

Background: DNA alkyltransferases repair mutagenic and carcinogenic alkylation DNA lesions.
Results: A thermophilic archaeal DNA alkyltransferase is degraded after alkylation in vivo. A novel assay is applied to study its activity in vitro.
Conclusion: The archaeal DNA alkyltransferase shows structure, activity, and in vivo regulation similar to its human homolog.
Significance: The function and regulation of DNA alkyltransferases might be conserved from archaea to humans.
Keywords: Archaea, DNA Methylation, DNA Methyltransferase, DNA Repair, Protein Degradation, DNA Alkylation
Abstract
Agents that form methylation adducts in DNA are highly mutagenic and carcinogenic, and organisms have evolved specialized cellular pathways devoted to their repair, including DNA alkyltransferases. These are proteins conserved in eucarya, bacteria and archaea, acting by a unique reaction mechanism, which leads to direct repair of DNA alkylation damage and irreversible protein alkylation. The alkylated form of DNA alkyltransferases is inactive, and in eukaryotes, it is rapidly directed to degradation. We report here in vitro and in vivo studies on the DNA alkyltransferase from the thermophilic archaeon Sulfolobus solfataricus (SsOGT). The development of a novel, simple, and sensitive fluorescence-based assay allowed a careful characterization of the SsOGT biochemical and DNA binding activities. In addition, transcriptional and post-translational regulation of SsOGT by DNA damage was studied. We show that although the gene transcription is induced by alkylating agent treatment, the protein is degraded in vivo by an alkylation-dependent mechanism. These experiments suggest a striking conservation, from archaea to humans, of this important pathway safeguarding genome stability.
Introduction
DNA alkyltransferases (called AGT,2 OGT, or MGMT, EC: 2.1.1.63) are conserved proteins that catalyze repair of alkylation damage by a one-step mechanism involving the transfer of the alkylic group from O6-alkyl-guanine or O4-alkyl-thymine to a cysteine residue in their own active site (for recent reviews, see Refs. 1 and 2). AGTs are called “suicide” or “kamikaze” proteins because the stoichiometry of the reaction is 1:1 and the alkylated form of the protein is irreversibly inactivated (2, 3). In human cells, alkylated AGT is rapidly directed to the degradation ubiquitin pathway due to alkylation-induced conformational change that turns the protein into a proteolysis-sensitive form (4).
Although AGT protects from mutagenic and carcinogenic effects of alkylating agents, in humans, it also plays a role in the onset of resistance to alkylating agents used in the treatment of several cancer types (5). Human AGT (hAGT) is strongly inhibited by O6-benzylguanine (O6-BG), whose benzylic group is transferred to the active site cysteine (2); this inhibitor is already used in clinical trials to increase the effectiveness of alkylation-based chemotherapy. Moreover, AGT overexpression is a possible strategy to protect normal cells against the effects of chemotherapy (6).
The resolution of the three-dimensional structure of hAGT in complex with DNA revealed a unique mechanism for DNA recognition and repair (4, 7). The protein contacts the DNA minor groove trough its helix-turn-helix domain. To access the alkyl group, which stands inside the double helix, the protein flips out the damaged base while an arginine residue forms interactions with the orphaned base. Despite the reported studies, however, details of the interaction with DNA and lesion recognition are not completely understood. In particular, it remains to be established whether to find the lesion, the protein needs to flip out every base or whether it is able to recognize some structural feature of the damaged base by simply scanning the double helix.
AGTs are present in organisms from the three living domains (eucarya, bacteria, archaea). For thermophilic organisms, alkylation lesions are particularly harmful because at high temperatures, unrepaired alkylated bases can be easily converted into abasic sites or DNA breaks that in turn can cause DNA fragmentation. Indeed, we have previously reported that the alkylating agent methyl methanesulfonate (MMS) is highly cytotoxic for the archaeon Sulfolobus solfataricus, which grows optimally at 80 °C (8). Limited information is available on thermophilic AGTs and their function and regulation. The three-dimensional structure of AGT homologs from two thermophilic archaea, Methanococcus jannaschii (9) and Sulfolobus tokodaii (Protein Data Bank (PDB) entry 1WRJ), has been determined, revealing overall similarities with the human protein. The MGMT from the archaeon Pyrococcus sp. KOD1, overexpressed in Escherichia coli, showed remarkable stability at high temperature and complemented the alkylating agent-sensitive phenotype of an E. coli ogt-deficient strain (10, 11). Other thermophilic AGTs were either not active (e.g. the AGT-like protein of Thermus thermophilus (12)) or poorly characterized because they were not soluble when heterologously expressed (e.g. the AGTs from the bacterium Aquifex aeolicus and the archaeon Archaeoglobus fulgidus (13)).
We report here in vitro and in vivo studies on the DNA alkyltransferase from the hyperthermophilic archaeon S. solfataricus (SsOGT). The protein was overexpressed in recombinant form in E. coli and characterized by using a novel method based on a fluorescent derivative of the competitive inhibitor O6-BG, showing that the protein is highly stable and active under a wide range of physical-chemical parameters. The assay also allowed the characterization of SsOGT mutant proteins with reduced or abolished DNA binding activity.
In S. solfataricus cells, the SsOGT RNA level was increased after exposure to MMS, confirming its involvement in repair of alkylation damage. Interestingly, the SsOGT protein was degraded in these cells in an MMS concentration-dependent fashion. We were able to demonstrate that degradation is a direct effect of the protein alkylation, and the degradation pathway is activated by MMS treatment. These experiments suggest a striking conservation, from archaea to humans, of the degradation mechanism that eliminates SsOGT after its action.
EXPERIMENTAL PROCEDURES
Reagents
Chemicals were purchased from Sigma, and E. coli strains and Pfu DNA polymerase were from Stratagene (La Jolla, CA). Synthetic oligonucleotides were from Primm (Milan, Italy) and are listed in supplemental Table I. DNA manipulation enzymes, as well as the SNAP-Vista GreenTM substrate, were from New England Biolabs (Ipswich, MA). The pQE31TM vector was from Qiagen (Hilden, Germany).
