

Background: Calcium signal induced by the environmental polycyclic aromatic hydrocarbon (PAH) benzo(a)pyrene (B(a)P) contributes to its aryl hydrocarbon receptor (AhR)-related toxic effects.
Results: β2-Adrenoreceptor (β2ADR) mediates B(a)P-induced calcium signal through a G protein/adenylyl cyclase/cAMP/Epac-1/inositol 1,4,5-trisphosphate pathway.
Conclusion: An important crosstalk between β2ADR and AhR pathways can be activated by PAHs.
Significance: β2ADR is potentially involved in deleterious effects of PAHs.
Keywords: Calcium, Camp, Cell signaling, Cell surface receptor, G protein-coupled receptors (GPCRs), benzo(a)pyrene, β2-adrenergic receptor, polycyclic aromatic hydrocarbon
Abstract
Polycyclic aromatic hydrocarbons (PAHs) such as benzo(a)pyrene (B(a)P) are widely distributed environmental contaminants, known as potent ligands of the aryl hydrocarbon receptor (AhR). These chemicals trigger an early and transient increase of intracellular calcium concentration ([Ca2+]i), required for AhR-related effects of PAHs. The mechanisms involved in this calcium mobilization were investigated in the present study. We demonstrated that B(a)P-mediated [Ca2+]i induction was prevented in endothelial HMEC-1 cells by counteracting β2-adrenoreceptor (β2ADR) activity using pharmacological antagonists, anti-β2ADR antibodies, or siRNA-mediated knockdown of β2ADR expression; by contrast, it was strongly potentiated by β2ADR overexpression in human kidney HEK293 cells. B(a)P was shown, moreover, to directly bind to β2ADR, as assessed by in vitro binding assays and molecular modeling. Pharmacological inhibition and/or siRNA-mediated silencing of various signaling actors acting downstream of β2ADR in a sequential manner, such as G protein, adenylyl cyclase, Epac-1 protein, and inositol 1,4,5-trisphosphate (IP3)/IP3 receptor, were next demonstrated to prevent B(a)P-induced calcium signal. Inhibition or knockdown of these signaling elements, as well as the use of chemical β-blockers, were finally shown to counteract B(a)P-mediated induction of cytochrome P-450 1B1, a prototypical AhR target gene. Taken together, our results show that B(a)P binds directly to β2ADR and consequently utilizes β2ADR machinery to mobilize [Ca2+]i, through activation of a G protein/adenylyl cyclase/cAMP/Epac-1/IP3 pathway. This β2ADR-dependent signaling pathway activated by PAHs may likely be crucial for PAH-mediated up-regulation of AhR target genes, thus suggesting a contribution of β2ADR to the health-threatening effects of these environmental pollutants.
Introduction
Polycyclic aromatic hydrocarbons (PAHs)3 constitute a major family of widely distributed environmental contaminants. They are usually generated through incomplete combustion of organic compounds and are thus notably found in diesel exhaust particles, cigarette smoke, charcoal-broiled foods, and industrial waste by-products. These pollutants are well known to promote various deleterious effects toward human health, including the development of cancers, cardiovascular pathologies, endocrine disruption, and inflammatory diseases (1–3). Most of these effects have been linked to the binding of PAHs to the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor primarily located in the cytosol (4). Such binding triggers AhR translocation into the nucleus and subsequent AhR interaction with specific genomic elements termed xenobiotic responsive elements, which are found in the promoter of PAH-responsive genes such as the carcinogen-bioactivating enzymes cytochrome P-450 (CYP) CYP1A1 and CYP1B1, or the pro-inflammatory chemokine CCL1 (5).
In addition, PAHs, including the prototypical PAH benzo(a)pyrene (B(a)P), have been shown to elicit an early and transient increase in intracellular calcium concentration ([Ca2+]i), that is required for up-regulation of AhR-mediated induction of PAHs target genes. For example, inhibition of the Ca2+ signal evoked by PAHs fully prevents their AhR-related inducing effects toward CYP1A1 or CCL1 expression (5). These data suggest that calcium is a major player of the AhR-signaling pathway activated by PAHs (5, 6). Early PAH-related calcium mobilization, however, occurs independently of AhR. Indeed, PAHs known to poorly activate AhR, such as benzo(e)pyrene or pyrene, are able to increase [Ca2+]i in a similar manner to that of PAH agonists of AhR such as B(a)P (7); in the same way, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), an halogenated aromatic hydrocarbon and potent AhR agonist, increases [Ca2+]i even in cellular models that do not express AhR (8). Moreover, knockdown AhR expression failed to counteract [Ca2+]i signal triggered by B(a)P (7).
Mechanisms that contribute to [Ca2+]i modulation by PAHs have been linked to inhibition of sarcoendoplasmic reticulum calcium ATPase (9) or to activation of protein-tyrosine kinases (10), ryanodine receptor (11), store-operated calcium channel, or inositol 1,4,5-trisphosphate (IP3) receptor (5, 12). However, the initial events that trigger calcium signaling in response to PAH exposure still remain unknown.
β-Adrenergic receptors (βADRs) belong to the family of G protein-coupled receptors and include the three isoforms β1, β2, and β3 ADR (13). These receptors participate to the control of many physiological processes, like the regulation of smooth muscle contraction, blood pressure, bronchodilation, and glycogenolysis. βADR stimulation by ligands, such as epinephrine, commonly leads to the activation of adenylyl cyclase (AC) via a Gs protein and, subsequently, increases the production of cAMP (14). This second messenger is a central player in intracellular signaling and is known to directly activate protein kinase A (PKA), special types of membrane ionic channels (cAMP-gated channel), and a family of guanine nucleotide exchange factors known as exchange protein factor directly activated by cAMP (Epac) and composed of two members, Epac-1 and Epac-2 (15, 16).
Signaling pathways, dependent on βADRs, especially β2ADR, are known to be modulated by PAHs and related AhR ligands, such as TCDD. For example, TCDD can decrease β-adrenergic responsiveness in cardiac muscle cells, with a concomitant increase in [Ca2+]i. This effect seems to be caused by the interaction of TCDD with βADRs upstream of AC (17–19). Accordingly, TCDD was shown to increase intracellular cAMP level via the activation of a G protein (20). In the same way, exposure to PAHs or to cigarette smoke (known to contain several PAHs) decreases the number of βADR, especially of β2ADR, on the surface of blood mononuclear cells, airway smooth muscle cells, or tracheal epithelial cells (21–23). Authors suggest that this decrease of βADR expression corresponds to a desensitization mechanism, a phenomenon usually triggered by βADR activation (24, 25).
The fact that PAHs interact with β2ADR function and/or expression and that signaling pathway related to this receptor can increase [Ca2+]i via cAMP-mediated Epac activation (26–28), indicates that β2ADR might play a role in B(a)P-induced [Ca2+]i increase. The present study was thus designed to gain insights into this hypothesis. Our data show that B(a)P binds to β2ADR and consequently utilizes β2ADR machinery to mobilize [Ca2+]i, through activation of a G protein/AC/cAMP/Epac-1/IP3 pathway. Furthermore, we observe that this β2ADR-dependent signaling pathway activated by PAHs is crucial for PAH-mediated up-regulation of AhR target genes such as CYP1B1, thus suggesting a contribution of β2ADRs to the deleterious effects of these environmental pollutants toward health.
EXPERIMENTAL PROCEDURES
Chemical and Reagents
2-Aminoethoxydiphenyl borate (2-APB), B(a)P, brefeldin A (BFA), carazolol, 2′-5′-dideoxyadenosine (dd-Ado), ICI-118,551, MDL-12,330A, salbutamol, propranolol, and 8-(diethylamino)octyl-3,4,5-trimethoxybenzoate hydrochloride (TMB-8) were provided by Sigma-Aldrich. Pluronic acid and fura-2 acetoxymethyl ester (Fura-2-AM) were provided from Invitrogen. Rabbit monoclonal antibody anti-β2ADR and control antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), whereas mouse monoclonal antibody anti-Epac-1 was obtained from Cell Signaling Technology (Beverly, MA). All other compounds were commercial products of the highest purity available. Chemicals were used as stock solutions in DMSO; the final concentration of this solvent in culture medium was always <0.2% (v/v), and control cultures received the same dose of vehicle as exposed cultures.
