

Background: The NMDA receptor mediates stretch-induced calcium influx and resulting neuronal excitotoxicity.
Results: Calcium influx through NMDA receptors following stretch is reduced in cultures expressing NR2B C-terminal mutations.
Conclusion: Mechanosensitivity of NMDA receptors is dependent on the NR2B subunit and PKC activity at the NR2B C terminus.
Significance: These data provide insight into NMDA receptor subtype-specific mechanisms that dictate response to neuronal stretch.
Keywords: Glutamate Receptors Ionotropic (AMPA, NMDA); Neurobiology; Neurotransmitter Receptors; Protein Kinase C (PKC); Receptor Regulation; NR2B; Mechanobiology
Abstract
N-Methyl-d-aspartate receptors (NMDARs), critical mediators of both physiologic and pathologic neurological signaling, have previously been shown to be sensitive to mechanical stretch through the loss of its native Mg2+ block. However, the regulation of this mechanosensitivity has yet to be further explored. Furthermore, as it has become apparent that NMDAR-mediated signaling is dependent on specific NMDAR subtypes, as governed by the identity of the NR2 subunit, a crucial unanswered question is the role of subunit composition in observed NMDAR mechanosensitivity. Here, we used a recombinant system to assess the mechanosensitivity of specific subtypes and demonstrate that the mechanosensitive property is uniquely governed by the NR2B subunit. NR1/NR2B NMDARs displayed significant stretch sensitivity, whereas NR1/NR2A NMDARs did not respond to stretch. Furthermore, NR2B mechanosensitivity was regulated by PKC activity, because PKC inhibition reduced stretch responses in transfected HEK 293 cells and primary cortical neurons. Finally, using NR2B point mutations, we identified a PKC phosphorylation site, Ser-1323 on NR2B, as a unique critical regulator of stretch sensitivity. These data suggest that the selective mechanosensitivity of NR2B can significantly impact neuronal response to traumatic brain injury and illustrate that the mechanical tone of the neuron can be dynamically regulated by PKC activity.
Introduction
N-Methyl-d-aspartate receptors (NMDARs)2 are glutamate receptors whose activation is important for the proper health and maintenance of neurons and neuronal networks (1–3). Functional NMDARs are usually comprised of two NR1 subunits and two subunits from the NR2 family (NR2A, NR2B, NR2C, and NR2D) (4, 5). The relative composition of expressed NMDARs varies throughout brain regions (6), changes over neural development (6–8), and directs the activation of selective signaling networks through the unique coupling of proteins to the C terminus of each NR2 subunit (9–12). Proper regulation of receptor activity is vital, because excessive activation of NMDARs is the primary mediator of excitotoxic cell death in numerous disease states (13–16), whereas the tonic synaptic activation of the receptor can stimulate key neuronal survival programs (3, 17). Although the competing roles of the NMDAR lead to a more complex view of how NMDAR stimulation directs physiological processes including synaptic plasticity (18, 19), dendritic growth (20), and recovery from injury (1, 13, 21), this more complete view provides new opportunities for developing directed NMDAR-targeted therapies. Several recent papers show that it is possible to use the molecular diversity of the NMDAR, including the selective action or blockade of specific receptor subpopulations, to promote necessary maintenance of neural circuits (22), minimize the overactivation of pathological pathways (16, 23), and effectively treat neurological disease (24–27).
The NMDAR is considered by many as one of the common mediators for the acute and progressive events that occur following traumatic brain injury (TBI) (21, 28–32). Based partly on studies showing an increased glutamate concentration in the extracellular space (28, 33–35), many consider TBI an extension of glutamate excitotoxicity. However, TBI has an important and unique mechanical etiology that can contribute to the heterogeneity of the disease in the clinical population. Moreover, the mechanical causality of TBI appears at the molecular level of the NMDAR. The NMDAR is mechanosensitive and expresses a unique switch in behavior following mechanical stimulation. Specifically, rapid neuronal stretch induces a persisting loss in the voltage-dependent Mg2+ block of NMDARs (36). Similar to a phosphorylation event or proteolysis, the mechanical event can function as a distinct regulator of NMDAR function. However, no data exist that describe how this mechanically initiated switch in NMDAR behavior is regulated among its receptor subunits. The diversity in the dynamic mechanosensing properties of NMDAR subtypes may prove important in understanding the post-acute regulation of neuronal homeostasis after traumatic mechanical injury, similar to how the diversity in NMDAR composition has led to a more complete understanding of signaling occurring after excitoxicity (15, 27, 37). Finally, identifying the potential domains of regulation across NMDAR subunits or, alternatively, within individual subunits will inform how normal physiological signaling acting upon the receptor can potentially augment the mechanical tone of NMDARs.
In this report, we systematically characterize the direct effects of mechanical stretch on calcium influx through the NMDAR. We examine key regulatory mechanisms of mechanosensitivity and test whether there are crucial domains on subunits that control this sensitivity to stretch. Together, our data reveal that dynamic mechanosensitivity of NMDARs is controlled by the intracellular domain of the NR2B subunit, in which a PKC-mediated phosphorylation site, Ser-1323, is critical for NR2B stretch sensitivity. Given the prominent role of NR1/NR2B NMDARs controlling neuronal fate in models of neurological disease, this points to a likely pathway whereby mechanical force is transferred into subunit-specific signaling cascades after mechanical trauma and influences cell fate after injury.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK 293T cells (ATCC) were cultured and maintained with DMEM with l-glutamine supplemented with 5% fetal bovine serum in a humidified incubator at 37 °C and 5% CO2. For experimentation, HEK 293 cells were plated onto a transparent silicone substrate (Sylgard 184 + 186 mix). These membranes were attached to stainless steel wells, leaving an exposed area (area − size of a single well from a 24-well plate) of membrane for plating. Following sterilization, the membranes were coated with poly-d-lysine (0.01 mg/ml) for 1 h, rinsed with sterile water, and coated with laminin (10 μg/ml) for 1 h. After the membranes were rinsed again, HEK 293 cells were plated at 1:20 dilution from a fully confluent flask.
For primary cortical cultures, cortical neurons were isolated from day 18 embryonic rats and plated on poly-d-lysine-coated silicone membranes at a density of 0.3 million/ml. The cultures were plated in minimum essential medium with glutamax + 10% horse serum, which was removed at 24 h and replaced with neurobasal (Invitrogen) + B-27 supplement (Invitrogen). At 24 h after plating, the cells were treated with AraC (1 μm) to prevent the growth of astrocytes. AraC was removed at 3 days in vitro, and the cells were cultured in a humidified incubator at 37 °C and 5% CO2. Primary cortical cultures were used at 12–15 days in vitro, an age that contains a diverse content of NMDAR subtypes.
Plasmids
NR1a, NR2A, and NR2B cDNA plasmids were obtained and subcloned from rat brain library as previously described (38–40). The NR2B-1036X and NR2B-1433X truncation mutants were generated by replacing fragments of wild type NR2B and introducing a stop codon at amino acid 1037 (41) and 1434, respectively, by PCR. Using wild type NR2B in the PRK7 vector as a template, the NR2B point mutants S1303A and S1323A were made using the PCR-based site-directed mutagenesis kit by Stratagene (Agilent Technologies). The mutations were verified by DNA sequencing through the Nucleic Acid/Protein Research Core at Children's Hospital of Philadelphia. Plasmid encoding GFP-PSD95 was a generous gift supplied by Dr. David Bredt.
Transfection
HEK 293 cells were transfected 24 h after plating with Lipofectamine 2000 according to the product instructions. All of the cultures were transfected with GFP-tagged PSD95 to provide a visual marker of successful transfection. The total amount of DNA transfected per well was held constant over all conditions (0.8 μg/well). Because of the tonic vesicular release of glutamate in HEK 293 cells, the cells were transfected in the presence of the NMDA antagonist APV (Sigma-Aldrich; 100 μm) throughout the transfection period and until the time of plating. For cultures transfected with NR1 and NR2 subunits, the ratio of transfected DNA was 2:1:1 (GFP-PSD95:NR1:NR2). This ratio did not vary for any of the mutant NR2 subunits.
