

Background: Expressing TDP-43 in yeast mimics several aspects of human TDP-43-based neurodegenerative diseases.
Results: A screen for compounds that rescued TDP-43 toxicity identified functionally distinct 8-hydroxyquinoline metal chelators.
Conclusion: Different 8-OHQs exhibit distinct modes of action and implicate multiple metal-dependent protective mechanisms against TDP-43, α-synuclein, and polyglutamine toxicity.
Significance: 8-Hydroxyquinolines may ultimately be tailored to modify diverse neurodegenerative diseases in man.
Keywords: Drug Screening, Metals, Neurodegenerative Diseases, Small Molecules, Yeast, TDP-43, α-Synuclein, Polyglutamine
Abstract
No current therapies target the underlying cellular pathologies of age-related neurodegenerative diseases. Model organisms provide a platform for discovering compounds that protect against the toxic, misfolded proteins that initiate these diseases. One such protein, TDP-43, is implicated in multiple neurodegenerative diseases, including amyotrophic lateral sclerosis and frontotemporal lobar degeneration. In yeast, TDP-43 expression is toxic, and genetic modifiers first discovered in yeast have proven to modulate TDP-43 toxicity in both neurons and humans. Here, we describe a phenotypic screen for small molecules that reverse TDP-43 toxicity in yeast. One group of hit compounds was 8-hydroxyquinolines (8-OHQ), a class of clinically relevant bioactive metal chelators related to clioquinol. Surprisingly, in otherwise wild-type yeast cells, different 8-OHQs had selectivity for rescuing the distinct toxicities caused by the expression of TDP-43, α-synuclein, or polyglutamine proteins. In fact, each 8-OHQ synergized with the other, clearly establishing that they function in different ways. Comparative growth and molecular analyses also revealed that 8-OHQs have distinct metal chelation and ionophore activities. The diverse bioactivity of 8-OHQs indicates that altering different aspects of metal homeostasis and/or metalloprotein activity elicits distinct protective mechanisms against several neurotoxic proteins. Indeed, phase II clinical trials of an 8-OHQ has produced encouraging results in modifying Alzheimer disease. Our unbiased identification of 8-OHQs in a yeast TDP-43 toxicity model suggests that tailoring 8-OHQ activity to a particular neurodegenerative disease may be a viable therapeutic strategy.
Introduction
Neurodegenerative diseases (ND)3 such as Parkinson disease, Alzheimer disease (AD), amyotrophic lateral sclerosis (ALS), and frontotemporal lobar degeneration (FTLD) exact a terrible personal and economic toll on both families and the health care system. This burden will continue to grow as the population ages over the coming decades. A major issue thwarting therapeutic advancement for ND is identifying compounds that target processes central to disease pathogenesis.
Neurodegenerative diseases are typically associated with the aggregation of disease-specific proteins. For example, α-synuclein (α-syn) aggregates can be observed in most cases of Parkinson disease, multiple systems atrophy, and dementia with Lewy bodies (1). The Aβ peptide (generated from amyloid precursor protein) is the primary component of amyloid plaques and soluble toxic oligomeric species in AD (2), whereas TDP-43 aggregates are found in certain neuronal subtypes of patients with FTLD-TDP and ALS-TDP (3). In most cases the aggregated protein is not mutated yet still constitutes a major fraction of the protein inclusion. Importantly, rare genetic mutations in these genes, including α-syn, amyloid precursor protein, and TDP-43, provide strong evidence for causal relationships between the misfolding of that particular protein and disease. As a result, these diseases are frequently modeled by overexpressing wild-type or disease mutant proteins in a variety of cells or organisms.
One model that might initially seem of unlikely benefit in studying complex human diseases is the budding yeast, Saccharomyces cerevisiae. However, the discoveries of cell-cycle regulation, checkpoints, and DNA damage repair in yeast all proved relevant to human cancers. Moreover, despite lacking the multicellular and physiological complexity of a nervous system, yeast cells have most of the major conserved cellular functions implicated in neurodegeneration. These include an elaborate protein homeostasis machinery (chaperones, proteasome, endoplasmic reticulum-associated degradation, etc), vesicle trafficking, mitochondria, autophagy, and apoptosis, making it possible to model the unique, multifaceted toxicities of different proteins at the cellular level (4). Indeed, the lack of intercellular connections eliminates the layers of complexity introduced by those interactions and provides focus on basic underlying proteotoxicity. Furthermore, the rapid, robust growth, ease of manipulation, and extensive genetic tools make yeast better suited to high throughput chemical and genetic screens than mammalian cells (4).
The most well developed ND yeast model is that of α-syn toxicity. Synucleinopathies, such as Parkinson disease, are quintessentially complex diseases with both genetic and environmental factors culminating in α-syn pathology. These diseases are poorly understood, even in cases where an underlying genetic lesion is known. Regardless of this complexity, overexpression of wild-type α-syn in yeast results in a dose-dependent toxicity that recapitulates several salient features of the synucleinopathies in humans (5, 6). Genetic and chemical screens of this model identified genes and compounds that also rescue α-syn toxicity in multiple neuronal models (5, 7). Remarkably, despite one billion years of evolutionary distance, yeast genetic screens revealed a direct role for α-syn in disrupting vesicle trafficking, and this connection was subsequently verified in neuronal experimental systems (5, 8–10). The α-syn yeast model also provided new, direct connections among α-syn, manganese toxicity, and PARK9, a manganese transporter that when mutated causes early-onset parkinsonism with pyramidal degeneration and dementia (Kufor-Rakeb syndrome) (11, 12). These results highlight the exceptionally conserved nature of basic protein trafficking and protein homeostasis machinery in eukaryotic cells.
The modeling of proteotoxicity in yeast has been extended to other neurodegenerative disease proteins, including polyglutamine (polyQ) expansion proteins (e.g. Huntingtin) and TDP-43. Expression of a polyQ-expanded exon I fragment of the Huntingtin protein in yeast recapitulates the polyQ length-dependent toxicity observed in diseases, such as multiple spinocerebellar ataxias (e.g. SCA1, SCA2), Huntington disease, and Kennedy disease (13–15). These polyQ expansion diseases are caused by both nuclear and cytoplasmic proteins whose only common feature is expansion of glutamine stretches past a similar toxic threshold (15). In yeast and mammalian models, the polyQ-expanded exon 1 fragment of Huntingtin severely impairs endoplasmic reticulum-associated degradation and causes endoplasmic reticulum stress (16). Moreover, the sequences flanking the polyQ stretch (14) as well as other Q-rich proteins in the cell (17) can affect polyQ aggregation and toxicity. These experiments provide insights into how different polyQ expansion proteins might affect different neurons and cause diseases depending on both the cis and trans content of that neuron proteome.
The yeast TDP-43 proteinopathy model, the impetus for the current study, similarly recapitulates several relevant disease features (18, 19). Wild-type TDP-43 was first identified in 2006 as the primary component of insoluble aggregates in sporadic ALS and FTLD with ubiquitin-positive inclusions (20). Since then, rare TDP-43 mutations have further supported a causal role for the protein in ALS and FTLD (21, 22). TDP-43 is an RNA-binding protein that functions at various stages of RNA metabolism and contains an aggregation-prone, “prion-like” C terminus (23, 24). In neurons affected by disease, full-length TDP-43 often translocates to the cytoplasm where it is partially cleaved to generate 35- and 25-kDa C-terminal fragments (20). Wild-type TDP-43 similarly localizes to cytoplasmic foci in yeast and induces cellular toxicity. As in humans, this toxicity is enhanced by disease mutations (18, 19). Pbp1, the yeast homolog of Ataxin-2, one of the aforementioned polyglutamine proteins, was identified as an enhancer of wild-type TDP-43 toxicity through a yeast genetic screen (25). This connection was subsequently validated in a neuronal TDP-43 model (25). Importantly, Ataxin-2 co-localizes with wild-type TDP-43, and the polyglutamine tract in Ataxin-2 is moderately expanded in a fraction of ALS patients, again directly linking yeast discoveries with human disease (26).
Here, we describe a screen for compounds that rescue the growth of cells expressing TDP-43, with both long term and short term goals in mind. Ultimately, such a screen may provide either lead compounds themselves or identify new potential drug targets. Indeed, several blockbuster drugs, including statins, velcade, cisplatin, methotrexate, and omeprazole work in yeast as they do in human cells because they target proteins conserved from yeast to man (27). More immediately, we have identified rescuing compounds that provide a set of biological probes for dissecting overlapping and distinct features of proteotoxicity.
