

Abstract
Obesogens are chemicals that predispose exposed individuals to weight gain and obesity by increasing the number of fat cells, storage of fats into existing cells, altering metabolic rates, or disturbing the regulation of appetite and satiety. Tributyltin exposure causes differentiation of multipotent stromal stem cells (MSCs) into adipocytes; prenatal TBT exposure leads to epigenetic changes in the stem cell compartment that favor the production of adipocytes at the expense of bone, in vivo. While it is known that TBT acts through peroxisome proliferator activated receptor gamma to induce adipogenesis in MSCs, the data in 3T3-L1 preadipocytes are controversial. Here we show that TBT can activate the RXR-PPARγ heterodimer even in the presence of the PPARγ antagonist GW9662. We found that GW9662 has a ten-fold shorter half-life in cell culture than do PPARγ activators such as rosiglitazone (ROSI), accounting for previous observations that GW9662 did not inhibit TBT-mediated adipogenesis. When the culture conditions are adjusted to compensate for the short half-life of GW-9662, we found that TBT induces adipogenesis, triglyceride storage and the expression of adipogenic marker genes in 3T3-L1 cells in a PPARγ-dependent manner. Our results are broadly applicable to the study of obesogen action and indicate that ligand stability is an important consideration in the design and interpretation of adipogenesis assays.
Keywords: obesogen, tributyltin, TBT, PPARγ, endocrine disrupter, adipogenesis
Introduction
The environmental obesogen model proposes that chemical exposure is a previously unappreciated risk factor for overweight and obesity [1]. Obesogens are functionally defined as chemicals, (dietary, endogenous, pharmaceutical, or xenobiotic), which, in combination with the more widely known and accepted factors of excess caloric input and reduced energy expenditure, predispose an exposed individual to subsequent weight gain and obesity [reviewed in 2, 3-6]. Obesogens can act by increasing the number of adipocytes or stem cells committed to the adipocyte lineage, or by altering basal metabolic rate, shifting energy balance to favor the storage of calories and by altering the hormonal control of appetite and satiety [reviewed in 2, 3-5, 7]. An increasing number of obesogens have been identified in recent years and this field of study is expanding rapidly.
One of the more well-understood obesogens is the organotin, tributyltin (TBT). We and others have shown that TBT exposure leads to increased differentiation of pre-adipocytes in vitro [8, 9], increased deposition of fat in vivo [8] and differentiation of multipotent stromal stem cells (MSCs) into adipocytes in vitro [10, 11]. TBT and the related compound triphenyltin are high affinity agonists for two nuclear receptors that are important for adipogenesis: the peroxisome proliferator activated receptor gamma (PPARγ) and the 9-cis retinoic acid receptor (RXR) [8, 9]. Prenatal exposure to TBT altered cell fate in the MSC compartment to favor the development of adipocytes at the expense of the bone lineage [10]. In accord with its molecular activity, we showed that TBT increased adipogenesis and adipogenic commitment in MSCs by activating PPARγ and that blocking PPARγ action with the potent and selective antagonist GW9662 strongly inhibited adipogenesis [10]. While it has not yet been demonstrated that TBT acts through PPARγ in the in vivo exposure model, it is clear that PPARγ activation is required for MSCs to enter the adipogenic pathway [reviewed in 12].
However, in contrast to what is known about the role of PPARγ in MSCs, the situation in murine 3T3-L1 pre-adipocytes is less clear. At least one group has shown that GW9662 is unable to inhibit TBT-mediated adipogenesis in these cells and they concluded that adipogenesis in 3T3-L1 cells might not be dependent on PPARγ, or any other nuclear receptor for that matter [13]. Spiegelman and colleagues showed that PPARγ activity is required for adipogenesis in 3T3-L1 cells using the very low affinity PPARγ antagonist bisphenol A diglycidyl ether (BADGE) [14]. They subsequently demonstrated that while PPARγ itself was required (together with a functional AF2 activation domain), the ability of PPARγ to be activated by ligand appeared to be dispensable for adipogenesis; although, the presence of an endogenous PPARγ ligand could not be excluded [15]. Since 3T3-L1 cells are a very commonly used and important model for adipocyte differentiation, we sought to understand these discrepancies and determine whether PPARγ activity was required for the induction of adipogenesis by TBT.
There are at least four possible reasons to explain the observation that TBT could cause adipogenesis in 3T3-L1 cells but that this induction could not be blocked by treatment with GW9662 [13]. The first and most obvious is that the process is not PPARγ mediated as has been suggested by other investigators [13]. We considered this possibility unlikely due to the well-established requirement for PPARγ in the adipogenesis of 3T3-L1 cells [14-16] and our results in MSCs [10]. A second possibility is that the RXR-PPARγ heterodimer is permissive for RXR activation even in the presence of a PPARγ antagonist such that pro-adipogenic genes normally targeted by this heterodimer are activated despite the antagonist. A third possibility is that the agonists and antagonists have different relative stabilities or persistence in culture; although, there are no data available on this point. Lastly, TBT might act through RXR homodimers to induce adipogenesis via RXR- PPARγ target genes as has been described for 9-cis retinoic acid, or the synthetic rexinoid LG100268 in cell culture and in PPARα knockout mice [17]. None of these possibilities has previously been explored.