Cell Culture
Human endothelial HMEC-1 cells, obtained from the Centers for Disease Control and Prevention (Atlanta, GA), and human embryonic kidney HEK293 cells were routinely maintained in MDCB131 and Dulbecco's modified Eagle's medium, respectively, containing penicillin (50 units/ml) and streptomycin (50 μg/ml) and supplemented with 10% fetal calf serum. HEK293 cells permanently expressing β2ADR were obtained by β2ADR cDNA transfection using Lipofectamine (Invitrogen). Briefly, HEK293 cells were seeded at 2.5 × 105 cells/well in 6-well plates, transfected with either 2.5 μg of empty pcDNA3.1(+)neo vector (HEKwt) or 2.5 μg of pcDNA3.1(+)neo vector containing HA-tagged human β2ADR ORF (HEKβ2), and subsequently selected with G418 sulfate (1 mg/ml).
Intracellular Calcium Concentration Measurements
Variations in [Ca2+]i were analyzed in PAH-exposed HMEC-1 or HEK293 cells by microspectrofluorometry using the Ca2+-sensitive probe Fura-2AM, as previously reported (12). Briefly, HMEC-1 or HEK293 cells were incubated at 37 °C in cell suspension buffer (134.8 mm NaCl, 4.7 mm KCl, 1.2 mm K2HPO4, 1 mm MgCl2, 1 mm CaCl2, 10 mm glucose, 10 mm HEPES, pH 7.4) supplemented with 1.5 μm Fura-2AM and 0.006% pluronic acid. Following probe loading, cells were placed in a continuously perfused recording chamber mounted on the stage of an epifluorescence microscope (Nikon), and trapped dye fluorescence was measured at 510 nm. The ratio of fluorescence intensities recorded after excitation at 340 nm (F340) and 380 nm (F380) was used to estimate [Ca2+]i. Results are presented as normalized calcium level, knowing that basal [Ca2+]i was arbitrary normalized to 1. The monochromator and the photometers, which allow emission and detection of fluorescence from three to five cells in the field of view, were part of a DeltaRAM system from Photon Technology International (PTI, Birmingham, UK), which also provided software systems to acquire and process data.
siRNA Transfection
Chemically synthesized, double-stranded, ON-TARGETplus SMARTpool siRNAs targeting β2ADR or Epac-1 were purchased from Dharmacon (Chicago, IL). ON-TARGETplus non-targeting siRNAs were used as a control. Semi-confluent cells were transfected with 100 nm siRNAs using Dharmafect-1 transfection reagent diluted in antibiotic-free culture medium. Forty-eight hours after transfection, cells were exposed to treatments. Transfection efficacy was verified by Western blotting analysis of β2ADR and Epac-1 expression.
Crude Membrane Preparation
Crude membranes were prepared by differential ultracentrifugation as previously reported (29). Briefly, after washing, cells were lysed in buffer containing 1 mm EDTA, 0.2 mm phenylmethylsulfonyl fluoride, and protease inhibitors in 10 mm Tris, pH 7.4, and centrifuged at 500 × g for 5 min to remove nuclei and unbroken cells. Supernatant was next ultracentrifuged at 40,000 × g for 30 min. Pellet, containing membranes, was resuspended in lysis buffer and centrifuged at 40,000 × g for 30 min. The resulting pellet was suspended in binding buffer (0.5 mm MgCl2, in 50 mm Tris, pH 7.4), aliquoted, and stored at −80 °C until use.
[3H]B(a)P Binding Assay
HEKβ2 crude membranes (1.5 μg of protein) were added to tubes containing [3H]B(a)P (American Radiolabeled Chemicals, St. Louis, MO; specific activity, 50 Ci/mmol). The final concentrations of the tritiated PAH were 0–100 nm. Total volume was adjusted to 200 μl with binding buffer, supplemented with 2% bovine serum albumin, and tubes were incubated for 30 min at 37 °C. Incubation was terminated by adding 5 ml of ice-cold binding buffer to the tube and rapidly filtering contents through a Whatman GF/C glass microfiber filter under low vacuum. The filter was rinsed twice with 10 ml of ice-cold washing buffer containing 10 mm Tris and 0.5 mm MgCl2 at pH 7.4. Radioactivity trapped in the filters was then measured by scintillation counting. “Nonspecific” binding was determined in parallel assay tubes that contained crude membranes of HEKwt cells instead of HEKβ2 cells. “Specific” binding was defined as the difference between the total and the nonspecific binding.
Molecular Modeling
The complete docking procedure consists of an initial docking/scoring step, followed by a pharmacophore filtering. Docking and scoring were performed by LigandFit, with CFF force field, a softened van der Walls parameter, and a dielectric constant of 1. The most probable binding site was the orthosteric one, with a total volume of 715 Å3, partitioned onto seven subsites for extensive screening. The conformers of flexible ligands were calculated before docking by using the catalyst diverse conformation generation method. The filtering was based on the thiadiazole part of crystallographic timolol (PDB: 3D4S) (30). The aromatic pharmacophore was defined as a centroid of 1-Å radius and two orthogonal and opposite projections from the aromatic plane of 1.5-Å radius. All the algorithms used are implemented in Discovery Studio 2.1 (Accelrys Inc., San Diego, CA). For calibration of the scoring function, a dataset of ten β2ADR ligands (Kd ranging from 0.1 to 4500 nm) (31) was used to calibrate and validate the docking/scoring protocol. Starting from the Ludi free energy expression form (32), we considered its five components as adjustable parameters. Hence, they were recalibrated by least-squares fitting to the experimental Kd.
Intracellular cAMP and IP3 Measurements
Cellular concentration of cAMP or IP3 was quantified by the chemiluminescent immunoassay cAMP-ScreenTM System (Applied Biosystems, Foster City, CA) or IP-One elisa (Cisbio, Bedford, MA), respectively, according to the manufacturer's instructions.
RNA Isolation and Analysis
Total RNA was isolated from cells using the TRIzol method (Invitrogen); it was then subjected to reverse transcription real-time quantitative PCR (RT-qPCR) analysis, as previously described (33). Primers were as follows: CYP1B1-forward: 5′-TGATGGACGCCTTTATCCTC-3′; CYP1B1-reverse: 5′-CCACGACCTGATCCAATTCT-3′; 18S-forward: 5′-CGCCGCTAGAGGTGAAATTC-3′; 18S-reverse: 5′-TTGGCAAATGCTTTCGCT-3′. The specificity of each gene amplification was verified at the end of qPCR reactions through analysis of dissociation curves of the qPCR products. Amplification curves were analyzed with ABI Prism 7000 SDS software using the comparative cycle threshold method. Relative quantification of the steady-state target mRNA levels was calculated after normalization of the total amount of cDNA tested to an 18 S RNA endogenous reference.
Immunoblotting Analysis
Immunoblotting was performed on crude membrane (for analysis of β2ADR protein level) or on total cellular extracts (for analysis of Epac-1 protein level) as previously described (34). Briefly, protein samples (40 μg) were subjected to electrophoresis in a 10% acrylamide gel and electrophoretically transferred to a nitrocellulose membrane (Bio-Rad). After blocking with Tris-buffered saline containing 4% bovine serum albumin and 0.1% Tween 20 at room temperature, membranes were incubated with specific primary antibody overnight at 4 °C and, subsequently, with appropriate horseradish peroxidase-conjugated secondary antibody for 1 h. Immunolabeled proteins were finally visualized by autoradiography using chemiluminescence.
Statistical Analysis
Results are usually presented as means ± S.D. They were statistically analyzed using the Student's t test. The criterion of significance was p < 0.05; data from binding assays were analyzed using SigmaPlot 11 software.
