

Abstract
Eukaryotic sulfite oxidase is a dimeric protein that contains the molybdenum cofactor and catalyzes the metabolically essential conversion of sulfite to sulfate as the terminal step in the metabolism of cysteine and methionine. Nitrate Reductase is an evolutionally related molybdoprotein in lower organisms that is essential for growth on nitrate. In this study we describe human and chicken sulfite oxidase variants in which the active site has been modified to alter substrate specificity and activity from sulfite oxidation to nitrate reduction. Based on sequence alignments and the known crystal structure of chicken sulfite oxidase two residues are conserved in nitrate reductases that align with residues in the active site of sulfite oxidase. Based on the crystal structure of yeast nitrate reductase, both positions were mutated in human sulfite oxidase and chicken sulfite oxidase. The resulting double mutant variants demonstrated a marked decrease in sulfite oxidase activity but gained nitrate reductase activity. An additional methionine residue in the active site was proposed to be important in nitrate catalysis, and therefore the triple variant was also produced. The nitrate-reducing ability of the human sulfite oxidase triple mutant was nearly three-fold higher than that of the double mutant. In order to obtain detailed structural data on the active site of these variants, the analogous mutations in chicken sulfite oxidase were generated in order to perform crystallographic analysis. The crystal structures of the Mo domains of the double and triple mutants were solved to 2.4 Å and 2.1Å resolution, respectively.
Sulfite oxidase (SO) (1) is a member of the family of proteins containing the molybdenum cofactor (Moco) that consists of a single Mo atom coordinated through a dithiolene group to molybdopterin (MPT)(2). The Mo atom is coordinated to the protein via a single cysteine residue (3). Eukaryotic SO is an essential protein that resides in the intermembrane space of the mitochondria and catalyzes the essential oxidation of sulfite to sulfate in the terminal step of the degradation of sulfur-containing compounds including the amino acids methionine and cysteine. Loss of the ability to oxidize sulfite can be due to either a defect in the Moco biosynthetic pathway or point mutations in the sox gene itself (4). Of the human enzymes containing Moco, only SO is essential, and a number of point mutations in the SO gene have been previously identified in SO-deficient patients (5). SO deficiency leads to severe effects including seizures, attenuated brain growth, mental retardation, and other neurological and metabolic problems, usually resulting in death during the first year of life (6). The sulfite oxidation reaction catalyzed by SO is summarized below and a scheme of the likely reaction cycle is provided in Figure 1.
Figure 1.

Reaction Cycle of SO.
| (7) | Eq. 1 |
In the first part of the cycle (reductive half-reaction), sulfite binds at the MoVI center and is oxidized to sulfate generating a two-electron reduced MoIVFeIII species (8). In the first intramolecular electron transfer (Fig. 1, IET #1) between the Mo and heme domains, one of the two reducing equivalents generated by sulfite oxidation is transferred from MoIV to the b5-type heme in the N-terminal domain yielding a transient MoVFeII species. The second part of the cycle (oxidative half-reaction) begins with an electron transfer from the heme to the exogenous electron acceptor, cytochrome c (cyt c). After a second IET from MoV to FeIII, the fully oxidized species is regenerated by electron transfer from FeII to a second molecule of cyt c. Eukaryotic SO purifies as a homodimer of ~52 kDa subunits, and each subunit consists of an N-terminal b5-type heme domain, a central domain containing the Moco, and a C-terminal region mediating dimerization (9, 10) (Figure 2).
Figure 2.

The Domain Structure of SO and NR proteins. The monomer of P. angusta NR is arranged with an N-terminal Moco domain, a dimerization domain, a heme domain and a NADPH/FAD domain. In comparison the CSO monomer contains an N-terminal heme domain, central Moco domain and C-terminal dimerization domain.
The structure of chicken SO
The crystal structure of chicken liver SO was solved at a resolution of 1.9 Ǻ (11) (Figure 3A). The structure revealed a sulfate molecule bound in the active site, allowing identification of the likely ligands and H-bonds to the substrate during catalysis. The coordination around the Mo atom in the active site consists of two oxo groups (equatorial and axial) and three sulfur ligands, including two dithiolene sulfurs and the thiol group of Cys 185 arranged in a square pyramidal geometry (12). Solvent access is via a positively charged channel that opens onto the equatorial oxo group of the Mo atom. The substrate-binding pocket is positively charged and is formed by five residues including three arginines at positions 138, 190, and 450, as well as Tyr 322 and Trp 204. All five of these residues are conserved in all eukaryotic SO proteins isolated to date. The importance of Arg 138 and 190 (Arg 160 and 211, respectively, in human SO) was demonstrated by the identification of the R160Q and R211Q mutations in human patients presenting with severe SO deficiency (11). The structural and kinetic characterization of the R160Q variant has been accomplished using a variety of techniques including flash photolysis (13) and EXAFS (14). Very recently, the molybdenum site of this clinical mutant was investigated by a combination of X-ray absorption spectroscopy and density functional theory calculations (14). Based on these studies, this variant has a six-coordinate pseudo-octahedral active site with coordination of the Oε of Gln160 to molybdenum (14). This is in contrast to the wild-type SO active site which is five-coordinate and exhibits approximately square-based pyramidal geometry (15). While mutations of the remaining substrate binding pocket residues have yet to be observed in human patients, the Tyr 322 (Tyr 343 in human SO) residue was proposed to be important in labilizing the oxo group of MoVI to allow oxygen transfer to sulfite based on the proximity of this residue to the equatorial oxo group in the crystal structure. Furthermore, this residue has been proposed to serve an additional role as a proton shuttle between water (or OH-) in the coupled electron-proton transfer (CEPT) mechanism during reduction and reoxidation of the Mo center (16). The human Y343F variant has been created and extensive kinetic and structural studies have indicated that the Tyr residue is critical for substrate binding (17), IET (18), and overall catalysis including serving as a proton shuttle between the Mo-oxo group and water (or hydroxo) in the coupled electron-proton transfer during reduction and reoxidation of the Mo center.
Figure 3.
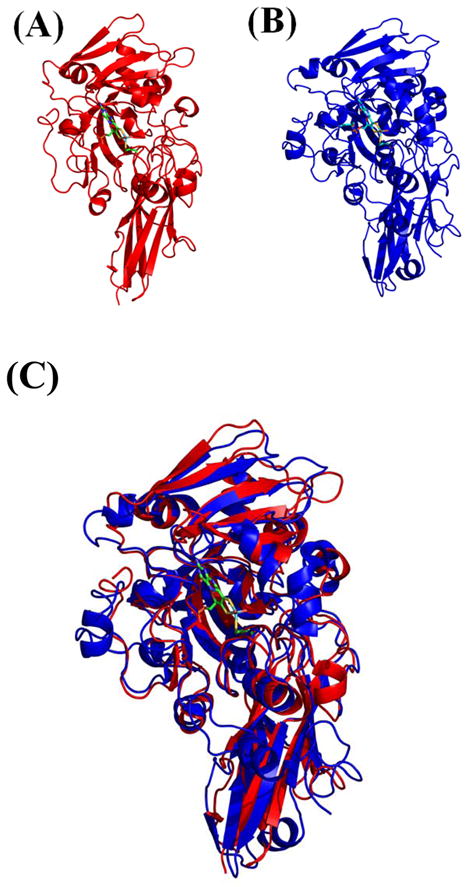
Model of active sites of SO/NR with substrates. The Structure of the Mo domains of (A) Sulfite Oxidase and (B) Nitrate Reductase (C) Alignment of chicken SO (red) and the P. angusta NR (blue) Mo domains showed excellent agreement. The Moco is shown as green sticks.
The structure of plant SO
In 2003, the structure of a second member of the SO superfamily, A. thaliana SO, was solved at a resolution of 2.3 Ǻ (19). Plant SO is homodimeric, but is devoid of the heme domain. In contrast to chicken SO, plant SO crystallized without a sulfate bound in the active site. While the five active-site residues mentioned above are strictly conserved between mammalian and plant SO, only 4 of them superimposed with the residues in the chicken SO active site. The exception was Arg 374 (equivalent to Arg450/472 in chicken/human SO), in which the side chain pointed toward the solvent access channel rather than toward bound sulfate seen in the chicken SO structure. Since there was no sulfate in the active site, based on these findings, Arg 374 was proposed to be essential for both substrate recruitment/binding and specificity due to its conformational change in the presence of bound substrate.