We show here that the RXR-PPARγ heterodimer can be activated by TBT in transient transfection assays in COS7 cells, even in the presence of saturating amounts of GW9662; although, GW9662 completely blocks activation through PPARγ. Using standard assay conditions, GW9662 was unable to block adipogenesis induced by TBT or by ROSI. We found that the half-life of GW9662 is extremely short in cell culture (~2 hours) compared with the PPARγ agonist ROSI (> 24 hours). When the cell culture regimen was altered to ensure an adequate supply of GW9662, we found that GW9662 strongly inhibited the adipogenic activities of ROSI and TBT as measured by cell morphology, triglyceride accumulation and expression of adipogenic genes. Therefore, PPARγ activity is required for TBT-induced adipogenesis in 3T3-L1 cells and we conclude that ligand stability is an important consideration when designing and interpreting the results of adipogenesis assays.
Results
The RXR-PPARγ heterodimer is permissive for RXR activation
It was previously shown that the RXR-PPARγ heterodimer is permissive for RXR activation in the absence of a PPARγ ligand and that activation of this heterodimer was synergistic in the presence of both RXR and PPARγ ligands [18]. However, the responsiveness of the RXR-PPARγ heterodimer in the presence of a PPARγ antagonist was unknown. We tested the permissiveness of the RXR-PPARγ heterodimer by employing chimeric receptors that enabled us to distinguish whether PPARγ or RXR was being activated in the heterodimer. This system relies on the heterodimerization interface in the ligand binding domains of RXR and its heterodimeric partners [19] and utilizes a fusion between the GAL4 DNA-binding domain and the PPARγ ligand binding domain (GAL-PPARγ) together with a construct containing only the ligand binding domain of RXRα (L- RXRα) [20]. When GAL-PPARγ is transiently transfected into cells together with a GAL4-dependent reporter gene, it is activated by the PPARγ ligand ROSI as expected and this activation is strongly inhibited by GW9662 (Figure 1a). Similarly, TBT activates GAL-PPARγ and this activation is blocked by GW9662 in a dose-dependent manner (Figure 1b). When L- RXRα alone is transfected with the reporter, it is not responsive to ligands since there is no DNA-binding domain (supplemental Figure 1). However, when L-RXRα is co-transfected with GAL-PPARγ and the GAL4-reporter, the heterodimer is responsive to the PPARγ ligand ROSI (Figure 1c), to TBT (Figure 1d) and to the RXR activator AGN195203 (supplemental Figure 1). GW9662 effectively blocks activation by ROSI on GAL-PPARγ in the presence of GAL-L-RXRα (Figure 1c). In contrast, GW9662 is almost completely unable to block activation by TBT on GAL4-PPARγ + L-RXRα (Figure 1d). This suggests that the RXR-PPARγ heterodimer is permissive for RXR activation even when PPARγ activation is blocked by GW9662, thereby providing one possible explanation for the inability of GW9662 to block TBT-mediated adipogenesis.
Figure 1. The RXR-PPARγ heterodimer is permissive for RXR activation in the presence of GW9662.
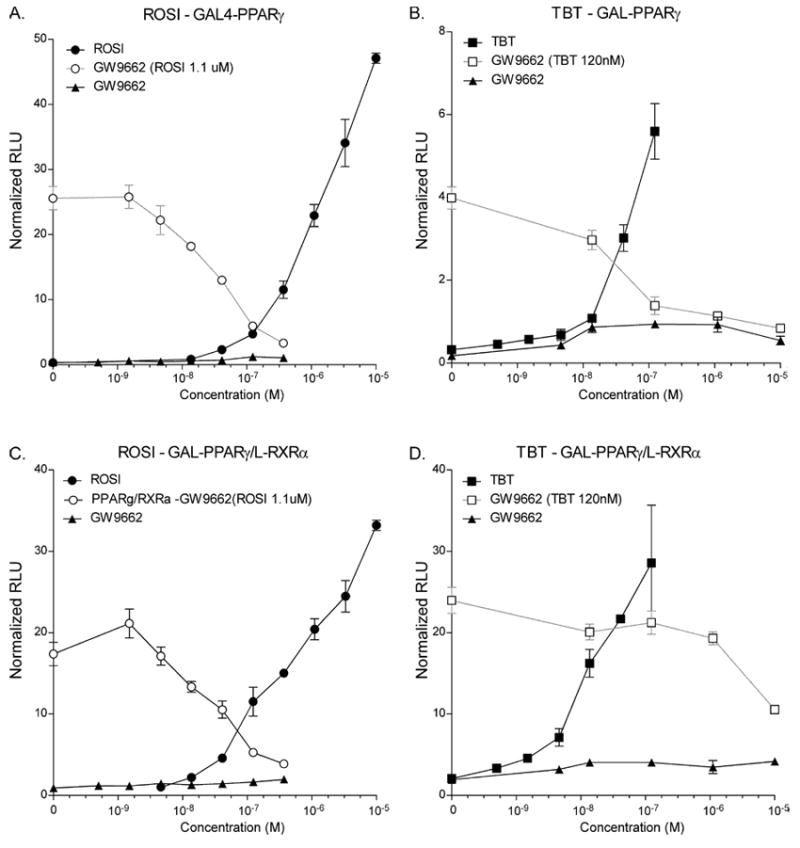
Cos-7 cells were transfected with the indicated receptor expression plasmids together with MH100x4-tk-luc reporter and CMX-β-galactosidase transfection control in 96-well plates and treated with a dilution series of the indicated test compounds or with a dilution series of GW9662 and a constant amount of ROSI (1.1 μM) or TBT (120 nM). A,B - Activation of CMX-GAL-PPARγ by ROSI or TBT. C,D – Activation of CMX-GAL-PPARγ+L-RXRα by ROSI or TBT. Data are expressed as relative light units normalized to ß-galactosidase transfection control and represent the average ± S.E.M. of triplicates. Experiments were performed at least 3 times.