EXPERIMENTAL PROCEDURES
Plasmids and Yeast Strains
TDP-43-GFP integrating constructs were generated by Gateway cloning. TDP-43-GFP (18) was transferred to pRS303 (304 or 305)-Gal-attR destination clones by LR reactions. Expression clones were confirmed by restriction digest and DNA sequencing. TDP-43-GFP yeast strains were generated by integrating these constructs by homologous recombination at the HIS3, TRP1, or LEU2 loci in the yeast strain, W303 (MATa his3, leu2, trp1, ura3, pdr1::Kan, pdr3::Kan, where PDR1 and PDR3 are transcription factors that regulate drug efflux). Expression was confirmed by galactose induction followed by fluorescence microscopy or Western blot analysis. α-syn and htt-72Q strains were described previously (7, 14). Toxic genes were obtained from the FLEXgene library (28) and transformed into the same W303 strain. Strains and plasmids are described (supplemental Tables S1 and S2).
Small Molecule Screen
The small molecule screen was carried out at the Harvard Institute for Chemical and Cell Biology. Briefly, three-copy TDP-43-GFP was cultured overnight in raffinose to late log (OD600 ∼ 1–2). Cultures were then diluted to an OD600 of 0.01 in complete synthetic media containing 2% galactose. Forty microliters of culture was dispensed to clear bottom 384-well plates, after which 100 nl of compounds were pinned at a final concentration of ∼15–30 μm in DMSO, all in duplicate. Plates were incubated at 30 °C for 48 h, and OD600 was read with an Envision plate reader. Z scores were calculated according to the equation, (well OD600 − plate average)/(plate S.D.). Hits (Z score >3) across all plates were ranked. 8-Hydroxyquinolines (8-OHQ) hits were identified for subsequent followup in the current study.
Compounds
The following compounds were reconstituted in DMSO at either 10 or 20 mm: HQ-161 (5511452, Chembridge), HQ-415 (5994-0105, ChemDiv), Clioquinol (Sigma).
Compound Treatment in Yeast Models
Growth assays were performed in either Bioscreen CTM instruments or in 384-well plates. For TDP-43, overnight raffinose cultures were diluted into 2% galactose and compound/DMSO at an OD600 of 0.01. HiTox α-syn was diluted to an OD600 of 0.05, and htt-72Q was diluted to an OD600 to 0.02. For dose-response curves, OD600 at 24 h was used as an end point for YFP and α-syn, and 36 h was used for htt-72Q and TDP-43. EC50 values were determined from dose-response curves at the 50% maximal effect for each compound. “Toxic genes” were grown in media lacking uracil to maintain the plasmid and diluted to an OD600 of 0.01 for growth curves. All drug concentrations are noted within figures and legends.
Synergy experiments were performed by Bioscreen CTM with growth conditions and time points as described above. Concentrations of each compound in combination are noted in the figure.
Antioxidant treatment of yeast models was performed by Bioscreen CTM. Rescue of H2O2 toxicity with antioxidants or 8-OHQs was performed in 384-well plates.
Metal-responsive transcription factor experiments were performed in 384-well plates. Each strain was diluted to an OD600 of 0.01 in galactose-containing media and treated with a 6-point by 2-fold dilution series for each 8-OHQ. Only the instructive concentrations are shown for clarity. Significance was determined with a Student's t test.
Metal competition experiments with 8-OHQs were performed with Bioscreen CTM assays in each toxicity model diluted as described above. Equimolar concentration of metals (copper sulfate, iron chloride, zinc sulfate; Sigma) or a water control was added simultaneously with HQ-161, HQ-415, or clioquinol (CQ), and the same points were taken as described above. The rescue of 8-OHQ toxicity by metals was performed in 384-well plates. Equimolar concentrations of each metal were added with a toxic concentration of 8-OHQs to a wild-type yeast strain.
Yeast Microscopy
Microscopy was performed on formaldehyde-fixed cells that were grown in the same manner as the growth assays. Multiple concentrations of each drug were set up and the most efficacious used for microscopy. A 16-h time point was used for each treatment. Samples were coded, randomized, and thus blinded to the researcher until after analysis to prevent any bias. Images were taken according to bright field views (not fluorescence) to eliminate bias toward any phenotype.
Quantitation of phenotypes was performed on three independent treatments and averaged across ∼150 cells per experiment. For TDP-43, cells were scored as having 0–1 focus, 2 foci, or 3 or more foci. For α-syn, cells were scored as dead, having large accumulations, or plasma membrane-localized α-syn. For htt-72Q, cells were scored as having diffuse/punctate htt-72Q, moderate aggregates, or large aggregates. Significance was determined by one-way ANOVA and Tukey's multiple comparison test.
Compound Treatment and Analysis of C. elegans
The treatment and analysis of α-syn-induced neuronal degeneration has been described (5, 7). Compound concentrations are provided in the figures. At least three independent experiments were performed, and significance was determined by one-way ANOVA and Tukey's multiple comparison test.
RNA Analysis
RNA analysis was performed on yeast treated for 4 h with either DMSO or 8-OHQ. RNA was extracted using the hot acid-phenol method and reverse-transcribed with oligo(dT) primers using SuperScript III (Invitrogen). Gene-specific primers for CTR1, FRE3, or ZTR1 were used for quantitative real time PCR (supplemental Table S3; 7900HT Fast Real-Time PCR, Applied Biosystems). Expression values are normalized to the ACT1 transcript. Experiments were performed three independent times. Statistical significance was determined by one-way ANOVA and Tukey's multiple comparison test.
RESULTS
Small Molecule Screening of Yeast TDP-43 Toxicity Model Identifies 8-Hydroxyquinolines
To screen for compounds that alleviate TDP-43 toxicity, TDP-43-GFP was placed under the control of a galactose-regulated promoter that enabled tight repression in glucose-containing media and strong expression upon shifting cells to galactose. Single copies of WT TDP-43-GFP were integrated at each of three genomic loci where insertions themselves have no detrimental effects on neighboring gene expression or cell physiology. Induction of TDP-43 expression produced both dose-dependent aggregation and toxicity (Fig. 1, A and B). Pilot experiments with the strain containing three copies of the TDP-43 gene demonstrated that spontaneous suppressors, which can cause false positives during screening, were rare, as they would require deletion of more than one copy of TDP-43. Finally, genes controlling the expression of drug pumps were deleted to minimize efflux of compounds during screening.
FIGURE 1.

Small molecule screen of TDP-43 toxicity identifies 8-hydroxyquinolines. A, three yeast strains expressing one, two, or three copies of TDP-43-GFP show a dose-dependent increase in foci size and number. Green, TDP-43-GFP; blue, DNA/DAPI. B, increased TDP-43-GFP aggregation observed in A correlates with reduced growth rate and increase in toxicity. C, HQ-161 structure and dose-response curve in YFP and TDP-43-GFP yeast strains are shown. The functional chelating regions are circled in red. Values reflect the difference in growth (OD600) between the compound treated and DMSO control-treated strains at 36 h of a Bioscreen CTM growth experiment. Concentrations are in μm. The EC100 and EC50 are noted to the right of the dose-response curve. D, HQ-415 structure and dose-response curve in YFP and TDP-43-GFP yeast strains are shown. E, CQ structure and dose-response curve in YFP and TDP-43-GFP yeast strains are shown. HQ-161, HQ-415, and CQ are represented in blue, red, and green, respectively, throughout all figures.
The screen was performed by diluting 3× TDP-43 yeast cells into galactose-containing media, distributing them to 384-well plates, and pinning compounds (∼15–30 μm final concentration) to duplicate plates. After 48 h, the OD600 was read and Z-scores, defined as (OD600 well − OD600 plate avg)/S.D.plate), were determined. Approximately 200,000 compounds were screened from various commercial chemical libraries with a 0.53% hit rate (Z score above 3). One group of six 8-OHQ reproducibly scored as highly active compounds (supplemental Fig. S1). Z scores from four plates in duplicate sets (supplemental Fig. S2A) show the reproducibility of the screen and the strength of the 8-OHQ hits. 8-OHQs were also among the top hits when ranked with total screen hits (supplemental Fig. S2B).
These compounds bear considerable resemblance to the archetypical 8-OHQ, CQ (Fig. 1E; supplemental Fig. S1). The 8-OHQs are known weak, bidentate chelators of copper, iron, and zinc where the hydroxyl group and the quinoline nitrogen coordinate metals (Fig. 1, C–E, red dashed ovals). Importantly, CQ and a structural analog (PBT2) have shown protection against neurodegeneration in several different disease contexts (29–34). We, therefore, focused further investigation on the activities of these compounds, comparing their effects with that of CQ. We chose two structurally distinct 8-OHQs of molecular weight 161 and 415 (herein named HQ-161 and HQ-415) for comparison to CQ. Both compounds lack the aromatic halogens present in CQ (Fig. 1E), instead containing a lone hydroxyl group (HQ-161; Fig. 1C) or a large, aromatic-containing side group (HQ-415; Fig. 1D) off the 8-OHQ core. Importantly, these compounds are commercially available, freeing them for further study in other systems.
