

Background: The cysteine synthase complex (CSC) plays a pivotal role in plant and bacterial sulfur metabolism.
Results: CSC formation involves at least three stable complexes.
Conclusion: Assembly begins with a weak tethering of components followed by two favorable isomerizations that lead to a cessation of cysteine biosynthesis.
Significance: How the CSC assembles is intimately linked to the regulation of sulfur biology.
Keywords: Allosteric regulation, Enzyme kinetics, Enzyme mechanisms, Protein assembly, Protein conformation, O-Acetyl-l-serine sulfhydrylase, Cysteine synthetase, Protein assembly, Protein complex, Serine O-acetyltransferase
Abstract
Control of sulfur metabolism in plants and bacteria is linked, in significant measure, to the behavior of the cysteine synthase complex (CSC). The complex is comprised of the two enzymes that catalyze the final steps in cysteine biosynthesis: serine O-acetyltransferase (SAT, EC 2.3.1.30), which produces O-acetyl-l-serine, and O-acetyl-l-serine sulfhydrylase (OASS, EC 2.5.1.47), which converts it to cysteine. SAT (a dimer of homotrimers) binds a maximum of two molecules of OASS (a dimer) in an interaction believed to involve docking of the C terminus from a protomer in an SAT trimer into an OASS active site. This interaction inactivates OASS catalysis and prevents further binding to the trimer; thus, the system exhibits a contact-induced inactivation of half of each biomolecule. To better understand the dynamics and energetics that underlie formation of the CSC, the interactions of OASS and SAT from Escherichia coli were studied at equilibrium and in the pre-steady state. Using an experimental strategy that initiates dissociation of the CSC at different points in the CSC-forming reaction, three stable forms of the complex were identified. Comparison of the binding behaviors of SAT and its C-terminal peptide supports a mechanism in which SAT interacts with OASS in a non-allosteric interaction involving its C terminus. This early docking event appears to fasten the proteins in close proximity and thus prepares the system to engage in a series of subsequent, energetically favorable isomerizations that inactivate OASS and produce the fully isomerized CSC.
Introduction
Bacterial sulfur metabolism is a complex, sensitive function of an organism's environment. Bacteria respond to changes in sulfur nutrients with coordinated shifts in the levels of hundreds of cellular components (1–5). Such shifting can play a vital role in bacterial pathogenesis. For example, as Mycobacterium tuberculosis invades the macrophage, it up-regulates pathways that produce the sulfur reductants, including cysteine and methionine, that enable it to survive in the harsh oxidative environment of the phagosome (6, 7). The Homo sapiens genome does not encode cysteine biosynthetic enzymes; hence, these proteins are targets for antibiotic development.
The cysteine synthase complex (CSC)2 is intricately involved in the control of sulfur metabolism in plants (8) and bacteria. The complex is composed of serine acetyltransferase (SAT) and O-acetyl-l-serine sulfhydrylase (OASS) (9, 10), which catalyze the final steps in cysteine biosynthesis (see Reactions 1 and 2, respectively) (11).
SAT is catalytically active in the complex and produces O-acetyl-l-serine (OAS), a cysteine precursor with two allosteric functions in Escherichia coli and Salmonella typhimurium. CysB is a transcriptional activator of the cysteine regulon (12, 13), a collection of 9 operons that together encode 24 open reading frames (14, 15). OAS and its isomer, N-acetyl-l-serine (16), up-regulate CysB expression by binding to and preventing CysB from binding to its own promoter, where it inhibits transcription (17). In addition, OAS regulates the balance of the bound and free forms of SAT and OASS by binding to and dissociating the CSC (9, 18, 19). OASS appears to be inactive in the complex (10, 19, 20); hence, dissociation stimulates both OAS consumption and cysteine synthesis. Completing this metabolic circuitry is a negative feedback contact to cysteine, a potent inhibitor of SAT (9, 21, 22). Early work in bacteria indicates that OASS levels could be considerably higher than those of SAT, raising questions regarding the metabolic functions of OASS and the role of the CSC (9, 11). Recent work reveals that the E. coli mRNA levels of cysE and cysK, which encode SAT and OASS, are comparable under a variety of growth conditions, suggesting that the protein levels may be more similar than originally thought (5).
Ultracentrifugation studies have shown that the CSC consists of six protomers of SAT and four of OASS (9). Structural work has shown that SAT is a hexamer (a dimer of homotrimers (21, 23)) and that OASS is a dimer (24–26); hence, the SAT:OASS stoichiometry in the complex is 1:2. While this stoichiometry has been called into question (23, 27–28), the consensus appears to be that the CSC consists of one OASS per SAT trimer (9, 29–31), a position well supported by the current work.
OASS catalyzes a classic PLP-dependent, ping-pong β-elimination reaction in which PLP, in Schiff-base linkage with a conserved lysine, is displaced from the enzyme by attack of OAS, forming an external Schiff base that rearranges to eliminate its β-substituent (acetate). Hydrogen sulfide then adds to the enzyme and attacks the β-carbon of the intermediate, forming a cysteine-PLP adduct that is cleaved to release cysteine during re-attachment of PLP to the conserved lysine. The SAT mechanism had long been considered to be ping pong (32, 33); however, recent revaluations suggest that it is random sequential (34, 35).
To better understand how the CSC assembles, formation of the complex was studied at equilibrium and in the pre-steady state. The system was probed for folding intermediates on the assembly pathway using an experimental design in which the irreversible disassembly of the CSC is initiated at various points during complex formation. Reaction-stage-dependent reversal of the forward process allows transient, optically silent intermediates to be detected as they return to their optically active forms. Two such intermediates were identified using this strategy. A single intermediate has been suggested for the CSC from Haemophilus influenza (36). Comparison of the kinetics and energetics of the binding of SAT with its 10-mer, C-terminal peptide supports a mechanism in which the C terminus of SAT attaches to OASS through a non-allosteric interaction that tethers the CSC proteins. The loosely coupled complex then engages in at least two energetically favorable isomerizations that lead to the optimally stabilized CSC and the inactivation of OASS.