The PPARγ antagonist GW9662 is unable to block adipogenesis mediated by TBT or ROSI
Considering that the RXR-PPARγ heterodimer is permissive for RXR activation in the presence of GW9662, we next tested the effects of TBT and ROSI on adipogenesis of 3T3-L1 cells in the presence or absence of GW9662 (Figure 2) using standard adipogenesis assay conditions [8]. Adipogenesis was inferred by staining the cultures with the triglyceride-specific dye, Oil Red O. In accord with published results [13] we were unable to block the adipogenic effects of TBT by co-treating the cultures with GW9662 (Figure 2). Importantly, however, GW9662 did not block the ability of ROSI to induce differentiation either (Figure 2), an essential control that was absent from the previously published study [13]. This suggested that either published studies from other laboratories demonstrating that ROSI acts through PPARγ to induce adipogenesis in 3T3-L1 cells [8, 9, 14] are incorrect, or that there might be a difference in the relative stability of ROSI and TBT compared with GW9662 that could account for the inability of GW9662 to overcome the effects of ROSI or TBT.
Figure 2. GW9662 does not inhibit adipogenesis mediated by ROSI or TBT in 3T3-L1 cells.
3T3-L1 cells were differentiated into mature adipocytes by the addition of an adipogenic cocktail (MDI) at 2 days post-confluence for 2 days, followed by treatment with 100 nM ROSI or TBT in the presence or absence of 1 μM GW9662. After 7 days of treatment, cells were fixed, stained with Oil Red O and the results quantified using Image J. 6 individual pictures were quantified and averaged for each sample and data are shown as means ± S.E.M for triplicate samples. Data are expressed as average area fraction for averages of 6 samples with n = 3 replicates.
GW9662 has a very short half-life in cell culture
To test the hypothesis that GW9662 might be short-lived in cell culture, we next tested the half-life of the various compounds in 3T3-L1 cells using mass spectrometric analysis. We treated 3T3-L1 cells with 1 μM ROSI or GW9662, collected samples of media or cell pellets at different time points and analyzed the levels of ROSI or GW9662 in the extracts by mass spectrometry. Figure 3 shows that whereas ROSI had a relatively long half-life in the supernatant and in the cell pellet (~26 and ~54 hours respectively), GW9662 has a very short half-life in both the media and in the cell pellet (~1.9 and ~2.2 hours respectively). Therefore, while GW9662 is sufficiently persistent to block PPARγ activity in the relatively short transfection experiments shown in Figure 1 (less than 24 hour treatment with ligands), it is unlikely to persist long enough in cell culture to inhibit adipogenic differentiation over the 7 day length of the adipogenesis assay. Thus, if one wishes to block PPARγ activity in adipogenesis assays, the supply of GW9662 must be replenished at regular intervals to maintain sufficient levels to block PPARγ activity.
Figure 3. GW9662 has a very short half-life compared with rosiglitazone.
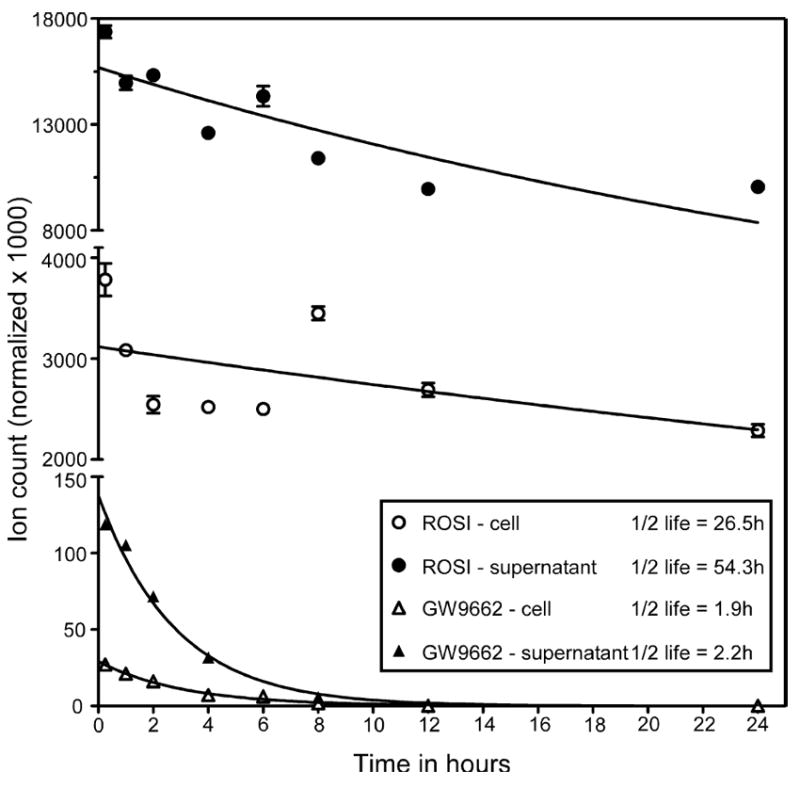
3T3-L1 cells were treated with 1 μM ROSI or 1 μM GW9662 and samples taken at 0, 1, 2, 4, 8, 12, 24, 36 and 48 hrs. After extraction of supernatants or cell pellets, ethyl acetate soluble materials were recovered and analyzed by tandem mass spectrometry and normalized to an internal dexamethasone standard. Data are expressed means ± S.E.M for triplicate samples.