DNA Constructs
The Ssogt gene (ORF SSO2487) was amplified from S. solfataricus P2 genomic DNA (14) by using the oligonucleotides Ogt-5′ and Ogt-3′, which possess an internal BamHI and HindIII site, respectively (supplemental Table I). These sites allowed the insertion of the BamHI/HindIII-amplified fragment in the pQE31TM vector, in the same frame and downstream of a hexahistidine tag, leading to the pQE-ogt plasmid. The SsOGT protein expressed from this plasmid contains four extra amino acid residues at its C terminus (152LQPG155). The SsOGT R102A mutant was obtained in the pQE-ogt plasmid using the GeneTailorTM site-directed mutagenesis system (Invitrogen) with the following oligonucleotides: R102Amut and R102Arev (supplemental Table I). The synthetic SsOGT-H5 gene was custom-made by GeneArt AG (Regensburg, Germany). It contains five point mutations (S100A, R102A, G105K, M106T, K110E) in the DNA binding region (supplemental Fig. S1A), a mutation in position 132 (S132E), and a small multicloning site before the stop codon. The SsOGT-H5 mutant protein contains six extra amino acid residues at its C terminus (152LQVPST157).
Proteins
All proteins were expressed in the E. coli ABLE C strain. Cultures were grown at 37 °C in 1.0–2.0 liters of Luria-Bertani (LB) medium supplemented with 50.0 μg/ml ampicillin and were induced for 16 h with 0.2–0.5 mm isopropyl-1-thio-β-d-galactopyranoside when an absorbance value of 0.8–1.0 A600 nm was reached. Cells were centrifuged for 15 min at 6000 rpm in a Sorvall GSA rotor, resuspended 1:3 (w/v) in buffer A (50 mm phosphate, 500 mm NaCl, pH 7.4), treated with lysozyme (1.0 mg/g of wet cells) and BenzonaseTM (Novagen, 25.0 units/g of wet cells) for 1 h at room temperature, and finally lysed sequentially by a French press and a sonicator while kept refrigerated. The lysate was clarified by centrifugation for 30 min at 60,000 × g in a Beckman 70 Ti rotor, and the cell-free extract was applied to a HisTrap HP 1.0-ml FPLC column (GE Healthcare). After two washing steps of 10 column volumes of buffer A and 10 column volumes of buffer A supplemented with 0.1 m imidazole, the elution was performed in 20 column volumes of buffer A by applying a linear gradient of 0.1–0.5 m imidazole. Fractions containing the expected protein band were pooled and dialyzed against 1× PBS (20.0 mm phosphate buffer, 150 mm NaCl, pH 7.3), concentrated with an Amicon Ultracel® 10K (Millipore), and finally stored at −20 °C with the addition of 20% glycerol. SDS-PAGE assessed the purity of the proteins, and their concentration was determined with a Bio-Rad protein assay kit (Bio-Rad Pacific).
Radioactive Assay for O6-Methyl-guanine DNA Methyltransferase Activity
This method is based on the use of a radiolabeled 38-bp dsDNA substrate (ds-UPm), obtained by annealing two complementary oligonucleotides (UPm and DOWN, supplemental Table I), which contains a single O6-methyl-guanine overlapping an MboI site. The MboI endonuclease is unable to digest the methylated site unless the methyl group is removed, giving two fragments of 23 and 19 bp (see Fig. 1A). After annealing, the fragment was 3′-labeled with [32P]αCTP using 5.0 units of Klenow fragment for 1 h at 37 °C. The mixture containing the labeled dsDNA was incubated with the appropriate amounts of SsOGT at 70 °C for 10 min in OGT reaction buffer (50.0 mm Tris-HCl, 1.5 mm DTT, 0.1 mm EDTA, 20% glycerol, pH 8.0), and the reaction was stopped on ice and incubated with 5.0 units of MboI supplemented with 10.0 mm MgCl2 and 0.1 m NaCl for 1 h at 37 °C. Samples were denatured after the addition of a stop solution (95% formamide, 20.0 mm EDTA, 0.05% bromphenol blue) and run on an 8% sequencing gel. This was analyzed by autoradiography with a Storm PhosphorImager system (GE Healthcare).
FIGURE 1.

O6-methyl-guanine DNA methyltransferase activity of SsOGT. A, scheme of the radioactive assay (see “Experimental Procedures” for details). B, autoradiography of the reactions with 50 ng of SsOGT, in the presence of a radiolabeled 38-bp dsDNA containing or not a single methyl-guanine (ds-UPm and ds-UP, respectively). Reactions were incubated at 70 °C for 1 h; then, 5.0 units of MboI endonuclease were added where indicated, and incubation continued at 37 °C (see Table 1 and “Experimental Procedures” for details).
Fluorescent Assay for O6-Alkyl-guanine alkyltransferase Activity
In each sample, about 5.0 μm of protein (0.1 μg/μl) was incubated at 25 °C with 5.0 μm of the SNAP-Vista GreenTM substrate in a total volume of 10.0 μl of 1× Fluo Reaction Buffer (50.0 mm phosphate, 100 mm NaCl, 1.0 mm DTT, pH 6.5) for 20 min. Samples were denatured and loaded on 15% acrylamide SDS-PAGE. Bands were visualized by direct gel imaging using the VersaDoc 4000TM system (Bio-Rad), applying a blue LED/530 bandpass filter as excitation/emission parameters, respectively; the intensity of the bands was determined using the Quantity OneTM software. Values were corrected for the actual amount of protein loaded in each lane, by measuring the intensity of bands after Coomassie Blue staining, and final data were plotted by using the GraFit 5.0 software (15). The reaction conditions described above were conventionally indicated as standard; any changes from these conditions are indicated. An activity assay at different pH conditions was carried out in 50.0 mm sodium citrate (pH 3.0, 4.0, and 5.0), 50.0 mm sodium phosphate (pH 6.0, 6.5, and 7.0), 50.0 mm Tris-HCl (pH 7.5 and 8.0), and 50.0 mm borate (9.0 and 10.0). For thermostability studies, 10-μl aliquots of reactions containing 5.0 μm of the protein in 1× Fluo Reaction Buffer, incubated at the appropriate temperatures for different time spans, were centrifuged for 2 min at 16,000 × g. Soluble fractions were then incubated in the presence of the substrate under standard conditions. Second-order rate constants of the labeling reactions were determined by incubation of purified SsOGT or SsOGT-H5 mutant (5.0 μm) in the presence of an excess of SNAP-Vista GreenTM (20.0 μm) in 1× Fluo Reaction Buffer at 25 and 70 °C, taking aliquots at different times. Final corrected data from gel imaging were used for the determination of constants as described (16).