NMDAR Mechanosensitivity
Transfected HEK 293 cells were used 16–20 h after transfection. The cultures were incubated with the fluorescent calcium indicator, Fura 2AM (5 μm; Invitrogen), in controlled saline solution (126 mm NaCl, 5.4 mm KCl, 2 mm MgCl2, 1.8 mm CaCl2, 10 mm HEPES, 25 mm glucose) supplemented with 100 μm APV for 40 min at 25 °C. Cultures were placed in an apparatus that would apply a brief pressure to the exposed cell culture, and the membrane would deform in proportion to the applied pressure (42). We used the amount of membrane deformation (40%) as a measure of the mechanical input delivered to the HEK 293 cells. In a separate group of cultures, we used a 100 μm NMDA application to evaluate the response of transfected cells to agonist. All of the cultures were imaged continuously before and after the mechanical or chemical insult, collecting images at the emission fluorescence of 510 nm that appeared when cultures were alternately excited at 340 and 380 nm every 3 s. A pair of emission images from each wavelength was used to generate a Fura ratio (340 nm/380 nm) image at each time point. The cultures were imaged for 30 s before stimulation and up to 3 min following stimulation. Following Fura imaging, the cells were excited at 488 nm to detect the presence of GFP within the cells. The GFP signal was not detected during the 340- or 380-nm excitation during Fura imaging.
For testing stretch sensitivity of primary cortical neurons, the test protocol remains the same, with one primary difference. Cortical neurons were incubated with Fura 2AM, prior to stimulation for 40 min at 37 °C in saline solution (51.3 mm NaCl, 5.4 mm KCl, 2 mm MgCl2, 1.8 mm CaCl2, 26 mm NaHCO3, 0.9 m NaH2PO4, 10 mm HEPES, 25 mm glucose) without the addition of APV. The cultures were alternatively left untreated or treated with APV (25 μm) to block all NMDARs, Ro-256981 (1 μm) to block NR1/NR2B, bafilomycin A1 (500 nm) to inhibit glutamate vesicle refilling, tamoxifen citrate (20 μm) to inhibit PKC, or PMA (500 nm) to activate PKC. To block only synaptic NMDARs, cultures were treated with bicuculline and MK801 as previously described (1, 29).
Data Analysis
Stimulation of HEK 293 cells and primary neurons were analyzed using MetaMorph to quantify the extent of calcium influx following injury or NMDA stimulation. Traces of Fura ratio (F340/F380) over time were collected for all GFP-positive cells. Cells with a baseline Fura ratio of greater than 0.95, indicating an elevated initial calcium level, were excluded from analysis. The response was quantified for each individual cell by calculating the peak fractional change in Fura ratio poststimulation over the average baseline ratio prestimulation ((Fpeak − Fbaseline)/Fbaseline). To normalize data, the peak response for each cell was normalized by the peak response for the NR1/NR2B group for the given stimulation, either stretch or NMDA. A minimum of four to six separate cultures was used for each experimental group. Significance between groups was determined with a one-way analysis of variance and post hoc Tukey's test.
RESULTS
Subunit Composition of NMDAR Influences Mechanosensitivity
Following dynamic 40% stretch of NMDAR transfected HEK 293 cells, we observed two different calcium responses: 1) a significant and gradual rise in cytosolic calcium, indicated by a relative increase in the Fura-2 fluorescence ratio, which occurred and plateaued within the first 2 min poststretch, and 2) no significant increase in the relative Fura-2 ratio. We did not observe any stretch-induced calcium increase in nontransfected cells, indicating that the stretch level was not sufficient to cause the formation of nonspecific, transient pores in the plasma membrane (data not shown). Moreover, cells transfected with only GFP-PSD95 showed little to no change in intracellular calcium, indicating that a functional NMDAR was necessary to elicit a response following mechanical stimulation.
Cells were transfected with GFP-PSD95 alone or along with NR1 and NR2A (NR1/NR2A) or with NR1 and NR2B (NR1/NR2B). NR1/NR2B NMDAR-expressing cells respond to the stretch stimulus with an immediate rise in intracellular calcium, whereas cells expressing NR1/NR2A do not show a stretch-induced calcium rise (Fig. 1). Quantified, the normalized peak percentage of change in the calcium signal was significantly greater in NR1/NR2B-transfected cells compared with GFP-PSD95 control (p < 0.05), whereas transfection of NR1/NR2A was not different from control (Fig. 1D) (means ± S.E.: NR1/NR2B, 1.0 ± 0.03; NR1/NR2A, 0.14 ± 0.01; GFP-PSD95, 0.19 ± 0.02). Although NR1/NR2A- and NR1/NR2B-transfected cells display differential response to stretch, cells transfected with the different subunits respond similarly to the application of 100 μm NMDA (Fig. 1E) (NR1/NR2B, 1.0 ± 0.03; NR1/NR2A, 0.90 ± 0.03; GFP-PSD95, 0.01 ± 0.01). These data suggest that, among the common diheteromeric forms of the NMDAR expressed in the cortex and hippocampus, the NR1/NR2B NMDARs are significantly more sensitive to mechanical stretch than NR1/NR2A NMDARs.
FIGURE 1.

NR1/NR2B NMDARs are more sensitive to stretch than NR1/NR2A. HEK 293 cells, plated on flexible membranes, were transfected with GFP-PSD95 alone, or along with NR1 and either NR2A or NR2B. A, representative images of transfected cells, pre- and poststimulation, that were stimulated by either 40% stretch (left) or 100 μm NMDA (right). B and C, average Fura ratio, representing intracellular calcium, following stretch (B) or NMDA stimulation (C), demonstrates that although NR1/NR2B- and NR1/NR2A-expressing cells have a similar response to NMDA, stretch-induced calcium influx is greater in NR1/NR2B-expressing cells. D, the average peak fractional change in Fura ratio, normalized to the NR1/NR2B response, following stretch demonstrates significant stretch sensitivity in NR1/NR2B-transfected cells (*, p < 0.05 compared with GFP-PSD95), whereas NR1/NR2A stretch responses are not different from control GFP-PSD95 responses. E, normalized response to NMDA stimulation was similar among NR1/NR2A and NR1/NR2B-transfected cells (*, p < 0.05 compared with GFP-PSD95).
With this new information on the subunit selectivity of the NMDAR response to mechanical stretch, we tested the relative contribution of NMDAR subtypes to stretch injury in cortical neurons. Expression of NMDAR subtypes changes over time in dissociated cortical neurons. NR1/NR2A, NR1/NR2B, and NR1/NR2A/NR2B NMDARs contribute to a mixed pool of NMDAR subtypes in synaptic locations, whereas NMDARs at extrasynaptic NMDARs are either NR1/NR2A/NR2B or NR1/NR2B NMDARs. For these studies we used a protocol that blocks the synaptic NMDAR pool using the sequential application of bicuculline to produce bursts of action potential firing in the culture network followed with MK-801 treatment to noncompetitively block the ion channel of NR1/NR2A, NR1/NR2B, and NR1/NR2A/NR2B NMDARs (1). In our cultures, bicuculline + MK801 pretreatment led to a significant reduction in calcium influx after 100 μm NMDA stimulation (0.14 ± 0.02 of untreated, NMDAR-stimulated cultures; p < 0.01). The remaining population of NMDARs after bicuculline + MK-801 pretreatment was dominated by NR1/NR2B NMDARs, because treatment of these cultures with the highly specific NR2B antagonist Ro 25-6981 treatment nearly eliminated the NMDA-mediated calcium influx (0.03 ± 0.01 relative to untreated controls; Fig. 2A), suggesting that in our cultures, the extrasynaptic NMDAR pool mostly consists of NR1/NR2B NMDARs. Thus, the bicuculline and MK801 pretreatment offers the advantage of pharmacologically isolating the most mechanically responsive NMDAR subtype (NR1/NR2B) directly in cortical neurons. When mechanically injuring cortical cultures, our data show that significant initial calcium influx occurs through extrasynaptic NR1/NR2B NMDARs (Fig. 2B). These extrasynaptic NR1/NR2B NMDARs have a larger contribution to the overall calcium influx following stretch compared with NMDA stimulation (Fig. 2, A and B) (stretch, 0.41 ± 0.02; NMDA, 0.17 ± 0.03, p < 0.05). However, synaptic NMDARs also contribute to the calcium influx following stretch, likely a result of stretch-induced presynaptic glutamate release. Further, the relative calcium influx in cortical neurons to stretch injury was completely blocked with APV pretreatment (0.06 ± 0.004; Fig. 2B), demonstrating that glutamate binding is necessary for stretch-induced calcium influx in neurons.