The structure of recombinant chicken SO
Since SO is the only Moco-containing protein essential for normal human development, structure-function studies on the enzyme are of significant relevance to human health. Unfortunately, human SO (with 68% sequence identity to chicken SO) remains resistant to crystallization, and the gene for chicken SO has never been isolated, so there are significant barriers to performing structure-function studies on mammalian SO or in determining disease etiology in mutants isolated from human patients with SO deficiency. However, recently our lab successfully created an artificial gene for chicken SO, allowing us to successfully introduce mutations into the chicken SO protein to begin structure-function studies (20). When recombinant chicken SO was crystallized in both sulfate-bound and unbound conformations, this represented the first time the same Moco enzyme was crystallized in the presence and absence of the product, and later in the presence and absence of substrate (12) verifying that the Arg 450 (human Arg 472) residue does indeed change conformation depending on the presence of substrate in the active site (17). The Arg 450 residue pointed toward the active site in the presence of substrate, coordinating the sulfite, but when no sulfite was present, the side chain pointed toward the solvent access channel. This change in conformation results in a much smaller binding pocket and shifts the position of the sulfur away from the Mo atom presumably preparing the pocket for product release, and possibly transmitting conformational changes to other parts of the protein, including the heme domain, allowing domain movement to occur during the reaction cycle. It should be noted that Arg 450 is located in the dimerization domain, so it has also been proposed that this residue could possibly transmit conformational changes from the active site to the dimer interface, thus transmitting information between the two subunits of SO. However, until the current study the importance of this third active site Arg in either substrate binding or in activity had never been directly verified by site-directed mutagenesis and kinetic analysis. In this study, we changed the Arg472 residue in human SO (analogous to Arg450 in chicken SO) to either Met or Gln to directly observe changes in the activity of the protein as a result of the change at this position from a positive to an uncharged residue.
The R138Q variant of chicken SO
The de novo creation of the chicken SO gene also allowed the structure of the R138Q chicken SO protein to be solved (20). As predicted from previous work on the analogous R160Q human protein (13), the replacement of a positively charged arginine with a neutral glutamine in the active site changed the surface potential of the active site and solvent access channel; however, this change was not due only to the loss of the positive charge of Arg138. The most significant structural change in the active site of the R138Q mutant was actually the orientation of the side chain of Arg 450 toward the active site due to the loss of the repulsive effect of the Arg 138 side chain on Arg 450 in the absence of sulfate in the active site. This resulted in a significant change in the position of the Arg 450 (human/plant Arg 472/374) residue, resulting in negative charges becoming exposed along the solvent access tunnel so that despite the absence of sulfate in the active site, the side chain of Arg 450 was pointing into the active site, creating a much smaller binding pocket. Thus this residue was found to occupy a similar position as in a wild type product-occupied pocket, albeit in the absence of product.
Assimilatory Nitrate Reductase (NR)
Assimilatory NR is a member of the SO family of Moco-containing enzymes that catalyzes the first step of nitrate assimilation in algae, fungi and plants (21–23). Assimilatory NR forms homodimers of approximately 120 kDa, and oligomerization is dependent upon MPT. NR catalyzes the overall reaction:
| Eq. 2 |
The reaction cycle of NR begins with the reductive half-reaction when NAD(P)H reduces FAD. The second step is IET via heme as in SO. The third step is the oxidative half reaction in which Mo transfers two electrons to nitrate, which serves as both the terminal 2-electron acceptor and as the substrate, yielding the reaction products nitrite and a hydroxyl ion(24). While NR and SO perform opposite reactions on their respective substrates, there is 31% identity between the heme domains of chicken SO and assimilatory NR from A. thaliana and 38% between their respective Mo domains (11), and in both cases, the electron flow is from the C to the N terminus of the multi-domain protein (Figure 2). The X-ray structure of the Mo domain of P. angusta NR was recently solved to 1.7 Å (25). The structure of the Moco in NR is identical to that found in chicken SO (Figure 3B), and four out of the six active site residues are conserved. The two variant residues are the Tyr 322 and Arg 450 of SO, which are replaced by Asn and Met/Leu or Val, respectively. In all NR sequences the Asn residue corresponding to the Tyr in SO is strictly conserved. There appears to be a division over the consensus sequence of the residue corresponding to Arg 450 in SO between the lower eukaryotic fungal NRs and higher eukaryotic plant NRs. A Met, Leu, or Thr replaces the chicken SO Arg 450 at the analogous position in fungal NRs, whereas in all plant NR, the Arg is replaced by Met. Given the difference in substrates (NO3− vs. SO3−2), it is proposed that the Arg 450 is replaced since nitrate has to be bound with one of its O atoms positioned toward the Mo atom for subsequent removal to produce the nitrite product. Stopped flow kinetic analysis of the plant NR revealed that the overall catalytic rate was not limited by any individual step, and unlike SO, electron transfer between domains appeared to be rate limiting in NR since the rate was much faster when an artificial electron donor such as methyl viologen or bromphenol blue was used rather than an NAD(P)H driven reaction (24).
Rational design of SO variants with NR activity
Enzyme specificity towards substrates is determined in part by the local encoding of a complementary surface to a given substrate by the active site residues (26). The following work describes the strategy by which the substrate specificity and activity of SO enzymes has been altered. Using predictions from the sequence alignments of SO and NR and analysis of the crystal structures of chicken SO and the NR Mo domain from P. angusta (Figure 3C) the predicted catalytic site residues of NR were introduced into chicken and human SO. We designed and characterized several SO variants. The resulting variants were assayed for the ability to oxidize sulfite to sulfate and for the acquisition of nitrate reduction. The double mutant exhibited decreased ability to oxidize sulfite and gained ability to reduce nitrate. Additionally, a third position in NR was identified near the edge of the active site that was hypothesized to play a role in substrate binding. This additional position was introduced into both double mutants to generate SO triple variants. The triple mutants of both human SO and chicken SO exhibited a higher affinity for nitrate, and moderately enhanced NR activity compared to the double mutants.
Finally, we were able to crystallize the chicken SO nitrate reductase variants CSO-2NR and CSO-3NR solving the structures to 2.4 Å and 2.1Å resolution, respectively, yielding detailed structural information about the active sites of these variants. The creation of these variant proteins has allowed unique insights into the varying functions of these two Moco-containing proteins. This information should be particularly valuable in understanding more about the nitrate reductase active site, as these proteins are generally less amenable to characterization than are sulfite oxidase proteins.
Experimental Procedures
Site-directed mutagenesis of eukaryotic SO enzymes
All mutations in human SO described were directly introduced into the pTG918 plasmid (27) using the Transformer Site-directed Mutagenesis kit (Clontech) as previously described (Clontech Laboratories Inc., Mountain View, CA) (17, 20). The CSO 2NR variant was generated in the wild-type CSO expression vector pTRC99a CLSO.2 with the R161G background. The Y322N mutation was introduced by overlapping primer extension PCR (28). The R450M mutation was introduced using the Transformer site directed mutagenesis kit as described above. The CSO 3NR variant Y322N/R450M/V452M mutations were introduced into the pTRC99a CLSO.2 vector as described above. All resulting constructs were verified by sequencing at the Duke University DNA analysis facility.
Expression and purification of SO
All recombinant forms of SO were expressed in the TP1000 strain of E. coli and purified as previously described (18, 27). For gel-filtration analysis of the oligomeric state of SO variants, purified protein (at 1 mg/ml) was injected onto a Superdex-200 FPLC column equilibrated with 50 mM potassium phosphate buffer and eluted using a flow rate of 1 ml/min. The FPLC system consisted of an AKTA Explorer system and protein was monitored at 280 nm. The concentration of purified SO was determined from the A413 using an extinction coefficient of 113 mM−1cm−1.
Molybdenum analysis
The Mo content of protein samples was quantified by atomic absorption spectroscopy using a Spectra AA-220 double beam atomic absorption spectrometer (Varian, Palo Alto, CA). Subsequent analyses were done using the conditions described previously by Johnson (3, 29).
