for the unhydrated structures and 1,609 spheres for the hydrated structures. The models were filtered according to N, followed by comparisons of their Rg and Rxs−1 values, and the models that remained were again ranked according to their R-factors.
Constrained Modeling of Trimeric and Tetrameric MBL
The MBL trimer and tetramer modeling was performed using the best-fit MBL monomer model. Search 1 for the MBL trimer utilized the best-fit MBL dimer model as a starting point, to which a third monomer was added and rotated about a dummy atom to yield 6,859 trial models as above. Other trimer and tetramer models were created by superimposing three or four best-fit monomer models, respectively. By holding monomer 1 fixed at 0, 0, and 0, about the x, y, and z axes, monomers 2 and 3 for the trimer or monomers 2–4 for the tetramer were then rotated about the three axes. To create MBL trimers in search 2, monomer 2 was rotated between 10 and 120° in 10° steps, and monomer 3 was rotated between 20 and 240° in 20° steps. In search 1 for the MBL tetramer, monomer 2 was likewise rotated between 10 and 90°, monomer 3 was rotated between 20° and 180°, and monomer 4 was rotated between 30° and 270°. This procedure resulted in 2,028 trial models for the MBL trimer and 900 trial models for the MBL tetramer. The x-ray scattering curves were calculated as above. The number of hydrated spheres N was calculated to be 2,413 and 3,217 for the MBL trimer and tetramer, respectively. The best-fit models were identified as above by ranking the models that passed the N, Rg, and Rxs−1 filters using the goodness-of-fit R-factor of the model.
The 10–12 best-fit monomer, dimer, trimer, and tetramer MBL models are currently available in the supplemental material named 3UJU (monomer), 3UJV (dimer), 3UJW (trimer), and 3UJX (tetramer).
RESULTS
Purification of Rat MBL Oligomers
Quantities of rat MBL sufficient to enable a detailed study of its structural organization were produced in Chinese hamster ovary cells and purified by affinity chromatography on mannose-Sepharose. The four MBL oligomers were first resolved by ion-exchange chromatography, and each oligomer was then purified by gel filtration. Previously, the collectins showed anomalous behavior on gel filtration because of their highly extended triple helical collagen-like structures (11, 33). Here, recombinant rat MBL, when analyzed by SDS-PAGE under nonreducing conditions, migrated as four major bands, one for each oligomer (data not shown). This is in agreement with previous SDS-PAGE images that reported predominant covalent monomer, dimer, trimer, and tetramer, alongside the low levels of noncovalent intermediate oligomers (4, 19). Under reducing conditions, MBL migrated as a single band with an apparent molecular mass of 29 kDa (data not shown). Thus, the major bands observed by nonreducing SDS-PAGE confirm the purifications of disulfide-linked MBL monomers, dimers, trimers, and tetramers as described previously (4, 19).
X-ray Scattering Data for Rat MBL Oligomers
X-ray scattering is a diffraction technique used to study the overall structure of biological molecules in random orientations in solution (34). Solution scattering was used to identify the structures of the MBL oligomers. The scattering data I(Q) were collected at concentrations between 0.23 and 0.92 mg/ml for the MBL oligomers with no radiation damage or x-ray-induced aggregation in the time frame analyses. The time-averaged runs were thus used for data analyses. Guinier analyses of the I(Q) data using three separate Q ranges gave the radius of gyration Rg and two cross-sectional radii of gyration Rxs.
The Rg value monitors the overall degree of elongation of the MBL oligomers. At the lowest Q values, Guinier analyses of I(Q) resulted in linear plots in Q ranges of 0.10–0.17 nm−1 for the monomer, 0.08–0.14 nm−1 for the dimer, 0.08–0.15 nm−1 for the trimer, and 0.06–0.10 nm−1 for the tetramer. The Rg values were measured within satisfactory Q·Rg limits (Fig. 2, A–D). The mean of two to four Rg values were 7.21 ± 0.22 nm for monomeric MBL, 10.42 ± 0.21 nm for dimeric MBL, 12.43 ± 0.11 nm for trimeric MBL, and 13.80 ± 0.11 nm for tetrameric MBL. As expected, the Rg values for the monomer, dimer, and trimer increased with the size of the oligomer. The Rg value of the tetramer is, however, comparable with that of the trimer, implying that both forms have related extended structures. As a control, the Guinier I(0)/c values (which are proportional to the molecular mass) increased in proportion to the size of each oligomer as expected. No change with protein concentrations in either the I(0)/c or Rg values for the four MBL oligomers was observed (data not shown). This indicated that neither self-association of MBL nor conformational change in MBL occurred as a result of increased protein concentration.
FIGURE 2.

X-ray Guinier Rg and Rxs analyses for four MBL oligomers. In the Guinier analyses, the open circles correspond to the data points, and the filled circles and lines correspond to the data points used to determine the Rg and Rxs values. The Q·Rg and Q·Rxs values for the fit ranges are denoted by arrows. The data are for concentrations of 0.31 and 0.46 mg/ml for monomeric MBL, 0.56 and 0.86 mg/ml for dimeric MBL, 0.46 and 0.69 mg/ml for trimeric MBL, and 0.43 and 0.70 mg/ml for tetrameric MBL. In all panels, the upper and lower plots correspond to the upper and lower concentrations respectively. For reason of clarity, the Guinier plots are displaced by 1.5 units on their vertical axes. A–D, Guinier plots of ln I(Q) versus Q2 to obtain the Rg values for MBL. The Q fit ranges were 0.10–0.17, 0.08–0.14, 0.08–0.15, and 0.06–0.10 nm−1 for the monomer (A), dimer (B), trimer (C), and tetramer (D), respectively. E–H, Guinier plots of ln I(Q)·Q versus Q2 to obtain the Rxs−1 values for MBL. The Q fit ranges were 0.41–0.65 nm−1 for the monomer (E), 0.42–0.58 nm−1 for the dimer (F) and trimer (G), and 0.41–0.56 nm−1 for the tetramer (H). I–L, Guinier plots to obtain the Rxs−2 values for MBL. The Q fit ranges were 0.65–0.92 nm−1 for the monomer (I), 0.65–0.85 nm−1 for the dimer (J) and trimer (K), and 0.68–0.85 nm−1 for the tetramer (L).
The x-ray cross-sectional Rxs−1 and Rxs−2 Guinier analyses monitored the shorter dimensions of the MBL oligomers. The ln I(Q)·Q versus Q2 plots showed two different linear regions (Fig. 2, E–L), in common with other extended proteins such as complement factor H (35). The first Rxs−1 region corresponded to a Q range of 0.41–0.65 nm−1 for the MBL monomer and a slightly lesser Q range of 0.42–0.58 nm−1 for the MBL dimer and trimer, and 0.42–0.56 nm−1 for the MBL tetramer (Fig. 2, E–H). The resulting Rxs−1 values for monomeric, dimeric, trimeric, and tetrameric MBL were 1.55 ± 0.05, 1.84 ± 0.08, 1.95 ± 0.06, and 2.20 ± 0.03 nm, respectively. The Rxs−1 values increased with the increasing size of the oligomer, showing that these monitor the spatial interactions between the monomeric subunits. The second Rxs−2 region corresponded to a larger Q range of 0.65–0.92 nm−1 for the monomer, 0.65–0.85 nm−1 for the dimer and trimer, and 0.68–0.85 nm−1 for the tetramer (Fig. 2, I–L). The Rxs−2 values were less variable than the Rxs−1 values, all being similar at 1.34 ± 0.09, 1.51 ± 0.03, 1.63 ± 0.04, and 1.67 ± 0.15 nm for monomeric, dimeric, trimeric, and tetrameric MBL, respectively. For only the monomer, the similarity of the Rxs−1 and Rxs−2 values is attributed to fitting two different regions of the same linear plot. Thus, the similar Rxs−2 values for all four MBL oligomers is attributed to the cross-sectional dimensions of the collagen triple helix, which is unaltered in the four MBL oligomers. This similarity shows that the triple helices are independent in MBL, i.e. they do not pack side-by-side in the dimer, trimer, and tetramer forms. In distinction, the Rxs−1 values for the dimer, trimer, and tetramer increase by up to 0.65 nm compared with the monomer. The increase is attributable to the existence of one, three, and six pairs of interactions between the monomers in the dimer, trimer, and tetramer, respectively. The existence of different Rxs−1 values shows that the monomers are positioned separately from each other but are in sufficient proximity to each other in the MBL oligomers to perturb the scattering curve.