Under appropriate conditions, antagonizing PPARγ blocks TBT- and ROSI-mediated adipogenesis in 3T3-L1 cells
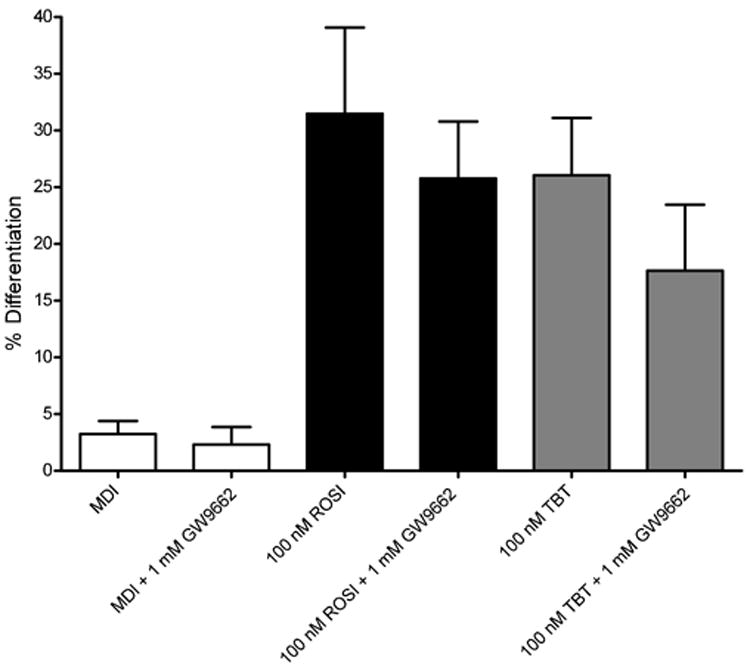
Considering these results, we redesigned the adipogenesis assays to include the addition of GW9662 at 8 hour intervals through the differentiation process. 3T3-L1 cells were seeded into 12-well plates, allowed to reach confluency, then induced to differentiate with a standard cocktail containing insulin, isobutylmethylxanthine, and dexamethasone for two days, followed by treatment with DMSO solvent control, 50 or 100 nM TBT, or 100 nM ROSI. Some wells were stained with Oil Red O after 7 days, whereas others were extracted for triglyceride analysis or RNA prepared for gene expression analysis. Figure 4a shows that under these conditions, 500 nM GW9662 strongly inhibited adipogenesis induced by 50 or 100 nM TBT or 100 nM ROSI. These results we confirmed by quantitative analysis of Oil Red O staining (Figure 4b) and by quantitation of triglycerides from extracted cells (Figure 4c).
Figure 4. Effect of TBT, ROSI and GW9662 on the differentiation of 3T3-L1 cells.
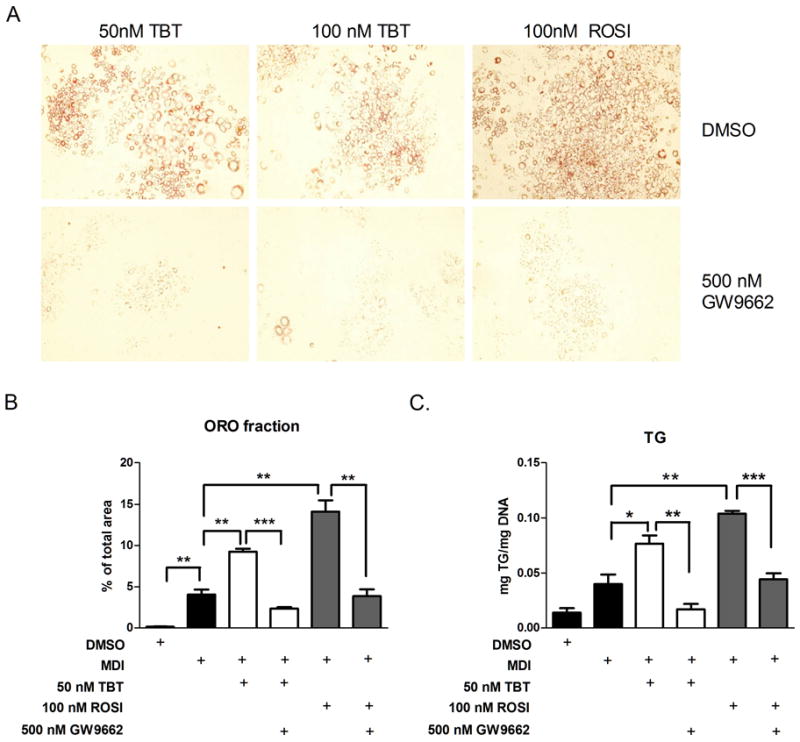
A. 3T3-L1 were differentiated into mature adipocytes by the addition of an adipogenic cocktail for 2 days, followed by supplementation with 50 or 100 nM TBT or 100 nM ROSI (top panels) or the same ligands plus 500 nM GW9662. Cells were stained with Oil red O 7 days after the differentiation cocktail was added. Representative pictures were presented for each treatment.
B. Lipid accumulation was quantified by Image J software and average area fraction was presented. Data were shown as means ± SEM for triplicate samples. 6 individual pictures were quantified and averaged for each sample. Data are expressed as average area fraction in n = 3 replicates ± SEM (n=6 per well).
C. Cellular triglycerides were extracted, quantitated and the results normalized to cellular DNA content. Data are expressed as mg triglyceride/mg cellular DNA and are presented as means ± SEM for triplicate samples.