HQ-161 and HQ-415 were retested in dose for their ability to rescue TDP-43. They were also tested for their toxicity to YFP-expressing control cells. The results are reported as the difference in growth between cells treated with DMSO alone and cells treated with compounds dissolved in DMSO. Both HQ-161 and HQ-415 rescued TDP-43 toxicity (Fig. 1, C and D). The effective concentration eliciting a 50% maximal response (EC50) for HQ-161 and HQ-415 were ∼25 and 15 μm, respectively. The maximal effective concentration (EC100) ranges for HQ-161 and HQ-415 were 30–40 and 15–20 μm, respectively. Note that both HQ-161 and HQ-415 become toxic at concentrations just above their EC100.
Surprisingly, CQ itself was completely inactive (Fig. 1E). In fact, it was highly toxic to both YFP and TDP-43-expressing cells even at concentrations ∼20-fold lower than that observed for HQ-415 (Fig. 1F; 30 versus 1.5 μm). Unlike HQ-415 and CQ, both of which demonstrated considerable cellular toxicity, HQ-161 was non-toxic up to 80 μm (Fig. 1C). All three 8-OHQs, therefore, have different properties with respect to TDP-43 rescue and cellular toxicity.
The high concentration of these compounds required for efficacy and a relatively narrow therapeutic window likely compromise their therapeutic potential; however, their distinct properties make them useful probes for evaluating mechanisms of proteotoxicity.
Effects of Compounds on Other Protein Toxicities
Given the previously reported broad activity of 8-OHQs as modulators of cellular toxicity in ND models (30, 32, 33), we assayed the rescuing activity of 8-OHQs in two additional yeast models of proteotoxicity, α-syn and polyglutamine (htt-72Q). TDP-43 was rescued by HQ-161 and HQ-415, yet not by CQ (Fig. 2, A and B). Both α-syn and htt-72Q were rescued by HQ-415 and CQ, yet not by HQ-161 (Fig. 2, C and D for α-syn and E and F for htt-72Q). Thus, although 8-OHQs can rescue diverse proteotoxicities, they also exhibit considerable selectivity. Importantly, because these models were constructed in genetically identical recipient strains and differ only in the toxic protein being expressed, these differences cannot be due to variations in drug uptake or cellular metabolism.
FIGURE 2.

8-Hydroxyquinolines rescue multiple models of proteotoxicity. 8-OHQs were tested in TDP-43 (A and B), α-syn (C and D), and htt-72Q (E and F) models of proteotoxicity. A, shown is representative growth curve of TDP-43 with most efficacious concentration of each 8-OHQ. B, shown is a dose-response curve of TDP-43 with each 8-OHQ where values reflect the difference in OD600 between compound-treated and DMSO control-treated cultures at 36 h of a Bioscreen CTM experiment. The x axis is log10, and concentrations are in μm. EC100 and EC50 are noted below the dose-response curves. C, shown is a representative growth curve of α-syn with the most efficacious concentrations of each 8-OHQ. D, shown is a dose-response curve of α-syn with each 8-OHQ. Values were taken from a 24-h time point. E, shown is a representative growth curve of htt-72Q with the most efficacious concentration of each 8-OHQ. F, shown is a dose-response curve of htt-72Q with each 8-OHQ. Values were taken from a 36-h time point.
As a control, we tested whether 8-OHQs were generically protective of other toxic proteins expressed from the same galactose-regulated promoter. Each 8-OHQ was assayed for rescuing activity of nine yeast genes. These have diverse functions and are known to create different levels of toxicity when overexpressed, including levels comparable with our disease models. None of the three 8-OHQs provided any protection against these other toxic proteins. Indeed, in some cases the 8-OHQs increased toxicity (supplemental Fig. S3). In addition to establishing the specificity of 8-OHQs, this experiment confirms that 8-OHQs do not act simply by decreasing expression from a galactose-regulated promoter or counteracting a generic toxicity due to protein overexpression.
8-OHQs Synergize to Rescue Proteotoxicity
Our results thus far support distinct activities for each 8-OHQ. To test this directly, we assayed for synergy between each compound in all three models. Synergy is defined as activity greater than is achieved by simple additivity and reflects distinct modes of action. Typically, synergy is assayed by comparing a pair of compounds mixed together at a fixed ratio across a range of concentrations and comparing that to each compound alone. A shift in efficacy toward lower concentrations indicates a synergistic interaction. In our case, however, the narrow window of efficacy for each compound and the fact that they become toxic above their most effective concentrations precludes this approach. We, therefore, tested for synergy in two different ways.
First, when two individual compounds were each active in a particular model (e.g. HQ-415 and CQ in α-syn), we tested multiple mixtures of the two compounds at concentrations near or below 50% of their most effective dose (EC50). We compared the resulting activity to that predicted by simply adding the individual responses (supplemental Fig. S4). Because compound effects were not linear, concentration pairs with greater activity than predicted from the added single dose effects were also compared with 2-fold higher doses of each compound to confirm that results were not additive (Fig. 3, A–C). All of the primary data in these analyses are shown in supplemental Fig. S4, with the most meaningful comparisons presented in Fig. 3, A–C.
FIGURE 3.

8-hydroxyquinolines synergize in proteotoxicity models. Compound synergy between 8-OHQs was investigated with active pairs of compounds and active/inactive pairs of compounds in each model. A, shown is TDP-43 with a potential synergistic combination with 1- and 2-fold doses of HQ-161 and HQ-415. Predicted and actual values for the combined dose are indicated. Concentrations (μm) of HQ-161 and HQ-415 are indicated below the graph. B, the α-syn model with a synergistic combination of CQ and HQ-415 exhibits activity greater than predicted by simple additivity and a 2-fold dose of either single compound. C, shown is a potential synergistic combination of HQ-415 and CQ in htt-72Q. The activity of a 2-fold dose of CQ exceeds the actual activity of the combination of single doses. D, shown are dose-response curves of TDP-43 treated with HQ-161 (top) and HQ-415 (bottom) in the presence or absence of two constant concentrations of CQ (noted in the legend). E, shown are dose-response curves of α-syn treated with HQ-415 (top) and CQ (bottom) in the presence or absence of two constant concentrations of HQ-161 (noted in the legend). F, shown are dose-response curves of htt-72Q treated with HQ-415 (top) and CQ (bottom) in the presence or absence of two constant concentrations of HQ-161 (noted in the legend). Identity and concentrations of inactive compounds are noted in the legend. The active compound is color-coded according to other figures and noted below the x axis. The y axis is a fraction of the maximal response for that compound.
HQ-415 strongly synergized with CQ at multiple concentrations in cells expressing α-syn. The activity of the most synergistic combination was much greater than twice the dose of either HQ-415 or CQ alone (Fig. 3B). This synergy is consistent with distinct modes of action. HQ-415 and CQ also initially appeared to synergize in the htt-72Q model. However, the 2-fold dose of CQ was actually greater than the activity of the combination, making synergy unclear (Fig. 3C). HQ-161 and HQ-415 did not convincingly synergy in cells expressing TDP-43 (Fig. 3A).
We used a second approach to assay for synergy between pairs of compounds where one was active in a particular model and the other was apparently inactive (e.g. as shown in Fig. 1, HQ-415 was active in the TDP-43 model, but CQ was not). Dose-response curves were generated for the active compound with or without a constant concentration of the inactive compound. We show the full dose-response curves for these types of compounds in Fig. 3, D–F, and report the EC50 in supplemental Fig. 5.
In TDP-43-expressing cells, CQ failed to synergize with HQ-161 and actually antagonized HQ-161 rescuing activity. In contrast, despite the fact that CQ was completely inactive against TDP-43, it strongly synergized with HQ-415, decreasing the EC50 from 16 to 5 μm. In cells expressing α-syn, HQ-161, despite being inactive on its own, synergized with HQ-415, reducing the EC50 from 21 to 14 μm. HQ-161 did not, however, synergize with CQ against α-syn toxicity. Finally, synergy was not detected in the htt-72Q model with any pair of compounds, and an antagonistic interaction was seen between HQ-161 and both HQ-415 and CQ.
Taken together, these data indicate that each compound has a unique mode of action. Furthermore, the non-additive interactions (both synergistic and antagonistic) observed with otherwise inactive compounds indicates they provide distinct activities that on their own do not affect growth.