RESULTS
β2ADR Is Involved in B(a)P-related [Ca2+]i Induction
Owing to the fact that endothelium constitutes one of the well known targets of PAHs (35–38), human microvascular endothelial cells HMEC-1 were mainly used in the present study. To investigate the potential role of β2ADR in the calcium signal triggered by PAHs, we first verified that β2ADR stimulation can modulate [Ca2+]i in this model. According to previous reports (26, 28), β2ADR activation by salbutamol (a β2ADR agonist) results in an increase in [Ca2+]i in HMEC-1 cells. This increase started after ∼5 min of exposure and was maximum at 30 min (Fig. 1A). As expected, this effect was strongly abolished by antagonizing β2ADR using a selective β2-blocker, ICI-118,551, or non-selective β-blockers, propranolol and carazolol (Fig. 1B). Interestingly, the prototypical PAH B(a)P was able to generate a [Ca2+]i increase with kinetics similar to that observed with salbutamol (Fig. 1A). Moreover, the B(a)P-induced [Ca2+]i increase was also diminished by β2ADR inhibition using ICI-118,551, propranolol, or carazolol (Fig. 1B), thus supporting an implication of β2ADR in B(a)P-mediated [Ca2+]i increase. This hypothesis was next confirmed by the fact that inhibiting β2ADR activity in HMEC-1 cells using specific antibodies targeting β2ADR, markedly prevented the increase of [Ca2+]i due to B(a)P (Fig. 2A); in the same way, transfection of siRNAs targeting β2ADR, which markedly reduced β2ADR expression in HMEC-1 cells as shown by Western blotting (Fig. 2B), counteracted the [Ca2+]i increase triggered by B(a)P (Fig. 2B). Finally, we used HEK293 cells, known to constitutively poorly express β2ADR (39), and transfected them either with control plasmid (HEKwt) or with a β2ADR plasmid (HEKβ2). As shown by Western blotting (Fig. 2C), HEKβ2 cells exhibited a very large overexpression of β2ADR when compared with HEKwt; they also displayed a marked increase of [Ca2+]i in response to B(a)P treatment, in contrast to HEKwt (Fig. 2C).
FIGURE 1.
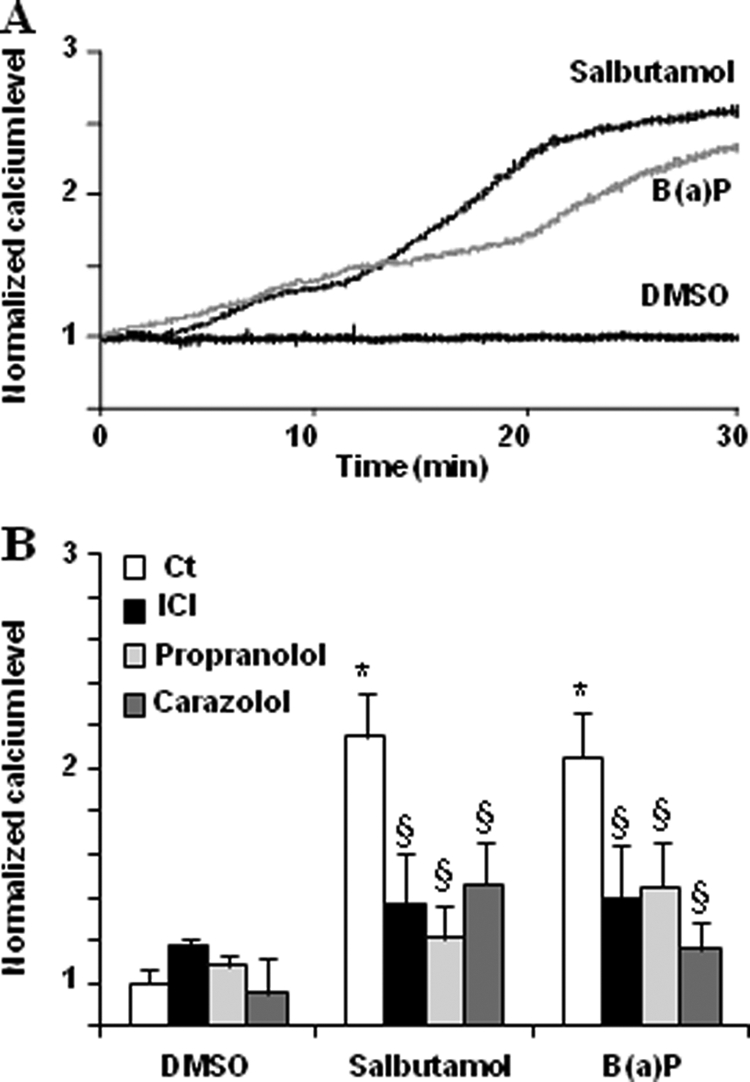
Effects of adrenergic β-blockers on salbutamol- or B(a)P-related [Ca2+]i increase in endothelial HMEC-1 cells. A, continuous recordings of normalized calcium level in HMEC-1 cells treated over 30 min by 10 μm salbutamol, 1 μm B(a)P, or vehicle (DMSO); the recordings shown are representative of three independent experiments. B, effect of 100 μm ICI-118,551 (ICI, a selective β2-blocker), 10 μm propranolol, or 10 μm carazolol (non-selective β-blockers) on the increase of normalized calcium level in HMEC-1 cells treated for 30 min by 10 μm salbutamol, 1 μm B(a)P, or vehicle (DMSO). Normalized calcium level was calculated as described under “Experimental Procedures.” Results are the means ± S.D. of at least three different experiments. *, p < 0.05 when compared with untreated cells (DMSO). §, p < 0.05 when compared with salbutamol or B(a)P-treated cells not exposed to β-blockers.
FIGURE 2.

β2ADR targeting impairs B(a)P-mediated [Ca2+]i induction. A, effect of β2ADR-targeting antibody (β2ADR Ab, 2 μg/ml) or control antibody (Ct Ab, 2 μg/ml) on calcium variations in HMEC-1 cells treated by 1 μm B(a)P or vehicle (DMSO). B, effect of β2ADR-targeting siRNAs (iβ2ADR) or non-targeting siRNAs (iCt) on calcium variations in HMEC-1 cells treated by 1 μm B(a)P or vehicle (DMSO), and on β2ADR or caveolin-1 (used as internal control) protein levels in HMEC-1 cells (inset, bottom right). C, effects of 1 μm B(a)P or vehicle (DMSO) on [Ca2+]i level in control HEK293 cells (HEKwt) and β2ADR-transfected HEK293 cells (HEKβ2). Inset (bottom right) shows β2ADR or caveolin-1 (used as internal control) protein levels in HEKwt and HEKβ2 cells. Normalized calcium level was calculated as described under “Experimental Procedures.” Traces (left) represent continuous recording of normalized [Ca2+]i changes over the 30-min period of treatment; the recordings shown are representative of three independent experiments. Histograms (right) represent normalized calcium level after 30 min of treatment. Results are the means ± S.D. of at least three different experiments. *, p < 0.05 when compared with untreated cells (DMSO).
Gs/AC/cAMP Pathway Is Involved in B(a)P-related [Ca2+]i Induction
It has been demonstrated that β2ADR-mediated [Ca2+]i increase requires Gs protein-mediated activation of AC as a first step and the subsequent production of cAMP (26, 28). In addition, it is well known that TCDD can rapidly increase cAMP level in C3H10T1/2 cells in a G protein-dependent manner (20). Therefore, we first explored if B(a)P exposure can also induce cAMP levels in HMEC-1 cells. As shown in Fig. 3A, B(a)P transiently enhanced cAMP level in HMEC-1 cells, with a rapid onset after 2 min of exposure and a maximum effect at 10 min. Moreover, this induction was significantly abolished in the presence of the selective β2-blocker ICI-118,551, or a competitive inhibitor of AC, dd-Ado (Fig. 3B), pointing to the involvement of β2ADR-related AC activation in the cAMP level increase by B(a)P. We noted that the induction peak of cAMP levels in B(a)P-exposed HMEC-1 cells occurred earlier than that of [Ca2+]i (10 min versus 30 min), which suggests that PAHs-related induction of [Ca2+]i occurs downstream of the activation of cAMP. We then investigated the impact of cAMP increase on B(a)P-related calcium signal. As shown in Fig. 3 (C and D), B(a)P failed to significantly increase [Ca2+]i in HMEC-1 cells when cAMP production was inhibited using a potent inhibitor of G protein (suramin, Fig. 3C) or competitive inhibitors of AC such as dd-Ado (Fig. 3D) or MDL-12,330A (data not shown).