FIGURE 2.
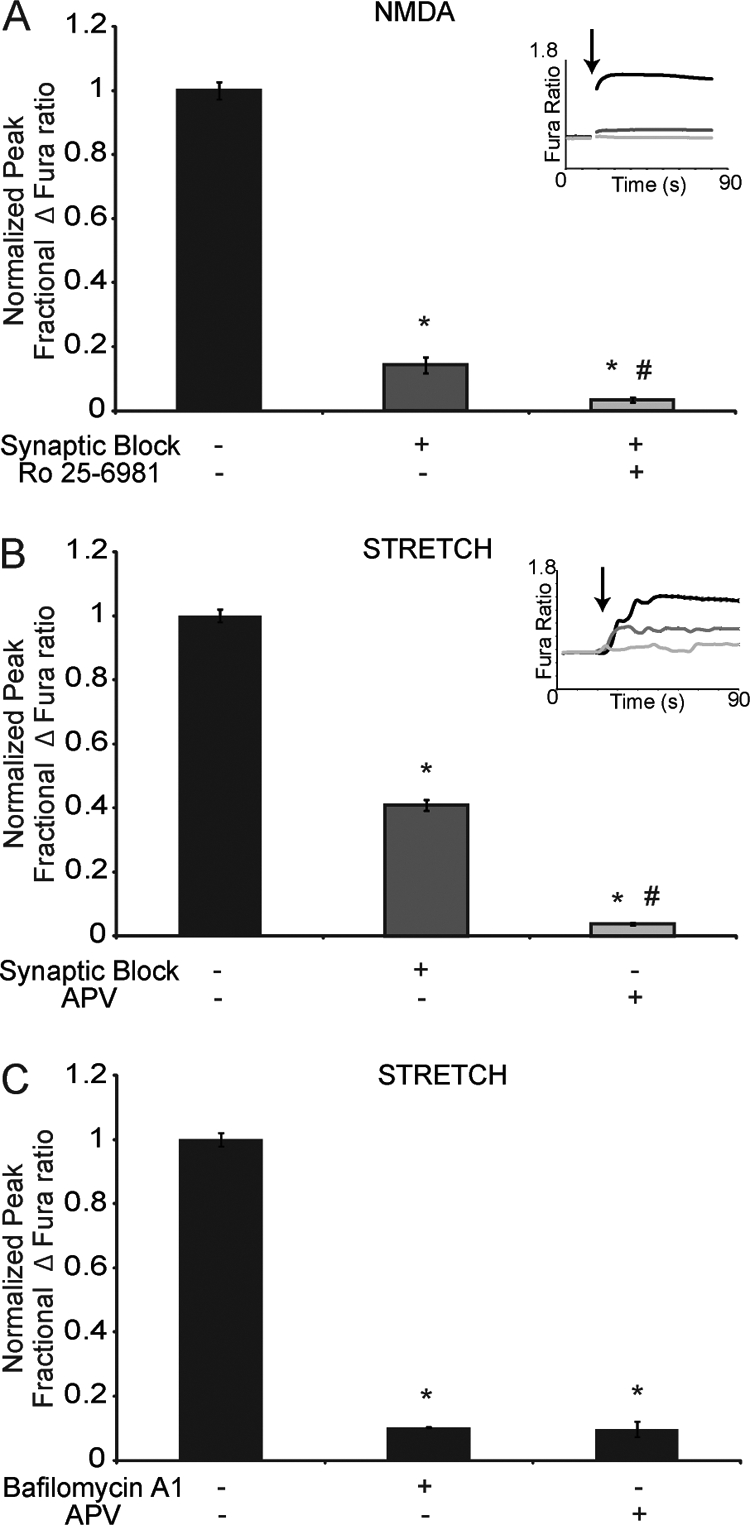
Mechanoactivation of NMDARs in cortical neurons. NR1/NR2B NMDARs in primary cortical neurons were isolated by pretreating cultures with bicuculline methiodide (10 μm) in the presence of MK-801 (50 μm), a technique to block NR1/NR2B, NR1/NR2A, and NR1/NR2A/NR2B NMDARs at the synapse. A, calcium response following 100 μm NMDA stimulation (inset, Fura traces immediately following NMDA stimulation (arrow)) in untreated neurons (black) and neurons pretreated with synaptic block protocol (dark gray) or synaptic block and Ro-256981 (1 μm) (light gray). The response was significantly reduced in cultures with synaptic block and abolished in synaptically blocked cultures that were concurrently treated with Ro 25-6981 (*, p < 0.05 compared with untreated; #, p < 0.05 compared with synaptic block alone), demonstrating that nearly all extrasynaptic NMDARs were of the NR1/NR2B subtype. B, calcium response following stretch (inset, Fura traces immediately following stretch (arrow)) was measured in untreated neurons (black) and neurons pretreated with synaptic block protocol (dark gray) or APV (25 μm) (light gray). The response was reduced, but not eliminated, in cultures with synaptic block (*, p < 0.05 compared with untreated) and was abolished in APV treated cultures (*, p < 0.05 compared with untreated; #, p < 0.05 compared with synaptic block), demonstrating that extrasynaptic NR1/NR2B NMDARs significantly contribute to the stretch response in neurons. C, cultures pretreated with bafilomycin A1 (500 nm) to block vesicular refilling showed no significant difference in calcium influx when compared with cultures pretreated with APV. Both APV- and bafilomycin-treated cultures were significantly different from untreated controls (*, p < 0.05).
To address the role of presynaptic release further, we blocked synaptic vesicular refilling with bafilomycin A1 pretreatment and observed a significant reduction in the stretch response (0.10 ± 0.003; Fig. 2C), where the response was not significantly different from APV-treated cultures. These data demonstrated a clear role for presynaptic glutamate release in initiating NMDAR-mediated calcium influx following stretch. Therefore, we conclude that injury-induced calcium influx in cortical neurons is mediated by glutamate release and the stretch-induced sensitivity of NR2B-containing NMDARs.
Triheteromeric NMDARs Express Intermediate Mechanosensitivity
To assess the mechanosensitivity of triheteromeric NR1/NR2A/NR2B NMDARs, HEK 293 cells were transfected with plasmids for GFP-PSD95, NR1, NR2A, and NR2B. Interestingly, the stretch response of cells expressing the combination of both the NR2A and NR2B subunits was significantly decreased from those expressing NR1/NR2B (NR1/NR2A/NR2B, 0.70 ± 0.03; NR1/NR2B, 1.0 ± 0.06; p < 0.05) but was not diminished to levels seen for NR1/NR2A (Fig. 3A). The response to mechanical stretch was distinct from the chemical agonist response, because 100 μm NMDA stimulation produced a calcium response in triheteromeric NMDARs that was not different from the NR1/NR2B receptor combination (Fig. 3B). One possibility that could lead to an uncertain interpretation of triheteromeric receptor experiments was that functional receptors in these transfected cultures could be composed of mixture of diheteromeric and triheteromeric receptor combinations. To address this uncertainty, we treated NR1/NR2A/NR2B-transfected cultures with Ro25–6981 (20 μm), an antagonist that blocks NR1/NR2B receptors but has minimal effect on triheteromeric receptors (43). In both mechanical and chemical stimulation experiments, pretreatment with Ro 25-6981 produced no significant differences in comparison with untreated triheteromeric cultures. Thus, we can conclude that NR1/NR2A/NR2B NMDARs display an intermediate form of mechanosensitivity, between that of NR1/NR2A and NR1/NR2B receptors.
FIGURE 3.