MATERIALS AND METHODS
The list of materials used in this work is as follows: OAS, dithiothreitol (DTT), EDTA, l-glutathione (reduced), glucose, imidazole, isopropyl-thio-β-d-galactopyranoside, LB media, lysozyme, maltose, β-mercaptoethanol, potassium phosphate, pyridoxine, SDS, and thiamin were the highest grade available from Sigma. Glutathione- and nickel-chelating resins were obtained from GE Healthcare. Primers and competent E. coli (BL21(DE3)) were purchased from Novagen. Ampicillin, guanidine hydrochloride, Hepes, KOH, KCl, MgCl2, NaCl, pepstatin A, phenylmethylsulfonyl fluoride, and Tris base were purchased from Fisher Scientific. Restriction enzymes and amylose resin were obtained from New England Biolabs. The P10 peptide was obtained from United Peptide Corp.
Cloning and Purification of OASS
OASS was cloned by PCR from E. coli K-12 genomic DNA using N- and C-terminal primers containing NdeI and XhoI restriction sites at the 5′- and 3′-ends of the coding region. The sense and antisense primer were: 5′-GACTACCATATGAGTAAGATTTTTGAAGATAAC and 5′-GACTACCTCGAGTTACTGTTGCAATTCTTTCTC, respectively. The coding region was subcloned into the pGEX-6P expression vector with an N-terminal His/GST tag (37). BL-21(DE3) containing the OASS expression plasmid was grown at 37 °C in LB medium containing ampicillin (100 μg/ml), pyridoxine (10 μm), and thiamin (15 μm) to an A600 of ∼0.7. The culture was then cooled to 18 °C, isopropyl-thio-β-d-galactopyranoside was added to 1.0 mm, and cells were harvested 16 h later. OASS was purified according to a published protocol (37). The resulting enzyme was >95% pure by SDS-PAGE. The kcat of purified enzyme (0.36 s−1) and its A412/A280 ratio (indicative of PLP content) were similar to those reported for the closely related enzyme from S. typhimurium, 0.56 s−1 and 3.5, respectively (38).
Cloning and Purification of SAT
The subcloning strategy and procedures are similar to those used to clone OASS with the exception that the growth medium did not include thiamin and pyridoxine. The sense and antisense primers (5′-GACTACCATATGTCAGATCCCATCACC and 5′-GACTACCTCGAGATGTCGTGTGCCGAACTG, respectively) position NdeI (5′) and XhoI (3′) restriction sites at the edges of the cysE coding region. Using these sites, the PCR product was inserted into a triple-tag pGEX-6P vector in which maltose-binding protein (39) is fused to the C terminus of GST (pGEX-His-GST-MBP). The induction protocol was identical to that of OASS. The SAT fusion was purified according to manufacturer's recommendations using Chelating Sepharose Fast Flow resin followed by amylose resin. The N-terminal His-GST-MBP tag was removed by digestion with PreScission protease during overnight dialysis against Hepes/K+ (25 mm, pH 7.5), KCl (0.10 m), and DTT (1.5 mm) at 4 °C and separated from SAT using a glutathione resin. Removal of the tag with PreScission protease leaves a small peptide (GPLH-) at the N terminus of the inserted coding region (40). The purity of SAT, assessed by SDS-PAGE, was >95%. The kcat of purified enzyme, 48 s−1, is similar to that determined in previous studies (11, 20).
Fluorescence Titrations
The fluorescence measurements were obtained using a Cary Eclipse fluorescence spectrophotometer with a 15-watt xenon lamp.
Stopped-flow Fluorescence Measurements
Pre-steady-state fluorescence experiments were performed using an Applied Photophysics SX20 stopped-flow spectrometer. The samples were excited at 414 nm, and light emitted above 455 nm was detected using a cutoff filter. The sequential mixing experiments were carried out with an SQ.1 sequential-mixing accessory. Typically, data from five to ten scans were averaged and fit using Applied Photophysics Pro-Data analysis software, which employs a Marquardt fitting algorithm.
RESULTS AND DISUCSSION
Monitoring OASS Complexes
The fluorescence properties of several OASS-bound forms of PLP have been characterized (29, 41–43). In the resting enzyme, PLP covalently attaches to a conserved active-site lysine and forms a protonated internal aldimine, which is fluorescent. The spectral features of the two protonated aldimine tautomers (the ketoenamine and enolimine) are distinct. The ketoenamine excitation λmax is near 414 nm, and its emission spectrum is centered near 510 nm (red spectrum, Fig. 1A). Substrate binding changes PLP fluorescence. In particular, the active site of OASS contains a subsite that binds the α-carboxyl of substrate. Binding at the subsite triggers partial closure of the active site of OASS (42, 44). This closure is believed to result in the observed ∼15 nm blue-shift in the PLP emission λmax and the increase in the emitted light intensity, which is due to a lengthening of the ketoenamine excited-state lifetime (42).
FIGURE 1.

The fluorescence of OASS-bound PLP. A, emission spectra of different OASS·PLP complexes. Spectra are given for OASS in complex with SAT (black), peptide (green), and free enzyme (red) and following reaction with OAS (blue). OASSdimer was 0.50 μm in all cases. The ligand concentrations were: SATtrimer (1.0 μm), or OAS (1.0 mm), or peptide (50 μm), or none. Solutions were buffered with Hepes/K+ (50 mm, pH 7.0), and spectra were acquired at T = 25 ± 2 °C. Samples were excited at λ = 414 nm. B, the structure of OASS bound to the C-terminal peptide of SAT. The figure was prepared using PyMOL, and the color scheme is as follows: peptide (blue), PLP (by atom), and OASS (wheat).