Determination of Kinetic Parameters for the Reaction of SsOGT and SsOGT-H5 Mutant
SsOGT samples (5.0 μm) were incubated with SNAP-Vista GreenTM in the range of 0.1–20.0 μm for 1 h at 50 °C and loaded on 15% acrylamide SDS-PAGE. Assuming the irreversible mechanism with 1:1 substrate/enzyme binding, corrected fluorescence intensity data were fitted by a linear equation, whose slope gave a direct reference value of fluorescence intensity/micromolar protein concentration, which was then used to estimate the amount of covalently modified protein (in pmol) in time-course experiments. These were performed at 50 °C using different protein/SNAP-Vista GreenTM molar ratios, taking 10.0-μl aliquots at specified intervals. Plots of the pmol of modified proteins versus time were fitted by exponential equations for the determination of the apparent rates for covalent modification (kobs). These values were plotted versus the SNAP-Vista GreenTM doses (VG) using the hyperbolic equation 1,
 |
where k and KS (referred to as KVG throughout) are the rate of covalent linkage and the dissociation constant for the free enzyme and free substrate during the first collision step, before covalent modification, respectively.
Competition Assay with ds-UPm
Time-course experiments similar to those described above were performed in the presence of increasing fixed doses of ds-UPm to calculate apparent KVG values (KVGapp) of proteins for SNAP-Vista GreenTM as a function of ds-UPm concentrations. Data were fitted according to the linear equation 2,
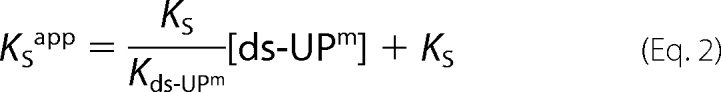 |
where Kds-UPm (referred to as KDNA throughout) is the affinity of the proteins for the ds-UPm substrate.
Labeling of SsOGT in E. coli Cells
An appropriate volume of culture of freshly transformed E. coli ABLE C cells was washed twice in the same volume of 1× Fluo Reaction Buffer and finally resuspended in 50.0 μl of the same buffer supplemented with 5.0 μm of the SNAP-Vista GreenTM substrate. After an incubation at 37 °C for 30 min, cells were washed twice with 1.0 ml of buffer and incubated for 30 min at 37 °C to allow the external diffusion of the unreacted substrate. Labeling was first verified by fluorescence imaging of whole cells extracts loaded on SDS-PAGE and then by fluorescence microscopy by using a DM-6000TM (Leica) microscope equipped with a 63× lens.
DNA Binding Assay
A tetramethylrhodamine-labeled dsDNA fragment was prepared by annealing the oligonucleotides A+ and D− (supplemental Table I). The fluorescent probe (0.2 μm) was incubated at 37 °C for 10 min with different amounts of proteins in a range of 0.0–25.0 μm, in a total volume of 10.0 μl of 1× binding buffer (20.0 mm Tris-HCl, 50.0 mm KCl, 0.1 mm DTT, 10% glycerol, pH 7.5). Samples were immediately loaded on a 5% polyacrylamide native gel in 1× TBE (90.0 mm Tris-HCl, 90.0 mm boric acid, 2.0 mm EDTA, pH 8.3). Signals were visualized by direct gel imaging using a green LED/605 bandpass filter as excitation/emission parameters, respectively.
Real Time-PCR
Culture growth and MMS treatment, RNA extraction, and cDNA preparation were performed as described previously (8). Real time PCR was performed by using the Ogt-left and Ogt-right oligonucleotides (supplemental Table I); normalization was with the 16 S rRNA (8).
Cell-free Extract Preparation and Analysis
Culture growth and MMS treatment were performed as described previously (8, 17). Normalization was with either the anti-DNA single-strand binding protein (18) or the anti-β-subunit of prefoldin (8) antibody.
RESULTS
Identification of SsOGT and Development of a New Assay for Its Activity
The S. solfataricus ORF SSO2487 encodes a putative 151-amino acid polypeptide homologous to DNA alkyltransferases, which we called SsOGT, with a predicted molecular mass of 17.0 kDa. Amino acid sequence alignment of SsOGT with hAGT and selected homologs from thermophilic archaea and bacteria showed conservation of sequence motives known to be involved in protein activity (supplemental Fig. S1A). Given the high (67.5%) amino acid sequence identity with the S. tokodaii homolog, we used the crystal structure of the latter (PDB entry 1WRJ) to obtain a reliable three-dimensional structure prediction for SsOGT. The model predicts high overall structural similarity between SsOGT and hAGT (supplemental Fig. S1B).
A His-tagged version of SsOGT, overexpressed and purified from E. coli cultures, revealed a monomeric structure in solution, as determined by size exclusion chromatography (data not shown). To test SsOGT activity, we first used a gel-based assay (3, 7). We designed a dsDNA oligonucleotide substrate (ds-UPm) containing a single O6-methylated guanine overlapping a restriction site for the methylation-sensitive MboI enzyme; removal of the methyl group by SsOGT would restore MboI cleavage assay (Fig. 1A). SsOGT turned out to be a bona fide DNA alkyltransferase (Fig. 1B).
This assay was quite laborious and time-consuming, as are all assays currently available to test DNA alkyltransferase activity (3, 7, 19–21). We thus set up an alternative, simpler, and faster method to analyze OGT activity, based on fluorescent derivatives of O6-BG. This molecule is known to inhibit hAGT by covalent transfer of the benzylic group to the active site cysteine (2), and its fluorescent conjugates are currently used to label derivatives of hAGT (SNAP-tagTM) (16, 22, 23). We reasoned that because of the irreversible nature and the 1:1 stoichiometry of the reaction, the amount of bound inhibitor could be used as a direct measure of the protein activity; in addition, thanks to the covalent protein-inhibitor bond, the complex could be analyzed even under denaturing conditions (Fig. 2A).