FIGURE 3.

Gs/AC/cAMP pathway is involved in B(a)P-related [Ca2+]i induction. A, cAMP levels in HMEC-1 cells treated by 1 μm B(a)P or vehicle (DMSO) during indicated times of exposure. B, cAMP levels in HMEC-1 cells after 10 min of treatment by 1 μm B(a)P in the presence or not of 100 μm ICI-118,551 (ICI, a selective β2-adrenergic antagonist) or 100 μm dd-Ado (an inhibitor of AC). C and D, effect of 10 μm suramin (a G protein inhibitor) (C) or 100 μm dd-Ado (D) on calcium variations in HMEC-1 cells treated by 1 μm B(a)P or vehicle (DMSO). Normalized calcium level was calculated as described under “Experimental Procedures.” Traces (left) represent continuous recording of normalized [Ca2+]i changes over the 30-min period of treatment; the recordings shown are representative of three independent experiments. Histograms (right) represent normalized calcium level after 30 min of treatment. Results are the means ± S.D. of at least three different experiments. *, p < 0.05 when compared with untreated cells (DMSO).
Epac-1 Is Involved in B(a)P-related [Ca2+]i Induction
An important target of cAMP is the family of Epac proteins, Epac-1 and -2. These guanine nucleotide exchanger factors were initially identified by De Rooji and co-workers to explain the PKA-independent activation of Rap by cAMP (40). There is now considerable evidence that these proteins play major roles in cellular physiology (41); they are notably involved in regulation of calcium channels and endothelium physiology and, according to Schmidt and co-workers (28), the increase of [Ca2+]i by β2ADR requires the cAMP-dependent activation of Epac-1. Two approaches were used to test the implication of Epac-1 in B(a)P-related [Ca2+]i variation in HMEC-1 cells. First, we used the Epac inhibitor BFA (42), which was found to markedly block B(a)P-related [Ca2+]i increase (Fig. 4A). Second, we down-regulated Epac-1 expression using siRNA transfection. As shown in Fig. 4B, the decrease of Epac-1 expression assessed by Western blotting was associated with an inhibition of B(a)P-related [Ca2+]i increase.
FIGURE 4.

Epac-1 is involved in B(a)P-related [Ca2+]i induction. Effect of 20 μm BFA (a nonspecific Epac inhibitor) (A), or of Epac-1-targeting siRNAs (iEpac-1) or non-targeting siRNAs (iCt) (B), on calcium variations in HMEC-1 cells treated by 1 μm B(a)P or vehicle (DMSO). Normalized calcium level was calculated as described under “Experimental Procedures.” Traces (left) represent continuous recording of normalized [Ca2+]i changes over the 30-min period of treatment; the recordings shown are representative of three independent experiments. Histograms (right) represent normalized calcium level after 30 min of treatment. Results are the means ± S.D. of at least three different experiments. Inset (bottom right) shows Epac-1 and total p-38 kinase (p-38 Total, used as internal control) protein levels in HMEC-1 cells transfected by iEpac-1 or iCt. *, p < 0.05 when compared with untreated cells (DMSO).
IP3 Is Involved in B(a)P-related [Ca2+]i Increase
IP3 is a universal intracellular messenger that mediates Ca2+ release from intracellular stores via the activation of IP3 receptor (IP3R) located at the endoplasmic reticulum membrane (43). This messenger usually originates from phospholipase C-dependent hydrolysis of phosphatidylinositol diphosphate after hormonal activation of the cell (28, 44). Because phospholipase C can be activated by Epac-1 following βADR stimulation (27), it is tempting to speculate that the βADR/AC/Epac-1-signaling cascade activated by B(a)P may result in IP3 up-regulation. Consistent with this hypothesis, B(a)P was found to significantly increase the IP3 level in HMEC-1 cells (Fig. 5A). This IP3 up-regulation was moreover inhibited in the presence of the β2ADR inhibitor ICI-118,551 or the AC inhibitor dd-Ado, thus likely indicating it was dependent on activation of the β2ADR/AC-signaling pathway in response to B(a)P. In agreement with this conclusion, IP3 accumulation elicited by exposure to B(a)P was observed in β2ADR-overexpressing HEKβ2 cells, but not in HEKwt cells (Fig. 5B).
FIGURE 5.

IP3 is involved in B(a)P-related [Ca2+]i induction. A, effect of 100 μm ICI-118,551 (ICI, a selective adrenergic β2-antagonist) or 100 μm dd-Ado (an inhibitor of AC) on IP3 induction in HMEC-1 cells treated for 1 h by 1 μm B(a)P or vehicle (DMSO). B, IP3 level in control HEK293 cells (HEKwt) and β2ADR-transfected HEK293 cells (HEKβ2) after exposure to 1 μm B(a)P or vehicle (DMSO) for 1 h. C–E, effect of 10 μm l-690.330 (L-690, an inhibitor of inositol-monophosphatase) of 50 μm 2-APB (an antagonist of IP3 receptor) (C) or of 50 μm TMB-8 (an inhibitor of intracellular calcium mobilization) (D) on calcium variations in HMEC-1 cells treated by 1 μm B(a)P or vehicle (DMSO) (E). Normalized calcium level was calculated as described under “Experimental Procedures.” Traces (left) represent continuous recording of normalized [Ca2+]i changes over the 30-min period of treatment; the recordings shown are representative of three independent experiments. Histograms (right) represent normalized calcium level after 30 min of treatment. Results are the means ± S.D. of at least three different experiments. *, p < 0.05 when compared with untreated cells (DMSO).
To determine whether B(a)P-mediated IP3 induction occurs upstream of B(a)P-related [Ca2+]i induction, cells were preincubated with IP3 recycling-blocking agents, namely l-690.330 and LiCl, known to disrupt IP3 metabolism by inhibiting inositol-monophosphatase, thus leading to the reduction of myo-inositol necessary for the building of neo-synthesized IP3, thereby finally down-regulating the IP3 level (45, 46). Calcium increase by B(a)P was found to be strongly repressed in the presence of l-690.330 (Fig. 5C) or LiCl (data not shown), suggesting an important contribution of IP3 to BaP-mediated [Ca2+]i variation. This conclusion was next fully confirmed by the fact that B(a)P-related [Ca2+]i increase was abolished by preincubation with 2-APB, a specific antagonist of IP3R (47) (Fig. 5D), or with TMB-8, frequently used as an inhibitor of calcium release from intracellular stores (48) (Fig. 5E).
Contribution of β2ADR Pathway to AhR-dependent Regulation of B(a)P Target Genes
To determine whether the different elements, acting in the β2ADR-related signaling cascade putatively activated by B(a)P and described above, may be implicated in the regulation of the AhR gene target CYP1B1, we next analyzed the effect of their chemical inhibition or down-regulation on B(a)P-mediated induction of CYP1B1 mRNA expression. The use of the βADR blockers propranolol and ICI-118,551 was found to markedly inhibit CYP1B1 up-regulation in response to B(a)P (Fig. 6A); similar results were observed with the G protein inhibitor suramin or the AC inhibitor dd-Ado (Fig. 6B). In the same way, BFA-mediated pharmacological inhibition of Epac-1 or the siRNA-related knockdown of Epac-1 expression prevented B(a)P-related CYP1B1 induction (Fig. 6, C and D). Finally, inhibiting calcium release from intracellular stores using TMB-8 or the IP3R antagonist 2-APB also counteracted CYP1B1 up-regulation (Fig. 6E).