We next tested the effects of TBT or ROSI on expression of adipogenic genes in the presence or absence of GW9662 using quantitative real time RT-PCR (QPCR). Both TBT and ROSI elicited significant increases in the expression of adipogenic marker genes such as aP2 (FABP4) (Figure 5a), adiponectin (Figure 5b), leptin (Figure 5c) and perilipin (Figure 5d). In all cases, GW9662 strongly inhibited TBT- or ROSI-mediated increases in adipogenic gene expression, although it did not reach statistical significance in all instances (e.g., perilipin and adiponectin for TBT + GW9662). Taken together, these results indicated that GW9662 is able to block adipogenesis, triglyceride accumulation and the expression of adipogenic marker genes induced by either ROSI or TBT.
Figure 5. Quantitative real time RT-PCR analysis of gene expression.
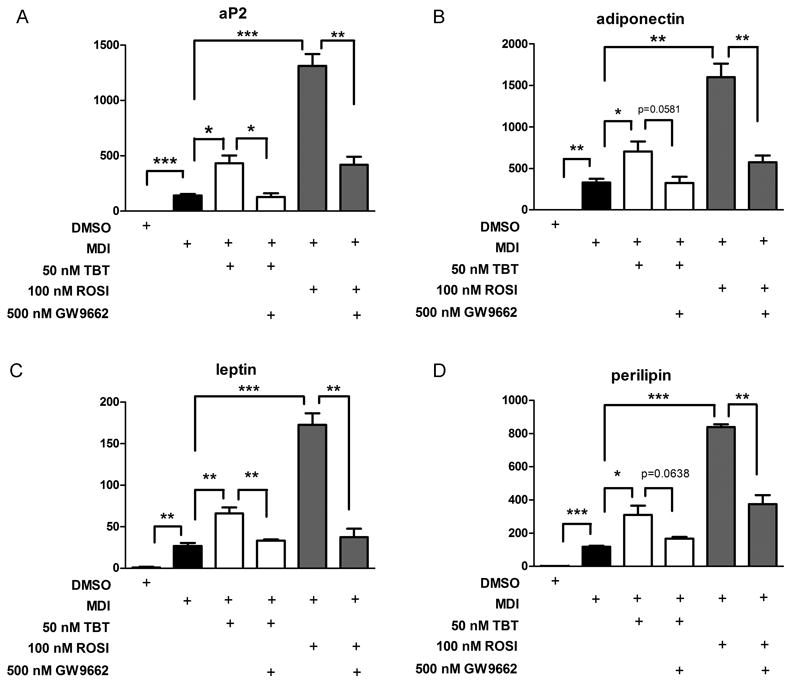
3T3-L1 cells were differentiated into mature adipocytes by the addition of an adipogenic cocktail for 2 days followed by treatment 50 nm TBT or 100 nM ROSI in the presence or absence of 500 nM GW9662 which was added every 8 hours during the differentiation process. RNA was collected from the cells on day 7, followed by real time RT-PCR for gene expression analysis. Data were presented as mean fold induction ± SEM compared with DMSO vehicle for triplicate samples. * = P < 0.5, ** = P< 0.01 and *** = P < 0.001. Experiments were repeated at least twice.
Discussion
Many investigations into adipocyte differentiation utilize pre-adipocyte cell lines such as 3T3-L1, or 3T3-F442A that are already committed to the adipocyte lineage, but which remain as fibroblasts in the absence of appropriate adipogenic stimulation [reviewed in 12]. PPARγ is one of the key genes in adipocyte differentiation and is considered by many to be the “master regulator” of adipogenesis [12, 16] and it is reasonable to hypothesize that treating susceptible cells with PPARγ ligands will induce adipogenesis [reviewed in 7]. As interest in the identity of chemical obesogens and their modes of action increases, it becomes critically important to have well-defined methods to determine which cellular pathways are being perturbed by obesogen action. Our study re-examined the molecular mechanisms through which TBT induced adipogenesis in 3T3-L1 cells since there had been an ongoing controversy about whether TBT regulates adipogenesis in these cells through a PPARγ-dependent mechanism [13].
One important test for whether a chemical acts through PPARγ is the ability of the adipogenic effect to be blocked by inhibiting the function of PPARγ using specific antagonists such as GW9662 [21] or T0070907 [22]. While it has been shown that the PPARγ antagonist T0070907 can block the ability of an adipogenic cocktail (containing isobutylmethylxanthine, insulin and dexamethasone) to induce adipogenesis in 3T3-L1 cells [22], relatively few studies have examined the effects of GW9662 on the induction of adipogenesis in these cells [13], particularly in response to treatment with candidate obesogens. In contrast, many studies have linked the induction of adipogenesis-related genes in mature adipocytes, with a requirement for PPARγ activation that can be blocked by GW9662 [23-26].
Spigelman and colleagues demonstrated that the synthetic compound bisphenol A diglycidyl ether was an antagonist of PPARγ at high levels (~100 μM) and could inhibit the induction of adipogenesis in 3T3-L1 cells by ROSI [14]. More recently, the same group showed that a mutated PPARγ that is unable to be activated by ligands still supported adipogenesis in 3T3-L1 cells, which could be interpreted to suggest that ligand activation of PPARγ, per se, is not required for adipogenesis [15]. They were careful to note that the level of PPARγ protein might be sufficient to obviate the need for ligand activation since this receptor has a significant amount of ligand-independent activation in transfection assays and that there might be an endogenous, albeit unidentified ligand for PPARγ [15]. They did show that the AF-2 transcriptional activation domain of PPARγ was required for adipogenesis, suggesting that PPARγ is acting as a transcriptional activator, irrespective of whether this is dependent on the presence of ligand [15]. This leaves open the question whether activation of PPARγ is required for adipogenesis in 3T3-L1 cells.