The distance distribution function P(r) curve is calculated from the full Q range of the I(Q) curve and represents all the distances between pairs of atoms within the macromolecule. This provides structural information in real space. The P(r) curve provides an alternative calculation of the Guinier Rg value and leads to maximum lengths (L) following an assumption of the maximum dimension (Dmax). The Rg and I(0)/c values from the P(r) curve agreed with those obtained from the Guinier analyses (Table 1). The maximum length L of the MBL oligomers was determined from the value of r when P(r) = 0 (Fig. 3). For monomeric MBL, L1 was found to be 22 nm. Since L2 increased to only 31 nm for dimeric MBL, this showed that the dimer forms a compact V-shaped solution structure with the CRD heads in proximity at the two extremities, as opposed to an extended end-to-end arrangement of two monomers that would have a length of 44 nm. For trimeric and tetrameric MBL, L3 and L4 were increased to 38 and 47 nm, respectively. These larger oligomers have a maximum length that is almost double that of the monomer. Their conformation is best explained by the successive addition of monomers in the same V-shape to build up wider structures. A double peak with maxima M1 and M2 was observed for all MBL oligomers (Fig. 3). M1 was located at a similar r value 4.0 ± 0.7 nm for all four oligomers, where this r value is low compared with those for L1–L4. M1 therefore represents the most common distance vectors within each MBL monomer, principally within the CRD region. The intensity of the broader peak M2 increased with the size of the MBL oligomer. It was located at similar r values of 19.1, 18.3, 20.3, and 19.8 nm for the monomer, dimer, trimer, and tetramer, respectively. Given the globular structure of each CRD, the intensity increases at 18–20 nm represent the distance between the CRD heads in each oligomer. Thus, the growth of the broad peak M2 with oligomer size provides further evidence for an assembly of V-shaped monomers to form the dimer, trimer, and tetramer structures in MBL. If MBL assembly corresponded to highly extended MBL structures or random V-shaped pairings in solution, peak M2 would not show this intensity increase.
TABLE 1.
Experimental scattering and sedimentation parameters for the MBL oligomers
| Monomer | Dimer | Trimer | Tetramer | |
|---|---|---|---|---|
| Rg (nm)a | 7.21 ± 0.22 | 10.42 ± 0.21 | 12.43 ± 0.11 | 13.80 ± 0.1 |
| 7.29 ± 0.21 | 10.48 ± 0.26 | 12.61 ± 0.19 | 14.82 ± 0.35 | |
| Rxs−1 (nm) | 1.55 ± 0.05 | 1.84 ± 0.08 | 1.95 ± 0.06 | 2.20 ± 0.03 |
| Rxs−2 (nm) | 1.34 ± 0.09 | 1.51 ± 0.03 | 1.63 ± 0.04 | 1.67 ± 0.15 |
| L (nm) | 22 | 31 | 38 | 47 |
| M1 (nm) | 3.5 | 3.7 | 4.1 | 4.7 |
| M2 (nm) | 19.1 | 18.3 | 20.3 | 19.8 |
| s20,w (S) | 2.8 ± 0.5 | 4.2 ± 0.4 | 5.7 ± 0.2 | 7.4 ± 0.1 |
a The first Rg value is from the Guinier fits, and the second Rg value is from the P(r) analyses.
FIGURE 3.

X-ray distance distribution function P(r) analyses for the four MBL oligomers. The P(r) curves correspond to concentrations of 0.46, 1.15, 0.92, and 0.70 mg/ml for the monomer, dimer, trimer, and tetramer, respectively. The peak maxima M1 and M2 depict the two most common distances within the MBL structures. The intensities of the four P(r) curves were normalized to peak M1 to clarify the dependence of M2 and L on oligomer size. The maximum length of the MBL oligomer is denoted by L at the r value where the P(r) curve reaches 0. Their values for the monomer, dimer, trimer, and tetramer (L1–L4) were determined to be 22, 31, 38, and 47 nm, respectively.
Analytical Ultracentrifugation of Rat MBL Oligomers
Analytical ultracentrifugation is used to study macromolecular structures in solution by monitoring the time course of sedimentation under high centrifugal force (36). This provides information on the structure and oligomerization of MBL, including an assessment of sample polydispersity. Sedimentation velocity experiments at 50,000 rpm were performed on the MBL dimer (0.37 mg/ml) and MBL trimer (0.15 mg/ml) samples that were used for x-ray scattering experiments (Fig. 4). The velocity data were analyzed using interference optics to generate the size distribution analyses c(s) of the MBL dimer and MBL trimer. The c(s) analyses identified the sedimenting species present in the samples. The analysis of 300 boundaries revealed excellent fits and satisfactory root mean square deviations (Fig. 4, A and C). For the MBL dimer, the major peak (72%) in the c(s) distribution corresponded to the MBL dimer, together with three other small peaks with integrated intensities of 10, 11, and 7% for the monomer, trimer, and tetramer (Fig. 4B). The presence of these minor species was also detected in nonreducing SDS-PAGE (4, 19) and is attributable to the elongated shapes of the MBL oligomers, which decreases the ability of gel filtration methods to separate the oligomers. The sedimentation coefficient s20,w provides an independent measure of macromolecular elongation to that from the Rg value. The s20,w values of the four resolved peaks in the MBL dimer sample were 3.2, 4.6, 5.9, and 7.3 S (Fig. 4B). The c(s) analyses also lead to molecular mass determinations. The molecular masses of the four peaks were 88, 168, 238, and 292 kDa. These molecular masses agree well with the sequence-predicted values of 74, 149, 222, and 297 kDa for the monomer, dimer, trimer and tetramer respectively. In addition, the s20,w values for the four peaks were similar to those of 3.7, 5.5, 6.3, and 7.7 S obtained previously for the individual oligomers (4), where previously these peaks were not resolved for reason of older data analysis software. Size distribution analyses of the MBL trimer also showed four resolved peaks (Fig. 4D), and integration showed that the major peak again represented 71% of the total, with 15, 10, and 4% corresponding to the monomer, dimer, and tetramer, respectively. The s20,w values of the MBL monomer, dimer, trimer, and tetramer were similar at 2.3, 3.8, 5.5, and 7.5 S, respectively. Variability in the individual s20,w values was attributable to the low proportions of the minor species. In addition to providing the s20,w structural data, it was concluded from analytical ultracentrifugation that the x-ray scattering data for each MBL oligomer corresponded to a major species with small amounts of other forms. The control calculations reported below show that this low polydispersity did not affect the outcome of the MBL modeling.
FIGURE 4.

Sedimentation velocity analyses of dimeric and trimeric MBL. The c(s) size distribution analyses correspond to the sedimentation velocity experiments performed at a rotor speed of 50,000 rpm. The concentrations of dimeric and trimeric MBL were 0.37 and 0.15 mg/ml, respectively. A and C, black outlines represent the experimental interference data, and the continuous white lines represent the boundary fits. For the dimer (A) and trimer (C), every 10th boundary of up to 300 scans was fitted, and only every 20th boundary is shown for clarity. B and D, c(s) analyses for the MBL dimer (B) and MBL trimer (D). The peaks corresponding to the monomer (M), dimer (D), trimer (Tri), and tetramer (Tet) are labeled.
Modeling of the Rat MBL Monomer
The rat MBL sequence and domain assignments are shown in Fig. 5. A structure for the rat MBL monomer (Fig. 1) was constructed starting from crystal structures for the trimeric CRD and neck region of rat MBL (PDB code 1RTM) (10) and short collagen peptides (37). The triple helix conformation of collagen consists of three polypeptide chains, each in an extended left-handed polyproline II-like helix and stabilized by interchain hydrogen bonds between C=O and N–H groups (38, 39). Triple helix sequence constraints are strict, where glycine residues occupy every third residue (37). Replacement of a Gly-Xaa-Yaa tripeptide by a Gly-Xaa dipeptide creates an interruption site, and this occurs in MBL. The crystal structure of a collagen sequence ((Pro-Hyp-Gly)4-Pro-Gly-(Pro-Hyp-Gly)5)3 is available (PDB code 1EI8) to show that this has a linear structure (27). Truncations and superimpositions were used to create a linear collagen triple helix with the same number of tripeptides in rat MBL, including the Gly-Xaa-Gly interruption in the correct position. The residue sidechains of this triple helix were replaced with those in rat MBL to form the triple helix structure used for scattering modeling.