Effects of 8-OHQs on Neurotoxic Protein Localization
We next examined the effects of 8-OHQs on the aggregation, localization, and accumulation of TDP-43, α-syn, and htt-72Q. The misfolding and aggregation of each protein is relevant to its toxicity, and each exhibits characteristic localization patterns in yeast cells. TDP-43-GFP typically forms an increasing number of foci that correlates with toxicity (Fig. 4A, left) (18, 19). α-Syn transitions from the plasma membrane to foci of stalled vesicles when it accumulates to toxic levels (Fig. 4B, left) (6, 9). This localization is partially reversed upon either genetic or chemical rescue (5, 7). Finally, htt-72Q-CFP localization ranges from diffuse or punctate to very large aggregates (Fig. 4C, left) (13). Quantified localization phenotypes for all treatments are depicted to the right of the corresponding images (Fig. 4, A–C, right).
FIGURE 4.

8-Hydroxyquinolines modify aspects of toxic protein accumulation and localization. A, representative cells expressing TDP-43-GFP are shown with different numbers of foci. Cells with three or more foci were quantified for TDP-43 cultures treated with DMSO or each 8-OHQ at its most efficacious concentration (right). Active compounds, HQ-161 and HQ-415, reduce the number of cells with three or more TDP-43 foci. B, representative cells with α-syn-GFP localized to the plasma membrane, cytoplasmic vesicle accumulations, or dead cells are shown. Quantitation of cells with plasma membrane-localized α-syn-GFP foci treated with DMSO or each 8-OHQ is shown (right). Active compounds, HQ-415 and CQ, increase the number of cells with plasma membrane localized α-syn-GFP. C, representative cells exhibiting diffuse/small punctuate htt-72Q-CFP, moderate aggregate loads, or high aggregates loads are shown with quantitation of large aggregates to the right. HQ-415 modestly reduced the number of cells with a high aggregate load, whereas CQ did not. Values reflect averages of three independent experiments, and error bars depict S.D. Significance was determined by one-way ANOVA with a Tukey test of multiple comparisons. *, p < 0.05; **, p < 0.01; ***, p < 0.001. CFP, cyan fluorescent protein.
Both HQ-161 and HQ-415, but not CQ, significantly reduced the number of cells with three or more TDP-43 foci (Fig. 4A). This reduction in foci correlated with their efficacy against TDP-43 toxicity. HQ-415 and CQ, but not HQ-161, also strongly reversed the α-syn foci accumulation associated with toxicity, resulting in nearly exclusive plasma membrane localization (Fig. 4B). Finally, HQ-415 trended toward a reduction in the number of cells with large htt-72Q aggregates. CQ, despite rescuing htt-72Q, did not decrease htt-72Q accumulation (Fig. 4C). In all cases inactive compounds had no effect on protein localization. Thus, 8-OHQ-dependent changes in protein localization largely correlated with the rescue of cell growth.
8-OHQs Are Protective in C. elegans Model of α-Syn Toxicity
We previously showed that compounds and genetic modifiers identified from yeast α-syn screens function in a C. elegans model of α-syn toxicity (5, 7, 9, 11). The activity of these modifiers and validation in mammalian neurons indicates that the proteotoxicity elicited by α-syn in yeast is fundamentally similar to that in the neurons of complex organisms. Despite a conserved toxic mechanism of TDP-43 among yeast, flies, and humans (25), C. elegans models of TDP-43 neuronal toxicity have thus far failed to recapitulate the cytoplasmic aggregation and toxicity of TDP-43 observed in yeast and mammalian cells (35, 36).4 Because HQ-415, CQ, and HQ-161 (in combination with HQ-415 and CQ) each rescued the α-syn model, we focused our efforts on assaying 8-OHQ activity in this well established and validated nematode model of α-syn neurotoxicity.
In this model GFP and α-syn are both under transcriptional control of the dat-1 promoter, which restricts their expression to dopaminergic (DA) neurons (5). Development of this organism is highly stereotyped, and wild-type worms invariably have six DA neurons in their head. These neurons can be readily visualized in intact, living animals with GFP due to the transparency of the C. elegans cuticle. Expressing α-syn causes degeneration of DA neurons (Fig. 5A, gray bars), which is quantified based on a decrease in the percentage of worms that retain the wild-type number of neurons. This is easily seen in a representative α-syn-expressing worm (Fig. 5B). After compound treatment, DA neurons can be counted and compared with mock-treated worms.
FIGURE 5.
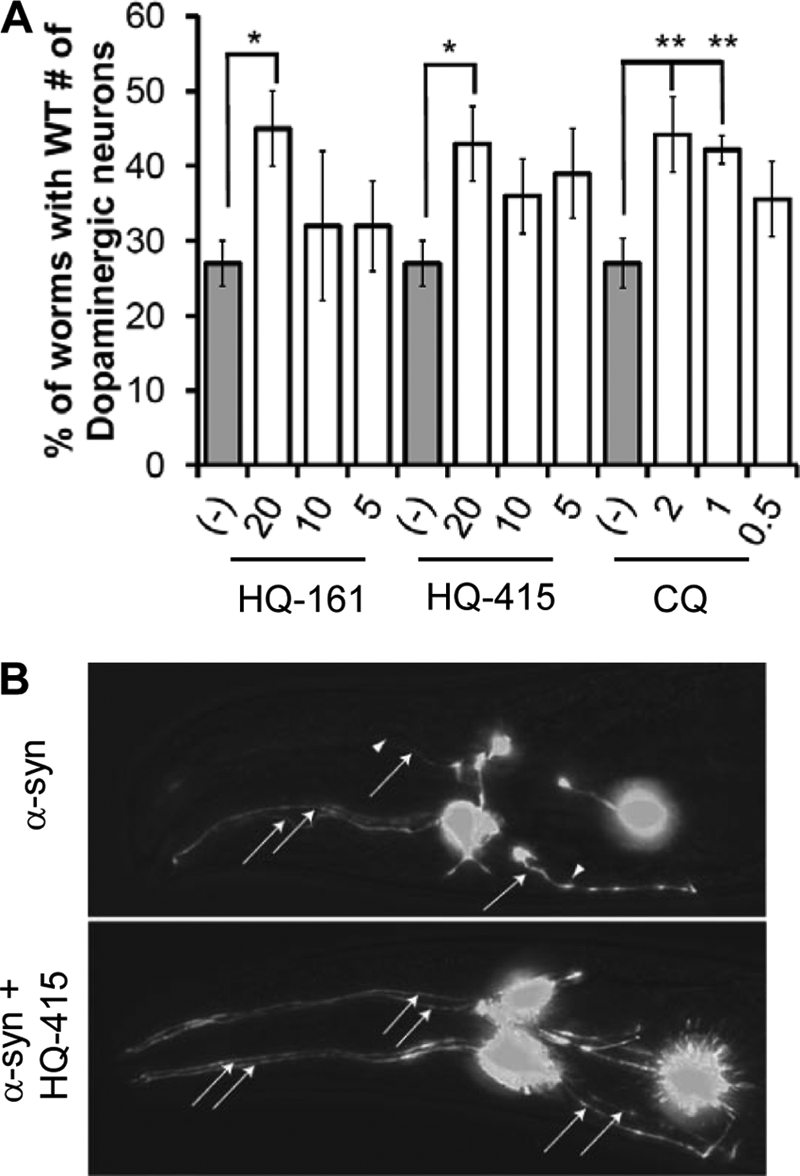
8-Hydroxyquinolines rescue a C. elegans model of α-syn toxicity. A, C. elegans expressing α-syn in dopaminergic neurons were rescued by treatment with HQ-161, HQ-415, and CQ. Reported values are the percentage of worms that retained the wild-type number (6) of DA neurons. Experiments were performed at least three independent times and are compared with animals exposed to 0.2% DMSO vehicle only. Significance was determined by one-way ANOVA with a Tukey test of multiple comparisons. *, p < 0.05; **, p < 0.01. B, representative images show DA neuronal loss by α-syn expression in the anterior (head) region of a nematode exposed to vehicle alone (top) and an α-syn-expressing nematode rescued by HQ-415 where all six DA neurons are intact (bottom). Neurons are imaged GFPs whose expression is restricted to DA neurons by transcriptional control of the DA neuron-specific dat-1 promoter. Arrows with lines show present neurons (specifically axons), and arrowheads alone show locations of lost neurons. The neuronal cell bodies are the brightly fluorescent structures.
HQ-415 and CQ, both of which demonstrated strong efficacy in the yeast α-syn model, partially rescued DA neuron loss, suggesting that the mechanisms of action are conserved between yeast and worms (Fig. 5, A and B). HQ-161 also rescued α-syn toxicity on its own (Fig. 5A), indicating that its activity against α-syn is stronger in nematodes than in yeast (Fig. 2A).