Although the CSC structure has not yet been determined, structures of OASS in complex with the C-terminal peptide of SAT provide valuable insight into the molecular basis of complex formation. Notably, these structures have provided the impetus for peptide library screens that aim to identifying novel antibiotics (25). The structures show the peptide in Van der Walls contact with PLP at the base of an active-site cleft (Fig. 1B) (26, 45). The C-terminal carboxylate of the peptide is docked into the acceptor subsite, suggesting that the fluorescence changes caused by peptide binding should mimic those of compounds that bind the subsite: this is precisely what is observed. The binding of either SAT or peptide to OASS increases the intensity of PLP fluorescence (6.1- and 3.7-fold, respectively) and blue-shifts the excitation λmax by ∼15 nm (black and green spectra, Fig. 1A). Unlike peptide or SAT, the addition of OAS causes OASS to move through its first half-reaction. This chemistry produces an external aldimine whose absorbance maximum is red-shifted ∼45 nm, causing a near cessation of fluorescence at λex 414 nm (blue spectrum, Fig. 1A). These spectral changes and interpretations are used extensively in the studies that follow.
Multiple Complexes during CSC Formation
To construct a quantitative model for SAT binding to OASS, the forward and reverse directions of the binding reaction were monitored via fluorescence. The reported CSC dissociation constants range from 2.6 nm to 150 pm (20, 29, 46) and are too low to measure by fluorescence. Fortunately, disassembly can be monitored by dissociating the complex using OAS. To simplify analysis of kinetic data, it is often valuable to acquire data under conditions where the reactions are irreversible. Toward this end, the extent of CSC dissociation at equilibrium was determined as a function of OAS concentration. These data reveal that the complex is quantitatively dissociated at OAS concentrations in excess of 50 μm (Fig. 2A).
FIGURE 2.

Isomerization during cysteine synthase formation. A, the OAS-induced dissociation of the CSC. OAS was added to an equilibrated solution of CSC, and the ensuing dissociation reaction was allowed to reach equilibrium. The normalized fluorescence (I/I0) reports the CSC concentration and indicates complete dissociation of the complex at OAS concentrations above 50 μm. The conditions were as follows: OASSdimer (1.0 μm), SATtrimer (1.0 μm), Hepes/K+ (50 mm, pH 7.0), and OAS concentrations are indicated in the figure; T = 25 ± 1 °C. B, the CSC dissociation progress curve. Dissociation was initiated by addition of OAS (0.10 mm final, from 60 mm stock) to a solution containing CSC (0.25 μm) that was formed by equilibration of OASSdimer (0.25 μm) and SATtrimer (2.0 μm) in Hepes/K+ (50 mm, pH 7.0); T = 25 ± 2 °C. Dissociation was monitored via fluorescence (λex 414 nm, λem 510 nm). The solid line represents the best-fit of the data to a single exponential decay, where koff = 0.0008 ± 0.0001 s−1. C, association of the CSC. The reaction was initiated by mixing a solution containing OASSdimer (0.10 μm) with an equal volume of a solution containing SATtrimer (8.0 μm). The solutions were equilibrated prior to mixing at 25 ± 2 °C in Hepes/K+ (50 mm, pH 7.0). Reactions were monitored by following fluorescence intensity (λex 414 nm, a cut-off filter was used to detected emitted light above 455 nm). The progress curve shown is the average of eight repetitions. The curve passing through the data represents the best-fit of a single-exponential model. D, kobs versus [SAT] for CS formation. kobs values were obtained from progress curves generated over a series of SAT concentrations using the methods and conditions described in B. The reactions were performed in triplicate and were pseudo first order with respect to OASS in all cases. kon and koff were obtained from the best fit straight line to the kobs versus [SAT] data; kon = 8.1 ± 0.2 ·106 m−1s−1, koff = 6.1 ± 0.9 s−1.
Having established conditions for irreversible dissociation of the CSC, its dissociation was initiated by addition of OAS (0.10 mm final concentration from 60 mm stock) to a solution containing CSC formed by equilibrating OASS (0.25 μm dimer) and SAT (2.0 μm trimer) (Fig. 2B). The progress of the reaction was monitored using fluorescence and is described well by a single-exponential model with an off-rate constant of 0.0008 ± 0.0001 s−1. The forward (or association) reaction was monitored with the aid of a stopped-flow fluorometer (see “Materials and Methods”). A representative stopped-flow progress curve for CSC formation is shown in Fig. 2C. The data are well described by a single-exponential equation. Progress curves were obtained over a series of SAT concentrations, and the observed rate constants (kobs) were plotted versus [SAT] to obtain values for kon and koff (Fig. 2D). The experimental conditions were such that [SAT]trimer ranged from 20 to 80 × [OASS]dimer; hence, the reactions were pseudo first order with respect to OASS. The kobs versus [SAT] plot is linear and predicts kon and koff values of 8.1 ± 0.2 · 106 m−1 s−1 and 6.1 ± 0.9 s−1, respectively.
Remarkably, the koff values obtained from the CSC association and dissociation experiments vary by a factor of 7600. The designs of these experiments differ considerably in the time interval over which the complex is allowed to form; dissociation experiments allow 5–10 min while association experiments provide tenths of seconds. Thus, it is plausible that a first complex, the intermediate, forms rapidly and is then converted to a second form of the complex, the isomerized form. Assuming this two-step model, the absence of a rapid phase in the dissociation experiment indicates that the isomerized form is the predominant form at equilibrium (its Gibbs potential is lower than that of the intermediate). The lack of a detectible second phase in the association reaction indicates that the environment of PLP does not change detectably following the first step; the subsequent isomerizations are optically “silent.”
A two-stage mixing experiment was designed to probe for the putative CSC folding intermediate. The first step in a two-step process produces the intermediate, and the rate of this step can be controlled with reactant concentrations. Given kon and koff for formation of the intermediate (8.1 ± 0.2 · 106 m−1s−1 and 6.1 ± 0.9 s−1) one can calculate that, under the conditions associated with Fig. 3A (0.20 μm and 8.0 μm OASSdimer and SATtrimer, respectively), the binding reaction will reach >99% completion after 0.10 s, at which point 92% of the OASS will have bound SAT. If binding occurs quickly relative to isomerization, a burst of the intermediate will occur and will taper as the intermediate shifts to the isomerized complex. If the irreversible dissociation is initiated during the pulse, by a second mix with OAS, the intermediate present at to of the second mix will either partition back into solution or forward toward the isomerized complex. Given that dissociation from the intermediate is 7600-fold faster than from the isomerized complex, the reaction is expected to show two phases: a rapid phase whose amplitude is proportional to the concentration of the intermediate and a slow phase that corresponds to release from the isomerized complex.