FIGURE 5.

Generation of the starting linear rat MBL monomer model. A, crystal structure of the CRD and neck regions of MBL was joined to the collagen region to create an initial MBL monomer. A total of 5,000 conformationally randomized models were created (see under “Experimental Procedures”). Vector 1 was defined by the Gly-36 pair and the Gly-86 pair on chains B and C of the collagen region, and vector 2 was defined by the Val-93 pair and the Thr-131 pair on chains B and C in the CRD and neck region. Models with an inter-vector angle close to 180° defined the starting MBL model. B, starting MBL model was completed by addition of the three N-terminal cysteine-rich peptides in β-strand conformations at the N-terminal residues of the collagen region and the addition of four glycosylgalactosyl disaccharides to residues Lys-44, Lys-47, Lys-79, and Lys-82 in each of the three collagen polypeptide chains (gray). At the bottom, the rat MBL sequence is aligned with the four regions of MBL. The N-terminal cysteine residues involved in oligomerization and the interruption in the 19 Gly-Xaa-Yaa triplets are highlighted in gray.
To model the four MBL oligomers, a linear starting MBL monomer model was created. The crystal structure of the CRD and neck region was first joined with the MBL triple helix model. Direct superimposition was not possible due to a sequence gap 89RA90 between the two models. To circumvent this, 5,000 conformationally randomized peptides 88SRAI91 were generated to bridge the gap. Superimposition of the first and last residues onto chain A in the two structures resulted in 5,000 randomized structures. Linear monomer models were identified from the angle between the vectors defining the long axis of the triple helix and the neck region (Fig. 5A). Models with angles close to 180° between these vectors were rapidly identified and were visually inspected to determine the most suitable model. The missing 89RA90 residues in chains B and C were subsequently attached. To complete the MBL model, extended N-terminal β-strand cysteine-rich polypeptides were superimposed onto chains A–C at the N terminus of the triple helix (Fig. 5B). Twelve glucosylgalactosyl disaccharides were added to the triple helix.
Constrained Modeling of the Rat MBL Monomer
The linear MBL monomer model showed divergence from the experimental scattering data. The experimental Rg value of 7.21 nm for the MBL monomer was less than the calculated Rg value of 7.69 nm for the linear model. It was concluded that the MBL monomer adopted a bent conformation in solution.
The MBL modeling searches are summarized in Table 2. For the monomer, a bend in the monomer was principally apparent at the swivel between the collagenous and neck regions (Fig. 1B). This follows the evidence from atomic force microscopy for large variations in the CRD region relative to the collagenous region and much less so at the collagen interruption (14). The collagen interruption resulted in a linear crystal structure for the collagen tripeptide (27), and this interruption is not expected to be flexible. Accordingly, conformationally randomized MBL monomer models were generated for trial-and-error searches based on variations at the swivel region. Given that there is usually some disorder at the end of triple helices, and it is not known how triple helices and α-helices are connected, a longer linker peptide 87DSRAIE92 was used to ensure the testing of a wider range of conformations at this location in the MBL model. Calculation of the scattering curves from the 5,000 models confirmed that a sufficiently wide range of conformations had been tested (supplemental Fig. S1A). Superimposition of the first 100 of the 5,000 models revealed visually that a wide range of CRD conformations had been generated (supplemental Fig. S2A). The best-fit models were identified by applying filters to reject models with too few Debye spheres or showed discrepant Rg values. This left 348 models, from which the best-fit 12 models were identified from their lowest R-factors (goodness of fit) (Table 3). All 12 best-fit models revealed that the CRD/neck region is bent relative to the collagen triple helix (supplemental Fig. S2B). The mean angle between the two regions is 133°. The best-fit model showed an Rg value of 7.14 nm and an Rxs value of 1.60 nm in good agreement with the experimental Guinier values of 7.21 ± 0.22 and 1.55 ± 0.05 nm, respectively. The R-factor was 4.0%. This agreement is better compared with the linear starting model of monomeric MBL (Rg value of 7.69 nm) that showed an R-factor of 5.9%. The visual comparison of the experimental and modeled I(Q) curves showed a very good fit from low Q out to a Q value of 1.2 nm−1 (Fig. 6A). The P(r) curve for the best-fit MBL monomer model (Fig. 6A) reproduced the experimental P(r) curve (Fig. 3). The length of the MBL model of 22 nm from the CRD to the collagen N terminus agreed well with the maximum length of 22 nm from the experimental P(r) analyses (Fig. 6B). As an independent check of the scattering modeling, the s20,w value of the best-fit MBL monomer model was calculated to be 2.9 S using HYDROPRO. This value was in good agreement with the mean experimental value of 2.8 ± 0.5 S (Table 1), being within an acceptable precision of ± 0.21 S (40). In conclusion, a bent MBL monomer structure (available in the supplemental material) accounted well for its scattering curve and sedimentation s20,w value.
TABLE 2.
Summary of modeling procedures for the MBL oligomers
| Oligomer | Search | Conformational variable | Conclusion |
|---|---|---|---|
| Monomer | 1 | Randomized CRD region at the swivel | 12 best-fit modelsa were identified from 5,000 trials |
| 2, 3, 4 | Randomized N terminus at the hub | Not used further | |
| Dimer | 1 | Two monomers rotated in 10° increments; bases separated by 0 nm | Not used further |
| 2 | Two monomers rotated in 10° increments; bases separated by 3.3 nm | 12 best-fit modelsa were identified from 6,859 trials | |
| 3 | Two monomers rotated in 10° increments; bases separated by 4.8 nm | Not used further | |
| 4 | Two monomers rotated in 10° increments; bases separated by 6.6 nm | Not used further | |
| Trimer | 1 | Rotated a monomer relative to the best-fit dimer which was kept fixed | Not used further |
| 2 | Monomer 1 fixed; monomers 2 and 3 subjected to 10° and 20° rotational increments respectively | 12 best-fit modelsa were identified from 2,028 trials | |
| Tetramer | 1 | Monomer 1 fixed; monomers 2–4 subjected to 10, 20, and 30° rotational increments, respectively | 10 best-fit modelsa were identified from 900 trials |
a PDB-formatted coordinates for the best-fit oligomer models are provided in the supplemental material.
TABLE 3.
X-ray modeling fits for the MBL monomer solution structure
| Search | Filter | Models | Hydrated spheres | Rg | Rxs | R-factor |
|---|---|---|---|---|---|---|
| nm | nm | % | ||||
| Search 1, CRD-collagen optimization | None | 5,000 | 602–844 | 4.53–7.36 | 0.69–1.73 | 4.0–11.1 |
| N, Rg, R | 12 | 815–833 | 6.89–7.15 | 1.59–1.68 | 4.0–4.1 | |
| Best-fit | 1 | 828 | 7.14 | 1.60 | 4.0 | |
| Linear modela | 1 | 824 | 7.70 | 1.40 | 5.9 | |
| Experimentala | NAb | 828 | 7.21 ± 0.22 | 1.55 ± 0.1 | NA | |
| Search 2, N-terminal chain A optimization | None | 5,000 | 782–885 | 6.80–7.38 | 1.48–1.73 | 4.1–6.6 |
| N, Rg, R | 12 | 816–853 | 7.05–7.31 | 1.57–1.66 | 4.1–4.2 | |
| Best-fit | 1 | 827 | 7.14 | 1.59 | 4.1 | |
| Search 3, N-terminal chain B optimization | None | 5,000 | 783–882 | 6.73–7.38 | 1.55–1.81 | 4.2–6.5 |
| N, Rg, R | 12 | 833–870 | 7.06–7.25 | 1.64–1.71 | 4.2–4.3 | |
| Best-fit | 1 | 833 | 7.20 | 1.64 | 4.2 | |
| Search 4, N-terminal chain C optimization | None | 5,000 | 784–882 | 6.75–7.33 | 1.50–1.75 | 4.1–7.1 |
| N, Rg, R | 12 | 825–859 | 7.08–7.21 | 1.58–1.70 | 4.1–4.3 | |
| Best-fit | 1 | 825 | 7.16 | 1.61 | 4.1 |
a The values for the linear model and the experimental values are the same in all four searches.
b NA means not applicable.