All Three 8-OHQs Bind Metals
Several possible activities might promote rescue by the three 8-OHQs. 8-OHQs are generally known to be weak chelators of copper, iron, and zinc. We verified that HQ-161, HQ-415, and CQ each bind each of these metals by the ability of the metals to change the UV absorbance spectra of the compounds (supplemental Fig. S6). With these activities, 8-OHQs could chelate metals to reduce metal-dependent reactive oxygen species (ROS) generation. Alternatively, 8-OHQs could act as ionophores, which are lipid-soluble compounds that bind to and facilitate import of extracellular metals. Finally, 8-OHQs could chelate intracellular metals to elicit protection. We, therefore, returned to yeast to distinguish among these possibilities.
Mechanism of Action; 8-OHQs as Antioxidants
In several different NDs, neurons show clear signs of oxidative stress. Evidence suggests that this is at least in part due to the metal-dependent conversion of H2O2 to free radicals through the Fenton reaction (37, 38). Chelation of metals by 8-OHQs could, therefore, reduce metal-dependent ROS, providing a mechanism of action for rescue. To test this, we first established a positive control, demonstrating that known antioxidants (ascorbic acid and N-acetylcysteine) rescued yeast cells from ROS generated by H2O2 (Fig. 6A). These same antioxidants, however, did not rescue any of the neurotoxic protein models (Fig. 6B). In a complimentary experiment, the 8-OHQs did not rescue wild-type yeast cells from H2O2 (Fig. 6C). Together, these data indicate that in our system rescue by 8-OHQs is not mediated by reducing metal-dependent ROS.
FIGURE 6.

8-Hydroxyquinolines do not function as antioxidants. A, the antioxidants ascorbic acid (left) and N-acetylcysteine (right) rescue H2O2 toxicity. Values represent growth of the model normalized to an untreated YFP control strain after 24 h of a Bioscreen CTM growth assay. B, antioxidants do not rescue TDP-43, α-syn, or htt-72Q toxicity. Values represent growth of the model normalized to an untreated YFP control strains after 24 h (α-syn) or 36 h (TDP-43 and htt-72Q) of a Bioscreen CTM growth assay. TDP-43, α-syn, and htt-72Q are shown in purple, blue, and orange, respectively. C, H2O2 toxicity was not rescued by HQ-161, HQ-415, or CQ. Increasing concentrations of H2O2 were treated with concentrations of 8-OHQs indicated in the legend. Values represent growth relative to the non-H2O2, non-compound-treated condition after 24 h growth in 384-well plates.
Mechanism of Action; 8-OHQs as Ionophores
8-OHQs might also function as ionophores to rescue toxicity in our yeast models in a manner similar to that reported for the compound PBT2, which imports copper and zinc in AD models (29). As ionophores, 8-OHQs would bind extracellular metals and transport them directly across membranes independent of active metal pumps and transporters. If metal depletion is the root cause of proteotoxicity, then ionophores might provide protection by acting to reestablish normal metal distribution.
To test ionophore activity, we assayed each 8-OHQ for its ability to rescue yeast strains whose growth is limited by the availability of copper, iron, or zinc. We employed yeast strains carrying individual deletions of the MAC1, AFT1, and ZAP1 genes. These genes encode the transcription factors that regulate the uptake of copper, iron, or zinc, respectively. Deletions of the genes reduce growth rates due to the disruption of the specific metal uptake pathways. In these strains the addition of exogenous metals rescues growth. Therefore, ionophore activity could restore metal levels and rescue growth rates. On the other hand, intracellular metal chelation by the 8-OHQs may cause synthetic toxicity.
We treated these deletion strains with several concentrations of each 8-OHQ and compared growth to an isogenic, wild-type parental strain. For each strain, growth in the presence of the 8-OHQs was normalized to growth in the absence of compound (Fig. 7, A–C; only the most instructive concentrations are shown for each). Data are also summarized in a heat map format for ease of comparison, where yellow reflects rescue (likely ionophore activity) and blue reflects toxicity (likely intracellular chelation; Fig. 7D).
FIGURE 7.

8-Hydroxyquinolines possess both intracellular chelation and ionophore activities. 8-OHQs can either function as ionophores or be toxic to strains deleted for metal-responsive transcription factors. Strains individually deleted for mac1 (copper), aft1 (iron), or zap1 (zinc) were treated with HQ-161 (A), HQ-415 (B), or CQ (C) and compared with wild-type yeast at multiple concentrations. Values reflect growth of yeast treated with 8-OHQs relative to growth of yeast treated with DMSO alone. The single most instructive concentrations are shown. Three independent experiments were averaged, and significance was determined by Student's t test. D, shown is a heat map representation of ionophore (top) or toxic (bottom) activities of each transcription factor deletion strain. The values are the difference in percent growth between the deletion strain and the wild-type strain. Yellow, increased growth relative to no compound; blue, decreased growth relative to no compound; black, no effect on growth relative to no compound.
HQ-161 partially restored growth in the strain deficient for copper, mac1 (Fig. 7A). It was toxic in aft1, the deletion strain that is deficient in iron, at concentrations >2-fold that used for rescuing TDP-43 toxicity (80 μm, Fig. 7A). HQ-415 was synthetically toxic with mac1 and zap1 strains (Fig. 7B). Interestingly, HQ-415 partially rescued aft1 at low concentrations (3.125 μm) yet was synthetically toxic with aft1 at concentrations used for rescuing toxicity models (25 μm; Fig. 7B). CQ strongly rescued mac1 and was synthetically toxic to aft1 and zap1 (Fig. 7C).
Rescue of deletion strains indicates that HQ-161 and CQ both facilitated copper import, whereas HQ-415 promoted iron import. However, the distinct toxicity for each 8-OHQ in each deletion strain indicates that they indeed also chelated intracellular metals at near therapeutic concentrations (Fig. 7D). In addition, exogenous metals had little effect on the rescue of toxicity models (supplemental Fig. S7). Each 8-OHQ demonstrated unique interactions with deletion strains, consistent with different bioactivities in yeast.
Mechanism of Action; 8-OHQs as Intracellular Chelators
The evidence for both ionophoric and intracellular chelation activities with these compounds prompted us to ask if therapeutic doses of 8-OHQs had intracellular consequences on the steady-state metal levels of wild-type yeast. To do so, we assayed expression of transcripts that report on the cellular depletion of copper (CTR1), iron (FRE3), or zinc (ZTR1). These transcripts encode proteins involved in metal uptake and are controlled by the MAC1, AFT1, and ZAP1 transcription factors, respectively (Fig. 7). Wild-type yeast were grown in the presence of each 8-OHQ at the indicated concentrations, and RNA levels were quantified by RT-PCR relative to the mRNA for actin (ACT1), a transcript whose expression was not altered in our conditions.
HQ-161 treatment of wild-type yeast had no effect on metal-responsive transcripts (Fig. 8A), indicating it did not overtly deplete cells of any metal. In contrast, HQ-415 induced CTR1, FRE3, and ZTR1 transcripts, reflecting depletion of copper, iron, and zinc (Fig. 8A). This broad effect is consistent with the toxicity produced by HQ-415 in all three transcription factor deletion strains relative to the wild-type strain (Fig. 7D).
FIGURE 8.

Intracellular metal chelation is the predominant protective activity of 8-hydroxyquinolines. A, RT-PCR analysis of 8-OHQ-treated WT cells revealed different metal depletion gene expression changes. Up-regulation of CTR1, FRE3, and ZTR1 transcripts served as indicators of copper, iron, and zinc depletion, respectively. Values are the average of three independent experiments and are reported normalized relative to an unchanging ACT1 control transcript. Significance was determined by one-way ANOVA with a Tukey test of multiple comparisons. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, the addition of copper and iron largely prevents rescue by 8-OHQs. Values are expressed as the difference in OD600 at 24 h (α-syn) or 36 h (TDP-43/htt-72Q) from DMSO control treated cultures. Yellow, rescue; blue, toxic; black, no effect. (−), C, F, and Z depict no metal, copper, iron, and zinc, respectively. C, metals rescued toxicity of 8-OHQs. Wild-type yeast were treated with toxic concentrations of each 8-OHQ and with two concentrations of each metal (either equimolar or a 2:1 8-OHQ:metal ratio). Yellow reflects rescue; black reflects no rescue.
Finally, CQ depleted cells of iron as evidenced by the up-regulation of FRE3, but did not deplete copper, as evidenced by the failure to induce CTR1 (Fig. 8A). This result is consistent with CQ-specific toxicity with aft1, the transcription factor that regulates iron uptake (Fig. 7B), and its rescue of and lack of toxicity toward mac1, the transcription factor regulating copper uptake (Fig. 7A).
These data further confirm that protective concentrations of HQ-415 and CQ can on their own induce expression of genes reflective of metal depletion. Thus, the metal depletion activities do not depend on expression of the toxic protein they rescue but are an inherent property of the compound.