FIGURE 3.

Reaction-stage-dependent dissociation of the CS complex reveals an additional isomerization. A, dissociation progress curves. Binding reactions were initiated by rapidly mixing equal volumes of OASSdimer (0.40 μm) and SATtrimer (16.0 μm). The reactions were allowed to proceed over a series of time intervals, the delay times, before the complex was irreversibly dissociated by mixing rapidly with an equal volume of OAS (0.40 mm). The dissociation reactions were monitored via fluorescence changes (λex 414 nm, λem 510 nm). The white lines represent the behavior predicted by double-exponential best fits of the data. All solutions were buffered using Hepes/K+ (50 mm, pH 7.0) and equilibrated at T = 25 ± 1 °C prior to mixing. B, exponential decay of the fast-phase amplitude. The fast-phase amplitudes predicted by double-exponential, best fits of the individual dissociation reactions are plotted versus the delay (or reaction) times to reveal the way in which the amplitude decays over time. The amplitude data were well fit using a single-exponential model, which yielded a best-fit rate constant of 4.1 ± 0.3 s−1.
The result of such a two-stage mixing experiment is shown in Fig. 3A. OASSdimer (0.40 μm) was mixed with an equal volume of SATtrimer (16 μm), and the reaction was aged for defined intervals before initiating dissociation of the complex by mixing with an equal volume of OAS (0.40 mm). The progress curves present two clear phases: a rapid phase whose amplitude decreases with reaction age, and a slow second phase with a rate constant of 0.001 ± 0.00012 s−1, which agrees well with the value of 0.0008 ± 0.00012 s−1 determined previously (Fig. 2B). Thus, the complex disassembles in at least two steps, and the existence of an intermediate has been confirmed. Release of OASS from the isomerized complex is extremely slow and thus will not contribute significantly to the fast-phase fluorescence. Consequently, a plot of the fast-phase amplitude versus delay time provides a progress curve for conversion of the intermediate to the isomerized complex from which the rate constant for this effectively irreversible conversion can be obtained. This plot (Fig. 3B) was fit to a single-exponential decay that yielded a best-fit rate constant of 4.9 ± 0.2 s−1.
Given a two-step mechanism, one expects the dissociation constant for the intermediate to equal koff obtained from the pre-steady-state binding experiments, 6.1 s−1 (Fig. 2C); this is not the case. Fitting the fast phase of the progress curves (Fig. 3A) yielded an average rate constant of 0.58 ± 0.032 s−1. This value is 11-fold smaller than that obtained from the single-mix experiments (6.1 s−1), which provide no delay prior to dissociation. Thus, given sufficient time, the first observable complex in the forming reaction (OASS·SAT) accumulates as a second intermediate (OASS·SAT′) whose dissociation rate constant falls between that of OASS·SAT (6.1 s−1) and the fully isomerized form, OASS·SAT″ (0.0008 s−1). These data demonstrate that CSC formation involves three or more species, two of which are optically “silent.” Note that 0.58 s−1 and 0.0008 s−1 represent composite constants that report on the net movement CSC-bound OASS from different points in the assembly reaction into bulk solvent (47).
The absence of three phases in the double-mixing experiment indicates that the non-isomerized complex (OASS·SAT) has decayed to the point that its dissociation is not detected. This complex decays both by dissociation and passage to the first isomerized form. Given kon and koff governing formation of this complex and the minimum double-mixing dead time (∼0.10 s), one can show by simulation that the isomerization rate constant needed to reduce the concentration of this species below the limits of detection (≤ 5% of its maximum value) must be ≥16 s−1.
The three-step CSC assembly mechanism predicted by the data is shown in Fig. 4. The complexes and the rate constants that govern their interconversion are shown. Where appropriate, the constants are assigned experimental values. Long arrows indicate composite rate constants and the reaction regions over which they apply. The dissociation of OS″ is extremely slow relative to the other species. Thus, the composite constant for dissociation of this complex (0.0008 s−1) should approximate k−3 well. Given this approximation, Keq for the OS′ to OS″ conversion can be estimated at 612, which suggests that this reaction stabilizes the complex by as much as −3.8 kcal/mole at 25 °C. The overall equilibrium constant for formation of the CSC from E. coli has been estimated between 2.6 nm and 150 pm (20, 29, 46). Using these values and setting k−3 = 0.0008 s−1, one can calculate that Keq for the second step lies between 0.5 and 8.3. In summary, the system moves through two isomerization reactions subsequent to initial docking, the second of which effectively locks OASS into a complex from which the half-life for escape is ∼14 min, roughly comparable to the doubling time of the organism in rich media.
FIGURE 4.
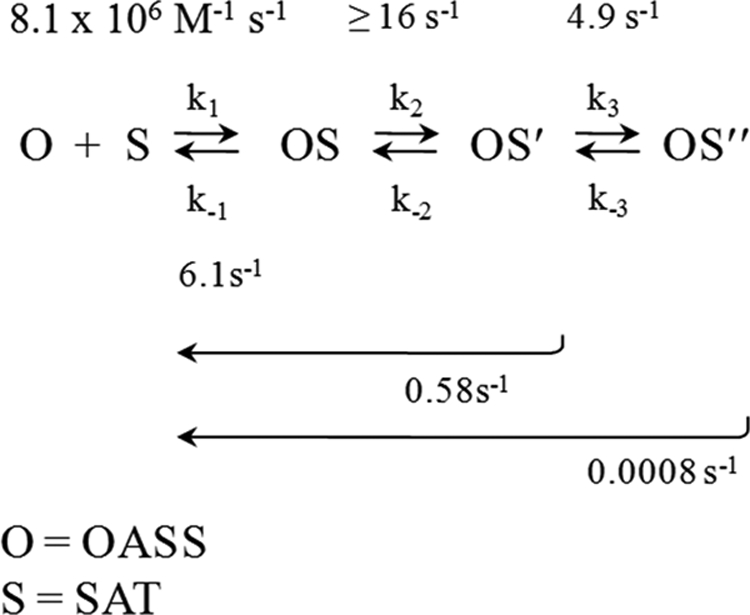
The CSC assembly mechanism. The complexes and their associated rate constants are shown. Rate constants are labeled with their experimentally determined values. Long arrows indicate regions of the reaction that are associated with composite rate constants. Isomerized forms are indicated with single and double prime symbols.