We and others have shown that organotins such as TBT [8, 13] and triphenyltin [9, 27] bind to PPARγ and RXRs [28] with nanomolar affinity. Moreover, organotins induce adipogenesis in 3T3-L1 cells [8, 9, 13, 27] and in adipose-derived [10] and bone-marrow derived [11] MSCs. Only in the case of the adipose derived MSCs has it been convincingly shown that PPARγ activation is required for the adipogenic activity of TBT and ROSI [10]. One group has suggested that TBT acts through a non-PPARγ mediated pathway to induce adipogenesis in 3T3-L1 cells [13]. However, considering the high affinity binding of TBT to RXR and PPARγ, it seems more likely that TBT is acting through one of these receptors to induce adipogenesis.
We sought to address the issue of whether TBT induces adipogenesis by a PPARγ-dependent or PPARγ-independent pathway by testing alternative possibilities. We found that the RXR-PPARγ heterodimer is permissive for RXR activation even in the presence of the PPARγ antagonist GW9662 (Figure 1), which suggested that TBT could act on the same target sites through the RXR half of the heterodimer. While this could be a plausible mechanism for TBT-induced adipogenesis in 3T3-L1 cells, it does not explain our observation (Figure 2) that GW9662 was unable to inhibit adipogenesis induced by treatment with ROSI. Considering this finding, we explored the relative stability of ROSI and GW9662 in the culture medium and cellular fraction. Surprisingly, we found that while the stability of ROSI was greater than 24 hours in either the medium or cells, GW9662 was quickly eliminated from both the cells and medium with a half-life of ~2 hours. When we increased the frequency of GW9662 addition to account for its short half-life, GW9662 was able to block adipogenesis elicited by ROSI or TBT (Figure 4) as well as the induction of PPARγ target genes such as aP2 (FABP4), adiponectin, leptin and perilipin (Figure 5). Taken together, we interpret these data to indicate that TBT acts through PPARγ in 3T3-L1 cells to induce adipogenesis and the expression of PPARγ target genes. We conclude that PPARγ activity is required for adipogenesis in 3T3-L1 cells and that ligand stability is an important consideration when designing and interpreting the results of adipogenesis assays.
Our results have important implications for the design and interpretation of adipogenesis assays that seek to test the pathways through which candidate obesogens might act. If GW9662 is not supplemented into the medium frequently enough, it is possible to make the erroneous inference that the candidate chemicals are acting through a non-PPARγ-dependent pathway. We suggest that in order to avoid such mistakes, it is essential to utilize ROSI (or another strong PPARγ activator) as a positive control in adipogenesis assays. Another indispensable control must be that GW9662 (or some other PPARγ antagonist) can block the induction of adipogenesis by ROSI before concluding that the antagonist does or does not inhibit the activity of another candidate compound. Considering the increasing interest in testing chemicals for their ability to induce adipogenesis, our results will be broadly applicable to the study of candidate obesogens and their mechanisms of action.
Methods
Transfection assays
pCMX-GAL4 and fusion constructs to nuclear receptor LBD [GAL4-hPPARγ] have been previously described [8]. The use of L-RXRα as a specific probe to assess the activation of RXR, independently of its heterodimeric partners was previously reported [20]. Transfections were performed in Cos7 cells essentially as described elsewhere using MH100-x4-TK-Luc as reporter and normalized to pCMX-β-galactosidase controls [29]. Briefly, Cos7 cells were seeded at 5000 cells per well in 96-well tissue culture plates in 10% fetal bovine serum/DMEM and transfected for 8 h with 11 μg/plate of DNA/calcium phosphate precipitate mix (MH100x4-TK-Luc:CMX-β-galactosidase:nuclear receptor effector(s) at a ratio of 5:5:1). Cells were washed free of precipitate with PBS and media were replaced with a serum free medium DMEM-ITLM (DMEM containing 5 μg/mL insulin, 5 μg/mL holo-transferrin, 5 μg/mL selenium, 0.5% defined lipid mix (Invitrogen), 0.12% w/v delipidated bovine serum albumin (Sigma)) plus ligands for an additional 24 h before assays for luciferase and ß-galactosidase activity {Grun, 2006 #569}. All transfection data points were performed in triplicate, and all experiments were repeated at least three times.
3T3-L1 cell culture
3T3-L1 cells were maintained in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 50 U/ml penicillin and 50 μg/ml streptomycin. For differentiation, 3T3-L1 cells were plated at a density of 5000 cells/cm2 on 12-well plates and cultured until 2 days post confluence. At this time, they were switched to a differentiation medium containing 10% FBS, MDI (5 μg/mL insulin, 0.25 μM dexamethasone and 0.5 mM 3-isobutyl-1-methylxanthine), 8 μg/mL biotin, 8 μg/mL pantothenate 50 U/ml penicillin and 50 μg/ml streptomycin. After 2 further days of incubation, cells were maintained in DMEM/10% FBS, biotin, pantothenate, penicillin, streptomycin containing TBT or ROSI at the concentrations indicated in the figure legends. For antagonist assays, GW9662 was added to 500 nM into the medium every 8 hours for 7 days. Cells were harvested for subsequent assays 7 days after addition of the differentiation cocktail. All experiments were repeated at least three times.