FIGURE 2.

Fluorescent assay for analysis of SsOGT activity. A, scheme of the assay with the fluorescent substrate/inhibitor SNAP-Vista GreenTM. B, 5.0 μm purified SsOGT was incubated with increasing concentrations of SNAP-Vista GreenTM, as indicated, under standard conditions and run on SDS-PAGE; a typical gel is shown where F indicates fluorescent protein-inhibitor bands and C indicates Coomassie Blue-stained bands. Histograms report the mean ± S.D. of the corrected data, obtained from gel imaging, from three independent experiments. C, plot of the first-order rate constants for covalent modification of SsOGT (kobs) as a function of SNAP-Vista GreenTM concentration. Rates values obtained from time-course experiments (inset) were fitted according to Equation 1. D, competition assay. 5.0 μm SsOGT were assayed under standard conditions at the indicated dsDNA/SNAP-Vista GreenTM molar ratios. Each lane contained ∼2.0 μg of protein. The histogram reports the quantification of data from two independent experiments, corrected as reported for panel B; symbols are as in panel B.
We first tested the SsOGT activity toward a fluorescein derivative of O6-BG (SNAP-Vista GreenTM) by incubation at 70 °C; activity was assessed by quantification of fluorescent protein band in SDS-PAGE. SNAP-Vista GreenTM was bound covalently, and the fluorescent band intensity showed a linear dependence on the inhibitor concentration (Fig. 2B), reaching a plateau at 1:1 protein/inhibitor ratio, thus confirming the predicted stoichiometry and suicide nature of this reaction. Using this technique, as little as 10 ng of protein could be detected (data not shown). Time-course experiments performed at different protein/fluorescent substrate/inhibitor ratios (Fig. 2C) led to the determination of kinetic constants for the covalent modification of SsOGT by SNAP-Vista GreenTM at 50 °C, which are listed in Table 1.
TABLE 1.
Kinetic parameters for the reaction of SsOGT with SNAP-Vista Green™ in the absence and in the presence of ds-UPm DNA
Constants are obtained by fitting the data to the hyperbolic Equation 1 shown under “Experimental Procedures.”
| ds-UPm | k | KVG |
|---|---|---|
| μm | s−1 | μm |
| 0 | 0.14 ± 0.01 | 10.25 ± 2.41 |
| 0.63 | 0.17 ± 0.01 | 21.14 ± 1.73 |
| 1.00 | 0.15 ± 0.02 | 25.68 ± 2.14 |
| 1.25 | 0.13 ± 0.01 | 27.73 ± 3.81 |
To compare the affinity of SsOGT for the inhibitor versus a natural substrate, we performed competition assays by adding increasing amounts of ds-UPm or its non-methylated counterpart (ds-UP (supplemental Table I)) to standard SsOGT labeling reactions (Fig. 2D). In the presence of ds-UPm, strong reduction of the fluorescent labeling efficiency was observed, which dropped to about 90% at 1.0:1.0 DNA/SNAP-Vista GreenTM ratio. No competition was observed with ds-UP, even at high DNA/inhibitor ratio, suggesting that inhibition is due to the presence of the methyl-guanine group and not to nonspecific binding to DNA. The kinetic constants determined in the presence of fixed increasing amounts of ds-UPm showed, as expected, similar k values but increase of the dissociation constant KVG with increasing DNA concentrations (supplemental Fig. 2A; Table 1). The reciprocal values of the experimental data were fitted to the linearized form of equation 1 (supplemental Fig. S2A, inset). All the plots reached a similar intercept but showed a rising slope with increasing concentration of ds-UPm, confirming that SNAP-Vista GreenTM and ds-UPm compete for the same active site. A linear plot of the KVG values as a function of the ds-UPm concentration (according to Equation 2; supplemental Fig. S2B) allowed the calculation of the affinity of SsOGT for ds-UPm (KDNA = 0.77 ± 0.01 μm), which was an order of magnitude lower than KVG (Table 1), confirming the data reported in Fig. 2D. Taken together, these experiments demonstrate that our assay can be used for the exact determination of the protein affinity for different natural as well as synthetic substrates and inhibitors.
Fluorescent labeling of SsOGT could be seen in intact E. coli cells incubated with SNAP-Vista GreenTM (Fig. 3), and gel imaging after SDS-PAGE of whole E. coli cell extracts expressing SsOGT revealed a single fluorescent band of the expected molecular weight (data not shown). From these experiments, we conclude that fluorescent labeling is a sensitive and specific method for qualitative and quantitative analysis of SsOGT activity, and we used it in all the subsequent experiments.
FIGURE 3.

In vivo labeling of SsOGT in E. coli ABLE C cells. Cultures transformed with either pQE31TM or pQE-ogt were incubated with SNAP-Vista GreenTM for 30 min at 37 °C and analyzed by fluorescence microscopy.
Biochemical Characterization of SsOGT
In agreement with the thermophilic nature of its natural source, the SsOGT optimal activity was at 80 °C; however, significant activity was found at temperatures as low as 25 °C (Fig. 4A), a property quite unusual for thermophilic proteins, which generally show very low or no activity at low temperatures. Second-order rate constant calculations of the reaction showed that at 70 °C, the SsOGT reaction is very fast and is completed in a few minutes. The values obtained at this temperature (5.33 ± 1.49 × 104 s−1 m−1) were in the same order of magnitude of that measured at 24 °C for the modified hAGT SNAP-tagTM (2.8 × 104 s−1 m−1) by using a similar substrate (16). When the same experiment was performed at 25 °C, SsOGT was found ∼20-fold less active (0.28 ± 0.03 × 104 s−1 m−1).
FIGURE 4.