FIGURE 6.
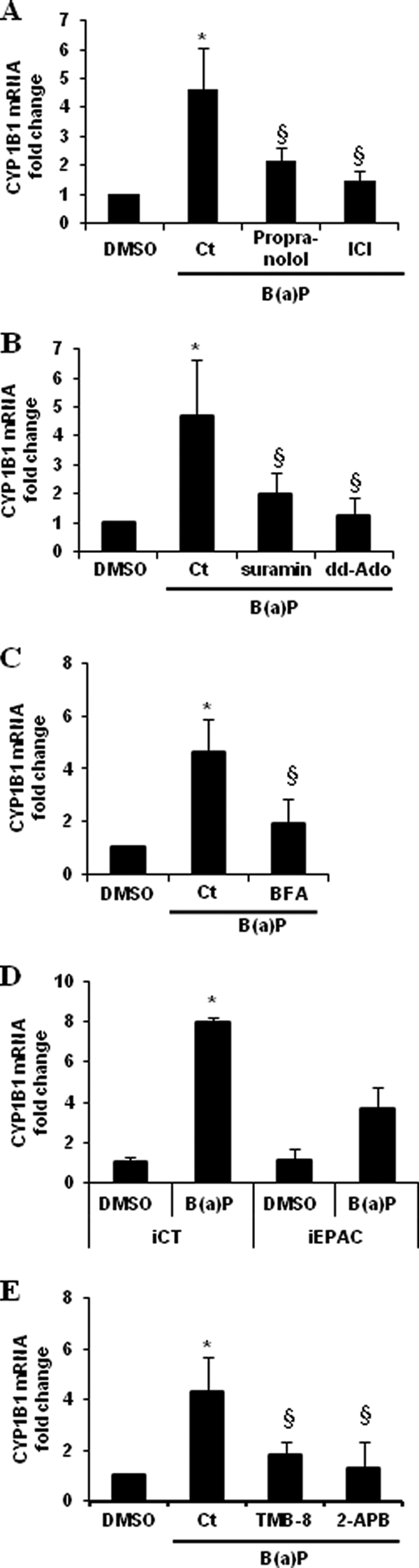
Implication of β2ADR pathway in PAHs-related CYP1B1 induction. A–D, CYP1B1 mRNA induction rates were measured in HMEC-1 cells treated for 1 h by 1 μm B(a)P or vehicle (DMSO), in the presence or not of β-blockers (10 μm propranolol, 10 μm carazolol, or 100 μm ICI-118,551 (ICI)) (A), a G protein inhibitor (10 μm suramin) or an inhibitor of AC (100 μm dd-Ado) (B), a nonspecific Epac inhibitor (20 μm BFA) (C), an antagonist of IP3 receptor (50 μm 2-APB) or an inhibitor of intracellular calcium mobilization (10 μm TMB-8) (E). D, effect of Epac-1-targeting siRNAs (iEpac-1) or non-targeting siRNAs (iCt) on CYP1B1 induction in HMEC-1 cells treated for 1 h by 1 μm B(a)P or vehicle (DMSO). mRNA expression of CYP1B1 was analyzed using RT-qPCR. Data are expressed relatively to mRNA levels of CYP1B1 found in corresponding control cells, arbitrarily set to 1 unit. Data are the means ± S.D. of four independent experiments. *, p < 0.05 when compared with untreated cells (DMSO). §, p < 0.05 when compared with B(a)P-treated cells.
B(a)P Directly Binds β2ADR
The implication of β2ADR in B(a)P-related [Ca2+]i induction may correspond to a direct activation of these receptors by the PAH, or it may alternatively reflect a positive and required cooperation between the β2ADR system and another yet unknown system activated by B(a)P. To gain insights into this issue, we first performed B(a)P saturation binding assays using β2ADR-positive crude membranes. Total binding was determined by incubating crude membranes from HEKβ2 cells with different concentrations of [3H]-B(a)P (from 0 to 100 nm), whereas nonspecific β2ADR-independent binding was concomitantly evaluated using crude membranes from HEKwt cells instead of HEKβ2 counterparts. Subtraction of nonspecific binding from total binding revealed a specific and saturable binding of B(a)P to HEKβ2 crude membranes, with a Bmax of ∼2.5 pmol/mg and a Kd of ∼10 nm (Fig. 7A). To further investigate the possibility of direct interaction between B(a)P and β2ADR, we then docked B(a)P into the x-ray structure of human β2ADR (PDB: 2RH1) (49). The most favorable binding domain for B(a)P was located between TM3, TM5, and TM6 segments of β2ADR (Fig. 7B). This binding mode most likely allows extensive interactions of B(a)P with Phe-193, Tyr-199, Phe-289, and Phe-290 on β2ADR and partially overlaps the carazolol binding site (Fig. 7C).
FIGURE 7.

Direct binding of B(a)P to β2ADR. A, binding study: crude membranes from β2ADR-overexpressing HEKβ2 cells were incubated with various concentrations of [3H]B(a)P for 30 min at 37 °C. Radioactivity bound to crude membranes was then determined through filtration and scintillation counting. “Nonspecific” binding was determined in parallel assays using crude membranes from control HEKwt cells instead of HEKβ2 cells. Data represent the means ± S.D. of six independent experiments. B and C, molecular modeling of β2ADR-B(a)P interaction. Location of B(a)P in the putative PAH binding site of human β2ADR. B(a)P was docked in the x-ray structure of β2ADR (PDB: 2RH1) B, docked B(a)P (yellow sticks) fully superimpose carazolol (gray sticks) location in crystallized structure of β2ADR (49) (C).
DISCUSSION
Exposure to PAHs is well known to trigger an early increase of [Ca2+]i, which is thought to participate to the up-regulation of various genes targeted by the PAHs-activated transcription factor AhR (5, 50). The aim of the present study was to characterize the initial events implicated in this calcium mobilization by PAHs. Taking B(a)P as an example of PAH, the present work strongly suggests that [Ca2+]i induction in endothelial HMEC-1 cells is initially caused by interaction of the PAH with β2ADR and subsequent activation of a G protein/AC/cAMP/Epac-1/IP3 pathway (Fig. 8).
FIGURE 8.

Schematic overview of the different signaling steps involved in B(a)P-induced β2ADR-mediated [Ca2+]i induction in endothelial HMEC-1 cells. B(a)P, benzo(a)pyrene; β2ADR, β2-adrenergic receptor; PGs, Gs protein; A.C, adenylyl cyclase; Epac-1, exchange protein factor directly activated by cAMP; PLC, phospholipase C; IP3, inositol 1,4,5-trisphosphate; and ER, endoplasmic reticulum.