Sulfite oxidase activity assays
Steady-state kinetic assays were performed aerobically at 25 °C using a 1.0 cm path length cuvette in a Shimadzu UV-1601PC spectrophotometer. Assays were carried out in 20 mM or 50mM buffers adjusted to the desired pH with acetic acid to minimize anion inhibition of SO as previously described (8, 17). The buffers used were as follows: Bis-Tris (pH 6.0–6.5), Bis-Tris propane (pH 6.5–7.5), Tris (pH 7.5–8.5), and glycine (pH 9.0–10.0). The glycine buffers were adjusted to the desired pH using NaOH. Steady-state pH profiles were obtained using 15–50 μM horse heart cyt c (Sigma), 0.50–2.5 μg/ml SO, and varying concentrations of sulfite in a final assay volume of 1 ml. The reduction of cyt c was monitored at 550 nm using an extinction coefficient of 19 mM−1cm−1. The steady-state reaction parameters kcat and Km were obtained by a direct fit of the concentration dependence to the Michaelis-Menten equation using Kaleidagraph (Synergy Software). Steady-state assays with ferricyanide as the electron acceptor were performed using 20 mM buffers as described above, 40 μM ferricyanide, 1.5–5.0 μg/ml SO, and varying concentrations of sulfite in a final assay volume of 1 ml. The reduction of ferricyanide was monitored at 420 nm, and enzyme activity was reported in U/mg, where one unit is equal to an absorbance change of 1.0 AU/min at 420 nm.
Nitrate reductase assays
Nitrate reductase assays were carried out anaerobically at 25 °C using dithionite-reduced methyl viologen as the electron donor and monitoring the oxidation of MV at 600nm using a Shimadzu UV-1601PC spectrophotometer. Nitrate reductase assays were carried out anaerobically at 25 °C using dithionite-reduced methyl viologen (MV) as the electron donor and monitoring its oxidation at 600 nm using the extinction coefficient of 8.25 mM−1cm−1 (30). Assay conditions include 20 μM MV, 50 μg enzyme and varying concentrations of potassium nitrate in a final volume of 2 ml. Measurement of nitrate reductase activity was carried out at pH 7.0 using 50 mM Bis-tris Propane, and at pH 8.5 using 50 mM Tris-HCl. The pH of the buffer was adjusted with acetic acid to minimize anion inhibition. Absorption change was monitored on a Shimadzu UV-1601PC spectrophotometer instrument. All buffer solutions were made anaerobic by bubbling with argon gas for at least 30 min prior to use. All assays were prepared in an anaerobic chamber (Coy Laboratory Products Inc. Grass Lake, Michigan), in 13 × 100 mm cuvettes and sealed with a rubber septum. The assay reagents methyl viologen, sodium dithionite, and potassium nitrate were brought into the anaerobic chamber as dry stocks and solutions were prepared using anaerobic buffer. The steady-state reaction parameters kcat and Km were obtained by a direct fit of the concentration dependence to the kinetic studies.
Stopped flow assays of HSO SO
Rapid reaction kinetic studies were performed using an SX.20MV Stopped-flow Reaction Analyzer (Applied Photophysics Ltd.) as previously described (8, 17). The dead-time of the instrument was determined to be <1.5 ms with a path length of 10 mm. To prepare the instrument for anaerobic operation, the sample tubing and valve lines were incubated with 250 mM sodium dithionite for several hours and then thoroughly rinsed with O2-free water. Additionally, the sample-handling unit of the stopped-flow apparatus was fitted with an anaerobic accessory that enclosed the drive syringe plungers and was continuously flushed with nitrogen gas. Reaction temperature was maintained at 25 °C using a circulating water bath connected to the thermostat bath housing the drive syringes. All solutions were made anaerobic by bubbling with argon gas for at least 30 min prior to use, and solutions of human SO were prepared by diluting <100 μg of a concentrated stock of the enzyme into a final volume of ~5 ml of anaerobic buffer. All dilutions were performed in an anaerobic chamber (Coy Laboratory Products Inc.), and 5 ml Hamilton gas-tight syringes were used to load samples into the stopped-flow spectrophotometer. Rapid kinetic assays of the reductive half-reaction were performed anaerobically at 25 °C using 0.3–0.8 μM of protein. Reduction of the SO b5-type heme was directly monitored by measuring the extinction change of the Soret peak at 425 or 430 nm. The kobs of each individual reaction were obtained by fitting individual kinetic traces to single or double exponential curves using a nonlinear least-squares Levenberg-Marquardt algorithm. The maximal rate parameter kred as well as the Kdsulfite for the reductive half-reactions were obtained by fitting the kobs at varying sulfite concentrations to a hyperbolic curve as shown below.
| Eq. 3 |
Crystallization of the nitrate reductase variants of CSO
After purification, the CSO-2NR variant was buffer exchanged using a PD-10 ion exchange column (GE Healthcare) into 20 mM Tris pH 7.8,100 mM NaCl, and concentrated to 25 mg/ml using a Vivaspin 30,000 concentrator (Vivascience). This sample served as a stock protein solution, and was stored at 4 °C prior to crystallization. From this stock solution the protein was diluted to a 10 mg/ml solution using a buffer solution of 20mM, Tris pH 7.8, containing 100 mM NaCl. Crystals of CSO-2NR variant were obtained by the hanging drop vapor diffusion method at 17 ° C, by adding 2 μl of protein solution to 2 μl of crystallization solution equilibrated against 1 ml of reservoir crystallization solution of 17% PEG 10,000 (w/v), 10 mM Barium Chloride dihydrate, 100 mM ammonium acetate, and 100 mM Bis-Tris propane pH 5.5. Red crystals were observed after three weeks and were grown for five to six weeks. Crystals were transferred to the crystallization solution containing 5% glycerol, which was increased step-wise to a final concentration of 30%, then flash frozen in liquid nitrogen. The crystals grew in the I41 space group with unit cell dimensions of a = 85.62, b = 85.62, c = 153.37 and a solvent content of 54.5%. The CSO-3NR variant crystal form was grown in crystallization conditions consisting of 15 % PEG 6000 (w/v), 5 % MPD (v/v), 100 mM MES pH 6.5, and 2% (w/v) benzamidine hydrochloride hydrate by the hanging drop method as mentioned above. The crystals were observed after three weeks reaching a final size by six weeks, and again, were red in color, grew in the I41 space group with unit cell dimensions of a = 85.6, b = 85.6, c = 152.8 and a solvent content of 54.3%.
X-ray diffraction data sets were collected for both the CSO-2NR and CSO-3NR variants at the South East Regional Collaborative Access Team (SER-CAT) BM-22 line at the Advanced Photon Source, Argonne National Laboratory. All data sets were scaled and indexed using DENZO and SCALEPACK (31). The structures were solved by molecular replacement using PHASER in the CCP4 suite of programs. The coordinates from the recombinant chicken SO residues 95-466 (PDB code 2A99) excluding the cofactor and solvent molecules were used for the molecular replacement search model (32). Iterative model building using COOT (33) with PROBE (34) for visualization of all-atom contacts and refinement was done with REFMAC (35). The stereochemistry of the structures was evaluated using MOLPROBITY (36) to assess the Ramachandran plots and atomic clash scores. For the structures reported in this study TLS refinement was performed at the final stages of refinement (37), and water molecules were added to complete the models using COOT.
Results and Discussion
HSO R472Q and R472M single substitution variants
When the crystal structure of plant SO was determined, all residues in the active site superimposed with the previously determined chicken SO structure with the exception of the side chain of the Arg 374 residue (analogous to Arg 450 and 472 in chicken and human SO, respectively). Since plant SO crystallized in the absence of sulfate, it was proposed that this residue plays a crucial role in substrate recruitment and binding (19). To directly test the role of this residue in substrate binding, we generated and purified the R472Q (for a similarly sized but uncharged substitution) and the R472M (the analogous residue in NR) variants of human SO. Both variants behaved similarly to wild type SO during purification, and purified as dimers with a full complement of Mo as measured by atomic absorption spectroscopy.