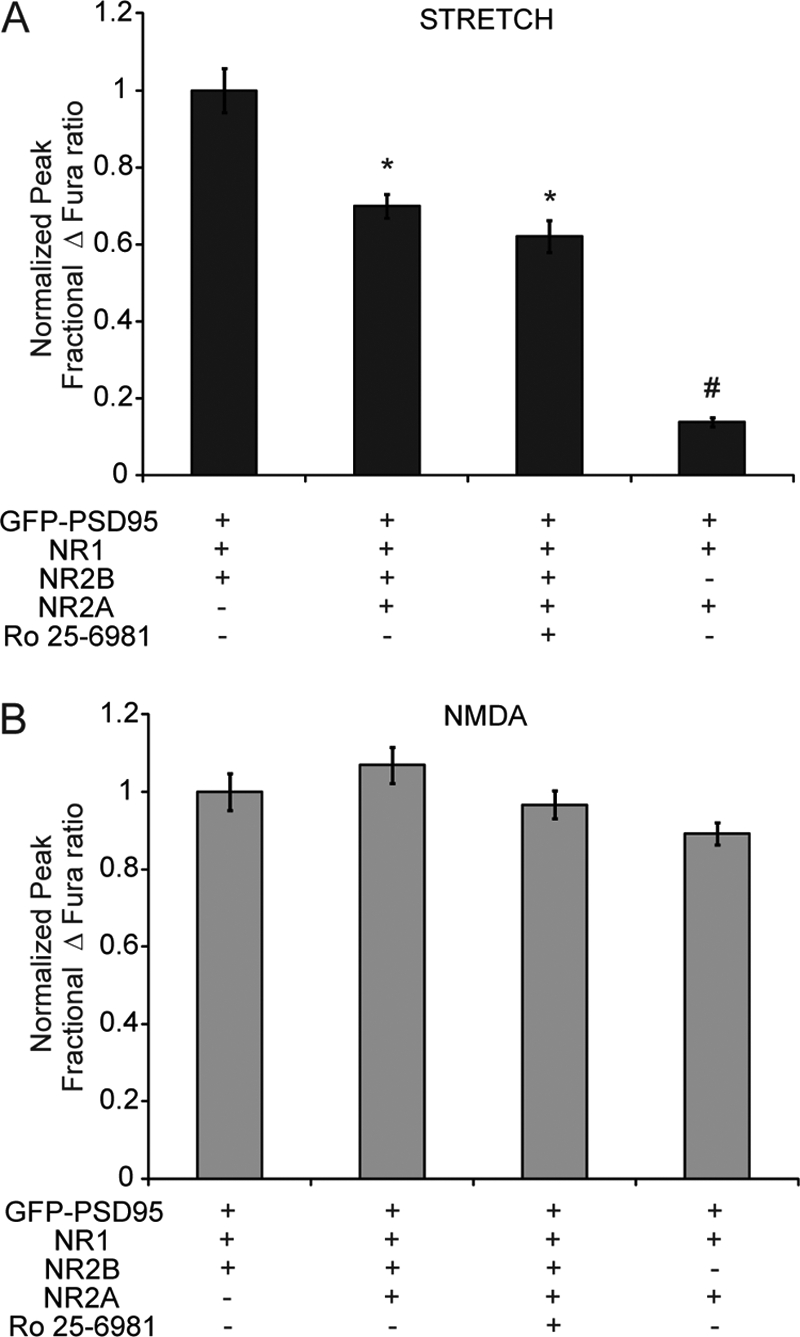
Intermediate mechanosensitivity in triheteromeric NR1/NR2A/NR2B receptors. The cells were transfected with GFP-PSD95, with either NR1 and NR2B, or with NR1, NR2A and NR2B. Cells transfected with all subunits were either left untreated or treated with NR1/NR2B-specific antagonist, Ro 25-6981. A, normalized response to stretch was significantly decreased, but not eliminated, in cells expressing NR1, NR2A, and NR2B, demonstrating that these cells exhibit intermediate mechanosensitivity (*, p < 0.05 compared with NR1/NR2B). B, response to 100 μm NMDA application was not different. Treatment with Ro 25-6981 did not alter the response of NR1/NR2A/NR2B-transfected cells, suggesting that expressed NMDARs in these cells are primarily triheteromeric.
We next tested whether the stretch sensitivity of NR1/NR2B NMDARs can be modulated by alternatively blocking two modes of receptor activity: glutamate binding or ionic flux through the channel pore. NR1/NR2B transfected cultures were stimulated with or without the pretreatment of NMDAR antagonists APV (200 μm) and MK801(50 μm). Pretreatment of cultures with APV, a competitive NMDAR antagonist that inhibits glutamate binding on the NR2 subunit, led to no significant stretch response. In comparison, pretreatment of cultures with MK801, which binds to and blocks the channel pore, resulted in a significant decrease in calcium response compared with untreated NR1/NR2B-transfected cells (Fig. 4A). Further, significant differences among the cumulative distributions of individual responses demonstrated that antagonism increases the amount of “nonresponding” cells after stretch (Fig. 4B). In comparison, APV pretreatment decreased the stretch response significantly more than MK801 (NR1/NR2B + APV, 0.07 ± 0.02; NR1/NR2B + MK801, 0.27 ± 0.04; p < 0.05) and further reduced the number of responding cells, as measured by the differential response distributions among antagonists. Both APV and MK801 completely eliminated the NMDA response to levels not different from responses from cells transfected with only GFP-PSD95 (Fig. 4, C and D).
FIGURE 4.

NR1/NR2B stretch response is dependent on glutamate activity. Cells transfected with GFP-PSD95, NR1, and NR2B were left untreated or pretreated with NMDAR competitive antagonist, APV, or NMDAR pore blocker, MK801. A, normalized peak change in Fura ratio after stretch shows that treatment with either antagonist significantly reduced stretch response (* p < 0.05 compared with NR1/NR2B). The response in NR1/NR2B-transfected cells treated with MK801 was significantly greater than those treated with APV (#, p < 0.05 compared with NR1/NR2B + APV), suggesting that the receptor pore cannot be fully blocked by MK801 after stretch. B, normalized response following 100 μm NMDA stimulation was completely blocked by both types of NMDAR antagonists (*, p < 0.05). Both APV and MK801 completely eliminated the response to NMDA treatment (D) and significantly reduced the response to stretch (C) across the population of NRI/NR2B-transfected cells.
NR2B C-terminal Tail Confers Mechanosensitivity
The NR2A and NR2B subunits share ∼70% homology (44) but contain important differences in the sequence and structure of their C-terminal tails. These differences are important in dictating subunit-specific functions of receptors, leading to differential cytoskeletal anchoring, protein binding, and association in signaling complexes (10–12, 45–48). We examined whether the C-terminal tail of the NR1/NR2B receptor confers the observed mechanosensitivity of the receptor by studying the response of recombinant receptors composed of NR2B truncation mutants. One key regulatory domain is the distal region of the C-terminal tail, which contains the binding domain for PSD95. However, cells expressing NR2B-1433X, an NR2B truncation mutant that eliminates only the distal portion of the C-terminal tail, did not display any difference in mechanosensitivity compared with wild type NR2B (Fig. 5A). Cells that express NR2B-1036X, which eliminates the majority of the C terminus including numerous cytoskeletal binding sites and phosphorylation sites, displayed a significantly reduced level of mechanosensitivity (NR1/NR2B, 1 ± 0.08; NR1/NR2B-1036X, 0.43 ± 0.05, p < 0.05) (Fig. 5C). Neither mutant displayed a difference in the chemical agonist response (100 μm NMDA) when compared with the NR1/NR2B wild type (Fig. 5, B and D). Importantly, the reduced mechanosensitivity of the truncation mutant compared with wild type NR2B was evident even in experiments where PSD-95 was not included (Fig. 5, E and F). These data suggest that the intermediate region of the NR2B C terminus between amino acids 1036 and 1433 confers NR2B mechanosensitivity but does not affect the normal agonist response of the receptor.
FIGURE 5.