Clearly, the compounds have both the ability to act as ionophores and to chelate intracellular metals. But which of these activities is involved in protecting against the toxic protein? To test this, we assayed the ability of exogenous metals to interfere with 8-OHQ activity in the proteotoxicity models. Indeed, equimolar copper and iron each significantly interfered with rescue by HQ-161 and HQ-415 in all models tested (Fig. 8B). In a complementary experiment, supplemental copper and iron each rescued wild-type yeast from toxic levels of each 8-OHQ (Fig. 8C, left and middle columns). In this second experiment, HQ-415, and to a lesser extent HQ-161, also were modestly rescued by zinc (Fig. 8C, right column).
In contrast to HQ-161 and HQ-415, the addition of copper and iron promoted the ability of CQ to rescue TDP-43 but interfered with CQ ability to rescue α-syn and htt-72Q (Fig. 8B). This dichotomy suggests that a critical balance between toxic and protective chelation by CQ is buffered by supplemental copper or iron in the TDP-43 strain. In addition, despite an apparent interaction between CQ and zap1 (Fig. 7C), zinc failed to rescue CQ toxicity (Fig. 8C).
The complex biological properties of these compounds are clearly not a direct consequence of the metal binding activities in isolation in vitro (supplemental Fig. S6). The ability of equimolar quantities of metals to interfere with 8-OHQ rescuing activities against toxic proteins indicates that intracellular chelation is more important in the rescuing activity of the 8-OHQs than ionophore activity. Importantly, the chelation-dependent rescuing activity was unique to 8-OHQs as penicillamine (copper chelator), deferoxamine (iron chelator), and TPEN (zinc chelator) all failed to rescue the toxicity of each model (supplemental Fig. S8).
DISCUSSION
We used a strain of yeast expressing toxic levels of TDP-43 to screen a large chemical library for suppressors of toxicity. The approach was completely unbiased, requiring only that compounds rescue growth. The most striking group of related compounds identified were 8-OHQs, a class of metal binding compounds with a strong previous connection to AD (29, 31). We then took advantage of an isogenic series of yeast strains expressing proteins involved in diverse neurodegenerative diseases to investigate the breadth of these compound activities. Surprisingly, each compound had a unique pattern of rescue. Exploiting the facility of genetic analysis in yeast, we next targeted different metal homeostasis pathways and employed a variety of defined growth conditions in these isogenic strains to investigate the compound mechanisms of action. We establish that none of the compounds rescue by reducing oxidative stress. Each of the compounds do, however, have unique ionophore properties and effects on intracellular metal chelation. Taken together, our data argue that intracellular metal chelation is particularly important for the ability of each of the 8-OHQs to rescue individual toxicity models (Table 1).
TABLE 1.
Summary of 8-OHQ activities
Data for each 8-OHQ are summarized to serve as a quick reference in comparing distinct 8-OHQ activities. The number before each parameter denotes the corresponding figure. Terms in parentheses add detail where a simple Yes or No is insufficient.
| Figure-Parameter | HQ-161 | HQ-415 | CQ |
|---|---|---|---|
| 1- Rescue TDP-43 | Yes | Yes | No |
| 2- Rescue α-syn | No | Yes | Yes |
| 2- Rescue htt-72Q | No | Yes | Yes |
| 3- Synergize with HQ-161 | n/a | Yes (α-syn) | No |
| 3- Synergize with HQ-415 | Yes (α-syn) | n/a | Yes (α-syn) |
| 3- Synergize with CQ | No | Yes (TDP-43 and α-syn) | n/a |
| 4- Reduce TDP-43 foci | Yes | Yes | No |
| 4- Reverse α-syn foci | No | Yes | Yes |
| 4- Reduce htt-72Q agg. | No | Yes | No |
| 5- Rescue α-syn C. elegans | Yes | Yes | Yes |
| 6- Antioxidant? | No | No | No |
| 7- Cu2+ ionophore | Yes (weak) | No | Yes (strong) |
| 7- Intracellular Cu2+ chelator | No | Yes (strong) | No |
| 7- Fe3+ Ionophore | No | Yes (low μm) | No |
| 7- Intracellular Fe3+ chelator | Yes (weak) | Yes (higher μm) | Yes |
| 7- Zn2+ Ionophore | No | No | No |
| 7- Intracellular Zn2+ chelator | No | Yes | Yes |
| 8- Induce Cu starvation | No | Yes | No |
| 8- Induce Fe starvation | No | Yes | Yes |
| 8- Induce Zn starvation | No | Yes | No |
| 8- Rescue reversed by Cu2+ | Yes (TDP-43) | Yes (all) | Yes (α-syn and htt-72Q); increased (TDP-43) |
| 8- Rescue reversed by Fe3+ | Yes (TDP-43) | Yes (all) | Yes (α-syn and htt-72Q); increased (TDP-43) |
| 8- Rescue reversed by Zn2+ | No | No | No |
| 8- 8-OHQ toxicity rescued by Cu2+ | Yes | Yes | Yes |
| 8- 8-OHQ toxicity rescued by Fe3+ | Yes | Yes | Yes |
| 8- 8-OHQ toxicity rescued by Zn2+ | No | Yes (mild) | No |
Different Activities of 8-OHQs in Yeast Proteotoxicity Models
What might be responsible for the significant differences in biological activity of 8-OHQs? Because structure determines function, the non-chelating regions of each 8-OHQ likely confer unique properties that alter activity. Hydrophobicity alone may affect bioactivity as HQ-161, HQ-415, and CQ have partition constants (log P) of ∼1.4, ∼4.5, and ∼2.9, respectively. Differences in hydrophobic character could alter subcellular distribution among cytosol, membranes, and organelles, thus causing significant effects in 8-OHQ activity and toxicity rescue. The aromatic halides of CQ, for example, promote membrane localization (39). In addition, biophysical parameters within different subcellular environments, such as the pH in the cytosol (neutral) versus the vacuole (acidic), may affect metal affinities. The pKa of the position-8 hydroxyl group or quinoline nitrogen can be affected by the neighboring substituents, resulting in altered resonance that might uniquely alter the charged state of 8-OHQs and, as a result, their chelating potential. Thus, it is not surprising that the biological activities are not a simple result of their in vitro metal binding activities (supplemental Fig. S6).
How do we reconcile the ionophore and metal chelation activities of these compounds in rescuing proteotoxicity? Because 8-OHQs are relatively weak chelators, they may facilitate some level of metal uptake (e.g. CQ and copper, Fig. 7) and subsequently dissociate inside the cell to promote chelation of other intracellular metal ions to rescue toxicity. This scenario would predict that the addition of metals would out-compete endogenous free or protein-bound metals for 8-OHQ binding, which indeed was observed and argues this activity is dominant over metal import (Fig. 8B).
Inside the cell, 8-OHQs could affect metal distribution between different cellular compartments, or they might directly impact metalloprotein activity. Metals are exceptionally common, spatially regulated protein co-factors predicted to modulate the activities of >40% of enzymes, with an estimated 80% of oxidoreductases predicted to require copper or iron (40). Numerous enzymes that function in respiration, amino acid metabolism, TCA cycle, kinase/phosphatase activity, nucleotide metabolism, fatty acid metabolism, and stress responses require copper or iron to function (41). These metals can facilitate the formation of protein structures, the ability of proteins to signal properly, and directly participate in catalytic functions. But metals are also in limiting abundance and are typically protein-bound (42).
Within this incredible wealth of potential targets, a few particular chelating activities of 8-OHQs have been described. CQ alters cytochrome cooxidase, aconitase, alkaline phosphatase, malate dehydrogenase, and SOD1 activity in yeast (39). CQ modifies the activity of a mitochondrial protein (CLK-1) to promote longevity in nematodes and mice (43). 8-OHQs inhibit histone demethylases (44). And CQ and PBT2 extract metals from Aβ (29, 30). Metal affinities and the access to metals in metalloproteins are likely strongly influenced by the non-chelating regions of 8-OHQs. This in turn would establish unique sets of affected metalloproteins that could restrict activity to certain models. The relative lack of specificity along with the sheer number and diverse functions of metalloproteins makes it difficult to determine the relevant targets (41). But the genetic tractability of yeast makes this difficult subject at least approachable.
The stronger chelation activities of HQ-415 and CQ compared with HQ-161 and perhaps a wider range of affected metalloproteins may initiate multiple independent responses that in turn produce unique patterns of rescue for each model. In other words, some alterations by HQ-415 or CQ may protect against α-syn, whereas others protect against htt-72Q or TDP-43 toxicity. Alternatively, a single consequence of HQ-415 or CQ chelation may rescue a common pathology among each model. The synergy among 8-OHQs and the rescue of each model by multiple, functionally distinct 8-OHQs establish that each model can benefit from at least two metal chelation-dependent protective pathways.