The direct involvement of β2ADR in calcium mobilization by PAHs is thus supported by (i) the specific binding of B(a)P to β2ADR as revealed by saturation binding assay and molecular docking, (ii) the inhibition of B(a)P-mediated [Ca2+]i induction by chemical βADR blockers, including the selective β2ADR blocker ICI-118,551, by a monoclonal antibody anti-β2ADR, or by siRNA-mediated knockdown of β2ADR expression, and (iii) the fact that the β2ADR-transfected HEKβ2 cells exhibited a marked increase of [Ca2+]i in response to B(a)P, in contrast to control HEKwt cells. Downstream events following β2ADR interaction with B(a)P likely successively involve Gs protein, AC, cAMP, Epac-1 and finally IP3 (Fig. 8). Indeed, the calcium signal generated by B(a)P is reduced by suramin, a G protein inhibitor. Involvement of the downstream factors AC/cAMP was further demonstrated by (i) the β2ADR- and AC-dependent induction of cAMP levels by B(a)P in HMEC-1 cells and (ii) the inhibition of B(a)P-triggered calcium mobilization by blocking AC activity and consequently cAMP production by dd-Ado or MDL-12,330A. Epac-1, well known to be activated by cAMP (40), was most likely the following actor in the signaling cascade contributing to B(a)P-mediated [Ca2+]i increase, because the Epac inhibitor BFA, as well as siRNAs-mediated down-regulation of Epac-1 expression, counteracted calcium mobilization due to the PAH. β2ADR-mediated Epac-1 activation is known to induce IP3 formation in a phospholipase C-dependent manner (27, 28), and such a cellular process is likely to also occur in B(a)P-exposed HMEC-1 cells. These cells were found to exhibit induction of IP3 levels, counteracted by chemical inhibition of β2ADR or AC. The fact that B(a)P treatment of β2ADR-overexpressing HEKβ2 cells, but not of control HEKwt cells, induced IP3 levels also supports a link between B(a)P-β2ADR interaction and IP3. Finally, IP3 is likely a major final actor involved in calcium mobilization triggered by PAHs, because the use of IP3 recycling-blocking agents like l-690.330 prevented [Ca2+]i increase in B(a)P-exposed HMEC-1 cells. Terminally, IP3 likely acts on IP3R found in membranes of endoplasmic reticulum to release calcium, because the IP3R antagonist 2-APB was found to abolish calcium mobilization in HMEC-1 cells treated by B(a)P. Similarly, 2-APB has been found to abolish calcium increase occurring in macrophages exposed to B(a)P (5).
It should be kept in mind that, besides the βADR machinery leading to IP3 formation, various mechanisms contributing to calcium modulation by PAHs have been previously reported, such as protein-tyrosine kinases (10), sarcoendoplasmic reticulum calcium ATPases (9), or ryanodine receptor (11). The exact relevance of these mechanisms with respect to calcium mobilization and their putative interplays with the βADR system remain to be determined. Nevertheless, they may be particularly operant in cells weakly expressing β2ADR.
Our study with B(a)P showed that β2ADR-related adrenergic-like effects are required for the up-regulation of AhR target gene by PAHs. Indeed, the pharmacological inhibition of various actors of the β2ADR/G protein/AC/cAMP/Epac-1/IP3-signaling pathway, as well as the down-regulation of Epac-1 expression, were shown to counteract B(a)P-mediated induction of CYP1B1 mRNA levels (Fig. 6). The crucial element implicated in this interaction between β2ADR- and AhR-signaling cascades is likely to be calcium. Indeed, inhibition of the terminal step of calcium release from intracellular stores through the use of 2-APB was sufficient for inhibiting CYP1B1 induction in HMEC-1 cells exposed to B(a)P. Similarly, 2-APB and the calcium chelating agent 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis have been shown to prevent the up-regulation of various AhR targets such as CYP1A1 and the chemokine CCL1 in response to AhR agonists (5, 6, 12). The exact way that calcium interacts with the AhR-signaling cascade remains to be determined, even if a role for calcium-activated signaling pathways such as calmodulin-dependent protein kinases may be suspected (6, 51). In addition to calcium, cAMP may also play a role in the AhR signaling cascade, because cAMP and its downstream kinase PKA have been reported to constitute players of the AhR pathway by themselves (20, 52).
The affinity of B(a)P (Kd = 10 nm) is among the highest reported for β2ADR ligands. Amazingly the neutral lipophilic nature of PAHs structure does not match the known β2ADR pharmacophore (53). Thus, the exact nature of B(a)P binding to β2ADR remains to be characterized. Nevertheless, the great accessible surface area of B(a)P is likely to favor large van de Walls contacts. Besides, docking analysis suggest extensive interactions of B(a)P with the amino acids Phe-193, Tyr-199, Phe-289, and Phe-290 on β2ADR. The aromatic nature of these amino acids combined with that of B(a)P, as well as the geometry of predicted complexes, argue strongly in favor of π-stacking. The reduced potency of 6-nitro-B(a)P for elevating [Ca2+]i when compared with that of B(a)P strongly supports this hypothesis (data not shown). Indeed, electron-withdrawing substituents, such as the nitro group found in 6-nitro-B(a)P, reduce the interaction energy of T-shaped aromatic dimers (54). Interestingly, π-stacking has already been emphasized for βADRs ligands (55, 56). Other target proteins of PAHs, such as α1-acid glycoprotein or the estrogen receptor (57, 58), also rely on aromatic interaction.
The low reported Kd (10 nm) for B(a)P binding to βADR is in the range of B(a)P concentrations to which humans may be commonly exposed through diet, air pollution, or cigarette smoke (59, 60). This suggests that environmental exposure to PAHs may result in deleterious interactions with the β-adrenergic system in humans. Such putative adrenergic-like effects of PAHs remain to be determined. However, administration of B(a)P at low doses has been demonstrated to impair β2ADR-mediated stimulation of adipose tissue lipolysis and cause weight gain in mice (61), supporting in vivo interactions of PAHs with βADRs. It should be kept in mind that the overall interplay between PAHs and the adrenergic system may be more complex than simply reflecting an activation of β2ADR by PAHs. Indeed, exposure of mice to the halogenated hydrocarbon TCDD decreases βADR responsiveness, especially in the heart (17, 18), whereas PAH-containing cigarette smoke has been shown to decrease expression of βADRs in blood lymphocytes (23). Such data may be consistent with a desensitization of the β-adrenergic system, which occurs in response to initial stimulation of the adrenergic system by agonists (25) and involves the down-regulation of βADR expression as well as the internalization of membrane βADRs. In agreement with this hypothesis, exposure to a mixture of PAHs for 24 h dramatically diminishes expression of β2ADR mRNAs and protein in airway smooth muscle cells (21).
In summary, the prototypical PAH B(a)P was found to bind to β2ADR in endothelial HMEC-1 cells and to consequently activate a G protein/AC/cAMP/Epac-1/IP3 pathway, which in turn resulted in a [Ca2+]i increase, required for B(a)P-mediated induction of the AhR gene target CYP1B1. Such data are therefore consistent with a previously unsuspected implication of the β2ADR system in deleterious effects of environmental PAHs.
This work was supported by the Programme National de Recherche sur les Perturbateurs Endocriniens from the French Ecology minister.
- PAH
- polycyclic aromatic hydrocarbon
- AhR
- aryl hydrocarbon receptor
- CYP1A1
- -1B1, cytochromes P-450 1A1 and 1B1
- B(a)P
- benzo(a)pyrene
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- βADR
- β2-adrenoreceptor
- β2ADR
- β2-adrenoreceptor
- AC
- adenylyl cyclase
- 2-APB
- 2-aminoethoxydiphenyl borate
- BFA
- brefeldin A
- dd-Ado
- 2′-5′-dideoxyadenosine
- TMB-8
- 8-(diethylamino)octyl-3,4,5-trimethoxybenzoate hydrochloride
- Fura-2AM
- fura-2 acetoxymethyl ester
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- IP3 receptor.