Triglyceride assays
3T3-L1 culture and induction of adipogenesis was performed in 12-well plates as described above. After 7 days of differentiation, cells were washed with PBS, the aqueous medium aspirated and total lipid extracted by adding 1 mL of hexanes:isopropanol (3:2 v/v) to the plates and shaking for 1 hr. The organic layer was collected to glass 13×100 mm test tubes, dried under a stream of argon gas and solubilized in 300 μL of chloroform containing 1% Triton-X100. Samples were dried again under argon, resuspended in 200 μL of pure water and incubated at 37°C for 1 h to ensure dissolution. Triglycerides were quantified using the Infinity Triglyceride kit (Thermo Scientific) and normalized to cellular DNA content. For DNA extraction, cells were homogenized in buffer containing 0.1 M NaOH, 0.1 M NaCl, 20 mM EDTA, followed by centrifugation. 100 μL of supernatant was collected and mixed with 1 ml of 10 mM MOPS (free acid), pH 4.6, and DNA concentration was determined by A260 measurement.
Oil red O staining and analysis
Cells were washed twice with PBS and fixed in 10% formalin in PBS for 15 min, rinsed with 60% isopropyl alcohol, and stained with 0.3% oil red O in 60% isopropanol for 30 min. Cells were then rinsed twice with 60% isopropyl alcohol and mounted in 30% glycerol. Lipid accumulation was quantified as previously described [10] using Image J (version 1.36b; Wayne Rasband). Data represent mean ± SEM from n = 3 wells per treatment and n = 6 pictures per well.
Analysis of mRNA by real-time reverse transcriptase polymerase chain reaction
Total RNA was extracted using the TRIzol reagent (GIBCO-BRL, Gaithersburg, MD). Complementary DNA was generated from 1 μg DNase-treated RNA using SuperScript™ III RNase H- Reverse Transcriptase (Invitrogen) after DNase I digestion (Ambion, Austin, TX) following the manufacturer recommended protocol. Real-time PCR was performed in the DNA Engine Opticon Thermal Cycler [MJ Research (Watertown, MA/Bio-Rad Laboratories (Hercules, CA)]. Quantitative PCR analyses for target genes (Listing in Supplemental Table 1) were performed with FastStart SYBR Green QPCR Master Mix (Roche, Nutley, NJ) and 100 nM of primers chosen with using PerlPrimer (v1.1.14 Copyright © 2003-2006 Owen Marshall). Relative quantification of the target gene transcript in comparison with β-actin (housekeeping gene) expression level in the same sample, was made following the Pfaffl method (2010) and modified as previously described [10].
Mass spectrometric analysis
3T3-L1 cells were seeded into 10 cM dishes and grown to 80% confluency in DMEM+10% FBS. Media was then aspirated and replaced with 15 mL of fresh medium containing 1 μM of ROSI or GW9662. After 5 minutes, a 1 mL aliquot of media was taken and frozen at -80 °C as T=0 hours. Plates were incubated in a CO2 incubator at 37°C in 5% CO2 in air for 1, 2, 4, 8, 12, 24, 36 or 48 hrs. Samples for analysis of media were taken from the same plate at successive intervals. For cellular fractions, plates were treated in parallel in the same manner. At the end of each time point, the medium was aspirated, the cells trypisinized and stored at -80°C until all samples were ready. Triplicate samples of 300 μL of media or cell pellets resuspended in 300 μL of distilled H2O were spiked with dexamethasone at 1 μM (as an internal standard) and extracted with an equal volume of ethyl acetate. After vigorous vortexing for 2 minutes, the phases were separated by centrifugation and the organic phase removed to an autosampler vial. The samples were evaporated to dryness in a SpeedVac concentrator, then resuspended in 100 μL of 50% methanol in distilled H2O. Samples were analyzed on a Waters Quattro Premier XE triple quadrupole tandem mass spectrometer and values obtained for ROSI or GW9662 normalized to dexamethasone to correct for extraction efficiency. All experiments were repeated at least twice.
Supplementary Material
Research Highlights.
The RXR-PPARγ heterodimer is permissive for RXR activation in the presence of the PPARγ antagonist GW9662.
GW9662 is unable to block adipogenesis in 3T3-L1 cells mediated by tributyltin (TBT) or rosiglitazone (ROSI).
GW9662 does not block adipogenesis in 3T3-L1 cells because it has a very short half life (~2 hours) compared with ROSI (> 24 hours).
When GW9662 is added every 8 hours, it inhibits TBT and ROSI-mediated adipogenesis.
PPARγ activity is necessary for either TBT or ROSI to induce adipogenesis in 3T3-L1 cells.