FIGURE 6.

X-ray curve fits for the best-fit models of MBL oligomers. A, C, E, and G, black open circles represent the experimental I(Q) scattering curves, and the gray continuous lines represent the best-fit MBL modeled I(Q) curves. The modeled P(r) curves in continuous gray lines are shown as insets in the upper right, with the experimental P(r) curves shown in black. A and B, best curve fit for the MBL monomer is shown, together with its best-fit structure, based on the CRD-collagen optimization search for the MBL monomer (Table 2). C and D, best curve fit for the MBL dimer is shown, together with its best-fit structure. The 2nd lower curve fit corresponds to 20% monomer and 80% dimer (see text), and the resulting P(r) curves is shown as a dashed gray line in the inset. The approximate angle between the two collagen helices is also shown. E and F, best curve fit for the MBL trimer is shown, together with its best-fit structure. The 2nd lower curve fit corresponds to 10% monomer, 10% dimer, and 80% trimer (see text). G and H, best curve fit for the MBL tetramer is shown, together with its best-fit structure.
Other searches 2–4 investigated the conformation of the extended N-terminal cysteine-rich peptides (Table 3). Starting from an arbitrary sequence 35CGRDGRD41, which was conformationally randomized, the scattering curve was calculated for all 5,000 monomer models. The best-fit conformation from search 2 (chain A) was used for search 3 (chain B), and the best-fit structure from search 3 was used in search 4 (chain C). No detectably improved MBL monomer fits were obtained. It was concluded that these N-terminal peptides were either too small in size to influence the curve fit or their conformations were ill-defined.
Constrained Modeling of the Rat MBL Dimer
Although it is possible that MBL is flexible, the existence of the broad peak M2 in the P(r) curves (Fig. 3) indicates that flexibility between the MBL monomers is limited. Previous structural proposals for MBL oligomers include “bouquet-like” arrangements or cruciform-like structures with several degrees of symmetry (10). Although the x-ray data indicate that the two monomers form a V-shaped structure, all possible arrangements of two monomers in the dimer were nonetheless tested (16). This utilized the best-fit MBL monomer model (Fig. 6B), which was assumed to be unchanged when incorporated into the dimer. Although the combination of monomer and dimer fitting would optimize the monomer conformation within the dimer, the required calculations were beyond our present computational capacities.
Four dimer searches were performed to evaluate a sufficient range of MBL models for comparison with the experimental x-ray scattering data (Table 2). In all four searches, the two MBL monomers were initially superimposed upon one another. Next, monomer 1 was held fixed, whereas monomer 2 was systematically rotated about monomer 1 to explore all possible orientations between monomers 1 and 2. To allow for the unknown separation between the MBL monomers, search 1 had no separation, although searches 2–4 had separations of 3.3, 4.8, and 6.6 nm, respectively, between the N-terminal Glu-46 α-carbon residues.
Search 2 with the 3.3 nm separation yielded the best results (Table 4). Superimposition of a selection of models in Search 2 visually revealed that a wide range of randomized MBL dimer conformations had been generated as required (supplemental Fig. S2C). A broad minimum was seen in the R-factor versus Rg analysis of all the models (supplemental Fig. S1B). Many good-fit models were identified. The 12 best-fit models were identified by comparison of the modeled Rg values with the experimental Rg values of 10.42 and 10.48 nm (supplemental Fig. S1B). These 12 models showed R-factors from 8.7 to 9.1% (Table 4), with these being lower than for the other three searches. The best-fit MBL dimer model of the 12 has an R-factor of 9.0%, being defined by x, y, and z axis rotations of 90, 60, and 10°, respectively (Fig. 6D). This model gave reasonable curve fits out to a Q value of at least 0.9 nm−1 (Fig. 6C), although there was a minor inflection at 0.25 nm−1. It also reproduced the double-peaked P(r) curve. Small discrepancies in the experimental and modeled P(r) curves in the region of peak M2 were best explained by either flexibility in the dimer structure at the hub region, similar to that proposed for complement C1q (41), flexibility in the swivel region (14), or sample polydispersity (4 and see below), any of which would cause peak M2 to become broader. Inspection of the 12 models revealed similar V-shaped dimer structures held together at the base of the collagen triple helix by a mean angle of 69 ± 17° (supplemental Fig. S2D). The maximum dimension of the 12 models was 28 nm, in fair agreement with that of 31 nm obtained experimentally. The 12 models gave a mean s20,w value of 4.14 ± 0.06 S, in good agreement with the experimental value of 4.2 ± 0.4 S (Table 1). In conclusion, good curve fits were obtained with a V-shaped dimer structure.
TABLE 4.
X-ray modeling fits for the MBL dimer, trimer, and tetramer solution structures
| Search | Filter | Models | Hydrated spheres | Rg | Rxs | R-factor |
|---|---|---|---|---|---|---|
| nm | nm | % | ||||
| MBL dimer search 1 (0 nm separation) | None | 6,859 | 1,471–1,755 | 7.40–12.30 | 0.05–2.88 | 10.9–15.6 |
| N, Rg, Rxs, R | 12 | 1,527–1,733 | 7.60–11.38 | 1.67–2.30 | 10.9–11.6 | |
| Best-fit | 1 | 1,695 | 9.88 | 2.11 | 11.9 | |
| Experimentala | NAb | 1,609 | 10.42 | 1.84 | NA | |
| MBL dimer search 2 (3.3 nm separation) | None | 6,859 | 1,596–1,756 | 7.39–11.83 | 0.13–2.69 | 8.7–16.6 |
| N, Rg, Rxs, R | 12 | 1,659–1,731 | 10.43–11.12 | 1.70–2.09 | 8.7–9.1 | |
| Best-fit | 1 | 1,710 | 10.43 | 2.09 | 9.0 | |
| MBL dimer search 3 (4.8 nm separation) | None | 6,859 | 1,592–1,748 | 7.42–12.10 | 0.02–2.76 | 9.5–16.3 |
| N, Rg, Rxs, R | 12 | 1,673–1,718 | 10.66–11.01 | 2.01–2.22 | 9.5–9.9 | |
| Best-fit | 1 | 1,717 | 10.79 | 2.10 | 9.5 | |
| MBL dimer search 4 (6.6 nm separation) | None | 6,859 | 1,416–1,747 | 7.35–13.80 | 0.01–2.86 | 9.9–16.0 |
| N, Rg, Rxs, R | 12 | 1,660–1,712 | 10.25–10.97 | 1.97–2.44 | 10.2–10.4 | |
| Best-fit | 1 | 1,684 | 10.46 | 2.32 | 10.2 | |
| MBL trimer search 1 | None | 6,859 | 2,269–2,554 | 10.74–14.79 | 0.61–2.99 | 7.1–17.2 |
| N, Rg, Rxs, R | 12 | 2,416–2,524 | 10.96–12.26 | 1.34–2.49 | 7.1–7.5 | |
| Best-fit | 1 | 2,518 | 12.25 | 2.03 | 7.4 | |
| MBL trimer search 2 | None | 2,028 | 2,253–2,591 | 0.34–15.80 | 0.04–3.17 | 7.1–79.0 |
| N, Rg, Rxs, R | 12 | 2,507–2,552 | 12.33–12.89 | 1.21–2.15 | 7.1–8.0 | |
| Best-fit | 1 | 2,530 | 12.60 | 1.87 | 7.1 | |
| Experimentala | NA | 2,413 | 12.43 | 1.95 | NA | |
| MBL tetramer search 1 | None | 900 | 3,033–3,411 | 1.20–44.28 | 0.01–7.50 | 14.2 to >100 |
| N, Rg, Rxs, R | 10 | 3,221–3,344 | 14.12–17.85 | 1.17–4.1 | 14.5–17.6 | |
| Best-fit | 1 | 3,296 | 17.08 | 1.43 | 14.5 | |
| Experimental | NA | 3,217 | 13.80 | 2.20 | NA |
a The experimental values are the same in the four dimer and two trimer searches.
b NA means not applicable.
Constrained Modeling of the Rat MBL Trimer and Tetramer
The modeling of the MBL trimer and tetramer solution structures required a different strategy (Table 2). In illustration of this, a first search for the MBL trimer based on the best-fit MBL dimer model, to which a third monomer was added in all rotations, was tested (Table 4). Even though a good fit was obtained, an insufficient range of trial MBL trimer conformations were generated, and it could not be assumed that the dimer was unchanged in conformation within the trimer.