Biochemical characterization of wild type SsOGT and SsOGT-H5. A, activity assay at different temperatures. Reactions were performed for 1.0 min under standard conditions except for the temperature used. The activity at the optimal temperature (80 and 70 °C for the wild type and H5 mutant, respectively) was referred to as 100%. Data are from two independent experiments for each assay. B, pH-dependent activity. Reactions were performed under standard conditions except for the pH and/or buffer used. The activity at the optimal pH (6.0) was referred to as 100%. Data are from two independent experiments for each assay. Error bars in panels A and B indicate mean ± S.D. C, assay under different reaction conditions. Reactions were in standard conditions, with the differences indicated in the histogram. Quantification was as described in the legend for Fig. 2. Data are from a single experiment for each condition. mut, mutant.
SsOGT showed significant activity in the pH 5.0–8.0 interval, with an optimum at pH 6.0 (Fig. 4B), and was tolerant to a number of different reaction conditions (Fig. 4C), such as ionic strength (0.0–1.0 m NaCl) and glycerol (up to 20%). The protein was also resistant to low concentrations of SDS and Triton X-100 (0.01% and 0.1–0.5%, respectively), whereas it was markedly more active in sarcosyl at all tested concentrations (0.1–1.0%). Interestingly, the protein was insensitive to EDTA up to 10.0 mm, suggesting that in contrast to hAGT, SsOGT does not contain any structural Zn2+ ion. SsOGT activity showed high thermal stability (supplemental Table II).
Analysis of SsOGT Mutants in DNA Binding Activity
Structural analysis has shown that in hAGT, DNA binding depends on residues forming a helix-turn-helix motif, which is involved in the direct interaction with the DNA minor groove (7); in particular, mutation of the arginine 128 residue was shown to reduce DNA binding (24). Sequence alignment and structural prediction showed strong conservation of these residues among AGT homologs (supplemental Fig. S1A). To test the involvement of these residues in DNA binding, we obtained the single mutant SsOGT-R102A (with Arg-102 corresponding to Arg-128 in hAGT) and a multiple mutant (SsOGT-H5) carrying, in addition to R102A, substitution of four conserved residues in the helix-turn-helix domain (S100A, G105K, M106T, K110E). The mutant proteins were expressed and purified as described for the wild type. DNA binding assays performed with a tetramethylrhodamine-labeled oligonucleotide (supplemental Table I) showed that the wild type SsOGT binds dsDNA in a cooperative manner, as reported for hAGT (24), with a plateau at a protein/DNA ratio of 15 (Fig. 5A). The wild type protein was also able to bind ssDNA with comparable efficiency (not shown). Although SsOGT-R102A showed reduced, but not completely abolished DNA binding, SsOGT-H5 was completely defective in both dsDNA and ssDNA binding, even at high protein/DNA ratio (Fig. 5A and data not shown). In competition experiments performed by adding increasing amounts of ds-UPm or ds-UP to standard reactions with SNAP-Vista GreenTM (Fig. 5B), moderate reduction of the fluorescent labeling efficiency was observed with SsOGT-R102A in the presence of the methylated oligonucleotide; no reduction was observed with the H5 mutant, showing that the amino acid substitutions introduced by mutagenesis completely abolished interaction with DNA. This latter mutant was thus used for further analysis. When this mutant was incubated for 1 h with saturating concentrations of SNAP-Vista GreenTM at 70 °C and the reaction end points were analyzed, SsOGT-H5 showed the same labeling efficiency as the wild type (supplemental Fig. S3). Kinetic analysis was performed as reported for the wild type protein, with the exception that, to calculate the KVG, the reciprocal values of the experimental data were fitted by linearization of Equation 1. This analysis showed reduced affinity of the mutant for SNAP-Vista GreenTM as compared with the wild type, with no reduction of the reaction rate, suggesting that the mutations affected, besides DNA, also the inhibitor binding. As reported in Table 2, identical KVG values were obtained in the absence and in the presence of 1.25 μm ds-UPm, indicating that the presence of this substrate does not affect the labeling efficiency of SsOGT-H5, confirming the data in Fig. 5B. These experiments suggest that our assay can also be applied to the characterization of proteins with impaired DNA binding. Using this assay, we found that the SsOGT-H5 protein shows pH dependence and activity under different conditions not significantly different from the wild type (Fig. 4, B and C). The temperature dependence was also similar to that of the wild type in the 25.0–80.0 °C range, whereas at 90.0 °C, the activity of the H5 mutant was reduced (Fig. 4A), likely due to lower stability at this temperature, as also suggested by thermal stability analysis (supplemental Table II).
FIGURE 5.

Effect of mutation of helix-turn-helix motif of SsOGT. A, DNA binding assay of wild type SsOGT and H5 mutant. Proteins were incubated in the presence of a tetramethylrhodamine-labeled dsDNA at different DNA/protein molar ratios. Native 5% polyacrylamide gel was analyzed by gel fluorescence imaging. B, competition assay for the SsOGT-R102A and H5 mutants. The conditions of the assay were identical to those used for the analysis of the wild type (see “Experimental Procedures” and legend for Fig. 2). Data are from two independent experiments. Error bars indicate mean ± S.D.
TABLE 2.
Comparison of kinetic parameters for the reaction of SsOGT and SsOGT-H5 mutant
Constants are obtained by fitting the reciprocal of data to the linearized Equation 1 shown under “Experimental Procedures.”
| ds-UPm | k | KVG | |
|---|---|---|---|
| μm | s−1 | μm | |
| SsOGT | 0 | 0.12 ± 0.01 | 4.66 ± 0.07 |
| 1.25 | 0.13 ± 0.01 | 23.16 ± 0.01 | |
| SsOGT-H5 | 0 | 1.1 ± 0.4 | 36.44 ± 7.96 |
| 1.25 | 1.2 ± 0.3 | 37.54 ± 5.85 |
SsOGT in DNA Damage Response
Treatment of S. solfataricus cultures with >0.25 to <0.7 mm MMS induce transient growth arrest lasting up to 24 h followed by growth resumption, suggesting efficient repair of alkylation damage. In contrast, MMS concentrations higher than 0.7 mm induce cell death, DNA fragmentation, and degradation of proteins involved in DNA damage response and tolerance (Fig. 6A) (8, 25).