NR2B C-terminal tail confers mechanosensitivity. The cells were transfected with NR2B or with truncation mutants of NR2B, NR2B-1036X, or NR1433X, which truncate the C-terminal tail distal to amino acid 1036 and 1433, respectively. A and B, normalized stretch response was significantly decreased in cells expressing NR2B-1036X (A), whereas response to 100 μm was not different (B). C and D, response to stretch was slightly increased in cells expressing NR2B-1433X (C), whereas response to NMDA stimulation was not different (D) among NR2B and NR2B-1433X. E and F, differential mechanosensitivity was also seen in cultures where PSD-95 was absent, where NR2B-1036X again demonstrated a reduced stretch response (E) and similar NMDA response (F), compared with wild type. This suggests that NR2B mechanosensitivity is conferred by the intracellular domain within amino acids 1036–1433 (*, p < 0.05 compared with NR1/NR2B response).
Regulation of NR2B Mechanosensitivity by PKC Phosphorylation Site on NR2B
The identified mechanoregulatory domain of the NMDAR (amino acids 1036–1433) contains numerous phosphorylation sites that can influence receptor function (16, 49). Among these phosphorylation sites are Ser-1303 and Ser-1323, which are phosphorylated by PKC. PKC has been shown in previous studies of NMDAR mechanoactivation to partially restore the injury-induced loss of Mg2+ block observed after injury (36). Thus, we examined the role of PKC activity in both primary cortical cultures and NR1/NR2B-transfected HEKs. In primary cortical neurons pretreated with 20 μm tamoxifen citrate, an inhibitor for PKC binding to its substrates, the response to mechanical stretch was nearly eliminated (Fig. 6, A and B). Pretreatment with a PKC activator, PMA (500 nm), did not change the injury mediated response in primary neurons. In response to 100 μm NMDA, PMA treatment slightly enhanced the calcium response, in agreement with previous reports, whereas PKC inhibition had no effect (Fig. 6C). Similar results were seen when PKC activity was modulated in NR1/NR2B-transfected HEKs. Tamoxifen citrate pretreatment significantly decreased the stretch response when compared with untreated cells (tamoxifen, 0.13 ± 0.01; untreated, 1 ± 0.04; p < 0.05), whereas pretreatment with PMA had no effect (Fig. 6D). In response to 100 μm NMDA in transfected HEKs, PMA-treated cells displayed a slightly reduced response, whereas tamoxifen produced no change (Fig. 6E). It is important to note that neither PKC activation nor inhibition significantly altered baseline calcium levels in transfected HEKs or cultured neurons. Thus, in agreement with previous reports of NMDAR mechanoactivation, PKC activity is observed to be crucial for the mechanical stimulation of NR1/NR2B NMDARs.
FIGURE 6.

PKC inhibition in recombinant NMDARs and primary neurons reduces receptor mechanosensitivity. Primary cortical cultures (day 15 in vitro) were treated left untreated or treated with tamoxifen or PMA. A, representative images of treated and untreated cortical cultures before and after stretch (left panels) or NMDA (right panels) stimulation. B, stretch response, normalized to response of untreated cultures, was significantly reduced in tamoxifen-treated cultures, whereas PMA had no effect (*, p < 0.05 compared with untreated). C, response to 100 μm NMDA stimulation was unchanged in tamoxifen-treated cells but increased in PMA-treated cells (*, p < 0.05 compared with untreated). HEK 293 cells transfected with GFP-PSD95, NR1, and NR2B were left untreated or treated with PKC inhibitor, tamoxifen, or PKC activator, PMA. D, tamoxifen treatment significantly decreased stretch response, whereas PMA had no effect (* p < 0.05 compared with NR1/NR2B). E, neither treatment produced significant change following NMDA stimulation in transfected HEKs.
To examine whether the observed PKC dependence of NR2B mechanoactivation was mediated through the PKC phosphorylation sites, two plasmids encoding NR2B point mutations were generated where each contains a serine to alanine point mutation at these residues. Expression of NR2B-S1303A in HEK cells produced no change in observed calcium influx, compared with cells expressing wild type NR2B, after stimulation with stretch or 100 μm NMDA (Fig. 7, A and B). However, expression of NR2B-S1323A significantly decreases the stretch response compared with wild type NR2B (Fig. 7C) (NR1/NR2B, 1 ± 0.04; NR1/NR2B-S1323A, 0.42 ± 0.03, p < 0.05). Response to 100 μm NMDA, however, was not different with the NR2B-S1323A recombinant receptor (Fig. 7D). These results establish that Ser-1323 on NR2B is a necessary determinant for NR2B mechanosensitivity, providing an intriguing mechanism for potentially augmenting the mechanical tone of NMDARs.
FIGURE 7.

NR2B mechanosensitivity is critically regulated by a single PKC phosphorylation site. The cells were transfected with NR2B or with NR2B point mutations, NR2B-S1303A or NR2B-S1323A, which contain serine to alanine point mutations at PKC phosphorylation sites Ser-1303 or Ser-1323, respectively. A and B, normalized stretch response (A) and NMDA response (B) was unchanged with the expression of NR2B-S1303A. C and D, response to stretch was significantly decreased in cells expressing NR2B-1433X (C), whereas response to NMDA stimulation was not different among NR2B and NR2B-S1323A (D). This suggests that NR2B mechanosensitivity is regulated by the PKC phosphorylation site, Ser-1323, on the NR2B C-terminal tail (*, p < 0.05 compared with NR1/NR2B response).
DISCUSSION
In this report, we examine the mechanisms regulating the dynamic mechanosensitivity of the NMDAR. Using a recombinant system, we showed that the mechanosensitivity of the NMDAR is prominently regulated by the NR2B subunit. Furthermore, we identified that the NR2B C-terminal tail and a known PKC phosphorylation site significantly controls mechanosensitivity. This suggests that PKC-mediated phosphorylation of NR2B can influence mechanical tone of the NMDAR, similar to how post-translation modifications can alter receptor activity.
Models Used to Evaluate NMDAR Mechanosensitivity
These data build on previous studies of the mechanical responsiveness of the NMDAR conducted by Casado, M., and Ascher (50) and Paoletti and Ascher (51) and are linked to studies conducted by Zhang et al. (36) in the same time period. Using a series of isolated membrane patch samples, Ascher and colleagues showed that membrane tension is a primary factor that influences NMDAR mechanosensitivity. Partly because the mechanical input in these past experiments was transferred through the plasma membrane by applying positive or negative pressures to the isolated patch, these past studies showed little role for the intracellular domains of either the NR2A or NR2B subunits in modulating the measured mechanosensitivity (50). Moreover, any effect of slowly applied membrane tension was reversible when the mechanical perturbation was removed. Rather than using slowly applied membrane expansion or contraction of membrane patch samples, our studies more closely resemble the work by Zhang et al. (36), who exposed cells adhered to a flexible membrane to a single dynamic stretch. We estimate that our model, along with that used by Zhang et al., is different from the Ascher model because mechanical forces are exerted on NMDARs through both the plasma membrane and indirectly through the intracellular domains coupled to the cytoskeleton. Unlike the Ascher studies, this dynamic perturbation causes an irreversible change in the physiological properties of the NMDAR, leading to a persisting calcium increase and, in primary neurons, an altered response to NMDA (36).
Our studies extend this past work by evaluating subunit specificity and regulation of NMDAR mechanosensitivity. We eliminated the potential complicating factors caused by studying mechanical stretch in cortical neurons by investigating mechanosensitivity on recombinant receptors in a well characterized expression system (41, 52–55). Potential confounding factors in cortical neurons include the enhanced or rapid release of glutamate vesicles in the presynaptic bouton after stretch, the transient impairment of glutamate uptake by astrocytes, or the physical widening of the synaptic cleft caused by mechanical stretch—all of which can mask the observation of the direct stretch effect on the NMDAR. Our recombinant receptor approach also avoids the complications from blocking different receptor populations in primary neurons with antagonists, some of which are known to have only modestly higher affinity for different subunits (56). Where appropriate, however, we exposed cortical neurons to mechanical injury and test specific conclusions from our recombinant receptor work.