Somewhat surprisingly, steady-state kinetics of both of these variants indicated that substitution at this position had very little effect on the Kmsulfite, and even at high pH, the values were very comparable to wild type SO (Table 1). In contrast to the Kmsulfite values, both mutations had drastic effects on catalysis as measured by the ability to reduce the exogenous electron acceptor cyt c in the presence of sulfite (Table 2). The impairment in catalysis was particularly pronounced at high pH values.
Table 1. Kmsulfite for wild-type, R472Q and R472M SO variants.
Steady state assays were carried out aerobically as described in Experimental Procedures.
| pH | Wild type Km(μ) |
R472Q Km(μ) |
R472M Km(μ) |
|---|---|---|---|
| 6.0 | 1.29 | 2.32 | 1.95 |
| 7.0 | 2.72 | 3.51 | 2.45 |
| 8.0 | 4.35 | 13.0 | 4.72 |
| 8.5 | 8.25 | 16.2 | 9.38 |
| 9.0 | 22.1 | 45.3 | 21.2 |
| 9.5 | 67.1 | 96.7 | 53.7 |
| 10.0 | 52.9 | 181.7 | 96.9 |
Table 2. kcat values for wild-type, R472Q and R472M SO variants.
Steady state assays were carried out aerobically as described in Experimental Procedures.
| pH | Wild type kcat(s−1) |
R472Q kcat(s−1) |
R472M kcat(s−1) |
|---|---|---|---|
| 6.0 | 13.2 | 8.17 | 5.02 |
| 7.0 | 24.2 | 5.77 | 3.60 |
| 8.0 | 25.9 | 9.34 | 3.48 |
| 8.5 | 26.9 | 4.58 | 3.77 |
| 9.0 | 25.7 | 4.24 | 3.44 |
| 9.5 | 26.3 | 1.67 | 1.81 |
| 10.0 | 13.0 | 0.52 | 0.46 |
Stopped flow analysis of the reductive half-reaction of R472Q and R472M
Stopped flow analysis of wild type, R472Q and R472M SO was performed in the absence of cyt c by measuring the spectral change of the Soret peak at 425 nm allowing direct observation of the reduction of heme during the reductive half reaction and obtaining a rate for IET #1 (Fig. 1). In contrast to the kcat values described above, the kred for both of these proteins was quite high at both pH 7.0 and 8.5 (Table 3). R472Q demonstrated nearly wild-type rates, and R472M had approximately 50% wild-type rates at both pH values. These results were somewhat unexpected since overall catalysis in both of these proteins is less than 10% of the wild type (Table 2). Another surprising result was the high Kdsulfite at pH 8.5 for both proteins, particularly R472Q; this was not anticipated in view of the nearly wild-type Kmsulfite values obtained in the steady state assays for both of these proteins (Table 1).
Table 3. Rapid reaction kinetics for wild-type, R472Q and R472M SO.
Stopped flow analysis was carried out anaerobically as described in Experimental Procedures
| kred(s−1) | Kdsulfite(μM) | |
|---|---|---|
| pH 8.5 | ||
| Wild type | 82.8 | 6.72 |
| R472Q | 71.2 | 227 |
| R472M | 38.9 | 73.4 |
| pH 7.0 | ||
| Wild type | 70.2 | 2.05 |
| R472Q | 83.4 | 14.5 |
| R472M | 39.0 | 3.23 |
These results form a direct contrast to the reductive half-reaction rates obtained in our laboratory for previously analyzed active-site variants, including R160Q (16) and Y343F (17). For both of these proteins we obtained very low kred values at pH 8.5 (5.3 s−1 and 4.8 s−1 for R160Q and Y343F respectively), as well as very high Kdsulfite values that were very close to the Kmsulfite values obtained for the steady-state reactions.
In order to rule out the possibility that the interaction of the heme domain with the exogenous electron acceptor cyt c was adversely affected in these variants, resulting in low overall catalysis but high reductive half-reaction rates, steady-state kinetics were performed in which the sulfite was held constant and cyt c was varied. The Kmcyt c obtained for R742Q was 4 μM and for R472M was 4.1 μM, which in both cases was very close to the wild-type value of 4.4 μM.
The Mo domains of R472Q and R472M SO
In order to measure the ability of R472Q and R472M to oxidize sulfite and transfer electrons to an artificial electron acceptor in the absence of the heme domain, the R472Q and R472M mutations were created in the Mo domain of SO. Mirroring the stopped-flow results described above, when the non specific electron acceptor ferricyanide (rather than cyt c) is used in steady state assays, the Vmax was not nearly as impaired as when cyt c is used for either protein (Table 4). Also similar to the stopped flow results in which the Kd for both protein significantly increased (Table 3), the Kmsulfite increased significantly in these assays as well (Table 4).
Table 4. Steady state activity of the Mo domains of wild-type, R472Q, and R742M SO using Ferricyanide as an artificial electron acceptor.
| Vmax (U/mg) | Kmsulfite(μM) | |
|---|---|---|
| pH 8.5 | ||
| Wild type | 99.8 | 14.9 |
| R472Q | 99.6 | 223 |
| R472M | 46.4 | 94.0 |
| pH 7.0 | ||
| Wild type | 78.0 | 4.56 |
| R472Q | 50.5 | 12.8 |
| R472M | 27.9 | 5.00 |
Again, the R472Q and R472 Mo domains variants are different from other previously characterized human SO variants (e.g. Y343F and R160Q) which have impaired catalysis, but also demonstrate a similarly low ability to reduce a nonspecific electron acceptor such as ferricyanide.
The human SO Y343N/R472M (HSO-2NR) variant
Of the five core residues forming the active site of SO, two are not conserved between SO and NR proteins. These two residues, Tyr 343 and Arg 472 (human numbering) were switched to the Asn and Met residues found in NR proteins. In examining the single substitution variant proteins at these two positions (Y343N and R472M), it is clear that these two residues play distinct roles. The Tyr 343 residue clearly plays an important role in mediating substrate specificity, since the single mutation Y343N demonstrates significant impairment in the ability to bind sulfite (Table 5) while retaining catalytic activity (~60% wild-type at pH 8.5, Table 6) while the R472M single mutant as discussed above has essentially wild-type Kmsulfite but only ~10% catalytic activity (Tables 1 and 2). The Y343N/R472M double mutant (HSO-2NR) behaved identically to wild-type HSO and purified as a dimer (10). The HSO-2NR variant lost essentially all ability to bind sulfite as demonstrated by a Kmsulfite of 42.9 mM at pH 8.5, 5000-fold higher than the wild-type value of 8.25 μM (Table 5). Furthermore, the HSO-2NR was severely impaired in the ability to oxidize sulfite (Table 6) across the range of pH values measured, resulting in a second-order rate constant (kcat/Kmsulfate) 7 orders of magnitude below wild-type SO (Table 7). However, this protein gained the ability to bind nitrate with a Kmnitrate of 568 μM as demonstrated by methyl-viologen mediated nitrate reduction assays performed at pH 8.5, while wild-type SO has negligible ability to either bind or reduce nitrate (Table 8). While this initial result was encouraging, the measured Kmnitrate of 568 μM at pH 8.5 was approximately 10-fold higher than observed in native NR proteins. However, when the assays were performed using standard nitrate reductase conditions at pH 7.0, the value of 24 μM was well within the range of native NR proteins (24, 38, 39), though the rate of catalysis remained quite low (Table 8).