OASS Interactions with the C-terminal Ten-residue Peptide of SAT (P10)
The binding of P10 (the SAT C-terminal 10-mer) to OASS is well described by a non-allosteric binding model (Fig. 5A). The dissociation constant predicted by the fit (0.32 ± 0.02 μm) is quite similar to that for the first step in the SAT binding reaction (koff/kon = 0.72 μm). The rate constants governing P10 binding to OASS (kon = 7.7 ± 0.1 × 106 m−1 s−1, koff = 3.6 ± 0.05 s−1) were obtained from a plot of kobs versus [P10] (Fig. 5B). These constants predict a Kd (0.47 ± 0.02 μm) that is in good agreement with the value determined by the equilibrium titration and are similar to kon and koff for the first step in the SAT binding reaction (kon = 8.2 × 106 m−1 s−1, koff = 6.1 s−1, Fig. 2D).
FIGURE 5.
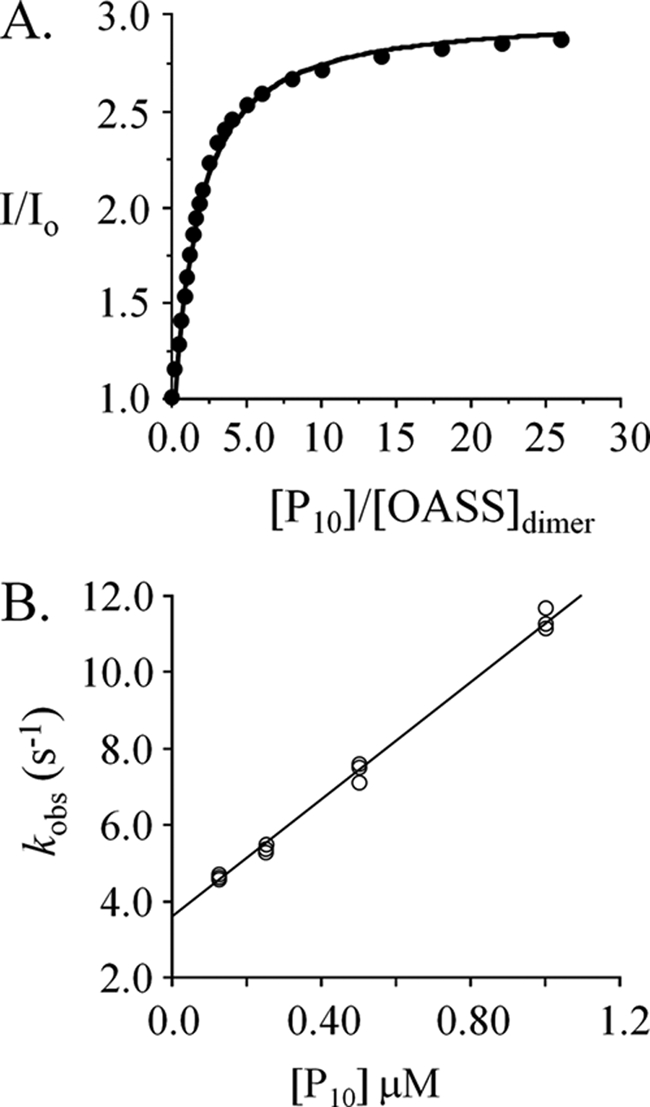
The interaction of peptide (P10) with OASS. A, the dissociation constant. Peptide binding to OASS was monitored via fluorescence changes at 510 nm (λex 414 nm). Titrations were carried out in Hepes/K+ (50 mm, pH 7.0) at 25 ± 2 °C. The concentration of OASSdimer was 0.50 μm. B, rate constants. Binding was initiated by rapidly mixing (1:1) OASSdimer (6.25 nm, final) with P10 (at the concentrations indicated). Solutions were buffered using Hepes/K+ (50 mm, pH 7.0) and equilibrated at 25 ± 2 °C prior to mixing. Reaction progress was monitored using fluorescence (λex 414 nm, λem > 455 nm). kobs values were determined in triplicate: each value was obtained by fitting the average of ten progress curves to a single-exponential decay. kon (7.7 × 106 m−1 s−1) and koff (3.6 s−1) were obtained by fitting the kobs versus [P10] data using the equation: kobs = kon · [pep] + koff.
The close agreement of the P10-binding constants determined by equilibrium and pre-steady-state methods reveals that, unlike SAT binding, in which cryptic isomerizations contribute to overall binding energetics, P10-binding does not lead to detectible isomerization. Thus, the peptide does not contain the structural determinants needed to move through the isomerization pathway traversed by SAT. The very similar values of kon and koff for the binding of P10 and SAT reveal that, in its early-stage interactions with OASS, SAT behaves very much like a simple peptide. The remainder of the 273-residue protein does not appear to interact significantly with OASS until its C terminus has docked into the OASS active site. The N terminus might then act as a linchpin to maintain SAT and OASS in close proximity, and to thereby provide an entropically advantaged position from which the system can search its conformational landscape until it accesses the isomerizations that lead ultimately to equilibrium.
P10 and SAT differ substantially in the extent to which their binding is linked to changes in function at the distal active site of OASS. This difference is evident in their OASS-binding stoichiometries (Fig. 6A). P10 is capable of binding both of OASS active sites, whereas SAT binds only one (similar stoichiometries are described for the H. influenzae system (29)). The fact that the OASS dimer binds only one SAT trimer suggests, but does not prove, that one of the OASS monomers remains unoccupied in the CSC. If OAS bound OASS in the complex, one expects a rapid change in PLP fluorescence caused by binding and/or chemistry. Instead, what is observed is a slow fluorescence change that is consistent with complex dissociation prior to reaction with OAS (Fig. 2B). Thus, both OASS active sites appear to be sequestered from substrate in the complex, an interpretation that is supported by the fact that OASS does not appear to turn over in the complex (28).