NR2B Mechanosensitivity Regulated by Intracellular C-terminal Tail
These data show that the NR2B intracellular domain is a critical regulator of mechanosensitivity. Past work demonstrates that cytoskeletal destabilization in cultured neurons significantly reduces the stretch response (29). Although both NR2A and NR2B contain cytoskeletal binding sites, NR2B is thought to be more strongly tethered through its binding of α-actinin (48) and spectrin (47). The most distal region of the NR2B subunit, containing the PSD-95 binding motif, only modestly affects the mechanical responsiveness of the recombinant NR1/NR2B receptor. Although this binding motif is critical for direct signaling of NR2B-containing NMDARs to different signaling networks, the predominant control of mechanosensitivity appears elsewhere. Although further studies are necessary to conclusively elucidate the role of PSD-95 during stretch, a potential role for PSD-95 to mitigate the stretch response and act as a “mechanical clutch” would provide additional insight on the potential differential mechanosensitivity among synaptic, extrasynaptic, and even presynaptic NMDARs.
Indeed, much of the control occurs within the intracellular domain (amino acids 1036–1433) of NR2B that we have identified as a critical region in defining NR2B mechanosensitivity. However, the loss of stretch sensitivity in truncated NR2B-expressing receptors was not complete and suggests that residual stretch sensitivity may be due to force transfer through the remaining C-terminal tail or through the plasma membrane. One intriguing possibility, for further study, is the potential regulation of mechanosensitivity by alternatively spliced cassettes in NR1. In our studies we utilized NR1a, which contains all alternatively spliced regions, but there is evidence that alternatively spliced cytoplasmic regions of NR1 are responsible for tethering to neurofilament (57) and microtubule filaments (58), providing a mechanism for different NR1 splice variants to exhibit differential stretch sensitivity. Unfortunately, our attempts to further examine the cytoskeletal role in mechanosensitivity were hampered by the inability to sufficiently destabilize the cytoskeleton network in HEK 293s without adversely affecting cell health.
Although the C terminus of the NR2B subunit is crucial for stretch sensitivity of the NMDAR, the mechanism that regulates the change in NMDAR physiology after stretch appears linked to the pore region of the NR2 subunit. Past work showed that dynamic stretch directly changes the efficiency of the magnesium block at normal resting membrane potential (36). We observed that MK801 pretreatment reduced, but did not eliminate, the proportion of cells responding to stretch, unlike its complete inhibitory effect in NMDA-stimulated cultures. MK801 and Mg2+ both block conductance through binding of a well defined region of the NMDAR pore (59), and thus stretch may induce a change in the pore region that could alter both the inherent Mg2+ block as well as MK801 effectiveness. It is important to note that glutamate binding is still necessary for the observed stretch response of NMDARs, because treatment with the competitive antagonist APV completely eliminated the stretch response. Moreover, blocking the refilling of synaptic vesicles also led to a significant reduction in the stretch response, suggesting that stretch initiates an instantaneous release of vesicles that contributes to the immediate calcium influx. In all, our data suggest a model in which glutamate released from vesicles during stretch initiates the calcium influx seen after injury, whereas stretch alters the normal regulation, via the partial loss of the native Mg2+ block, in NR2B-containing receptors.
We have no data measuring any potential change in affinity for either glutamate or NMDA to activate these recombinant receptors after stretch, which may be key information to collect in the future to understand whether stretch will also selectively alter this physiological feature of the receptor. In many ways, these studies point to the possibility that mechanical force can selectively modulate the physiology of the NMDAR by enhancing currents, differentially controlling activation of different subtypes, and altering the affinities of agonists and modulatory agents. Thus, stretch-induced receptor modulation can potentially act as a post-translational modification, similar to phosphorylation events that are known to significantly alter receptor activity (49).
PKC Regulation of NMDAR Mechanosensitivity
Pinpointing a single residue on NR2B that controls a majority of the NMDAR mechanosensitivity is potentially important in the regulation of neuronal injury. Both serine/threonine and tyrosine phosphorylation sites on NR2 subunits are well known regulatory mechanisms that can augment NMDAR current (49, 55, 60, 61). PKC has phosphorylation sites on both NR2A and NR2B and its activity is known to both directly and indirectly potentiate NMDAR current (55, 60–62). The specific site that we found to regulate mechanosensitivity of NR2B, Ser-1323, is one of two PKC phosphorylation sites known to be directly linked to PKC- and insulin-mediated enhancement of NMDAR currents (62). Although it is unknown how this site functionally relates to changes in NR1/NR2B NMDAR activity, PKC potentiation of NMDAR current mediates a reduction in the normal magnesium block of the receptor (63). The role of PKC in NMDAR mechanosensitivity is less clear. In their observation of stretch-induced loss in Mg2+ block, Zhang et al. (36) showed that PKC inhibition partially restored the block. However, results from our own lab have shown that PKC activity, acting upon the NR1 subunit, can reduce NMDAR cytoskeletal anchoring and decrease stretch induced calcium influx (29). Here, we demonstrate that mutation of Ser-1323 and PKC inhibition through tamoxifen treatment significantly reduced stretch sensitivity of NR2B. Because PKC stimulation was not required for mechanosensitivity, it suggests that some basal level of PKC activity is sufficient to induce stretch sensitivity. There is only a slight overlap, however, among the PKC regulation sites influencing NMDAR current regulation and the sites regulating the mechanosensitivity of the receptor. The Ser-1303 site on the NR2B subunit, also known to potentiate NMDAR current, does not have an effect on NR2B mechanosensitivity. Furthermore, the NR2A subunit has two analogous PKC phosphorylation sites at Ser-1291 and Ser-1312, and we found that the NR2A subunit does not confer mechanosensitive properties to the receptor. It thus remains an interesting question as to why one, but not all, of these similar sites has a role in NMDAR mechanosensitivity. Our data add to the debate over how PKC and its targets play a role in traumatic brain injury, where PKC inhibition can reduce mechanosensitivity (36), but PKC activation has recently been reported to improve learning and memory after mild TBI (64). Certainly, much of these disparate findings stem from the promiscuous actions of PKC, which have multiple direct and indirect roles in cellular signaling (65). Systematically introduced mutations in NR2B now provide a template for future studies to test how the control of this mechanosensitivity can affect the post-traumatic consequences of mechanical injury to primary neurons in networks.
Implications of selective NR2B mechanosensitivity
Robust NR2B-based mechanosensitivity, coupled with the absence of sensitivity for NR1/NR2A diheteromeric receptors, is in direct contrast with traditional chemical agonist activation of the NMDAR, where NR1/NR2A NMDARs are more readily activated than NR1/NR2B NMDARs (66–68). Interpreted strictly, this would suggest that stretch will preferentially activate NR1/NR2B NMDARs. Importantly, extrasynaptically located NMDARs, which are primarily of the NR1/NR2B subtype, are linked to pro-death signaling in models of excitoxicity through its actions on nitric oxide production (69), mitochondrial dysfunction (1), and inhibition of pro-survival transcription (1, 14). Certainly, NMDARs are well established as mediators of the pathology seen after TBI (13, 70, 71). The rapid blockade of NR1/NR2B NMDARs appears an especially attractive option for treating the consequences of TBI, because it would aid in mitigating calcium influx and resultant signaling from NR2B NMDARs altered by injury, and this approach is supported by past studies (21, 26, 72). Additionally, glutamate spillover and glutamate release from glia may provide a means to enhance the activation of nonsynaptic (NR1/NR2B) NMDARs on mechanically injured neurons, pointing to glia as a second potential therapeutic target for controlling the effects of mechanical injury on neurons in vivo. However, triheteromeric NMDARs also display mechanosensitive behavior. There remains some debate within the literature on the timing and relative distribution of triheteromeric NMDARs, with some suggesting that these receptors comprise a significant fraction of only synaptic NMDARs, whereas others suggest that these receptors also extend into extrasynaptic locations (73–75). Our past work shows that synaptic NMDARs represent a majority of the immediate stretch-induced calcium flux in primary cortical neurons (29). These past data, when combined with our current results, suggest that synaptic triheteromeric NMDARs could represent a significant fraction of the calcium influx in primary neurons after stretch. Synaptic signaling through NMDARs is receiving attention for its ability to stimulate prosurvival programs, suggesting that mechanoactivation of NR1/NR2B and triheteromeric NMDARs may provide competing signals for neuronal survival. Given our results, it is likely that mechanical activation of the receptor will lead to biochemical signaling profiles that are distinct from chemical NMDA activation profiles. Future work in this area can help elucidate distinct injury consequences that may prove to be better therapeutic targets and therefore warrant new treatment strategies.