Therapeutic Implications of 8-OHQ Activities in Yeast
The ability of different 8-OHQs to impinge on diverse proteotoxicities further links metal homeostasis to neurodegenerative diseases. Several NDs, including AD, Parkinson disease, and HD, are characterized by one or more metal-dependent pathological phenomena, including elevated metal concentrations, metal-dependent ROS, and metal-dependent protein aggregation (32, 33, 37, 38, 45–50). Indeed, PBT2, a therapeutic analog of CQ, has produced positive results in both mice (29) and humans (31, 34). The therapeutic potential of CQ or PBT2 has been described for these diseases; however, the potential of tailoring specificity by phenotypic screening of multiple diverse models is completely unexplored. Other disease models might greatly benefit from rigorously screening 8-OHQ libraries to fine-tune activity toward a particular metal-related dysfunction, metalloprotein activity, or combination of the two. Our yeast models are particularly well suited for such analyses. Both cell culture systems and animal models often suffer from complex phenotypes that are negatively impacted by heterogeneous genetic backgrounds and phenotypes that vary in strength, penetrance, and onset. These attributes make it difficult to clearly define the activities of related compounds and are the cause for discrepancies among different laboratories. In contrast, the yeast models of multiple disease proteins have robust, stereotyped toxicity profiles, and the genetic backgrounds are homogeneous, thereby allowing for the discrimination of distinctly acting compounds.
The broad therapeutic potential of 8-OHQs has considerable implications because of the heterogeneous nature of neurodegenerative diseases. Brains of patients often present diverse, overlapping pathologies. TDP-43 aggregates, for example, are observed at relatively high frequencies in sporadic AD patient brains (25–30%) as well as in Lewy Body diseases, and Huntington disease (51–53). Whether or not co-pathologies are central or peripheral to the primary disease pathogenesis is unclear. However, because NDs are so multifaceted, 8-OHQs may be positioned uniquely to break from the “one drug one target” dogma to modify clinically and pathologically diverse diseases. Either broadly active single agents or multi-drug combinations of selective 8-OHQs may increase the breadth of therapeutic potential.
Metal chelators are frequently dismissed as therapeutic agents because of the expectation that they lack mechanistic specificity. However, the fact remains that despite the daunting complexity of their potential biological effects, we and others have shown that they are protective against the proteins that cause neurodegenerative disease from yeast to nematodes to humans. Importantly, PBT2 is both well tolerated and effective in AD patients (31, 34). In addition, chelation activity can be limited to neurons by merging 8-OHQs with known neuronal enzyme inhibitors as prochelators (54). By this means, systemic chelation is only possible after proper localization and enzyme inhibition.
Although existing evidence demonstrates that clioquinol can modulate amyloidogenic proteins and AD, our data emphasize that structural alterations to 8-OHQs can have significant and biologically meaningful effects on their activities. We might, therefore, harness these diverse activities to ameliorate distinct proteotoxicities in a metal chelation-dependent manner. This principle, revealed in yeast, should be exploited to survey the protective activities of structurally diverse 8-OHQs in both yeast and neuronal proteotoxicity models to identify either disease-specific or broadly acting therapeutic 8-OHQs.
Supplementary Material
Acknowledgments
We thank members of the Lindquist laboratory for helpful discussions and comments on the manuscript, Joshua Kritzer (Tufts University) for comments on the manuscript, and Caroline Shamu and staff at the Harvard Institute for Cell and Chemical Biology Longwood small molecule screening center.
This work was supported, in whole or in part, by National Institutes of Health Grant 5 P50 NS038372. This work was also supported by a grant from the Robert P. and Judith N. Goldberg Foundation (RJG) (to S. L.).

This article contains supplemental Figs. S1–S8 and Tables S1–S3.
M. L. Tucci and K. A. Caldwell, unpublished data.
- ND
- neurodegenerative disease
- ALS
- amytrophic lateral sclerosis
- FTLD
- frontotemporal lobar degeneration
- 8-OHQ
- 8-hydroxyquinoline
- α-syn
- α-synuclein
- polyQ
- polyglutamine
- CQ
- clioquinol
- ROS
- reactive oxygen species
- DA
- dopaminergic
- AD
- Alzheimer disease
- ANOVA
- analysis of variance.
REFERENCES
- 1. Goedert M. (2001) α-Synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2, 492–501 [DOI] [PubMed] [Google Scholar]
- 2. Selkoe D. J. (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen-Plotkin A. S., Lee V. M., Trojanowski J. Q. (2010) TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 6, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khurana V., Lindquist S. (2010) Modeling neurodegeneration in Saccharomyces cerevisiae. Why cook with baker's yeast? Nat. Rev. Neurosci. 11, 436–449 [DOI] [PubMed] [Google Scholar]
- 5. Cooper A. A., Gitler A. D., Cashikar A., Haynes C. M., Hill K. J., Bhullar B., Liu K., Xu K., Strathearn K. E., Liu F., Cao S., Caldwell K. A., Caldwell G. A., Marsischky G., Kolodner R. D., Labaer J., Rochet J. C., Bonini N. M., Lindquist S. (2006) α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Outeiro T. F., Lindquist S. (2003) Yeast cells provide insight into α-synuclein biology and pathobiology. Science 302, 1772–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Su L. J., Auluck P. K., Outeiro T. F., Yeger-Lotem E., Kritzer J. A., Tardiff D. F., Strathearn K. E., Liu F., Cao S., Hamamichi S., Hill K. J., Caldwell K. A., Bell G. W., Fraenkel E., Cooper A. A., Caldwell G. A., McCaffery J. M., Rochet J. C., Lindquist S. (2010) Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models. Dis. Model Mech. 3, 194–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thayanidhi N., Helm J. R., Nycz D. C., Bentley M., Liang Y., Hay J. C. (2010) α-Synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 21, 1850–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gitler A. D., Bevis B. J., Shorter J., Strathearn K. E., Hamamichi S., Su L. J., Caldwell K. A., Caldwell G. A., Rochet J. C., McCaffery J. M., Barlowe C., Lindquist S. (2008) The Parkinson's disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Ham T. J., Thijssen K. L., Breitling R., Hofstra R. M., Plasterk R. H., Nollen E. A. (2008) C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLoS Genet 4, e1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gitler A. D., Chesi A., Geddie M. L., Strathearn K. E., Hamamichi S., Hill K. J., Caldwell K. A., Caldwell G. A., Cooper A. A., Rochet J. C., Lindquist S. (2009) α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 41, 308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramirez A., Heimbach A., Gründemann J., Stiller B., Hampshire D., Cid L. P., Goebel I., Mubaidin A. F., Wriekat A. L., Roeper J., Al-Din A., Hillmer A. M., Karsak M., Liss B., Woods C. G., Behrens M. I., Kubisch C. (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191 [DOI] [PubMed] [Google Scholar]
- 13. Krobitsch S., Lindquist S. (2000) Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1589–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duennwald M. L., Jagadish S., Muchowski P. J., Lindquist S. (2006) Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. U.S.A. 103, 11045–11050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orr H. T., Zoghbi H. Y. (2007) Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621 [DOI] [PubMed] [Google Scholar]
- 16. Duennwald M. L., Lindquist S. (2008) Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 22, 3308–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duennwald M. L., Jagadish S., Giorgini F., Muchowski P. J., Lindquist S. (2006) A network of protein interactions determines polyglutamine toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 11051–11056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson B. S., McCaffery J. M., Lindquist S., Gitler A. D. (2008) A yeast TDP-43 proteinopathy model. Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 6439–6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson B. S., Snead D., Lee J. J., McCaffery J. M., Shorter J., Gitler A. D. (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 21. Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., Miller C. C., Nicholson G., Shaw C. E. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rutherford N. J., Zhang Y. J., Baker M., Gass J. M., Finch N. A., Xu Y. F., Stewart H., Kelley B. J., Kuntz K., Crook R. J., Sreedharan J., Vance C., Sorenson E., Lippa C., Bigio E. H., Geschwind D. H., Knopman D. S., Mitsumoto H., Petersen R. C., Cashman N. R., Hutton M., Shaw C. E., Boylan K. B., Boeve B., Graff-Radford N. R., Wszolek Z. K., Caselli R. J., Dickson D. W., Mackenzie I. R., Petrucelli L., Rademakers R. (2008) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 4, e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buratti E., Baralle F. E. (2010) The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 7, 420–429 [DOI] [PubMed] [Google Scholar]