REFERENCES
- 1. Korashy H. M., El-Kadi A. O. (2006) The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab. Rev. 38, 411–450 [DOI] [PubMed] [Google Scholar]
- 2. Lewtas J. (2007) Air pollution combustion emissions. Characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat. Res. 636, 95–133 [DOI] [PubMed] [Google Scholar]
- 3. Phillips D. H. (1999) Polycyclic aromatic hydrocarbons in the diet. Mutat. Res. 443, 139–147 [DOI] [PubMed] [Google Scholar]
- 4. Hankinson O. (2005) Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 433, 379–386 [DOI] [PubMed] [Google Scholar]
- 5. N'Diaye M., Le Ferrec E., Lagadic-Gossmann D., Corre S., Gilot D., Lecureur V., Monteiro P., Rauch C., Galibert M. D., Fardel O. (2006) Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J. Biol. Chem. 281, 19906–19915 [DOI] [PubMed] [Google Scholar]
- 6. Monteiro P., Gilot D., Le Ferrec E., Rauch C., Lagadic-Gossmann D., Fardel O. (2008) Dioxin-mediated up-regulation of aryl hydrocarbon receptor target genes is dependent on the calcium/calmodulin/CaMKIα pathway. Mol. Pharmacol. 73, 769–777 [DOI] [PubMed] [Google Scholar]
- 7. Mayati A., Le Ferrec E., Lagadic-Gossmann D., Fardel O. (2011) Aryl hydrocarbon receptor-independent up-regulation of intracellular calcium concentration by environmental polycyclic aromatic hydrocarbons in human endothelial HMEC-1 cells. Environ. Toxicol. doi: 10.1002/tox.20675 [DOI] [PubMed] [Google Scholar]
- 8. Kobayashi D., Ahmed S., Ishida M., Kasai S., Kikuchi H. (2009) Calcium/calmodulin signaling elicits release of cytochrome c during 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced apoptosis in the human lymphoblastic T-cell line, L-MAT. Toxicology 258, 25–32 [DOI] [PubMed] [Google Scholar]
- 9. Zhao M., Lytton J., Burchiel S. W. (1996) Inhibition of sarco-endoplasmic reticulum calcium ATPases (SERCA) by polycyclic aromatic hydrocarbons. Lack of evidence for direct effects on cloned rat enzymes. Int. J. Immunopharmacol. 18, 589–598 [DOI] [PubMed] [Google Scholar]
- 10. Krieger J. A., Born J. L., Burchiel S. W. (1994) Persistence of calcium elevation in the HPB-ALL human T cell line correlates with immunosuppressive properties of polycyclic aromatic hydrocarbons. Toxicol. Appl. Pharmacol. 127, 268–274 [DOI] [PubMed] [Google Scholar]
- 11. Gao J., Voss A. A., Pessah I. N., Lauer F. T., Penning T. M., Burchiel S. W. (2005) Ryanodine receptor-mediated rapid increase in intracellular calcium induced by 7,8-benzo(a)pyrene quinone in human and murine leukocytes. Toxicol. Sci. 87, 419–426 [DOI] [PubMed] [Google Scholar]
- 12. Le Ferrec E., Lagadic-Gossmann D., Rauch C., Bardiau C., Maheo K., Massiere F., Le Vee M., Guillouzo A., Morel F. (2002) Transcriptional induction of CYP1A1 by oltipraz in human Caco-2 cells is aryl hydrocarbon receptor- and calcium-dependent. J. Biol. Chem. 277, 24780–24787 [DOI] [PubMed] [Google Scholar]
- 13. Kobilka B. K. (2011) Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol. Sci. 32, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deupi X., Kobilka B. K. (2011) Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology 25, 293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karpen J. W., Rich T. C. (2004) Resolution of cAMP signals in three-dimensional microdomains using novel, real-time sensors. Proc. Western Pharmacol. Soc. 47, 1–5 [PubMed] [Google Scholar]
- 16. Rich T. C., Karpen J. W. (2002) Review article: Cyclic AMP sensors in living cells: what signals can they actually measure? Ann. Biomed. Engineer. 30, 1088–1099 [DOI] [PubMed] [Google Scholar]
- 17. Canga L., Levi R., Rifkind A. B. (1988) Heart as a target organ in 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity. Decreased β-adrenergic responsiveness and evidence of increased intracellular calcium. Proc. Natl. Acad. Sci. U.S.A. 85, 905–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Canga L., Paroli L., Blanck T. J., Silver R. B., Rifkind A. B. (1993) 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases cardiac myocyte intracellular calcium and progressively impairs ventricular contractile responses to isoproterenol and to calcium in chick embryo hearts. Mol. Pharmacol. 44, 1142–1151 [PubMed] [Google Scholar]
- 19. Sommer R. J., Hume A. J., Ciak J. M., Vannostrand J. J., Friggens M., Walker M. K. (2005) Early developmental 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure decreases chick embryo heart chronotropic response to isoproterenol but not to agents affecting signals downstream of the β-adrenergic receptor. Toxicol. Sci. 83, 363–371 [DOI] [PubMed] [Google Scholar]
- 20. Vogel C. F., Sciullo E., Park S., Liedtke C., Trautwein C., Matsumura F. (2004) Dioxin increases C/EBPβ transcription by activating cAMP/protein kinase A. J. Biol. Chem. 279, 8886–8894 [DOI] [PubMed] [Google Scholar]
- 21. Factor P., Akhmedov A. T., McDonald J. D., Qu A., Wu J., Jiang H., Dasgupta T., Panettieri R. A., Jr., Perera F., Miller R. L. (2011) Polycyclic aromatic hydrocarbons impair function of β2-adrenergic receptors in airway epithelial and smooth muscle cells. Am. J. Resp. Cell Mol. Biol. 45, 1045–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laustiola K. E., Kotamäki M., Lassila R., Kallioniemi O. P., Manninen V. (1991) Cigarette smoking alters sympathoadrenal regulation by decreasing the density of β2-adrenoceptors. A study of monitored smoking cessation. J. Cardiovasc. Pharmacol. 17, 923–928 [DOI] [PubMed] [Google Scholar]
- 23. Laustiola K. E., Lassila R., Kaprio J., Koskenvuo M. (1988) Decreased β-adrenergic receptor density and catecholamine response in male cigarette smokers. A study of monozygotic twin pairs discordant for smoking. Circulation 78, 1234–1240 [DOI] [PubMed] [Google Scholar]
- 24. Collins S., Bouvier M., Lohse M. J., Benovic J. L., Caron M. G., Lefkowitz R. J. (1990) Mechanisms involved in adrenergic receptor desensitization. Biochem. Soc. Transact. 18, 541–544 [DOI] [PubMed] [Google Scholar]
- 25. Lefkowitz R. J., Pitcher J., Krueger K., Daaka Y. (1998) Mechanisms of β-adrenergic receptor desensitization and resensitization. Adv. Pharmacol. 42, 416–420 [DOI] [PubMed] [Google Scholar]
- 26. Keiper M., Stope M. B., Szatkowski D., Böhm A., Tysack K., Vom Dorp F., Saur O., Oude Weernink P. A., Evellin S., Jakobs K. H., Schmidt M. (2004) Epac- and Ca2+-controlled activation of Ras and extracellular signal-regulated kinases by Gs-coupled receptors. J. Biol. Chem. 279, 46497–46508 [DOI] [PubMed] [Google Scholar]
- 27. Oestreich E. A., Wang H., Malik S., Kaproth-Joslin K. A., Blaxall B. C., Kelley G. G., Dirksen R. T., Smrcka A. V. (2007) Epac-mediated activation of phospholipase Cϵ plays a critical role in b-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J. Biol. Chem. 282, 5488–5495 [DOI] [PubMed] [Google Scholar]
- 28. Schmidt M., Evellin S., Weernink P. A., von Dorp F., Rehmann H., Lomasney J. W., Jakobs K. H. (2001) A new phospholipase-C-calcium signaling pathway mediated by cyclic AMP and a Rap GTPase. Nat. Cell Biol. 3, 1020–1024 [DOI] [PubMed] [Google Scholar]
- 29. Rousseau G., Guilbault N., Da Silva A., Mouillac B., Chidiac P., Bouvier M. (1997) Influence of receptor density on the patterns of b2-adrenocepter desensitization. Eur. J. Pharmacol. 326, 75–84 [DOI] [PubMed] [Google Scholar]