This work was supported, in whole or in part, by National Institutes of Health Grants HD41699, NS35712, and NS015212. This work was also supported by a U.S. Department of Defense Grant W911NF-10-1-0526.
- NMDAR
- N-methyl-d-aspartate receptors
- TBI
- traumatic brain injury
- PMA
- phorbol 12-myristate 13-acetate
- APV
- (2R)-amino-5-phosphonovaleric acid.
REFERENCES
- 1. Hardingham G. E., Fukunaga Y., Bading H. (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414 [DOI] [PubMed] [Google Scholar]
- 2. Hetman M., Kharebava G. (2006) Survival signaling pathways activated by NMDA receptors. Curr. Top Med. Chem. 6, 787–799 [DOI] [PubMed] [Google Scholar]
- 3. Ikonomidou C., Bosch F., Miksa M., Bittigau P., Vöckler J., Dikranian K., Tenkova T. I., Stefovska V., Turski L., Olney J. W. (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283, 70–74 [DOI] [PubMed] [Google Scholar]
- 4. Cull-Candy S. G., Leszkiewicz D. N. (2004) Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, re16. [DOI] [PubMed] [Google Scholar]
- 5. Dingledine R., Borges K., Bowie D., Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61 [PubMed] [Google Scholar]
- 6. Monyer H., Burnashev N., Laurie D. J., Sakmann B., Seeburg P. H. (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540 [DOI] [PubMed] [Google Scholar]
- 7. Liu X. B., Murray K. D., Jones E. G. (2004) Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J. Neurosci. 24, 8885–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams K., Russell S. L., Shen Y. M., Molinoff P. B. (1993) Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron 10, 267–278 [DOI] [PubMed] [Google Scholar]
- 9. Jin S. X., Feig L. A. (2010) Long-term potentiation in the CA1 hippocampus induced by NR2A subunit-containing NMDA glutamate receptors is mediated by Ras-GRF2/Erk Map kinase signaling. PLoS One 5, e11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim M. J., Dunah A. W., Wang Y. T., Sheng M. (2005) Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46, 745–760 [DOI] [PubMed] [Google Scholar]
- 11. Köhr G., Jensen V., Koester H. J., Mihaljevic A. L., Utvik J. K., Kvello A., Ottersen O. P., Seeburg P. H., Sprengel R., Hvalby Ø. (2003) Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J. Neurosci. 23, 10791–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S., Tian X., Hartley D. M., Feig L. A. (2006) Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J. Neurosci. 26, 1721–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arundine M., Tymianski M. (2004) Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol. Life Sci. 61, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hardingham G. E., Bading H. (2010) Synaptic versus extrasynaptic NMDA receptor signalling. Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lynch D. R., Guttmann R. P. (2002) Excitotoxicity. Perspectives based on N-methyl-d-aspartate receptor subtypes. J. Pharmacol. Exp. Ther. 300, 717–723 [DOI] [PubMed] [Google Scholar]
- 16. Waxman E. A., Lynch D. R. (2005) N-Methyl-d-aspartate receptor subtypes. Multiple roles in excitotoxicity and neurological disease. Neuroscientist 11, 37–49 [DOI] [PubMed] [Google Scholar]
- 17. Ikonomidou C., Stefovska V., Turski L. (2000) Neuronal death enhanced by N-methyl-d-aspartate antagonists. Proc. Natl. Acad. Sci. U.S.A. 97, 12885–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu L., Wong T. P., Pozza M. F., Lingenhoehl K., Wang Y., Sheng M., Auberson Y. P., Wang Y. T. (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304, 1021–1024 [DOI] [PubMed] [Google Scholar]
- 19. Massey P. V., Johnson B. E., Moult P. R., Auberson Y. P., Brown M. W., Molnar E., Collingridge G. L., Bashir Z. I. (2004) Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci. 24, 7821–7828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ewald R. C., Van Keuren-Jensen K. R., Aizenman C. D., Cline H. T. (2008) Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J. Neurosci. 28, 850–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DeRidder M. N., Simon M. J., Siman R., Auberson Y. P., Raghupathi R., Meaney D. F. (2006) Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol. Dis. 22, 165–176 [DOI] [PubMed] [Google Scholar]
- 22. Kinney J. W., Davis C. N., Tabarean I., Conti B., Bartfai T., Behrens M. M. (2006) A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J. Neurosci. 26, 1604–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou M., Baudry M. (2006) Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J. Neurosci. 26, 2956–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen M., Lu T. J., Chen X. J., Zhou Y., Chen Q., Feng X. Y., Xu L., Duan W. H., Xiong Z. Q. (2008) Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 39, 3042–3048 [DOI] [PubMed] [Google Scholar]
- 25. Gogas K. R. (2006) Glutamate-based therapeutic approaches. NR2B receptor antagonists. Curr. Opin. Pharmacol. 6, 68–74 [DOI] [PubMed] [Google Scholar]
- 26. Okiyama K., Smith D. H., White W. F., Richter K., McIntosh T. K. (1997) Effects of the novel NMDA antagonists CP-98,113, CP-101,581 and CP-101,606 on cognitive function and regional cerebral edema following experimental brain injury in the rat. J. Neurotrauma 14, 211–222 [DOI] [PubMed] [Google Scholar]
- 27. von Engelhardt J., Coserea I., Pawlak V., Fuchs E. C., Köhr G., Seeburg P. H., Monyer H. (2007) Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology 53, 10–17 [DOI] [PubMed] [Google Scholar]
- 28. Faden A. I., Demediuk P., Panter S. S., Vink R. (1989) The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800 [DOI] [PubMed] [Google Scholar]
- 29. Geddes-Klein D. M., Serbest G., Mesfin M. N., Cohen A. S., Meaney D. F. (2006) Pharmacologically induced calcium oscillations protect neurons from increases in cytosolic calcium after trauma. J. Neurochem. 97, 462–474 [DOI] [PubMed] [Google Scholar]
- 30. McIntosh T. K., Vink R., Soares H., Hayes R., Simon R. (1990) Effect of noncompetitive blockade of N-methyl-d-aspartate receptors on the neurochemical sequelae of experimental brain injury. J. Neurochem. 55, 1170–1179 [DOI] [PubMed] [Google Scholar]
- 31. Shapira Y., Yadid G., Cotev S., Niska A., Shohami E. (1990) Protective effect of MK801 in experimental brain injury. J. Neurotrauma 7, 131–139 [DOI] [PubMed] [Google Scholar]
- 32. Smith D. H., Okiyama K., Gennarelli T. A., McIntosh T. K. (1993) Magnesium and ketamine attenuate cognitive dysfunction following experimental brain injury. Neurosci. Lett. 157, 211–214 [DOI] [PubMed] [Google Scholar]
- 33. Bullock R., Zauner A., Myseros J. S., Marmarou A., Woodward J. J., Young H. F. (1995) Evidence for prolonged release of excitatory amino acids in severe human head trauma. Relationship to clinical events. Ann. N.Y. Acad. Sci. 765, 290–297 [DOI] [PubMed] [Google Scholar]
- 34. Nilsson P., Ronne-Engström E., Flink R., Ungerstedt U., Carlson H., Hillered L. (1994) Epileptic seizure activity in the acute phase following cortical impact trauma in rat. Brain Res. 637, 227–232 [DOI] [PubMed] [Google Scholar]
- 35. Palmer A. M., Marion D. W., Botscheller M. L., Swedlow P. E., Styren S. D., DeKosky S. T. (1993) Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 61, 2015–2024 [DOI] [PubMed] [Google Scholar]
- 36. Zhang L., Rzigalinski B. A., Ellis E. F., Satin L. S. (1996) Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science 274, 1921–1923 [DOI] [PubMed] [Google Scholar]
- 37. Liu Y., Wong T. P., Aarts M., Rooyakkers A., Liu L., Lai T. W., Wu D. C., Lu J., Tymianski M., Craig A. M., Wang Y. T. (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 27, 2846–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boeckman F. A., Aizenman E. (1994) Stable transfection of the NR1 subunit in Chinese hamster ovary cells fails to produce a functional N-methyl-d-aspartate receptor. Neurosci. Lett. 173, 189–192 [DOI] [PubMed] [Google Scholar]