Table 5. Kmsulfite values of active site HSO variants.
| pH | Wild type Km (μ) |
Y343N Km (μM) |
V474M Km (μM) |
Y343F/R472Q Km (μM) |
Y343N/R472M Km (μM) |
Y343N/R472M/V474M Km (μM) |
|---|---|---|---|---|---|---|
| 6.0 | 1.29 | 95.3 | 0.46 | 61.4 | 16800 | 1100 |
| 7.0 | 2.72 | 94.7 | 1.34 | 87.7 | 4640 | 1420 |
| 8.0 | 4.35 | 297 | 3.19 | 282.7 | 19280 | 2140 |
| 8.5 | 8.25 | 850 | 3.54 | 712.5 | 42990 | 14000 |
| 9.0 | 22.1 | 2460 | 14.4 | 3340 | 85640 | 55500 |
| 9.5 | 67.1 | 9370 | 31.9 | 39900 | 208000 | 111000 |
| 10.0 | 52.9 | 12013 | 40.2 | 59660 | ND* | 418000 |
Not Determined
Table 6. kcat values of active site mutants.
| pH | Wild type kcat (s−1) |
Y343N kcat (s−1) |
V474M kcat (s−1) |
Y343F/R472Q kcat (s−1) |
Y343N/R472M kcat (s−1) |
Y343N/R472M/V47M kcat (s−1) |
|---|---|---|---|---|---|---|
| 6.0 | 13.2 | 3.17 | 5.96 | 1.44 | 1.35 | 1.90 |
| 7.0 | 24.2 | 12.77 | 11.4 | 1.73 | 1.13 | 3.37 |
| 8.0 | 25.9 | 13.75 | 17.8 | 2.05 | 1.33 | 3.58 |
| 8.5 | 26.9 | 16.91 | 15.8 | 2.25 | 1.42 | 3.90 |
| 9.0 | 25.7 | 15.54 | 19.6 | 2.31 | 1.13 | 5.60 |
| 9.5 | 26.3 | 8.11 | 17.1 | 4.94 | 0.97 | 5.23 |
| 10.0 | 13.0 | 4.38 | 12.4 | 2.96 | ND* | 2.80 |
Not Determined
Table 7. Second order rate constants (kcat/Kmsulfite) of active site mutants.
| pH | Wild-type | Y343N | V474M | Y343F/R472Q | Y343N/R472M | Y343N/R472M/V474M |
|---|---|---|---|---|---|---|
| 6.0 | 1.02 × 107 | 3330 | 1.30 × 106 | 2350 | 8.04 | 173 |
| 7.0 | 8.90 × 106 | 1.35 × 104 | 8.51 × 105 | 1970 | 24.4 | 237 |
| 8.0 | 5.95 × 106 | 4630 | 5.58 × 105 | 725 | 6.90 | 167 |
| 8.5 | 3.26 × 106 | 1990 | 4.46 × 105 | 316 | 3.30 | 27.9 |
| 9.0 | 1.16 × 106 | 632 | 1.36 × 105 | 69.2 | 1.32 | 10.1 |
| 9.5 | 3.92 × 105 | 86.6 | 5.36 × 104 | 12.4 | 0.47 | 4.71 |
| 10.0 | 2.46 × 105 | 36.5 | 3.08 × 104 | 4.96 | ND* | 0.67 |
Not Determined
Table 8. Nitrate-reducing activity of the HSO variants and wild-type NR proteins.
| Kmnitrate (μM) | kcat (s−1) | kcat/Kmnitrate (M−1s−1) | Reference | |
|---|---|---|---|---|
| pH 8.5: | ||||
| wild type SO | 43000 | <0.10 | ND* | This work |
| HSO-2NR | 568 | 0.65 | 1.1×103 | This work |
| HSO-3NR | 192 | 0.67 | 3.5×103 | This work |
| pH 7.0: | ||||
| HSO-2NR | 24 | 0.82 | 3.4×104 | This work |
| HSO-3NR | 30 | 1.07 | 3.5×104 | This work |
| S. oleracea | 12 | 180 | 1.5×107 | Pollock et al. 2002(39) |
| A. thaliana | 90 | 100 | 1.1×106 | Skipper et al. 2001(24) |
| P. pastoris | 30 | 159 | 5.3×106 | Barbier et al. 2004(38) |
Not Determined
The human SO Y343N/R472M/V474M (HSO-3NR) variant
Based on sequence analysis, the importance of an additional Met residue found in the active site of all NR proteins rather than the Val residue found in that position in SO proteins was not obvious. However, based on the recent crystal structure of yeast NR (25), the Met residue found in the active site of NR was proposed to be important in catalysis of nitrate reduction, due to the fact that the side chain of this residue changed conformation depending on the presence or absence of nitrate similarly to Arg 472 which changes position in SO. Therefore, the homologous Val residue in SO was changed to a Met to create a triple variant SO protein (Y343N/R472M/V474M). This triple mutant was expressed and its behavior during purification was identical to that of wild-type HSO (10). Interestingly, while the sulfite oxidation ability of this protein remained very low as expected, it was less impaired than HSO-2NR particularly at higher pH values, with higher kcat and lower Kmsulfite at all pH values (Tables 5–7). When nitrate reductase assays were performed at pH 8.5, the Kmnitrate decreased from 568 to 192μM and at pH 7.0 the kcat increased a little over 2 fold from 0.82 s−1 to 2.13 s−1 (Table 8).
The role of the heme domain in the NR activity of HSO-2NR and HSO-3NR
Previous studies using the native NR Mo domain fragment have demonstrated that the heme domain and the FAD domain are not necessary for activity in the presence of a ready supply of electron donors such as reduced viologens (38). Purification of the Mo domains of both HSO-2NR and 3NR allowed us to answer the question of whether the heme domain is necessary for nitrate reduction in this system (Table 9). The Mo fragments of both HSO_2NR and HSO_3NR retained low Kmnitrate and similar kcat to the full length proteins (Table 8) demonstrating that the heme domain is not needed as long as there is a source of electrons from dithionite and reduced viologens.
Table 9. Nitrate reducing activity of the Mo domains of human wild type SO, 2NR-SO and 3NR-SO.
| Kmnitrate (μM) | kcat (s−1) | |
|---|---|---|
| pH 8.5: | ||
| SO Modom | 2270 | 0.13 |
| Mo HSO-2NR | 872 | 0.47 |
| Mo HSO-3NR | 371 | 0.63 |
| pH 7.0: | ||
| SO Modom | 1410 | 0.06 |
| Mo HSO-2NR | 42 | 0.62 |
| Mo HSO-3NR | 37 | 1.93 |
Absorption spectra of the Mo domains of human NR-SO variants
Generating the Mo domains of these variants in the absence of the heme domain also allowed us to measure absorption spectra and to visualize the Mo center in a way that is not possible in the presence of the strongly absorbing heme chromophore (Figure 4). While the spectrum of the single active site mutants R472M and V474M appeared identical to wild type (data not shown), absorption spectroscopy revealed significant differences in the 480 and 350 nm absorption bands for the Y343N variant (Figure 4.), indicating altered geometry in the enedithiolate-to-Mo charge transfer bond at 360nm and in the Cys 207 to Mo charge transfer as demonstrated by a blue shift in the 480nm band.
Figure 4.

Absorption spectra of SO variants.
—, Wild type; ……, Y343N; -·-, 2NR SO; - - -, 3NR SO.
The chicken SO Y322F/R450M (2NR) variant
In the crystal structure of chicken SO, the active site residue Arg 450 (equivalent to HSO Arg 472) was observed to change conformation dependant on the presence of the sulfate. This suggested that Arg 450 was involved in substrate recruitment. Further examination of the crystal structure of the active site of chicken SO suggested that Tyr 322, due to its proximity to the molybdenum of the Moco, might play a role in electron transport and substrate recruitment. This supposition was supported by studies done on human SO variant Y343F (17, 18). When the Mo domain of assimilatory NR was crystallized, the active site was found to super-impose with the active site of SO at four of six positions (25). Interestingly the two positions that were not identical were the CSO positions Tyr 322 and Arg 450, which are strongly conserved in all SO proteins. The Tyr 322 corresponds to an Asn in NR and the Arg 450 corresponds to a Met in plant NRs or to a Leu/Thr in fungal NR proteins. This observation suggested that it might be possible to modulate the substrate affinity and convert the enzymatic activity of SO from sulfite oxidation to nitrate reduction. Since the HSO variants discussed above (HSO-2NR and HSO-3NR) were not amenable to crystallization, the equivalent double mutant of chicken SO, Y322N/R450M (CSO-2NR was generated, cloned, expressed and behaved identically to wild-type CSO during purification (9), and was characterized for sulfite oxidase and nitrate reductase activity. The variant exhibited a markedly decreased ability to bind sulfite at pH 8.5 with a Kmsulfite of 11730 μM and decreased ability to oxidize sulfite to sulfate with a kcatsulfite of 2.52 s−1 for a second order rate constant kcat/Kmsulfite of 2.14 × 102 compared to wild-type CSO with a Kmsulfite of 8.43 μM and a kcatsulfite of 73.3 s−1 for a kcat/Kmsulfite of 8.69 × 106. At pH 7.0 the 2NR variant exhibited an increase in the ability to bind sulfite with a Kmsulfite of 561 and an improved kcatsulfite of 4.83 s−1 for a second order rate constant of 8.61 × 103 that was still considerably lower than that of wild type SO with a Kmsulfite of 1.28 μM and a kcatsulfite of 35.9 s−1 for a kcat/Kmsulfite of 8.69 × 107 (Table 10).