FIGURE 6.

SsOGT in DNA damage response. A, S. solfataricus P2 cultures were grown at 80 °C until the exponential phase (time 0). At this time, cultures were split, and MMS was added to each aliquot at the indicated concentrations; one aliquot was mock-treated. Absorbance at 600 nm was measured at the indicated time points. Values are from five independent experiments. O.D., optical density. B, real time RT-PCR. Quantification of the ogt RNA in S. solfataricus in the absence and after 1 h of treatment with MMS at the indicated concentrations is shown. Data are from four independent experiments. C, Western blot analysis of total and soluble protein extracts (50 μg/lane) from S. solfataricus cultures prepared 2 h after MMS or mock treatment at the indicated concentrations. R, 300 ng of purified recombinant SsOGT. a-βpref., anti-β-prefoldin. D, Western blot of soluble protein extracts (50 μg/lane) from cultures treated with the indicated MMS doses for the indicated times. R, 20 ng of purified SsOGT. In the lane with the asterisk, only 10 μg of control extract was loaded. All filters were stripped and probed sequentially with anti-SsOGT (to follow the endogenous SsOGT) and either anti-DNA single-strand binding protein or anti-β-prefoldin (to control for protein loading). The histogram shows the quantification of SsOGT level normalized to the level of the reference protein in each lane; data are from four independent experiments. Error bars in panels A, B, and D indicate mean ± S.D. a.u., arbitrary units.
Quantitative RT-PCR experiments showed that the SsOGT steady-state RNA level increases 1 h after the addition of MMS in a drug concentration-dependent fashion (Fig. 6B), thus confirming that the protein is involved in alkylation damage repair. An antibody raised against the recombinant SsOGT detected a single protein band of the expected molecular weight in total extracts as well as soluble fractions prepared from exponentially growing S. solfataricus cultures (Fig. 6C); no protein band was detected in insoluble fractions (not shown). Surprisingly, despite the observed transcriptional induction, the intracellular SsOGT protein level was significantly reduced 2 h after treatment with 0.35 mm MMS and was almost absent if 1.4 mm MMS was used (Fig. 6C), suggesting that the protein is degraded after the alkylating agent treatment. Because methylated AGT undergoes specific degradation in human cells (26), we hypothesized that a similar mechanism may function in S. solfataricus. The analysis of SsOGT content in extracts from cells treated with different MMS conditions for different time spans showed different kinetics of SsOGT disappearance, depending on the drug concentration. 0.25 mm MMS induced slight reduction of the protein, detectable 6 h after treatment (Fig. 6D). At 0.35 mm, the protein level reduction was seen 2 h after the addition of the drug, reached the minimum at 6 h (when the protein content dropped to about one-fifth of that found in untreated cells), and started increasing again after 24 h (Fig. 6D). Finally, at 0.7 mm, SsOGT reduction was detectable 30 min after MMS addition and was more marked after 2 h. No increase in the protein content was seen at later times (Fig. 6D and data not shown). Thus, the protein content showed inverse correlation with MMS concentrations, which was consistent with alkylation-dependent degradation.
To test whether SsOGT degradation is dependent on its alkylation, we incubated normal or alkylated recombinant SsOGT with extracts from either controls or 0.35 mm MMS-treated cultures (Fig. 7A). The same level of His-tagged SsOGT was observed when the protein was incubated with both extracts; however, if the protein had been previously alkylated in vitro by incubation with ds-UPm, its level was reduced in extracts from MMS-treated, but not control, cultures. Thus, SsOGT alkylation triggers its degradation, but only in extracts from MMS-treated cells. In addition, when extracts from control cells were incubated with either 1.0 mm MMS or the ds-UPm oligonucleotide, endogenous SsOGT was not degraded (Fig. 7B), showing that not only the recombinant, but also the endogenous, SsOGT is not degraded in extracts from untreated cells, regardless of its alkylation status. Taken together, these experiments suggest that SsOGT degradation depends on its alkylation and is a process activated in vivo in response to alkylation damage.
FIGURE 7.

A, purified recombinant SsOGT was incubated for 1 h at 37 °C with 4-fold molar excess of either ds-UP or ds-UPm oligonucleotides, to obtain SsOGT or the methylated form (SsOGTm), respectively. 50 or 100 ng of either SsOGT or SsOGTm was mixed and incubated for 30 min at 70 °C with extracts from control or 0.35 mm MMS-treated cultures (200 μg/lane). Filters were probed with the anti-His-tag antibody. B, soluble extracts (80 μg/lane) from control cultures were incubated for 1 h at 70 °C in the presence of MMS (1.0 mm) and the ds-UPm or ds-UP oligonucleotides. R, 200 ng of purified SsOGT. Filters were stripped and probed sequentially with anti-SsOGT (to follow the endogenous SsOGT) and anti-β-prefoldin (a-βpref.) (to control for protein loading).
DISCUSSION
To fully characterize SsOGT activity, we have developed a novel method based on fluorescent derivatives of the inhibitor O6-BG, which are currently used to follow derivatives of hAGT, called SNAPTM, used as protein tag (16, 22, 23). We report here for the first time that such inhibitors are convenient analytical tools to assay AGT activity. The method, which can be applied to all AGTs sensitive to benzyl-guanine inhibitions, including the human protein, allows the determination of kinetics parameters for the covalent modification reaction as well as determination of the protein affinity for different substrates, is simple and reliable, is highly sensitive, specific, and quantitative, and allows analysis under both native and denaturing conditions.
In hAGT, the benzyl group of the O6-BG interacts with the Pro-140 residue, which facilitates the correct orientation of the inhibitor in the active site (4); AGTs lacking this residue are resistant to the inhibitor. Interestingly, despite the absence of the corresponding proline residue, SsOGT was significantly inhibited by O6-BG derivatives, suggesting that other interactions stabilize the inhibitor in the active site pocket.
The careful biochemical characterization of SsOGT showed that despite its thermophilic character and its high thermal stability, it also shows considerable activity at low temperature. In addition, it is active under a wide range of reaction conditions.