FIGURE 6.
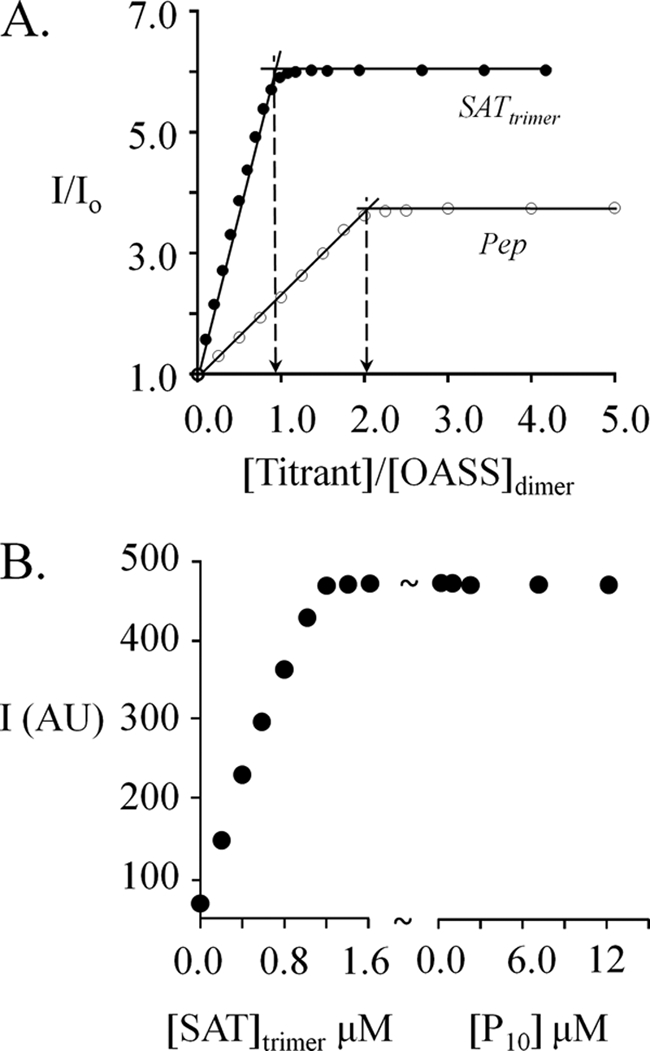
SAT binding prevents access to the unoccupied subunit of the OASS dimer. A, the stoichiometries of the peptide·OASS and SAT·OASS complexes. Equilibrium-binding titrations were carried out in Hepes/K+ (50 mm, pH 7.0) at 25 ± 2 °C, and binding was monitored via fluorescence changes (λex 414 nm, λem 510 nm). The OASS dimer concentrations used in the SAT and peptide titrations were 1.25 μm and 80 μm, respectively. B, SAT binding prevents P10 binding at the unoccupied site of OASS. OASSdimer (1.0 μm) was titrated to saturation with SAT and peptide was then added at the concentrations indicated. Conditions were otherwise identical to those described in A.
To determine whether the SAT-induced conformational change in the empty subunit of OASS also prevents binding of the peptide, OASS (1.0 μm dimer) was first saturated with SAT and the half-occupied dimer was then titrated with P10. At concentrations ranging as high as 12.5 μm (40 Kd), P10 did not influence the fluorescent intensity of the SAT-occupied OASS (Fig. 6B). Thus, OASS in the CSC is inaccessible to peptide. Findings similar to these have been discussed, but not presented, for the H. influenzae system (29).
The SAT:OASS binding stoichiometry and inaccessibility of the OASS in the CSC are consistent with a variety of interaction scenarios. It is plausible that both sites are occupied, each by a different C terminus of an SAT trimer (29). Alternatively, access to the putative open site may be prevented sterically by “burying” the site in the trimer. Finally, allosteric interactions may gate access to the open site. Neither the data presented here, nor existing structures can distinguish among these models. The allosteric model is strongly favored by modeling studies of the complex (30). The modeled structure places the unoccupied OASS active site at a distance that precludes its binding a second C terminus and predicts that the unoccupied site is allosterically “sealed” by the docking of SAT.
Assuming an allosteric mechanism, the simplest interpretation of findings presented here is that addition of SAT alters the structure of the unliganded site such that its affinity for peptide has been weakened beyond the point of detection: the OASS active site has been closed. Alternatively, the peptide could bind at the second site without altering PLP fluorescence. In either case, the effects of binding of SAT or peptide on function at the unoccupied site are very different. The fact that the early-stage interactions of SAT and peptide with OASS are virtually identical suggests that closure of the unoccupied site happens subsequent to docking of the SAT C terminus and might well be associated with the isomerizations that occur in the later stages of complex formation.
CONCLUSIONS
The pre-steady-state experiments presented here demonstrate that the CSC passes through at least three stable conformations in achieving its most stable configuration. A comparison of the kinetics and energetics of the binding of SAT with its C-terminal peptide reveals that, unlike SAT, peptide binding is monophasic, and binding at one OASS active site does not prevent, or otherwise influence, binding at the second. The rate constants governing the first phase of the SAT binding reaction are remarkably similar to those for the binding of peptide, suggesting that early docking of SAT occurs primarily through the its C terminus. The inability of the peptide to either induce isomerization or “close” the distal site suggests that SAT structure beyond its C terminus is required to engage in isomerization and that closure of the unoccupied OASS active site may be coupled to the one or more isomerizations.
This work was supported, in whole or in part, by National Institutes of Health Grant GM54469 (to T. S. L.).
- CSC
- cysteine synthase complex
- OASS
- O-acetyl-l-serine sulfhydrylase
- OAS
- O-acetyl-l-serine
- PLP
- pyridoxal phosphate
- SAT
- serine acetyltransferase.