- 24. Cushman M., Johnson B. S., King O. D., Gitler A. D., Shorter J. (2010) Prion-like disorders. Blurring the divide between transmissibility and infectivity. J. Cell Sci. 123, 1191–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elden A. C., Kim H. J., Hart M. P., Chen-Plotkin A. S., Johnson B. S., Fang X., Armakola M., Geser F., Greene R., Lu M. M., Padmanabhan A., Clay-Falcone D., McCluskey L., Elman L., Juhr D., Gruber P. J., Rüb U., Auburger G., Trojanowski J. Q., Lee V. M., Van Deerlin V. M., Bonini N. M., Gitler A. D. (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bonini N. M., Gitler A. D. (2011) Model organisms reveal insight into human neurodegenerative disease. Ataxin-2 intermediate-length polyglutamine expansions are a risk factor for ALS. J. Mol. Neurosci. 45, 676–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giaever G., Flaherty P., Kumm J., Proctor M., Nislow C., Jaramillo D. F., Chu A. M., Jordan M. I., Arkin A. P., Davis R. W. (2004) Chemogenomic profiling. Identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. U.S.A. 101, 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu Y., Rolfs A., Bhullar B., Murthy T. V., Zhu C., Berger M. F., Camargo A. A., Kelley F., McCarron S., Jepson D., Richardson A., Raphael J., Moreira D., Taycher E., Zuo D., Mohr S., Kane M. F., Williamson J., Simpson A., Bulyk M. L., Harlow E., Marsischky G., Kolodner R. D., LaBaer J. (2007) Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 17, 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adlard P. A., Cherny R. A., Finkelstein D. I., Gautier E., Robb E., Cortes M., Volitakis I., Liu X., Smith J. P., Perez K., Laughton K., Li Q. X., Charman S. A., Nicolazzo J. A., Wilkins S., Deleva K., Lynch T., Kok G., Ritchie C. W., Tanzi R. E., Cappai R., Masters C. L., Barnham K. J., Bush A. I. (2008) Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron 59, 43–55 [DOI] [PubMed] [Google Scholar]
- 30. Cherny R. A., Atwood C. S., Xilinas M. E., Gray D. N., Jones W. D., McLean C. A., Barnham K. J., Volitakis I., Fraser F. W., Kim Y., Huang X., Goldstein L. E., Moir R. D., Lim J. T., Beyreuther K., Zheng H., Tanzi R. E., Masters C. L., Bush A. I. (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30, 665–676 [DOI] [PubMed] [Google Scholar]
- 31. Lannfelt L., Blennow K., Zetterberg H., Batsman S., Ames D., Harrison J., Masters C. L., Targum S., Bush A. I., Murdoch R., Wilson J., Ritchie C. W. (2008) Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer's disease. A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786 [DOI] [PubMed] [Google Scholar]
- 32. Kaur D., Yantiri F., Rajagopalan S., Kumar J., Mo J. Q., Boonplueang R., Viswanath V., Jacobs R., Yang L., Beal M. F., DiMonte D., Volitaskis I., Ellerby L., Cherny R. A., Bush A. I., Andersen J. K. (2003) Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo. A novel therapy for Parkinson's disease. Neuron 37, 899–909 [DOI] [PubMed] [Google Scholar]
- 33. Nguyen T., Hamby A., Massa S. M. (2005) Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington's disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 102, 11840–11845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Faux N. G., Ritchie C. W., Gunn A., Rembach A., Tsatsanis A., Bedo J., Harrison J., Lannfelt L., Blennow K., Zetterberg H., Ingelsson M., Masters C. L., Tanzi R. E., Cummings J. L., Herd C. M., Bush A. I. (2010) PBT2 rapidly improves cognition in Alzheimer's disease. Additional phase II analyses. J. Alzheimers Dis. 20, 509–516 [DOI] [PubMed] [Google Scholar]
- 35. Ash P. E., Zhang Y. J., Roberts C. M., Saldi T., Hutter H., Buratti E., Petrucelli L., Link C. D. (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet 19, 3206–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liachko N. F., Guthrie C. R., Kraemer B. C. (2010) Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 30, 16208–16219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dong J., Atwood C. S., Anderson V. E., Siedlak S. L., Smith M. A., Perry G., Carey P. R. (2003) Metal binding and oxidation of amyloid-β within isolated senile plaque cores. Raman microscopic evidence. Biochemistry 42, 2768–2773 [DOI] [PubMed] [Google Scholar]
- 38. Dexter D. T., Wells F. R., Lees A. J., Agid F., Agid Y., Jenner P., Marsden C. D. (1989) Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J. Neurochem. 52, 1830–1836 [DOI] [PubMed] [Google Scholar]
- 39. Li C., Wang J., Zhou B. (2010) The metal chelating and chaperoning effects of clioquinol. Insights from yeast studies. J. Alzheimers Dis. 21, 1249–1262 [DOI] [PubMed] [Google Scholar]
- 40. Andreini C., Bertini I., Cavallaro G., Holliday G. L., Thornton J. M. (2008) Metal ions in biological catalysis. From enzyme databases to general principles. J. Biol. Inorg. Chem. 13, 1205–1218 [DOI] [PubMed] [Google Scholar]
- 41. De Freitas J., Wintz H., Kim J. H., Poynton H., Fox T., Vulpe C. (2003) Yeast, a model organism for iron and copper metabolism studies. Biometals 16, 185–197 [DOI] [PubMed] [Google Scholar]
- 42. Waldron K. J., Rutherford J. C., Ford D., Robinson N. J. (2009) Metalloproteins and metal sensing. Nature 460, 823–830 [DOI] [PubMed] [Google Scholar]
- 43. Wang Y., Branicky R., Stepanyan Z., Carroll M., Guimond M. P., Hihi A., Hayes S., McBride K., Hekimi S. (2009) The anti-neurodegeneration drug clioquinol inhibits the aging-associated protein CLK-1. J. Biol. Chem. 284, 314–323 [DOI] [PubMed] [Google Scholar]
- 44. King O. N., Li X. S., Sakurai M., Kawamura A., Rose N. R., Ng S. S., Quinn A. M., Rai G., Mott B. T., Beswick P., Klose R. J., Oppermann U., Jadhav A., Heightman T. D., Maloney D. J., Schofield C. J., Simeonov A. (2010) Quantitative high throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One 5, e15535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bishop G. M., Robinson S. R., Liu Q., Perry G., Atwood C. S., Smith M. A. (2002) Iron, a pathological mediator of Alzheimer disease? Dev. Neurosci. 24, 184–187 [DOI] [PubMed] [Google Scholar]
- 46. Adlard P. A., Bush A. I. (2006) Metals and Alzheimer's disease. J. Alzheimers Dis. 10, 145–163 [DOI] [PubMed] [Google Scholar]
- 47. Deibel M. A., Ehmann W. D., Markesbery W. R. (1996) Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer's disease. Possible relation to oxidative stress. J. Neurol. Sci. 143, 137–142 [DOI] [PubMed] [Google Scholar]
- 48. Bush A. I., Pettingell W. H., Multhaup G., d Paradis M., Vonsattel J. P., Gusella J. F., Beyreuther K., Masters C. L., Tanzi R. E. (1994) Rapid induction of Alzheimer A β amyloid formation by zinc. Science 265, 1464–1467 [DOI] [PubMed] [Google Scholar]
- 49. Kaur D., Andersen J. (2004) Does cellular iron dysregulation play a causative role in Parkinson's disease? Ageing Res. Rev. 3, 327–343 [DOI] [PubMed] [Google Scholar]
- 50. Fox J. H., Kama J. A., Lieberman G., Chopra R., Dorsey K., Chopra V., Volitakis I., Cherny R. A., Bush A. I., Hersch S. (2007) Mechanisms of copper ion mediated Huntington's disease progression. PLoS One 2, e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson A. C., Dugger B. N., Dickson D. W., Wang D. S. (2011) TDP-43 in aging and Alzheimer's disease. A review. Int. J. Clin. Exp. Pathol 4, 147–155 [PMC free article] [PubMed] [Google Scholar]
- 52. Nakashima-Yasuda H., Uryu K., Robinson J., Xie S. X., Hurtig H., Duda J. E., Arnold S. E., Siderowf A., Grossman M., Leverenz J. B., Woltjer R., Lopez O. L., Hamilton R., Tsuang D. W., Galasko D., Masliah E., Kaye J., Clark C. M., Montine T. J., Lee V. M., Trojanowski J. Q. (2007) Co-morbidity of TDP-43 proteinopathy in Lewy Body related diseases. Acta Neuropathol. 114, 221–229 [DOI] [PubMed] [Google Scholar]
- 53. Schwab C., Arai T., Hasegawa M., Yu S., McGeer P. L. (2008) Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 67, 1159–1165 [DOI] [PubMed] [Google Scholar]
- 54. Youdim M. B., Fridkin M., Zheng H. (2004) Novel bifunctional drugs targeting monoamine oxidase inhibition and iron chelation as an approach to neuroprotection in Parkinson's disease and other neurodegenerative diseases. J. Neural Transm. 111, 1455–1471 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.