- 39. Boeckman F. A., Aizenman E. (1996) Pharmacological properties of acquired excitotoxicity in Chinese hamster ovary cells transfected with N-methyl-d-aspartate receptor subunits. J. Pharmacol. Exp. Ther. 279, 515–523 [PubMed] [Google Scholar]
- 40. Gallagher M. J., Huang H., Pritchett D. B., Lynch D. R. (1996) Interactions between ifenprodil and the NR2B subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 271, 9603–9611 [DOI] [PubMed] [Google Scholar]
- 41. Wu H. Y., Hsu F. C., Gleichman A. J., Baconguis I., Coulter D. A., Lynch D. R. (2007) Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J. Biol. Chem. 282, 20075–20087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lusardi T. A., Rangan J., Sun D., Smith D. H., Meaney D. F. (2004) A device to study the initiation and propagation of calcium transients in cultured neurons after mechanical stretch. Ann. Biomed. Eng. 32, 1546–1558 [DOI] [PubMed] [Google Scholar]
- 43. Hatton C. J., Paoletti P. (2005) Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron 46, 261–274 [DOI] [PubMed] [Google Scholar]
- 44. Monyer H., Sprengel R., Schoepfer R., Herb A., Higuchi M., Lomeli H., Burnashev N., Sakmann B., Seeburg P. H. (1992) Heteromeric NMDA receptors. Molecular and functional distinction of subtypes. Science 256, 1217–1221 [DOI] [PubMed] [Google Scholar]
- 45. Foster K. A., McLaughlin N., Edbauer D., Phillips M., Bolton A., Constantine-Paton M., Sheng M. (2010) Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J. Neurosci. 30, 2676–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krapivinsky G., Krapivinsky L., Manasian Y., Ivanov A., Tyzio R., Pellegrino C., Ben-Ari Y., Clapham D. E., Medina I. (2003) The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40, 775–784 [DOI] [PubMed] [Google Scholar]
- 47. Wechsler A., Teichberg V. I. (1998) Brain spectrin binding to the NMDA receptor is regulated by phosphorylation, calcium and calmodulin. EMBO J. 17, 3931–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wyszynski M., Lin J., Rao A., Nigh E., Beggs A. H., Craig A. M., Sheng M. (1997) Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature 385, 439–442 [DOI] [PubMed] [Google Scholar]
- 49. Chen B. S., Roche K. W. (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Casado M., Ascher P. (1998) Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid. Common features with mechanosensitivity. J. Physiol. 513, 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paoletti P., Ascher P. (1994) Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron 13, 645–655 [DOI] [PubMed] [Google Scholar]
- 52. Grant E. R., Bacskai B. J., Anegawa N. J., Pleasure D. E., Lynch D. R. (1998) Opposing contributions of NR1 and NR2 to protein kinase C modulation of NMDA receptors. J. Neurochem. 71, 1471–1481 [DOI] [PubMed] [Google Scholar]
- 53. Guttmann R. P., Baker D. L., Seifert K. M., Cohen A. S., Coulter D. A., Lynch D. R. (2001) Specific proteolysis of the NR2 subunit at multiple sites by calpain. J. Neurochem. 78, 1083–1093 [DOI] [PubMed] [Google Scholar]
- 54. Kendrick S. J., Lynch D. R., Pritchett D. B. (1996) Characterization of glutamate binding sites in receptors assembled from transfected NMDA receptor subunits. J. Neurochem. 67, 608–616 [DOI] [PubMed] [Google Scholar]
- 55. Lynch D. R., Guttmann R. P. (2001) NMDA receptor pharmacology. Perspectives from molecular biology. Curr. Drug Targets 2, 215–231 [DOI] [PubMed] [Google Scholar]
- 56. Neyton J., Paoletti P. (2006) Relating NMDA receptor function to receptor subunit composition. Limitations of the pharmacological approach. J. Neurosci. 26, 1331–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ehlers M. D., Fung E. T., O'Brien R. J., Huganir R. L. (1998) Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J. Neurosci. 18, 720–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matsuda S., Hirai H. (1999) The clustering of NMDA receptor NR1 subunit is regulated by the interaction between the C-terminal exon cassettes and the cytoskeleton. Neurosci. Res. 34, 157–163 [DOI] [PubMed] [Google Scholar]
- 59. Kashiwagi K., Masuko T., Nguyen C. D., Kuno T., Tanaka I., Igarashi K., Williams K. (2002) Channel blockers acting at N-methyl-d-aspartate receptors. Differential effects of mutations in the vestibule and ion channel pore. Mol. Pharmacol. 61, 533–545 [DOI] [PubMed] [Google Scholar]
- 60. Jones M. L., Leonard J. P. (2005) PKC site mutations reveal differential modulation by insulin of NMDA receptors containing NR2A or NR2B subunits. J. Neurochem. 92, 1431–1438 [DOI] [PubMed] [Google Scholar]
- 61. Salter M. W., Kalia L. V. (2004) Src kinases. A hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328 [DOI] [PubMed] [Google Scholar]
- 62. Liao G. Y., Wagner D. A., Hsu M. H., Leonard J. P. (2001) Evidence for direct protein kinase-C mediated modulation of N-methyl-d-aspartate receptor current. Mol. Pharmacol. 59, 960–964 [DOI] [PubMed] [Google Scholar]
- 63. Chen L., Huang L. Y. (1992) Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature 356, 521–523 [DOI] [PubMed] [Google Scholar]
- 64. Zohar O., Lavy R., Zi X., Nelson T. J., Hongpaisan J., Pick C. G., Alkon D. L. (2011) PKC activator therapeutic for mild traumatic brain injury in mice. Neurobiol. Dis. 41, 329–337 [DOI] [PubMed] [Google Scholar]
- 65. Nelson T. J., Sun M. K., Hongpaisan J., Alkon D. L. (2008) Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 585, 76–87 [DOI] [PubMed] [Google Scholar]
- 66. Erreger K., Dravid S. M., Banke T. G., Wyllie D. J., Traynelis S. F. (2005) Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J. Physiol. 563, 345–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Santucci D. M., Raghavachari S. (2008) The effects of NR2 subunit-dependent NMDA receptor kinetics on synaptic transmission and CaMKII activation. PLoS Comput. Biol. 4, e1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Singh P., Hockenberry A. J., Tiruvadi V. R., Meaney D. F. (2011) Computational investigation of the changing patterns of subtype specific NMDA receptor activation during physiological glutamatergic neurotransmission. PLoS Comput. Biol. 7, e1002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sattler R., Xiong Z., Lu W. Y., Hafner M., MacDonald J. F., Tymianski M. (1999) Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284, 1845–1848 [DOI] [PubMed] [Google Scholar]
- 70. Rao V. L., Dogan A., Todd K. G., Bowen K. K., Dempsey R. J. (2001) Neuroprotection by memantine, a non-competitive NMDA receptor antagonist after traumatic brain injury in rats. Brain Res. 911, 96–100 [DOI] [PubMed] [Google Scholar]
- 71. Spaethling J. M., Geddes-Klein D. M., Miller W. J., von Reyn C. R., Singh P., Mesfin M., Bernstein S. J., Meaney D. F. (2007) Linking impact to cellular and molecular sequelae of CNS injury. Modeling in vivo complexity with in vitro simplicity. Prog. Brain Res. 161, 27–39 [DOI] [PubMed] [Google Scholar]
- 72. Dempsey R. J., Bakaya M. K., Doan A. (2000) Attenuation of brain edema, blood-brain barrier breakdown, and injury volume by ifenprodil, a polyamine-site N-methyl-d-aspartate receptor antagonist, after experimental traumatic brain injury in rats. Neurosurgery 47, 399–406 [DOI] [PubMed] [Google Scholar]
- 73. Al-Hallaq R. A., Conrads T. P., Veenstra T. D., Wenthold R. J. (2007) NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J. Neurosci. 27, 8334–8343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rauner C., Köhr G. (2011) Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-d-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem. 286, 7558–7566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tovar K. R., Westbrook G. L. (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 19, 4180–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]