Table 10. Sulfite oxidizing activity of the CSO variants and wild-type NR proteins.
| Kmsulfite (μM) | kcatsulfite (s−1) | kcat/Kmsulfite (M−1s−1) | Reference | |
|---|---|---|---|---|
| pH 8.5: | ||||
| CSO | 8.43 | 73.3 | 8.7×106 | Karakas et al. 2005 |
| CSO | 8.53 | 71.4 | 8.3×106 | This work |
| CSO-2NR | 11730 | 2.52 | 2.1×102 | This work |
| CSO-3NR | 6750 | 4.1 | 6.1×102 | This work |
| pH 7.0: | ||||
| CSO | 1.28 | 35.9 | 2.8×107 | Karakas et al. 2005 |
| CSO | 1.33 | 36.1 | 2.7×107 | This work |
| CSO-2NR | 561 | 4.83 | 8.6×103 | This work |
| CSO-3NR | 570 | 2.18 | 3.8×103 | This work |
When assayed for nitrate reduction activity at pH 8.5 wild type CSO exhibited negligible ability to reduce or bind nitrate, whereas the CSO-2NR variant exhibited a Kmnitrate of 1134 μM and kcatnitrate of 0.751 s−1 for a kcat/Kmnitrate of 6.62 × 102. When the 2NR variant was assayed at standard conditions at pH 7.0 the 2NR variant exhibited a Kmnitrate of 445 μM and kcatnitrate of 0.65 s−1 for a kcat/Kmnitrate of 1.46 × 103. While it was encouraging to observe modified substrate affinity and catalytic activity from sulfite oxidation to nitrate reduction in CSO, the Michaelis constant values were much higher than that of native NR enzymes (Table 11).
Table 11. Nitrate-reducing activity of the CSO variants and wild-type NR proteins.
| Kmnitrate (μM) | kcat (s−1) | kcat/Kmnitrate (M−1s−1) | Reference | |
|---|---|---|---|---|
| pH 8.5: | ||||
| CSO* | >20000 | <0.15 | ND | This work |
| CSO-2NR | 1134 | 0.751 | 6.6×102 | This work |
| CSO-3NR | 151.1 | 0.377 | 2.4×103 | This work |
| pH 7.0: | ||||
| CSO* | >20000 | <0.10 | ND | This work |
| CSO-2NR | 445 | 0.65 | 1.4×103 | This work |
| CSO-3NR | 72.7 | 0.96 | 1.3×104 | This work |
| S. oleracea | 12 | 180 | 1.5×107 | Pollock et al. 2002(39) |
| A. thaliana | 90 | 100 | 1.1×106 | Skipper et al. 2001(24) |
| P. pastoris | 30 | 159 | 5.3×106 | Barbier et al. 2004(38) |
ND Not determined
The chicken SO Y322F/R450M/R452M (3NR) variant
While the results of the double variants of SO were intriguing, the rate of catalysis was very low and the Michaelis constant of the enzyme was considerably higher than native NR enzymes. Based on the results of the double mutant variant, the active site of NR and SO were reevaluated and an additional Met residue was identified as potentially being involved in catalysis as this residue changed conformation dependant on the presence of sulfate in NR similar to Arg 450 in SO, additionally this Met residue is conserved in native NR enzymes and aligned with a Val residue in SO. This Val residue in SO was mutated to Met to create an SO triple mutant Y322N/R450M/V452M (CSO-3NR) variant. This CSO-3NR variant behaved identically to wild-type CSO during purification (9). The CSO-3NR variant exhibited decreased ability to oxidize sulfite at pH 8.5 with a Kmsulfite of 6750 μM and kcatsulfite of −4.1 s−1 for a kcat/Kmsulfite of 6.07 × 102. At pH 7.0 the variant had a Kmsulfite of 570 μM and kcatsulfite of 2.18 s−1 for a kcat/Kmsulfite of 3.82 × 103 (Table 10). When assayed for nitrate reductase activity at pH 7.0, the CSO-3NR exhibited a Kmnitrate of 72.7 μM, which is close to the Kmnitrate observed for native NR proteins, though the kcatnitrate of 0.96 s−1 for a kcat/Kmsulfite of 1.32 × 104 is relatively low (Table 11).
Crystallographic studies of Chicken 2NR and 3NR variants: structural determination
The structures of CSO-2NR and CSO-3NR variants were solved by molecular replacement (Fig. 7). The first attempt to solve the structure of CSO-2NR used the coordinates from the full-length natively purified CSO (PDB code 1SOX), as a search model, in PHASER. Unfortunately, this approach did not yield a search solution with all components. We then used the wild-type rCSO coordinates (rCSO PDB code 2a99 chain A monomer residues 95-466) as the search model. Despite the fact that the full-length proteins were used for crystallization and the crystals exhibited the characteristic red color of the heme prosthetic group, no electron density was observed for the N-terminal b5-type cytochrome domain in any of the structures we obtained, presumably because of the heme-domain adopting multiple stable orientations within the crystal. These results were consistent with previously published results (20). Both the CSO-2NR and CSO-3NR crystallized in the I41 space group, and all crystals had one monomer in the asymmetric unit as calculated by Matthews coefficient (40). Attempts were made to obtain a substrate bound crystal of both CSO-2NR and CSO-3NR by soaking and cocrystallizing with nitrate at 1 mM, but data sets collected from both methods indicated the absence of substrate in the active site, (Figure 7). This result is consistent with previously published results on the crystallization of the central Moco catalytic fragment of P. angusta NR (25). The structures CSO-2NR and CSO-3NR had good stereochemistry with all residues in the most favored and additionally allowed regions of the Ramachandran diagram as determined by MOLPROBITY (36). After several cycles of least squares minimization and model building the resulting crystallographic values Rcryst and Rfree are reported in Table 12.
Figure 7.

Structures of the active sites of A.) CSO 2NR active site residues shown in magenta (left) and with electron density around active site residues (right) and B.) CSO 3NR active site shown in orange (left) and with electron density around active site residues (right).
Table 12. Crystallographic data and refinement statistics for CSO-2NR and CSO-3NR.
| Data set | CSO 2NR | CSO 3NR |
|---|---|---|
| Resolution (Å) | 50-2.4 | 50-2.1 |
| Wavelength | 1.00 | 1.00 |
| Space group | I41 | I41 |
| Cell constants (Å) | a = 85.6 | a = 85.6 |
| b = 85.6 | b = 85.6 | |
| c= 153.4 | c= 152.8 | |
| Molecules asymmetric unit | 1 | 1 |
| Total observations | 102928 | 209711 |
| Unique reflections | 21499 | 31937 |
| Mean redundancy | 4.8 (4.1) | 6.6 (5.0) |
| Rsyma (%) | 9.9 (34.5) | 10.3 (28.8) |
| Completeness (%) | 99.6 (96.9) | 99.7 (97.2) |
| Mean I/σ | 23.2 (3.2) | 39.7 (4.3) |
| Rcrytb (%) | 16.9 | 17.6 |
| Rfreec (%) | 21.7 | 20.4 |
| Mean B factor | 21.9 | 20.7 |
| Number of atoms used in refinement | 3084 | 3083 |
| Number of waters | 182 | 189 |
| R.M.S. deviations bond lengths (Å) | 0.013 | 0.011 |
| R.M.S. deviations bond angles (°) | 1.407 | 1.27 |
| Ramachandran statistics* | 98.4/1.6 | 99.2/0.8 |
Rsym = Σ |I−<I>|, where I is the observed intensity and <I> is the average intensity of multiple symmetry-related observations of that reflection.
Rcryst = Σ (|Fo | − |Fc |) / Σ |Fo | where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree is the R-factor based on the data with withheld at random from structural refinement.
Ramachandran represent favored and allowed regions, with no outliers.