- 30. Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V. P., Chien E. Y., Velasquez J., Kuhn P., Stevens R. C. (2008) A specific cholesterol binding site is established by the 2.8 A structure of the human b2-adrenergic receptor. Structure 16, 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baker J. G. (2005) The selectivity of b-adrenoceptor antagonists at the human b1, b2, and b3 adrenoceptors. Br. J. Pharmacol. 144, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bohm H. J. (1998) Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Computer-aided Mol. Des. 12, 309–323 [DOI] [PubMed] [Google Scholar]
- 33. Sparfel L., Pinel-Marie M. L., Boize M., Koscielny S., Desmots S., Pery A., Fardel O. (2010) Transcriptional signature of human macrophages exposed to the environmental contaminant benzo(a)pyrene. Toxicol. Sci. 114, 247–259 [DOI] [PubMed] [Google Scholar]
- 34. Lecureur V., Ferrec E. L., N'diaye M., Vee M. L., Gardyn C., Gilot D., Fardel O. (2005) ERK-dependent induction of TNFα expression by the environmental contaminant benzo(a)pyrene in primary human macrophages. FEBS Lett. 579, 1904–1910 [DOI] [PubMed] [Google Scholar]
- 35. Gong Z., Yang J., Yang M., Wang F., Wei Q., Tanguay R. M., Wu T. (2006) Benzo(a)pyrene inhibits expression of inducible heat shock protein 70 in vascular endothelial cells. Toxicol. Lett. 166, 229–236 [DOI] [PubMed] [Google Scholar]
- 36. Kang J. J., Cheng Y. W. (1997) Polycyclic aromatic hydrocarbons-induced vasorelaxation through activation of nitric oxide synthase in endothelium of rat aorta. Toxicol. Lett. 93, 39–45 [DOI] [PubMed] [Google Scholar]
- 37. Omori N., Fukata H., Sato K., Yamazaki K., Aida-Yasuoka K., Takigami H., Kuriyama M., Ichinose M., Mori C. (2007) Polychlorinated biphenyls alter the expression of endothelial nitric oxide synthase mRNA in human umbilical vein endothelial cells. Hum. Exp. Toxicol. 26, 811–816 [DOI] [PubMed] [Google Scholar]
- 38. van Grevenynghe J., Monteiro P., Gilot D., Fest T., Fardel O. (2006) Human endothelial progenitors constitute targets for environmental atherogenic polycyclic aromatic hydrocarbons. Biochem. Biophys. Res. Commun. 341, 763–769 [DOI] [PubMed] [Google Scholar]
- 39. von Zastrow M., Kobilka B. K. (1992) Ligand-regulated internalization and recycling of human b2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J. Biol. Chem. 267, 3530–3538 [PubMed] [Google Scholar]
- 40. de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., Bos J. L. (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477 [DOI] [PubMed] [Google Scholar]
- 41. Gloerich M., Bos J. L. (2010) Epac. Defining a new mechanism for cAMP action. Annu. Rev. Pharmacol. Toxicol. 50, 355–375 [DOI] [PubMed] [Google Scholar]
- 42. Zhong N., Zucker R. S. (2005) cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J. Neurosci. 25, 208–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berridge M. J., Irvine R. F. (1989) Inositol phosphates and cell signaling. Nature 341, 197–205 [DOI] [PubMed] [Google Scholar]
- 44. Roscioni S. S., Elzinga C. R., Schmidt M. (2008) Epac: Effectors and biological functions. Naunyn-Schmiedeberg's Arch. Pharmacol. 377, 345–357 [DOI] [PubMed] [Google Scholar]
- 45. Atack J. R., Cook S. M., Watt A. P., Fletcher S. R., Ragan C. I. (1993) In vitro and in vivo inhibition of inositol monophosphatase by the bisphosphonate L-690,330. J. Neurochem. 60, 652–658 [DOI] [PubMed] [Google Scholar]
- 46. Phiel C. J., Klein P. S. (2001) Molecular targets of lithium action. Annu. Rev. Pharmacol. Toxicol. 41, 789–813 [DOI] [PubMed] [Google Scholar]
- 47. Xiao J., Liang D., Zhao H., Liu Y., Zhang H., Lu X., Liu Y., Li J., Peng L., Chen Y. H. (2011) 2-Aminoethoxydiphenyl borate, a inositol 1,4,5-triphosphate receptor inhibitor, prevents atrial fibrillation. Exp. Biol. Med. 235, 862–868 [DOI] [PubMed] [Google Scholar]
- 48. Malcolm C. S., Ritchie L., Grieve A., Griffiths R. (1996) A prototypic intracellular calcium antagonist, TMB-8, protects cultured cerebellar granule cells against the delayed, calcium-dependent component of glutamate neurotoxicity. J. Neurochem. 66, 2350–2360 [DOI] [PubMed] [Google Scholar]
- 49. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human b2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burchiel S. W., Davis D. A., Ray S. D., Archuleta M. M., Thilsted J. P., Corcoran G. B. (1992) DMBA-induced cytotoxicity in lymphoid and nonlymphoid organs of B6C3F1 mice. Relation of cell death to target cell intracellular calcium and DNA damage. Toxicol. Appl. Pharmacol. 113, 126–132 [DOI] [PubMed] [Google Scholar]
- 51. Han E. H., Kim H. G., Im J. H., Jeong T. C., Jeong H. G. (2009) Up-regulation of CYP1A1 by rutaecarpine is dependent on aryl hydrocarbon receptor and calcium. Toxicology 266, 38–47 [DOI] [PubMed] [Google Scholar]
- 52. Oesch-Bartlomowicz B., Oesch F. (2009) Role of cAMP in mediating AHR signaling. Biochem. Pharmacol. 77, 627–641 [DOI] [PubMed] [Google Scholar]
- 53. Wishart D. S., Knox C., Guo A. C., Cheng D., Shrivastava S., Tzur D., Gautam B., Hassanali M. (2008) Nucleic Acids Res. 36, D901-D906; database issue [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sinnokrot M. O., Sherrill C. D. (2004) Substituent effects in pi-pi interactions. Sandwich and T-shaped configurations. J. Am. Chem. Soc. 126, 7690–7697 [DOI] [PubMed] [Google Scholar]
- 55. Herm M., Molt O., Schrader T. (2002) Towards synthetic adrenaline receptors. Shape-selective adrenaline recognition in water. Chemistry 8, 1485–1499 [DOI] [PubMed] [Google Scholar]
- 56. Kaszuba K., Rog T., Bryl K., Vattulainen I., Karttunen M. (2011) Molecular dynamics simulations reveal fundamental role of water as factor determining affinity of binding of beta-blocker nebivolol to beta(2)-adrenergic receptor. J. Phys. Chem. 114, 8374–8386 [DOI] [PubMed] [Google Scholar]
- 57. Bader A. N., van Dongen M. M., van Lipzig M. M., Kool J., Meerman J. H., Ariese F., Gooijer C. (2005) The chemical interaction between the estrogen receptor and monohydroxybenzo[a]pyrene derivatives studied by fluorescence line-narrowing spectroscopy. Chem. Res. Toxicol. 18, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 58. Zsila F., Matsunaga H., Bikádi Z., Haginaka J. (2006) Multiple ligand-binding properties of the lipocalin member chicken α1-acid glycoprotein studied by circular dichroism and electronic absorption spectroscopy. The essential role of the conserved tryptophan residue. Biochim. Biophys. Acta 1760, 1248–1273 [DOI] [PubMed] [Google Scholar]
- 59. Aygun S. F. (2005) Determination of some polycyclic aromatic hydrocarbons in filter tar of Turkish cigarettes. J. Separation Sci. 28, 2370–2373 [DOI] [PubMed] [Google Scholar]
- 60. Aygün S. F., Kabadayi F. (2005) Determination of benzo[a]pyrene in charcoal grilled meat samples by HPLC with fluorescence detection. Int. J. Food Sci. Nutr. 56, 581–585 [DOI] [PubMed] [Google Scholar]
- 61. Irigaray P., Ogier V., Jacquenet S., Notet V., Sibille P., Méjean L., Bihain B. E., Yen F. T. (2006) Benzo[a]pyrene impairs b-adrenergic stimulation of adipose tissue lipolysis and causes weight gain in mice. A novel molecular mechanism of toxicity for a common food pollutant. FEBS J. 273, 1362–1372 [DOI] [PubMed] [Google Scholar]