Search 2 for the MBL trimer was based on equivalent rotations for the second and third monomers while holding the first monomer fixed (see under “Experimental Procedures”). This strategy ensured that all trimer conformations were investigated. Superimposition of every 36th model from the 2,028 created confirmed visually that a broad range of trial trimer conformations were formed as required (supplemental Fig. S2E). The observation of a minimum in the R factor versus Rg analysis for the 2028 models also showed that a sufficient number of models were tested (supplemental Fig. S1C). Twelve best-fit models were identified that had the lowest R-factor values between 7.1 and 8%. Interestingly, the 12 best-fit models consistently showed near-planar conformations, and five best models were mirror images of the other seven models (green and purple in supplemental Fig. S2F). The Rg values of the best-fit models were consistent with the experimental Guinier Rg value of 12.43 nm (Table 4). The best-fit trimer MBL model has the lowest R-factor of 7.1% (Fig. 6F). This near-planar structure is defined by monomer 1 with a rotation of 0, 0, and 0° about the x, y, and z axes, respectively, and monomer 2 with a rotation of 110, 70, and 10°, and monomer 3 with a rotation of 220, 140, and 20°. This best-fit model gave visually good curve fits out to a Q value of 1.6 nm−1 (Fig. 6E). The modeled P(r) curve reproduced the major M1 and M2 peaks (Fig. 6E); note that the experimental M2 peak is broader than the modeled one, probably for reason of flexibility or polydispersity as for the MBL dimer. The L value of the best-fit model (40 nm) was in good agreement with the experimental value (38 nm). In the 12 best-fit models, the mean angle between the collagen helices in monomers 1 and 2 was 36 ± 7°, whereas that between monomers 2 and 3 was 88 ± 11°. Both angles are close to that of 69 ± 17° for the MBL dimer. The mean experimental s20,w value of 5.7 ± 0.4 S for the MBL trimer was in good agreement with the mean s20,w value of 5.26 ± 0.04 S for the 12 best-fit MBL trimer models. In conclusion, near-planar best-fit trimer models were determined that resembled the MBL dimer structure.
The search for the MBL tetramer structure used the same rotational strategy used for the MBL trimer to give 900 models (see under “Experimental Procedures”). Superimposition of every 18th model (50 in total) showed that a broad range of trial conformations had been generated as required (supplemental Fig. S2G). A minimum in the R-factor versus Rg analysis confirmed that best-fit structures could be identified (supplemental Fig. S1D). The 10 best-fit models all showed near-planar structures with similar arrangements of the four MBL monomers within each tetramer (supplemental Fig. S2H). The best-fit MBL tetramer model had an R-factor of 14.5% (Fig. 6H). The best-fit model was defined by monomer 1 with a rotation of 0, 0, and 0° about the x, y, and z axes, monomer 2 with a rotation of 20, 40, and 10°, monomer 3 with a rotation of 40, 80, and 20°, and monomer 4 with a rotation of 60, 120, and 30°. This best-fit model gave a good curve fit to a Q value of 1 nm−1 (Fig. 6G). Its P(r) curve could not be determined for reason of instabilities in the I(Q) transformation. The longest dimension in the best-fit structure was 40 nm, which is comparable with the experimental L value of 47 nm (Fig. 6H). In the 10 best-fit near-planar structures (Table 4), the mean angle between monomers 1 and 2 was 43 ± 7°, that for monomers 2 and 3 was 46 ± 6°, and that for monomers 3 and 4 was 58 ± 6° (Fig. 6H). These angles are similar to those for the dimer and trimer (above) and also with atomic force microscopy that reported the average angle between the MBL monomers to be 40° (14). The s20,w value of the 10 best-fit tetramer models was 6.52 ± 0.12 S, which is comparable with the experimental s20,w value of 7.4 S ± 0.1 S for the MBL tetramer. In conclusion, despite the reduced quality of this curve fit, the modeling indicated that this tetramer structure is also near-planar and related in appearance to the dimer and trimer.
Effect of Polydispersity on the X-ray Fits
In the above modeling of the four oligomer solution structures, it was assumed that each scattering curve was monodisperse, i.e. free from small amounts of the other MBL oligomers. The three minor peaks and one major peak observed in the sedimentation c(s) analyses showed low polydispersity with as much as 10% of each of the three minor species being present when either the dimer or trimer was observed as the major species (Fig. 4). Accordingly, each curve fit in Fig. 6 was re-evaluated by allowing for the presence of all four oligomers in each scattering curve. The proportions of each oligomer was optimized in 5% increments to follow previous scattering procedures used to analyze mixtures of proteins (42). For the MBL monomer, the R-factor was not improved from 4.0% when 5–10% of dimer or trimer was included in the curve fits. For the dimer, the R-factor was improved from 9.0 to 8.0%, and the curve fits were visually much improved by optimizing a fit based on 20% monomer and 80% dimer (Fig. 6C). For the trimer, the R-factor of 7.1% was unchanged either with no other oligomers present or by adding 5% monomer and 5% dimer or 10% monomer and 10% dimer, although the visual agreement with the P(r) curve was improved. It was concluded that the reliability of the x-ray modeling fits was unaffected by the low polydispersity seen in the c(s) analyses of Fig. 4.
DISCUSSION
By this study, we have identified for the first time the molecular solution structures of the four major oligomeric forms of rat MBL. The experimental x-ray data showed that the MBL oligomers formed V-shaped structures, for which the evidence in the length L values and the observation of peak M2 (Fig. 3) showed that the CRD/neck regions were on average separated by about 20 nm in the oligomers. Next, constrained molecular modeling was performed to identify molecular structures for the four MBL oligomers. The MBL monomer was readily modeled using the combination of crystal structures for the CRD/neck region and a linear MBL-like collagen triple helix (10, 27). In accord with atomic force microscopy (14), the modeling of the monomer showed that the bend in MBL could be readily located at the swivel between the CRD/neck and triple helix. In further modeling curve fits (Table 2), this best-fit MBL monomer structure accounted for the scattering curves for the MBL dimer, trimer, and tetramer. These oligomer models showed near-planarity and structural similarity with each other in terms of V-shaped structures. Some flexibility in the MBL oligomers at the hub or swivel regions is possible, given the breadth of peak M2 in Fig. 3. These MBL structures are termed sertiform, in which they show a central hub from which the two, three, or four monomers fan outward in a near-planar arrangement, and the CRD heads are in proximity to each other (14). The experimental sedimentation coefficients were well explained by these models. Small imperfections in the x-ray curve fits for the dimer, trimer, and tetramer were explained in terms of low amounts of the other oligomers that could not be removed by gel filtration. The outcome of near-planar MBL structures for the oligomers is clear, and this provides strong support for an activation mechanism in which dimeric MASP2 binds to at least two sites in a near-planar rat MBL structure (43).
Protein crystallography to date has provided high resolution atomic structures for smaller parts of MBL, namely a single CRD/neck region and a related triple helix structure for a single MBL monomer. Up to now, electron microscopy and atomic force microscopy have been major techniques that provided structural insights on the MBL oligomers (14). The atomic force microscopy study in 2009 (14) studied MBL as a hydrated protein on a mica surface and showed that human MBL existed mostly as tetramers, but dimer to octamer were also present. Our solution structural model for rat MBL readily accounts for the addition of further monomers at the side of the hub to create wider sertiform structures with the CRD/neck regions positioned on one side of this structure. The 2009 (14) study also reported the most common angle between human MBL monomers to be 47 ± 24°. This is in good agreement with our value of 63° for the dimer, 36 and 86° for the trimer, and 54, 47, and 51° for the tetramer (Fig. 6). The length of the human MBL monomer was reported to be 19 ± 4 nm in the 2009 study (14). Although this is shorter than our value of 24–25 nm from the P(r) curve and the best-fit monomer model, the difference is explained by the 2009 measurement (14) being made between the center of the hub to the center of the CRD, and not between the extremities of the MBL monomer as in our study. We conclude that many features of our solution structure are in agreement with the 2009 atomic force microscopy study (14). Unlike microscopy, our solution structures show that the MBL oligomers are near-planar in appearance. Unlike microscopy, we are able to deposit MBL molecular structures in Protein Data Bank format to facilitate further investigations (see supplemental material).