Comparison of the crystal structure of CSO-2NR and wild-type CSO
The structure of CSO-2NR was refined to 2.4 Å resolution with a Rcryst of 16.7 and an Rfree of 20.7 The topology of the 2NR variant is very similar to that of the natively purified enzyme, consisting of the central Moco domain and the C-terminal dimerization domain as indicated by a RMSD of 0.142 Å for back-bone atoms. As reported previously, the overall topology of the enzyme is a unique mixed αβ fold (11, 20). The structure was solved by molecular replacement without solvent or cofactor atoms in the search model. A large difference density peak was observed in the active site of the enzyme that was determined to be the cofactor. The Moco was then modeled into the structure and found to be in good agreement with the density. The Moco is in virtually identical position to that of the wild type cofactor and bound deep inside the core of the domain held by an extensive network of hydrogen bonds and van der Waals interactions within the Moco binding pocket. The observed bond length of the Mo to the axial oxygen the bond distance was 1.72 Å, and the equatorial oxygen bond distance was ~ 1.98 Å. The bond distance of the equatorial oxygen ligand is between the previously determined EXAFS studies indicate single bond distance of 2.27 for the reduced molybdenum and the double bonded oxygen to molybdenum distance of ~1.7 Å (15, 41, 42). This suggests a partially reduced form of the molybdenum to MoV species. It is possible that the molybdenum of the cofactor is partially reduced either by the cryoprotectant glycerol or by photoreduction while in the synchrotron X-ray beam (11), however at a resolution of 2.4 Å it is difficult to assign the exact oxidation state of the molybdenum based solely on the crystallographic data. Comparison of the active site residues of CSO-2NR to wild type CSO indicates that several active site residues adopt similar conformations to wild type SO. An overlay of the two active sites reveals a RMSD of 0.091 Å between the wild type and the 2NR variant residues, it is apparent that in the 2NR variant the active site residues Arg 138, Cys 185, Arg 190, and Trp 204 all adopt virtually identical conformations to those in wild type SO (Figure 8a), indicating that these residues were not rearranged in the 2NR variant. The Asn 322 mutation’s side chain appears to have moved closer to the Moco that the Tyr 322 found in the wild-type active site. The more polar side chain of the asparagine may have entered deeper into the active site than the predominately hydrophobic tyrosine. The Met 450 side chain found in the 2NR and 3NR variants appears to be in a more extended conformation compared to the wild type CSO Arg 450 side chain.
Figure 8.

Comparison of the active site of CSO 2NR with A.) wild type CSO shown in gray and the 2NR variant shown in blue. Only the Moco of the 2NR is shown in green. B.) The active site of P. angusta NR shown in orange while the active site of the CSO 2NR variant shown in blue. Only the Moco of the 2NR is shown in green.
Comparison of the crystal structure of CSO-2NR and wild-type P. angusta NR
The structure of CSO 2NR is similar to the Mo domain structure of P. angusta wild-type NR (25) with a RMSD of 1.05 Å compared to the backbone atoms of NR (PDB code 2BIH). The overall fold of the CSO-2NR variant is similar to that of the P. angusta wild-type NR. As mentioned above, the bond length of the oxygen ligands of the molybdenum to axial oxygen the bond distance is 1.7 Å, and the equatorial oxygen bond distance equal to ~ 1.9 Å. In the crystal structure of wild-type NR the axial oxygen ligand of the molybdenum was at a distance of ~1.9 Å while the equatorial oxygen ligand was at a distance of ~2.1 Å, which would suggest the molybdenum of NR, is in the reduced form. As NR reduces nitrate to nitrite the enzyme is itself oxidized, so it is reasonable to suggest that molybdenum atom of NR exists in the reduced form prior to catalysis. The distance of the oxygen ligands to the molybdenum of Moco in the 2NR structure suggests that the Mo is in an intermediate redox state. The structure of the CSO-2NR active site is similar to that of wild type NR with a RMSD of 0.623 Å. The CSO 2NR active site residues Arg 138, Arg 190, Trp 204 and Cys 185 adopt similar conformations to that of the equivalent positions in wild type NR. (Figure 8b) The notable exception is the Met 450 of CSO 2NR compared to Met 427 in wild-type NR, the position of Met 450 in 2NR is farther away from the other active site residues compared to the wild type Met, in addition the side chain adopts a different orientation. In the 2NR structure the Met 450 side chain is pointed away from the Mo atom whereas the Met side chain of P. angusta NR is pointed towards the Mo atom of the active site of the enzyme.
Comparison of the crystal structure of CSO-3NR and wild-type CSO
The structure of CSO-3NR was refined to 2.1 Å resolution, with an Rcryst of 16.9 and an Rfree of 19.0. The structure of the 3NR variant of CSO has overall topography similar to wild type SO as indicated by a RMSD of 0.121 Å for all Cα backbone atoms. A positive difference density peak was observed in the active site of the enzyme that was determined to be the cofactor. The Moco was then was modeled into the structure in good agreement with the density. The Moco of CSO-3NR is in essentially the same position as the Moco in the wild type CSO enzyme. In the 3NR variant of CSO the bond length for the axial oxygen ligand of the molybdenum is ~1.70 Å, and the equatorial oxygen bond distance is equal to ~ 2.11 Å. These bond lengths would suggest that the enzyme is closer to the reduced form of the molybdenum than the 2NR variant. This equatorial oxygen distance is closer to the single bond hydroxyl molybdenum distance of 2.27Å. The active site residues of CSO-3NR and wild type CSO have adopted similar conformations as indicated by RMSD 0.064 Å. Inspection of the active site residues reveals that four residues; Arg 138, Arg 190, and Trp 204 and Cys 185 superimposed almost perfectly between the CSO-3NR variant and the wild type active site (Figure 9A.). The Asn mutation at position 322 appears to have adopted a similar position as the Tyr it replaces when comparing the Cβ positions of the two side chains. The Met 450 side chain appears to be directed towards the solvent, as is the Arg 450 of wild type SO in the absence of substrate. The Met 452 mutation appears to adopt a conformation that attempts to fill the volume of the wild-type Val side chain and the sulfur of the Met adopts a position close to the Cγ of the Val closest to the Moco, such that the sulfur is pointing into the active site of the enzyme.
Figure 9.

Comparison of the active sites of CSO 3NR with A.) wild type CSO shown in gray and the 3NR variant shown in blue. Only the Moco of the 3NR is shown in green. B.) The active site of P. angusta NR shown in orange while the active site of the CSO 3NR variant shown in blue. Only the Moco of the 3NR is shown in green.
Comparison of the crystal structure of CSO-3NR and wild-type NR
The structures of the CSO-3NR variant and the Mo domain of wild type P. angusta NR (PDB code 2BIH) share overall similarity as indicated by a RMSD of 1.08 Å when the Cα backbones are aligned. The Moco of the CSO 3NR variant is in virtually identical position as the Moco of the wild-type NR structure. As discussed above, in the CSO-3NR variant, the distance of the axial oxygen ligand to the molybdenum atom is 1.7 Å, and the distance from the equatorial oxygen ligand to the molybdenum is 2.1 Å. In the structure of the wild type NR Mo domain the distance from the axial oxygen to the molybdenum of the Moco is ~1.98 Å, and the distance from the equatorial oxygen to the molybdenum is ~2.08 Å. As stated previously, it is possible that the molybdenum of the cofactor is partially reduced either by the cryoprotectant glycerol or by photoreduction while in the synchrotron X-ray beam (11). The active site residues of 3NR and wild type NR have adopted similar conformations as indicated by a RMSD of 0.658 Å. The Arg 138, Cys 185, Arg 190, and Asn 322 of CSO-3NR superimpose well with the Arg 89, Cys 139, Arg 144, and Asn 272 of NR. The Trp 204 of CSO-3NR is in approximately in the same position as Trp 158 NR (Figure 9b.). The Met 450 does not align well with the Thr side chain found in the analogous position in wild type NR. The Met residue at position 452 adopts an alternate conformation from the wild type NR Met. However, the sulfur atom of the Met 452 is directed into the active site and is in approximately the same position as the sulfur of the wild type NR Met 427. The addition of the non-polar Met at 452 may also account for the Asn side chains adopting a conformation closer to that of wild type NR than does the Asn 322 found in CSO-2NR.