We have shown that our assay can be used, in combination with mutational analysis, to study the biochemical and kinetic properties of mutants impaired in DNA binding. We showed that both the Arg-102 residue and the helix-turn-helix domain are involved in DNA binding of SsOGT and that a multiple mutant completely defective for DNA binding can still be labeled efficiently by SNAP-Vista GreenTM.
Methylating agents are among the most commonly occurring mutagens in nature. Thermophilic organisms may be exposed to these agents, as by-products of biomass decay, as well as to alkylation damage generated by endogenous methylating agents, such as S-adenosylmethionine and N-nitroso compounds, which may be highly reactive at high temperatures. We reported previously that treatment of S. solfataricus cultures with high doses of MMS induces coordinate degradation of the complex formed by two proteins playing key roles in DNA damage response, namely reverse gyrase and the translesion polymerase PolY (8); this occurs in turn in concomitance with DNA fragmentation and cell death (25). We suggested that this apoptotic-like phenomenon might act, in the presence of irreparable DNA damage, to specifically eliminate proteins involved in lesion repair/tolerance. We now report that SsOGT is also degraded in response to MMS treatment, which might be part of the same phenomenon. However, several lines of evidence suggest that this is not the case and that rather, SsOGT is degraded by a different pathway. Indeed, in contrast to reverse gyrase/PolY, SsOGT is degraded even after treatment with low, not lethal, MMS concentrations, although the protein level is inversely related to MMS doses. In addition, degradation requires alkylation of SsOGT and some pathway activated in vivo in response to alkylation damage. Indeed, alkylated SsOGT, either endogenous or recombinant, is degraded by some factor(s) in extracts from MMS-treated, but not control cells; the same result is not obtained by direct addition of the alkylating agent or methylated DNA to extracts from untreated cells. In similar experiments, endogenous or recombinant reverse gyrase was also degraded by incubation in vitro with extracts from MMS-treated cells, but in that case, direct alkylation of the protein was not required (8).3
Alkylated AGT undergoes rapid degradation in human and yeast cells (27, 28); thus, our results suggest once more a striking evolutionary conservation, from archaea to higher eukaryotes, of an important pathway dealing with the maintenance of the genetic information, as repeatedly reported for all DNA metabolic processes (29).
Degradation of human AGT is triggered by a conformational modification caused by the addition of the alkyl group, leading to recognition by ubiquitin ligases and channeling to the proteasome pathway. All archaea contain 20 S core proteasome particles (30), suggesting that SsOGT might be degraded by this pathway. In addition, most archaea also encode ubiquitin-like proteins called UBl and small archaeal modifier proteins (SAMPs) (31, 32). Recently, small archaeal modifier proteins were found to be covalently linked to proteins, suggesting that they might function like ubiquitin in eukaryotes in degradation pathways (32). However, no such evidence is available so far for S. solfataricus. Moreover, we did not find evidence of post-translational modifications of SsOGT before degradation; thus, either the alkylated protein is targeted to degradation without further modification, or degradation follows ubiquitin-like modification very rapidly. Further studies are underway to unravel the mechanism and factors involved in degradation of OGT in archaea.
Acknowledgments
We are grateful to Giuseppe Di Tullio (Mario Negri Sud Institute, Chieti, Italy) for the anti-SsOGT antibody and to Vito Carratore (Institute of Protein Biochemistry, Consiglio Nazionale delle Ricerche, Naples, Italy) for N-terminal protein sequencing.
This work was partially supported by Merit Grant RBNE08YFN3 and Ministero degli Affari Esteri (Grant L. 401/1990).

This article contains supplemental Tables I and II and Figs. S1–S3.
A. Valenti, unpublished results.
- AGT/OGT
- O6-alkyl-guanine-DNA alkyltransferases
- hAGT
- human AGT
- SsOGT
- OGT from S. solfataricus
- MMS
- methyl methanesulfonate
- O6-BG
- O6-benzylguanine.
REFERENCES
- 1. Pegg A. E. (2011) Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 24, 618–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang C. G., Garcia K., He C. (2009) Damage detection and base flipping in direct DNA alkylation repair. ChemBioChem 10, 417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fang Q., Kanugula S., Pegg A. E. (2005) Function of domains of human O6-alkyl-guanine-DNA alkyltransferase. Biochemistry 44, 15396–15405 [DOI] [PubMed] [Google Scholar]
- 4. Daniels D. S., Mol C. D., Arvai A. S., Kanugula S., Pegg A. E., Tainer J. A. (2000) Active and alkylated human AGT structures: a novel zinc site, inhibitor, and extrahelical base binding. EMBO J. 19, 1719–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rabik C. A., Njoku M. C., Dolan M. E. (2006) Inactivation of O6-alkyl-guanine DNA alkyltransferase as a means to enhance chemotherapy. Cancer Treat. Rev. 32, 261–276 [DOI] [PubMed] [Google Scholar]
- 6. Margison G. P., Povey A. C., Kaina B., Santibáñez Koref M. F. (2003) Variability and regulation of O6-alkyl-guanine-DNA alkyltransferase. Carcinogenesis 24, 625–635 [DOI] [PubMed] [Google Scholar]
- 7. Daniels D. S., Woo T. T., Luu K. X., Noll D. M., Clarke N. D., Pegg A. E., Tainer J. A. (2004) DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 11, 714–720 [DOI] [PubMed] [Google Scholar]
- 8. Valenti A., Napoli A., Ferrara M. C., Nadal M., Rossi M., Ciaramella M. (2006) Selective degradation of reverse gyrase and DNA fragmentation induced by alkylating agent in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 34, 2098–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roberts A., Pelton J. G., Wemmer D. E. (2006) Structural studies of MJ1529, an O6-methylguanine-DNA methyltransferase. Magn. Reson. Chem. 44, S71–S82 [DOI] [PubMed] [Google Scholar]