We believe that the present x-ray scattering and analytical ultracentrifugation study is improved compared with an earlier x-ray scattering and microscopy study in 2007 (15) of oligomeric human MBL. We have studied the separated monomer, dimer, trimer, and tetramer forms of rat MBL, and the 2007 study analyzed human MBL on the assumption that this was a single trimer. In fact, the 2009 atomic force microscopy study (14) reported that tetramers were more abundant in the same human MBL preparation used for the 2007 scattering study (15) than trimers. The reliability of the synchrotron x-ray scattering data for rat MBL at 0.23–1.15 mg/ml is indicated by the very short data acquisition times of 2.5–5 s and extensive checks for the absence of radiation damage, including the use of flow cells. The 2007 x-ray data (15) used a laboratory-based x-ray source for human MBL at higher 2.12–4.25 mg/ml concentrations with very long data acquisition times, and precautions against radiation damage were not described. Our Rg and L values for rat MBL trimer were 12.5 and 38 nm respectively (Table 1), which differ from the 2007 study (15) that reported 11.0 and 32 nm, respectively, for the human MBL trimer. The present MBL structural modeling utilized crystal structures for the triple helix that included the Gly-Xaa-Gly interruption and the CRD in a constrained modeling procedure that assumed that these structures were preserved in solution. This constrained modeling revealed near-planar sertiform MBL structures that were consistent with the 2009 structures from atomic force microscopy and our sedimentation coefficients. The 2007 study (15) performed structural modeling with nonconstrained approaches using DAMMIN, GASBOR, and SASREF, in which no account was taken of these prior known crystal structures within MBL. Consequently the lack of restraints in the 2007 modeling produced curved collagen triple helix structures from a scattering curve that was assumed to correspond to only MBL trimer; such a model appears to be implausible(15). The 2007 modeling also resulted in nonplanar structures with a 3-fold rotational symmetry that were at variance with both the 2009 structures from atomic force microscopy and our present scattering modeling.
The constrained modeling of MBL resulted in sertiform-shaped molecular models for its monomer, dimer, trimer, and tetramer. The reliability of the scattering modeling assumes that the two crystal structures for collagen and the CRD are unchanged in solution. Previous studies have sometimes assumed that the Gly-Xaa-Gly interruption will lead to a major kink in the MBL triple helix. Because the same sequence interruption is present in all three MBL polypeptides, there is no reason to expect the disruption of a linear MBL triple helix. This was confirmed by the collagen crystal structure (27). Most MBL molecules studied by atomic force microscopy do not show a kink, and the 30% that do show such a kink exhibit a modest bend of only 40° (14). Mutation studies have shown that restoration of the Gly-Xaa-Gly motif to Gly-Xaa-Yaa does not affect the biological activity of MBL (44). In contrast, the interruption in the triple helix of complement C1q occurs in only one of its three polypeptide chains, thus facilitating the bending of the C1q triple helix at this point (41). The satisfactory outcome of the MBL modeling is best illustrated by the limited range of conformations found in the best-fit structures (supplemental Fig. S2). All 12 best-fit models for the MBL monomer showed that the CRD/neck region is bent relative to the triple helix region, although there is a limited range of bent structures that fit the scattering curve equally well. All 12 best-fit MBL dimer models show similar V-shaped structures within a narrow angular range. The trimer and tetramer modeling consistently showed similar V-shaped arrangements of monomers. A strength of scattering modeling is its ability to exclude poor-fit models; this MBL study has ruled out MBL structures that are cruciform-like or linear end-to-end assemblies of monomers.
The reliability of the MBL modeling is supported by our previous constrained scattering modeling analyses of other multidomain proteins. Thus, the scattering modeling of the three-domain SCR-6/8 domains of complement factor H predated the crystal structure of the same fragment; the scattering modeling showed the same inter-SCR domain arrangement found in the subsequent crystal structure (45, 46). The J-shaped scattering structure of the five-domain SCR-1/5 fragment of complement factor H was used to help solve the crystal structure of its complex with complement C3b (47, 48). The J-shaped structure of the five-domain SCR-1/5 rat CrrY protein was likewise confirmed by crystallography (29, 49). The scattering modeling of the compacted folded back five-domain arrangement of complement factor I was confirmed by its recent crystal structure (50, 51). At present, constrained scattering modeling has been applied to a broad spectrum of over 30 antibody and complement proteins, as well as two polyanionic oligosaccharides. The resulting molecular structures are available in the Protein Data Bank as a record (37, 40).
The four MBL solution structures provide new insights on the MBL interactions with mannose-coated pathogen surfaces, followed by the interaction of MBL with the MASP homodimer that leads to the activation of MASP. Although necessarily speculative in the absence of detailed structures for the MBL-pathogen and MBL-MASP complexes, three points can be made as follows.
(i) First, the flexibility at the hub and swivel regions of the MBL tetramer will facilitate the ability of the 12 CRD regions in the tetramer to interact with mannose residues at pathogen surfaces. Each single CRD-mannose interaction has a low affinity with a dissociation constant KD of about 1 mm. The multiple binding of CRD regions to a pathogen surface will increase the affinity by several orders of magnitude. In theory, two separate binding events with weak millimolar affinities will become a stronger interaction with a micromolar affinity (the product of the two KD values) if both weak sites bind simultaneously to the same cell surface (52). A third binding event is theoretically able to attain nanomolar affinity levels, and 2.6–3.2 nm affinities have been observed experimentally (53, 54). Accordingly, the availability of multiple CRD domains presented in flexible orientations within a near-planar and flexible MBL tetramer is considered to be key for optimal binding interactions.
(ii) Second, the near-planar MBL structures will facilitate the binding of the MASP dimer between two MBL monomers. A MASP monomer is composed of six CUB1-EGF-CUB2-SCR1-SCR2-SP domains in that order. Sequence alignment of MBLs and ficolins from a variety of species and subsequent mutagenesis experiments identified the 61Hyp-Gly-Lys-Leu-Gly-Pro66 motif within the collagen triple helix of rat MBL to form the binding site for MASP-2 (55). Crystal structures of the MASP fragments reveal a head-to-tail arrangement for the intact MASP dimer (Fig. 7). In this, the CUB1-EGF region is necessary for both MASP-2 dimer formation and the interaction of MASP-2 with MBL (56). There are four MBL collagen-binding sites on each MASP-2 (or MASP-1) dimer, and one MBL site within each CUB domains that is near a Ca2+ site (57, 58). The CUB1-EGF-CUB2 domains form an elongated X-shaped structure at their center, with an MBL-binding site at the end of each stem. Although not all four MBL sites are necessarily involved in the MBL-MASP interaction, the comparison of the MASP dimer with the near-planar MBL tetramer may account for their interaction (Fig. 7). The paired CUB1-EGF domains of the MASP dimer may lie between two MBL collagenous regions, given that their binding sites for MBL are on opposite sides of the MASP dimer. This interaction would make the long axis of the MASP dimer perpendicular to the plane of the MBL tetramer (Fig. 7).
FIGURE 7.

Model of the MASP-2 interaction with the rat MBL tetramer. The best-fit MBL tetramer model is shown in dark blue with the MASP-binding motif Hyp-Gly-Lys-Leu-Gly-Pro highlighted in pink on the collagen triple helix. MASP-2 is constructed from six domains in the order CUB1-EGF-CUB2-SCR-1-SCR-2-SP (CUB, C1r/C1s, Uefg, and bone morphogenetic protein-1; EGF, epidermal growth factor; SCR, short complement regulator; SP, serine protease). The MASP-2 model was created from crystal structures for the CUB1-EGF-CUB2 dimer, with each monomer shown in yellow or purple (PDB code 1NT0) (56). The remaining SCR-1-SCR-2-SP domain pair is shown in red and green (PDB code 1ZJK) (58). A, face-on view of the near-planar MBL tetramer in which the four CRD regions CRD-1 to CRD-4 are visible, and the linear MASP-2 dimer is shown end-on. B, view of A is turned by 90° about the x axis so that the full length of the MASP-2 dimer is seen together with its two serine protease and four short complement regulator domains. C, in an auto-activation model for MASP-2, the two serine protease domains of the MASP-2 dimer are folded back relative to the CUB-EGF domains that are positioned on the triple helical collagen regions. The two serine protease domains form contacts with each other in the plane of the MBL tetramer, as required for an auto-activation mechanism. D, view of C is turned by 90° about the x axis so that the postulated contact between the green and red serine protease domains is seen more clearly.