REFERENCES
- 1. Auger S., Danchin A., Martin-Verstraete I. (2002) Global expression profile of Bacillus subtilis grown in the presence of sulfate or methionine. J. Bacteriol. 184, 5179–5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forquin M. P., Hébert A., Roux A., Aubert J., Proux C., Heilier J. F., Landaud S., Junot C., Bonnarme P., Martin-Verstraete I. (2011) Global regulation of the response to sulfur availability in the cheese-related bacterium Brevibacterium aurantiacum. Appl. Environ. Microbiol. 77, 1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gyaneshwar P., Paliy O., McAuliffe J., Jones A., Jordan M. I., Kustu S. (2005) Lessons from Escherichia coli genes similarly regulated in response to nitrogen and sulfur limitation. Proc. Natl. Acad. Sci. U.S.A. 102, 3453–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gyaneshwar P., Paliy O., McAuliffe J., Popham D. L., Jordan M. I., Kustu S. (2005) Sulfur and nitrogen limitation in Escherichia coli K-12. Specific homeostatic responses. J. Bacteriol. 187, 1074–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zare H., Sangurdekar D., Srivastava P., Kaveh M., Khodursky A. (2009) Reconstruction of Escherichia coli transcriptional regulatory networks via regulon-based associations. BMC Syst. Biol. 3, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhave D. P., Muse W. B., 3rd, Carroll K. S. (2007) Drug targets in mycobacterial sulfur metabolism. Infect. Disord. Drug Targets 7, 140–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hatzios S. K., Bertozzi C. R. (2011) The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog. 7, e1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hirai M. Y., Fujiwara T., Awazuhara M., Kimura T., Noji M., Saito K. (2003) Global expression profiling of sulfur-starved Arabidopsis by DNA macroarray reveals the role of O-acetyl-l-serine as a general regulator of gene expression in response to sulfur nutrition. Plant J. 33, 651–663 [DOI] [PubMed] [Google Scholar]
- 9. Kredich N. M., Becker M. A., Tomkins G. M. (1969) Purification and characterization of cysteine synthetase, a bifunctional protein complex, from Salmonella typhimurium. J. Biol. Chem. 244, 2428–2439 [PubMed] [Google Scholar]
- 10. Kredich N. M., Tomkins G. M. (1967) in Organizational Biosynthesis (Vogel H. J., Lampen J. O., Bryson V., eds) pp. 189–198, Academic Press, New York [Google Scholar]
- 11. Kredich N. M., Tomkins G. M. (1966) The enzymic synthesis of L-cysteine in Escherichia coli and Salmonella typhimurium. J. Biol. Chem. 241, 4955–4965 [PubMed] [Google Scholar]
- 12. Kredich N. M. (1992) The molecular basis for positive regulation of cys promoters in Salmonella typhimurium and Escherichia coli. Mol. Microbiol. 6, 2747–2753 [DOI] [PubMed] [Google Scholar]
- 13. Ostrowski J., Kredich N. M. (1989) Molecular characterization of the cysJIH promoters of Salmonella typhimurium and Escherichia coli. Regulation by cysB protein and N-acetyl-L-serine. J. Bacteriol. 171, 130–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keseler I. M., Collado-Vides J., Santos-Zavaleta A., Peralta-Gil M., Gama-Castro S., Muñiz-Rascado L., Bonavides-Martinez C., Paley S., Krummenacker M., Altman T., Kaipa P., Spaulding A., Pacheco J., Latendresse M., Fulcher C., Sarker M., Shearer A. G., Mackie A., Paulsen I., Gunsalus R. P., Karp P. D. (2011) EcoCyc. A comprehensive database of Escherichia coli biology. Nucleic Acids Res. 39, D583–D590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kredich N. M. (1987) in Escherichia Coli and Salmonella Typhimurium Cellular and Molecular Biology (Neidhardt F. C., ed) pp. 419–428, American Society for Microbiology, Washington, D. C [Google Scholar]
- 16. Flavin M., Slaughter C. (1965) Synthesis of the succinic ester of homoserine, a new intermediate in the bacterial biosynthesis of methionine. Biochemistry 4, 1370–1375 [DOI] [PubMed] [Google Scholar]
- 17. Ostrowski J., Kredich N. M. (1991) Negative autoregulation of cysB in Salmonella typhimurium. In vitro interactions of CysB protein with the cysB promoter. J. Bacteriol. 173, 2212–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cook P. F., Wedding R. T. (1977) Salmonella typhimurium/enzymol. Arch. Biochem. Biophys. 178, 293–302 [DOI] [PubMed] [Google Scholar]
- 19. Droux M., Ruffet M. L., Douce R., Job D. (1998) Interactions between serine acetyltransferase and O-acetylserine (thiol) lyase in higher plants. Structural and kinetic properties of the free and bound enzymes. Eur. J. Biochem. 255, 235–245 [DOI] [PubMed] [Google Scholar]
- 20. Mino K., Yamanoue T., Sakiyama T., Eisaki N., Matsuyama A., Nakanishi K. (2000) Effects of bienzyme complex formation of cysteine synthetase from Escherichia coli on some properties and kinetics. Biosci. Biotechnol. Biochem. 64, 1628–1640 [DOI] [PubMed] [Google Scholar]
- 21. Olsen L. R., Huang B., Vetting M. W., Roderick S. L. (2004) Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Biochemistry 43, 6013–6019 [DOI] [PubMed] [Google Scholar]
- 22. Hindson V. J. (2003) Serine acetyltransferase of Escherichia coli. Substrate specificity and feedback control by cysteine. Biochem. J. 375, 745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pye V. E., Tingey A. P., Robson R. L., Moody P. C. (2004) The structure and mechanism of serine acetyltransferase from Escherichia coli. J. Biol. Chem. 279, 40729–40736 [DOI] [PubMed] [Google Scholar]
- 24. Chattopadhyay A., Meier M., Ivaninskii S., Burkhard P., Speroni F., Campanini B., Bettati S., Mozzarelli A., Rabeh W. M., Li L., Cook P. F. (2007) Structure, mechanism, and conformational dynamics of O-acetylserine sulfhydrylase from Salmonella typhimurium. Comparison of A and B isozymes. Biochemistry 46, 8315–8330 [DOI] [PubMed] [Google Scholar]