Acknowledgments
Supported by a grant from the NIH R01-ES015849 to (B.B.). J.Y. was supported by fellowships from the University of California Toxic Substances Research and Training Program, the UCI Minority Biomedical Research Support Program (NIH GM-55246) and NSF GK-12 Grant 0638751. J.Y. wishes to thank Dr. Felix Grün for help with the design and implementation of mass spectrometric measurements. We thank A. Janesick and R. Kaigh for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Grun F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147(6 Suppl):S50–55. doi: 10.1210/en.2005-1129. [DOI] [PubMed] [Google Scholar]
- 2.Blumberg B. Obesogens, stem cells and the maternal programming of obesity. Journal of Developmental Origins of Health and Disease FirstView. 2010:1–6. doi: 10.1017/S2040174410000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grun F, Blumberg B. Endocrine disrupters as obesogens. Mol Cell Endocrinol. 2009;304(1-2):19–29. doi: 10.1016/j.mce.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grun F, Blumberg B. Minireview: the case for obesogens. Mol Endocrinol. 2009;23(8):1127–1134. doi: 10.1210/me.2008-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janesick A, Blumberg B. The Role of Environmental Obesogens in the Obesity Epidemic. In: Lustig RH, editor. Obesity Before Birth. Springer; US: 2011. pp. 383–399. [Google Scholar]
- 6.Janesick A, Blumberg B. Adipocytes as Target Cells for Endocrine Disruption. In: Diamanti-Kandarakis E, Gore AC, editors. Endocrine Disrupters and Puberty. Humana Press/Springer; New York, NY: 2011. in press. [Google Scholar]
- 7.Janesick A, Blumberg B. PPARg as the target of obesogens. Journal of Steroid Biochemistry and Molecular Biology. 2011 doi: 10.1016/j.jsbmb.2011.01.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, Gardiner DM, Kanno J, Iguchi T, Blumberg B. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20(9):2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- 9.Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, Nishikawa J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol. 2005;67(3):766–774. doi: 10.1124/mol.104.008409. [DOI] [PubMed] [Google Scholar]
- 10.Kirchner S, Kieu T, Chow C, Casey S, Blumberg B. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol. 2010;24(3):526–539. doi: 10.1210/me.2009-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carfi M, Croera C, Ferrario D, Campi V, Bowe G, Pieters R, Gribaldo L. TBTC induces adipocyte differentiation in human bone marrow long term culture. Toxicology. 2008;249(1):11–18. doi: 10.1016/j.tox.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 12.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7(12):885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 13.Inadera H, Shimomura A. Environmental chemical tributyltin augments adipocyte differentiation. Toxicol Lett. 2005;159(3):226–234. doi: 10.1016/j.toxlet.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Wright HM, Clish CB, Mikami T, Hauser S, Yanagi K, Hiramatsu R, Serhan CN, Spiegelman BM. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J Biol Chem. 2000;275(3):1873–1877. doi: 10.1074/jbc.275.3.1873. [DOI] [PubMed] [Google Scholar]
- 15.Walkey CJ, Spiegelman BM. A functional peroxisome proliferator-activated receptor-gamma ligand-binding domain is not required for adipogenesis. J Biol Chem. 2008;283(36):24290–24294. doi: 10.1074/jbc.C800139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 17.IJpenberg A, Tan NS, Gelman L, Kersten S, Seydoux J, Xu J, Metzger D, Canaple L, Chambon P, Wahli W, Desvergne B. In vivo activation of PPAR target genes by RXR homodimers. EMBO J. 2004;23(10):2083–2091. doi: 10.1038/sj.emboj.7600209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulman IG, Shao G, Heyman RA. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimers: intermolecular synergy requires only the PPARgamma hormone-dependent activation function. Mol Cell Biol. 1998;18(6):3483–3494. doi: 10.1128/mcb.18.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perlmann T, Umesono K, Rangarajan PN, Forman BM, Evans RM. Two distinct dimerization interfaces differentially modulate target gene specificity of nuclear hormone receptors. Mol Endocrinol. 1996;10(8):958–966. doi: 10.1210/mend.10.8.8843412. [DOI] [PubMed] [Google Scholar]
- 20.Forman BM, Umesono K, Chen J, Evans RM. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 1995;81(4):541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 21.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43(4):527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 22.Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277(22):19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- 23.Choi SS, Cha BY, Lee YS, Yonezawa T, Teruya T, Nagai K, Woo JT. Magnolol enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Life Sci. 2009;84(25-26):908–914. doi: 10.1016/j.lfs.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Kershaw EE, Schupp M, Guan HP, Gardner NP, Lazar MA, Flier JS. PPARgamma regulates adipose triglyceride lipase in adipocytes in vitro and in vivo. Am J Physiol Endocrinol Metab. 2007;293(6):E1736–1745. doi: 10.1152/ajpendo.00122.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao Y, Yuan T, Yao W, Liao K. 3T3-L1 adipocyte apoptosis induced by thiazolidinediones is peroxisome proliferator-activated receptor-gamma-dependent and mediated by the caspase-3-dependent apoptotic pathway. FEBS J. 2010;277(3):687–696. doi: 10.1111/j.1742-4658.2009.07514.x. [DOI] [PubMed] [Google Scholar]
- 26.Yang JY, Della-Fera MA, Rayalam S, Park HJ, Ambati S, Hausman DB, Hartzell DL, Baile CA. Regulation of adipogenesis by medium-chain fatty acids in the absence of hormonal cocktail. J Nutr Biochem. 2009;20(7):537–543. doi: 10.1016/j.jnutbio.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 27.Hiromori Y, Nishikawa J, Yoshida I, Nagase H, Nakanishi T. Structure-dependent activation of peroxisome proliferator-activated receptor (PPAR) gamma by organotin compounds. Chem Biol Interact. 2009;180(2):238–244. doi: 10.1016/j.cbi.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 28.le Maire A, Grimaldi M, Roecklin D, Dagnino S, Vivat-Hannah V, Balaguer P, Bourguet W. Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 2009;10(4):367–373. doi: 10.1038/embor.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milnes MR, Garcia A, Grossman E, Grun F, Shiotsugu J, Tabb MM, Kawashima Y, Katsu Y, Watanabe H, Iguchi T, Blumberg B. Activation of steroid and xenobiotic receptor (SXR, NR1I2) and its orthologs in laboratory, toxicologic, and genome model species. Environ Health Perspect. 2008;116(7):880–885. doi: 10.1289/ehp.10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.