(iii) Finally, the activation of MASP after binding to MBL is clarified by our model. Trimers and tetramers of MBL are more effective at activating complement than dimers. This observation may be explained by the improved chance that two adjacent MBL triple helices bind to the pathogen at an angle best suited for the self-activation of MASP. Auto-activation of the serine protease domains of the MASP may occur through their ability to approach each other in such a MBL-MASP complex (Fig. 7, C and D) that is not possible outside this interaction, thereby triggering the lectin pathway of complement activation.
Supplementary Material
Acknowledgments
We thank Dr. T. Narayanan (European Synchrotron Radiation Facility, Grenoble, France) for excellent x-ray instrumental support and C. Bryant and K. Ihebunezie for additional support.

This article contains supplemental Figs. S1 and S2 and monomer_3UJU, dimer_3UJV, trimer_3UJW, and tetramer_3UJX model files.
J. T. Eaton and S. J. Perkins, unpublished software.
- MBL
- mannose-binding lectin
- MASP
- MBL-associated serine protease
- CRD
- carbohydrate-recognition domain
- PDB
- Protein Data Bank.
REFERENCES
- 1. Thiel S., Vorup-Jensen T., Stover C. M., Schwaeble W., Laursen S. B., Poulsen K., Willis A. C., Eggleton P., Hansen S., Holmskov U., Reid K. B., Jensenius J. C. (1997) A second serine protease associated with mannan-binding lectin that activates complement. Nature 386, 506–510 [DOI] [PubMed] [Google Scholar]
- 2. Drickamer K., Dordal M. S., Reynolds L. (1986) Mannose-binding proteins isolated from rat liver contain carbohydrate-recognition domains linked to collagenous tails. Complete primary structures and homology with pulmonary surfactant apoprotein. J. Biol. Chem. 261, 6878–6887 [PubMed] [Google Scholar]
- 3. Drickamer K., Taylor M. E. (1993) Biology of animal lectins. Annu. Rev. Cell Biol. 9, 237–264 [DOI] [PubMed] [Google Scholar]
- 4. Wallis R., Drickamer K. (1999) Molecular determinants of oligomer formation and complement fixation in mannose-binding proteins. J. Biol. Chem. 274, 3580–3589 [DOI] [PubMed] [Google Scholar]
- 5. Garred P., Pressler T., Lanng S., Madsen H. O., Moser C., Laursen I., Balstrup F., Koch C., Koch C. (2002) Mannose-binding lectin (MBL) therapy in an MBL-deficient patient with severe cystic fibrosis lung disease. Pediatr. Pulmonol. 33, 201–207 [DOI] [PubMed] [Google Scholar]
- 6. Haurum J. S., Thiel S., Jones I. M., Fischer P. B., Laursen S. B., Jensenius J. C. (1993) Complement activation upon binding of mannan-binding protein to HIV envelope glycoproteins. AIDS 7, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 7. Chong W. P., To Y. F., Ip W. K., Yuen M. F., Poon T. P., Wong W. H., Lai C. L., Lau Y. L. (2005) Mannose-binding lectin in chronic hepatitis B virus infection. Hepatology 42, 1037–1045 [DOI] [PubMed] [Google Scholar]
- 8. Fidler K. J., Hilliard T. N., Bush A., Johnson M., Geddes D. M., Turner M. W., Alton E. W., Klein N. J., Davies J. C. (2009) Mannose-binding lectin is present in the infected airway. A possible pulmonary defense mechanism. Thorax 64, 150–155 [DOI] [PubMed] [Google Scholar]
- 9. Sheriff S., Chang C. Y., Ezekowitz R. A. (1994) Human mannose-binding protein carbohydrate recognition domain trimerizes through a triple α-helical coiled-coil. Nat. Struct. Biol. 1, 789–794 [DOI] [PubMed] [Google Scholar]
- 10. Weis W. I., Drickamer K. (1994) Trimeric structure of a C-type mannose-binding protein. Structure 2, 1227–1240 [DOI] [PubMed] [Google Scholar]
- 11. Lu J. H., Thiel S., Wiedemann H., Timpl R., Reid K. B. (1990) Binding of the pentamer/hexamer forms of mannan-binding protein to zymosan activates the proenzyme C1r2C1s2 complex, of the classical pathway of complement, without involvement of C1q. J. Immunol. 144, 2287–2294 [PubMed] [Google Scholar]
- 12. Jensen P. H., Weilguny D., Matthiesen F., McGuire K. A., Shi L., Højrup P. (2005) Characterization of the oligomer structure of recombinant human mannan-binding lectin. J. Biol. Chem. 280, 11043–11051 [DOI] [PubMed] [Google Scholar]
- 13. Mohs A., Popiel M., Li Y., Baum J., Brodsky B. (2006) Conformational features of a natural break in the type IV collagen Gly-X-Y repeat. J. Biol. Chem. 281, 17197–17202 [DOI] [PubMed] [Google Scholar]
- 14. Jensenius H., Klein D. C., van Hecke M., Oosterkamp T. H., Schmidt T., Jensenius J. C. (2009) Mannan-binding lectin. Structure, oligomerization, and flexibility studied by atomic force microscopy. J. Mol. Biol. 391, 246–259 [DOI] [PubMed] [Google Scholar]
- 15. Dong M., Xu S., Oliveira C. L., Pedersen J. S., Thiel S., Besenbacher F., Vorup-Jensen T. (2007) Conformational changes in mannan-binding lectin bound to ligand surfaces. J. Immunol. 178, 3016–3022 [DOI] [PubMed] [Google Scholar]
- 16. Bonner A., Furtado P. B., Almogren A., Kerr M. A., Perkins S. J. (2008) Implications of the near-planar solution structure of human myeloma dimeric IgA1 for mucosal immunity and IgA nephropathy. J. Immunol. 180, 1008–1018 [DOI] [PubMed] [Google Scholar]
- 17. Okemefuna A. I., Nan R., Gor J., Perkins S. J. (2009) Electrostatic interactions contribute to the folded-back conformation of wild type human factor H. J. Mol. Biol. 391, 98–118 [DOI] [PubMed] [Google Scholar]
- 18. Wallis R., Drickamer K. (1997) Asymmetry adjacent to the collagen-like domain in rat liver mannose-binding protein. Biochem. J. 325, 391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C. B., Wallis R. (2001) Stoichiometry of complexes between mannose-binding protein and its associated serine proteases. Defining functional units for complement activation. J. Biol. Chem. 276, 25894–25902 [DOI] [PubMed] [Google Scholar]
- 20. Perkins S. J. (1986) Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering match points, and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur. J. Biochem. 157, 169–180 [DOI] [PubMed] [Google Scholar]
- 21. Narayanan T., Diat O., Bösecke P. (2001) SAXS and USAXS on the high brilliance beamline at the ESRF. Nucl. Instrum. Methods Phy. Res. A 467–468, 1005–1009 [Google Scholar]
- 22. Gilbert H. E., Eaton J. T., Hannan J. P., Holers V. M., Perkins S. J. (2005) Solution structure of the complex between CR2 SCR 1–2 and C3d of human complement. X-ray scattering and sedimentation modeling study. J. Mol. Biol. 346, 859–873 [DOI] [PubMed] [Google Scholar]
- 23. Glatter O., Kratky O. (1982) Small-angle X-ray Scattering, Academic Press, New York [Google Scholar]
- 24. Semenyuk A. V., Svergun D. I. (1991) GNOM–a program package for small-angle scattering data processing. J. Appl. Crystallogr. 24, 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schuck P. (1998) Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75, 1503–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schuck P. (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bella J., Liu J., Kramer R., Brodsky B., Berman H. M. (2006) Conformational effects of Gly-X-Gly interruptions in the collagen triple helix. J. Mol. Biol. 362, 298–311 [DOI] [PubMed] [Google Scholar]
- 28. Boehm M. K., Woof J. M., Kerr M. A., Perkins S. J. (1999) The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG. A study by x-ray and neutron solution scattering and homology modeling. J. Mol. Biol. 286, 1421–1447 [DOI] [PubMed] [Google Scholar]
- 29. Aslam M., Guthridge J. M., Hack B. K., Quigg R. J., Holers V. M., Perkins S. J. (2003) The extended multidomain solution structures of the complement protein Crry and its chimeric conjugate Crry-Ig by scattering, analytical ultracentrifugation, and constrained modeling. Implications for function and therapy. J. Mol. Biol. 329, 525–550 [DOI] [PubMed] [Google Scholar]