- 10. Nishikori S., Shiraki K., Okanojo M., Imanaka T., Takagi M. (2004) Equilibrium and kinetic stability of a hyperthermophilic protein, O6-methylguanine-DNA methyltransferase under various extreme conditions. J. Biochem. 136, 503–508 [DOI] [PubMed] [Google Scholar]
- 11. Leclere M. M., Nishioka M., Yuasa T., Fujiwara S., Takagi M., Imanaka T. (1998) The O6-methylguanine-DNA methyltransferase from the hyperthermophilic archaeon Pyrococcus sp. KOD1: a thermostable repair enzyme. Mol. Gen. Genet. 258, 69–77 [DOI] [PubMed] [Google Scholar]
- 12. Morita R., Nakagawa N., Kuramitsu S., Masui R. (2008) An O6-methylguanine-DNA methyltransferase-like protein from Thermus thermophilus interacts with a nucleotide excision repair protein. J. Biochem. 144, 267–277 [DOI] [PubMed] [Google Scholar]
- 13. Kanugula S., Pegg A. E. (2003) Alkylation damage repair protein O6-alkyl-guanine-DNA alkyltransferase from the hyperthermophiles Aquifex aeolicus and Archaeoglobus fulgidus. Biochem. J. 375, 449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. She Q., Singh R. K., Confalonieri F., Zivanovic Y., Allard G., Awayez M. J., Chan-Weiher C. C., Clausen I. G., Curtis B. A., De Moors A., Erauso G., Fletcher C., Gordon P. M., Heikamp-de Jong I., Jeffries A. C., Kozera C. J., Medina N., Peng X., Thi-Ngoc H. P., Redder P., Schenk M. E., Theriault C., Tolstrup N., Charlebois R. L., Doolittle W. F., Duguet M., Gaasterland T., Garrett R. A., Ragan M. A., Sensen C. W., Van der Oost J. (2001) The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc. Natl. Acad. Sci. 98, 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leatherbarrow R. J. (2004) GraFit, version 5.0, Erithacus Software Ltd., Staines, United Kingdom [Google Scholar]
- 16. Gautier A., Juillerat A., Heinis C., Corrêa I. R., Jr., Kindermann M., Beaufils F., Johnsson K. (2008) An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 15, 128–136 [DOI] [PubMed] [Google Scholar]
- 17. Napoli A., Valenti A., Salerno V., Nadal M., Garnier F., Rossi M., Ciaramella M. (2004) Reverse gyrase recruitment to DNA after UV light irradiation in Sulfolobus solfataricus. J. Biol. Chem. 279, 33192–33198 [DOI] [PubMed] [Google Scholar]
- 18. Wadsworth R. I., White M. F. (2001) Identification and properties of the crenarchaeal single-stranded DNA-binding protein from Sulfolobus solfataricus. Nucleic Acids Res. 29, 914–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishiguro K., Shyam K., Penketh P. G., Sartorelli A. C. (2008) Development of an O6-alkyl-guanine-DNA alkyltransferase assay based on covalent transfer of the benzyl moiety from [benzene-3H]O6-benzylguanine to the protein. Anal. Biochem. 383, 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishiguro K., Zhu Y. L., Shyam K., Penketh P. G., Baumann R. P., Sartorelli A. C. (2010) Quantitative relationship between guanine O6-alkyl lesions produced by OnriginTM and tumor resistance by O6-alkyl-guanine-DNA alkyltransferase. Biochem. Pharmacol. 80, 1317–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tintoré M., Aviñó A., Ruiz F. M., Eritja R., Fàbrega C. (2010) Development of a novel fluorescence assay based on the use of the thrombin binding aptamer for the detection of O6-alkyl-guanine-DNA alkyltransferase activity J. Nucleic Acids Article ID 632041, 9 pp [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K. (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89 [DOI] [PubMed] [Google Scholar]
- 23. Keppler A., Kindermann M., Gendreizig S., Pick H., Vogel H., Johnsson K. (2004) Labeling of fusion proteins of O6-alkyl-guanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods 32, 437–444 [DOI] [PubMed] [Google Scholar]
- 24. Lim A., Li B. F. (1996) The nuclear targeting and nuclear retention properties of a human DNA repair protein O6-methylguanine-DNA methyltransferase are both required for its nuclear localization: the possible implications. EMBO J. 15, 4050–4060 [PMC free article] [PubMed] [Google Scholar]
- 25. Rasimas J. J., Pegg A. E., Fried M. G. (2003) DNA binding mechanism of O6-alkyl-guanine-DNA alkyltransferase. Effects of protein and DNA alkylation on complex stability. J. Biol. Chem. 278, 7973–7980 [DOI] [PubMed] [Google Scholar]
- 26. Valenti A., Perugino G., Nohmi T., Rossi M., Ciaramella M. (2009) Inhibition of translesion DNA polymerase by archaeal reverse gyrase. Nucleic Acids Res. 37, 4287–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu-Welliver M., Pegg A. E. (2002) Degradation of the alkylated form of the DNA repair protein, O6-alkyl-guanine-DNA alkyltransferase. Carcinogenesis 23, 823–830 [DOI] [PubMed] [Google Scholar]
- 28. Hwang C. S., Shemorry A., Varshavsky A. (2009) Two proteolytic pathways regulate DNA repair by cotargeting the Mgt1 alkyl-guanine transferase. Proc. Natl. Acad. Sci. U.S.A. 106, 2142–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelman Z., White M. F. (2005) Archaeal DNA replication and repair. Curr. Opin. Microbiol. 8, 669–676 [DOI] [PubMed] [Google Scholar]
- 30. Maupin-Furlow J. A., Humbard M. A., Kirkland P. A., Li W., Reuter C. J., Wright A. J., Zhou G. (2006) Proteasomes from structure to function: perspectives from archaea. Curr. Top. Dev. Biol. Rev. 75, 125–169 [DOI] [PubMed] [Google Scholar]
- 31. Makarova K. S., Koonin E. V. (2010) Archaeal ubiquitin-like proteins: functional versatility and putative ancestral involvement in tRNA modification revealed by comparative genomic analysis. Archaea 2010, pii: 710303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Humbard M. A., Miranda H. V., Lim J. M., Krause D. J., Pritz J. R., Zhou G., Chen S., Wells L., Maupin-Furlow J. A. (2010) Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii. Nature 463, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]