- 25. Salsi E., Bayden A. S., Spyrakis F., Amadasi A., Campanini B., Bettati S., Dodatko T., Cozzini P., Kellogg G. E., Cook P. F., Roderick S. L., Mozzarelli A. (2010) J. Med. Chem. 53, 345–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schnell R., Oehlmann W., Singh M., Schneider G. (2007) Structural insights into catalysis and inhibition of O-acetylserine sulfhydrylase from Mycobacterium tuberculosis. Crystal structures of the enzyme α-aminoacrylate intermediate and an enzyme-inhibitor complex. J. Biol. Chem. 282, 23473–23481 [DOI] [PubMed] [Google Scholar]
- 27. Kumar S., Raj I., Nagpal I., Subbarao N., Gourinath S. (2011) Structural and biochemical studies of serine acetyltransferase reveal why the parasite Entamoeba histolytica cannot form a cysteine synthase complex. J. Biol. Chem. 286, 12533–12541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mino K., Yamanoue T., Sakiyama T., Eisaki N., Matsuyama A., Nakanishi K. (1999) Purification and characterization of serine acetyltransferase from Escherichia coli partially truncated at the C-terminal region. Biosci. Biotechnol. Biochem. 63, 168–179 [DOI] [PubMed] [Google Scholar]
- 29. Campanini B., Speroni F., Salsi E., Cook P. F., Roderick S. L., Huang B., Bettati S., Mozzarelli A. (2005) Interaction of serine acetyltransferase with O-acetylserine sulfhydrylase active site. Evidence from fluorescence spectroscopy. Protein Sci. 14, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feldman-Salit A., Wirtz M., Hell R., Wade R. C. (2009) A mechanistic model of the cysteine synthase complex. J. Mol. Biol. 386, 37–59 [DOI] [PubMed] [Google Scholar]
- 31. Mino K., Imamura K., Sakiyama T., Eisaki N., Matsuyama A., Nakanishi K. (2001) Increase in the stability of serine acetyltransferase from Escherichia coli against cold inactivation and proteolysis by forming a bienzyme complex. Biosci. Biotechnol. Biochem. 65, 865–874 [DOI] [PubMed] [Google Scholar]
- 32. Leu L. S., Cook P. F. (1994) Kinetic mechanism of serine transacetylase from Salmonella typhimurium. Biochemistry 33, 2667–2671 [DOI] [PubMed] [Google Scholar]
- 33. Mino K., Hiraoka K., Imamura K., Sakiyama T., Eisaki N., Matsuyama A., Nakanishi K. (2000) Characteristics of serine acetyltransferase from Escherichia coli deleting different lengths of amino acid residues from the C terminus. Biosci. Biotechnol. Biochem. 64, 1874–1880 [DOI] [PubMed] [Google Scholar]
- 34. Hindson V. J., Shaw W. V. (2003) Random-order ternary complex reaction mechanism of serine acetyltransferase from Escherichia coli. Biochemistry 42, 3113–3119 [DOI] [PubMed] [Google Scholar]
- 35. Johnson C. M., Huang B., Roderick S. L., Cook P. F. (2004) Kinetic mechanism of the serine acetyltransferase from Haemophilus influenzae. Arch. Biochem. Biophys. 429, 115–122 [DOI] [PubMed] [Google Scholar]
- 36. Salsi E., Campanini B., Bettati S., Raboni S., Roderick S. L., Cook P. F., Mozzarelli A. (2010) A two-step process controls the formation of the bienzyme cysteine synthase complex. J. Biol. Chem. 285, 12813–12822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andreassi J. L., 2nd, Leyh T. S. (2004) Molecular functions of conserved aspects of the GHMP kinase family. Biochemistry 43, 14594–14601 [DOI] [PubMed] [Google Scholar]
- 38. Tai C. H., Nalabolu S. R., Jacobson T. M., Minter D. E., Cook P. F. (1993) Kinetic mechanisms of the A and B isozymes of O-acetylserine sulfhydrylase from Salmonella typhimurium LT-2 using the natural and alternative reactants. Biochemistry 32, 6433–6442 [DOI] [PubMed] [Google Scholar]
- 39. Kapust R. B., Waugh D. S. (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8, 1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cordingley M. G., Callahan P. L., Sardana V. V., Garsky V. M., Colonno R. J. (1990) Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J. Biol. Chem. 265, 9062–9065 [PubMed] [Google Scholar]
- 41. Benci S., Vaccari S., Mozzarelli A., Cook P. F. (1999) Time-resolved fluorescence of O-acetylserine sulfhydrylase. Biochim. Biophys. Acta 1429, 317–330 [DOI] [PubMed] [Google Scholar]
- 42. Schnackerz K. D., Tai C. H., Simmons J. W., 3rd, Jacobson T. M., Rao G. S., Cook P. F. (1995) Identification and spectral characterization of the external aldimine of the O-acetylserine sulfhydrylase reaction. Biochemistry 34, 12152–12160 [DOI] [PubMed] [Google Scholar]
- 43. Strambini G. B., Cioni P., Cook P. F. (1996) Tryptophan luminescence as a probe of enzyme conformation along the O-acetylserine sulfhydrylase reaction pathway. Biochemistry 35, 8392–8400 [DOI] [PubMed] [Google Scholar]
- 44. Rabeh W. M., Cook P. F. (2004) Structure and mechanism of O-acetylserine sulfhydrylase. J. Biol. Chem. 279, 26803–26806 [DOI] [PubMed] [Google Scholar]
- 45. Huang B., Vetting M. W., Roderick S. L. (2005) The active site of O-acetylserine sulfhydrylase is the anchor point for bienzyme complex formation with serine acetyltransferase. J. Bacteriol. 187, 3201–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao C., Moriga Y., Feng B., Kumada Y., Imanaka H., Imamura K., Nakanishi K. (2006) On the interaction site of serine acetyltransferase in the cysteine synthase complex from Escherichia coli. Biochem. Biophys. Res. Commun. 341, 911–916 [DOI] [PubMed] [Google Scholar]
- 47. Cleland W. W. (1975) Partition analysis and the concept of net rate constants as tools in enzyme kinetics. Biochemistry 14, 3220–3224 [DOI] [PubMed] [Google Scholar]