- 30. Ashton A. W., Boehm M. K., Gallimore J. R., Pepys M. B., Perkins S. J. (1997) Pentameric and decameric structures in solution of serum amyloid P component by x-ray and neutron scattering and molecular modeling analyses. J. Mol. Biol. 272, 408–422 [DOI] [PubMed] [Google Scholar]
- 31. Perkins S. J. (2001) X-ray and neutron scattering analyses of hydration shells. A molecular interpretation based on sequence predictions and modeling fits. Biophys. Chem. 93, 129–139 [DOI] [PubMed] [Google Scholar]
- 32. García De La Torre J., Huertas M. L., Carrasco B. (2000) Calculation of hydrodynamic properties of globular proteins from their atomic level structure. Biophys. J. 78, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lipscombe R. J., Sumiya M., Summerfield J. A., Turner M. W. (1995) Distinct physicochemical characteristics of human mannose-binding protein expressed by individuals of differing genotype. Immunology 85, 660–667 [PMC free article] [PubMed] [Google Scholar]
- 34. Perkins S. J., Okemefuna A. I., Fernando A. N., Bonner A., Gilbert H. E., Furtado P. B. (2008) X-ray and neutron scattering data and their constrained molecular modeling. Methods Cell Biol. 84, 375–423 [DOI] [PubMed] [Google Scholar]
- 35. Aslam M., Perkins S. J. (2001) Folded-back solution structure of monomeric factor H of human complement by synchrotron x-ray and neutron scattering, analytical ultracentrifugation, and constrained molecular modeling. J. Mol. Biol. 309, 1117–1138 [DOI] [PubMed] [Google Scholar]
- 36. Cole J. L., Lary J. W., P Moody T., Laue T. M. (2008) Analytical ultracentrifugation. Sedimentation velocity and sedimentation equilibrium. Methods Cell Biol. 84, 143–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bella J., Eaton M., Brodsky B., Berman H. M. (1994) Crystal and molecular structure of a collagen-like peptide at 1.9-Å resolution. Science 266, 75–81 [DOI] [PubMed] [Google Scholar]
- 38. Fraser R. D., MacRae T. P., Suzuki E. (1979) Chain conformation in the collagen molecule. J. Mol. Biol. 129, 463–481 [DOI] [PubMed] [Google Scholar]
- 39. Okuyama K., Xu X., Iguchi M., Noguchi K. (2006) Revision of collagen molecular structure. Biopolymers 84, 181–191 [DOI] [PubMed] [Google Scholar]
- 40. Perkins S. J., Okemefuna A. I., Nan R., Li K., Bonner A. (2009) Constrained solution scattering modeling of human antibodies and complement proteins reveals novel biological insights. J. R. Soc. Interface 6, S679–S696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perkins S. J. (1985) Molecular modelling of human complement subcomponent Clq and its complex with Clr2 Cls2 derived from neutron-scattering curves and hydrodynamic properties. Biochem. J. 228, 13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okemefuna A. I., Stach L., Rana S., Buetas A. J., Gor J., Perkins S. J. (2010) C-reactive protein exists in an NaCl concentration-dependent pentamer-decamer equilibrium in physiological buffer. J. Biol. Chem. 285, 1041–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wallis R. (2007) Interactions between mannose-binding lectin and MASPs during complement activation by the lectin pathway. Immunobiology 212, 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kurata H., Cheng H. M., Kozutsumi Y., Yokota Y., Kawasaki T. (1993) Role of the collagen-like domain of the human serum mannan-binding protein in the activation of complement and the secretion of this lectin. Biochem. Biophys. Res. Commun. 191, 1204–1210 [DOI] [PubMed] [Google Scholar]
- 45. Fernando A. N., Furtado P. B., Clark S. J., Gilbert H. E., Day A. J., Sim R. B., Perkins S. J. (2007) Associative and structural properties of the region of complement factor H encompassing the Tyr402His disease-related polymorphism and its interactions with heparin. J. Mol. Biol. 368, 564–581 [DOI] [PubMed] [Google Scholar]
- 46. Prosser B. E., Johnson S., Roversi P., Herbert A. P., Blaum B. S., Tyrrell J., Jowitt T. A., Clark S. J., Tarelli E., Uhrín D., Barlow P. N., Sim R. B., Day A. J., Lea S. M. (2007) Structural basis for complement factor H linked age-related macular degeneration. J. Exp. Med. 204, 2277–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Okemefuna A. I., Gilbert H. E., Griggs K. M., Ormsby R. J., Gordon D. L., Perkins S. J. (2008) The regulatory SCR-1/5 and cell surface-binding SCR-16/20 fragments of factor H reveal partially folded-back solution structures and different self-associative properties. J. Mol. Biol. 375, 80–101 [DOI] [PubMed] [Google Scholar]
- 48. Wu J., Wu Y. Q., Ricklin D., Janssen B. J., Lambris J. D., Gros P. (2009) Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 10, 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roversi P., Johnson S., Caesar J. J., McLean F., Leath K. J., Tsiftsoglou S. A., Morgan B. P., Harris C. L., Sim R. B., Lea S. M. (2011) Structures of the rat complement regulator CrrY. Acta Crystallogr Sect. F Struct. Biol. Cryst. Commun. 67, 739–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chamberlain D., Ullman C. G., Perkins S. J. (1998) Possible arrangement of the five domains in human complement factor I as determined by a combination of x-ray and neutron scattering and homology modeling. Biochemistry 37, 13918–13929 [DOI] [PubMed] [Google Scholar]
- 51. Roversi P., Johnson S., Caesar J. J., McLean F., Leath K. J., Tsiftsoglou S. A., Morgan B. P., Harris C. L., Sim R. B., Lea S. M. (2011) Structural basis for complement factor I control and its disease-associated sequence polymorphisms. Proc. Natl. Acad. Sci. U.S.A. 108, 12839–12844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jencks W. P. (1981) On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U.S.A. 78, 4046–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wallis R., Dodd R. B. (2000) Interaction of mannose-binding protein with associated serine proteases. Effects of naturally occurring mutations. J. Biol. Chem. 275, 30962–30969 [DOI] [PubMed] [Google Scholar]
- 54. Thielens N. M., Cseh S., Thiel S., Vorup-Jensen T., Rossi V., Jensenius J. C., Arlaud G. J. (2001) Interaction properties of human mannan-binding lectin (MBL)-associated serine proteases-1 and -2, MBL-associated protein 19, and MBL. J. Immunol. 166, 5068–5077 [DOI] [PubMed] [Google Scholar]
- 55. Wallis R., Shaw J. M., Uitdehaag J., Chen C. B., Torgersen D., Drickamer K. (2004) Localization of the serine protease-binding sites in the collagen-like domain of mannose-binding protein. Indirect effects of naturally occurring mutations on protease binding and activation. J. Biol. Chem. 279, 14065–14073 [DOI] [PubMed] [Google Scholar]
- 56. Feinberg H., Uitdehaag J. C., Davies J. M., Wallis R, Drickamer K., Weis W. I. (2003) Crystal structure of the CUB1-EGF-CUB2 region of mannose-binding protein-associated serine protease-2. EMBO J. 22, 2348–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Teillet F., Gaboriaud C., Lacroix M., Martin L., Arlaud G. J., Thielens N. M. (2008) Crystal structure of the CUB1-EGF-CUB2 domain of human MASP-1/3 and identification of its interaction sites with mannan-binding lectin and ficolins. J. Biol. Chem. 283, 25715–25724 [DOI] [PubMed] [Google Scholar]
- 58. Gál P., Harmat V., Kocsis A., Bián T., Barna L., Ambrus G., Végh B., Balczer J., Sim R. B., Náray-Szabó G., Závodszky P. (2005) A true autoactivating enzyme. Structural insight into mannose-binding lectin-associated serine protease-2 activations. J. Biol. Chem. 280, 33435–33444 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.