Comparison of CSO-2NR and CSO-3NR structures
The structures of the CSO-2NR and CSO-3NR variants are closely related as indicated by the RMSD of 0.129 Å between all aligned Cα backbone atoms. The active site Moco, and residues Arg 138, Cys 185, Arg 190, and Trp 204 adopt virtually identical conformations. The additional Met substitution at position 452 appears to slightly adjust the conformation of the Asn substitution at position 322 as seen in Figure 9. The slightly larger Met appears to push the Asn side chain closer to the conformation seen in the wild type NR active site whereas in the 2NR the Asn was further from the Moco. The additional Met 452 substitution appears to shift the conformation of the adjacent Met 450 side chain. In the CSO-2NR structure the Met 450 sulfur is pointed away from the active site. In the CSO-3NR variant, the Met 450 is turned around, such that the sulfur is pointed into the active site.
Conclusions
The results described in this work show that while the HSO R472Q and R472M variants have very low overall steady state catalytic rates (Table 2), they are not significantly impaired in the ability to oxidize sulfite, or to transfer electrons from Mo to the heme during the reductive half reaction (Table 3). The mutants are much less impaired in their ability to transfer electrons to the artificial electron donor ferricyanide (Table 4). Taken together, these results provide evidence that these proteins are impaired in some aspect of the overall catalytic mechanism subsequent to the initial IET (see Fig. 1).
In contrast to eukaryotic SO proteins in which the Mo and heme domains are flexibly linked and move relative to each other during the reaction cycle, a bacterial counterpart of mammalian SO, SDH from Starkeya novella, consists of Mo- and heme-domains that are products of two separate genes that purify as a stable heterodimer (43). Unlike mammalian SO, SDH does not undergo any conformational changes or domain movement during the catalytic cycle and the IET rates in bacterial SDH are unaffected by altering the viscosity of the buffer solution (44, 45). In addition, the crystal structure of bacterial SDH demonstrated that the Mo and heme centers are locked in a conformation that maintains the metal centers close enough to each other to be in an IET-competent conformation throughout the catalytic cycle (46). Superposition of the Mo-domains of SDH and chicken SO shows a major difference in the orientation of the heme domains in the two enzymes. In SDH the Mo-Fe distance is 16.6 Å, in contrast to the 32 Å separation observed in chicken SO. Furthermore, bacterial SDH lacks an arginine in the analogous position as Arg 472 in human SO (Arg 450 in chicken SO), containing an alanine (Ala 358) instead (Figure 5). It is likely that SDH follows a different pathway for IET than mammalian SO proteins, based on the lack of domain movement during the reaction cycle.
Figure 5.

Overlay of chicken SO (black) and human SO (black in parentheses) with bacterial SDH (blue) active site residues.
In these studies, the native CSO proteins were mutated away from the naturally evolved SO active site sequence towards the NR active site. All the resulting single and multiple mutants were found to retain the dimeric structure, but to have decreased SO activity and have gained the ability to bind and reduce nitrate to nitrite. Wild type NR proteins can enhance the rate of nitrate reduction kcat/Kmnitrate by 1.5 × 107 (S. oleracea) to 5.3 × 106 (P. pastoris) times over the un-catalyzed reaction. It is remarkable that the nitrate reductase activity of CSO 3NR variant, with a kcat/Kmnitrate of 1.3 × 104, is two orders of magnitude of wild type P. pastoris NR. The difference in kcat/Kmnitrate between the 3NR variant is largely in part due to the lower kcat for nitrate compared to wild type NRs.
This study has also demonstrated that both human and chicken SO can bind and reduce nitrate with a minimum of 2 residues changed in the active site HSO Y343N/R472M, CSO Y322N/R450M, and the heme domain is not necessary for full activity in the presence of a ready supply of electrons from an electron donor such as reduced methyl viologen. Based on both structural data and the current study, Met 427 in nitrate reductase may perform an analogous role to Arg 472/450/374 in human/chicken/plant sulfite oxidases in mediating conformational changes of the active site upon substrate binding. It is also possible that Met 427 alters the redox properties of the Mo center to favor nitrate reduction.
The data presented in this paper should provide the basis for future comparative studies on SO and Assimilatory NR. For instance it would be interesting to fuse the double or triple mutant of SO to the heme and flavin domains of NR to examine whether the hybrid protein exhibits overall NADPH to nitrate activity. It would also be important to carry out detailed comparative EPR studies on NR and the several variants of SO described here, including signal shapes and relative reduction potentials.
Figure 6.

Overlay of yeast NR (black) and chicken SO (blue) active sites in the presence (A) and absence (B) of sulfate.
Acknowledgments
We thank Mr. Graham Alexander and Ms. Ashley Carpenter for technical assistance with cloning and protein purification. This work was supported by National Institute of Health (NIH) grant GM00091 to K.V.R.
The abbreviations used are
- SO
sulfite oxidase
- NR
nitrate reductase
- Moco
Molybdenum cofactor
- MPT
molybdopterin
- EXAFS
Extended X-ray absorption fine structure
- CEPT
coupled electron-proton transfer
- IET
intramolecular electron transfer
- HPLC
high performance liquid chromatography
- cyt c
cytochrome c
- HSO-2NR
Y343N+R472M human SO double variant with NR activity
- CSO-2NR
Y322N+R450M chicken SO double variant with NR activity
- HSO-3NR
Y343N+R472M+V474M human SO triple variant with NR activity
- CSO-3NR
Y322F+R450M+V452M chicken SO triple variant with NR activity
Footnotes
Coordinates: Coordinates and structure factors for CSO-2NR and CSO-3NR have been deposited in the Protein Data Bank (accession codes 3R18 and 3R19 respectively).
References
- 1.Kessler DL, Rajagopalan KV. Hepatic sulfite oxidase. Identification of the molybdenum center as the site of irreversible inactivation by ferricyanide. Biochim Biophys Acta. 1974;370:399–409. doi: 10.1016/0005-2744(74)90101-6. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JL, Hainline BE, Rajagopalan KV. Characterization of the molybdenum cofactor of sulfite oxidase, xanthine oxidase, and nitrate reductase. Identification of a pteridine as a structural component. J Biol Chem. 1980;255:1783–1786. [PubMed] [Google Scholar]
- 3.Garrett RM, Rajagopalan KV. Site-directed mutagenesis of recombinant sulfite oxidase: identification of cysteine 207 as a ligand of molybdenum. J Biol Chem. 1996;271:7387–7391. [PubMed] [Google Scholar]
- 4.Rupar CA, Gillett J, Gordon BA, Ramsay DA, Johnson JL, Garrett RM, Rajagopalan KV, Jung JH, Bacheyie GS, Sellers AR. Isolated sulfite oxidase deficiency. Neuropediatrics. 1996;27:299–304. doi: 10.1055/s-2007-973798. [DOI] [PubMed] [Google Scholar]
- 5.Shih VE, Abroms IF, Johnson JL, Carney M, Mandell R, Robb RM, Cloherty JP, Rajagopalan KV. Sulfite oxidase deficiency. Biochemical and clinical investigations of a hereditary metabolic disorder in sulfur metabolism. N Engl J Med. 1977;297:1022–1028. doi: 10.1056/NEJM197711102971902. [DOI] [PubMed] [Google Scholar]
- 6.Johnson JL, Rajagopalan KV. Human sulfite oxidase deficiency. Characterization of the molecular defect in a multicomponent system. J Clin Invest. 1976;58:551–556. doi: 10.1172/JCI108500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey JL, Cole RD. Studies on the reaction of sulfite with proteins. J Biol Chem. 1959;234:1733–1739. [PubMed] [Google Scholar]
- 8.Brody MS, Hille R. The kinetic behavior of chicken liver sulfite oxidase. Biochemistry. 1999;38:6668–6677. doi: 10.1021/bi9902539. [DOI] [PubMed] [Google Scholar]
- 9.Kessler DL, Rajagopalan KV. Purification and properties of sulfite oxidase from chicken liver. Presence of molybdenum in sulfite oxidase from diverse sources. J Biol Chem. 1972;247:6566–6573. [PubMed] [Google Scholar]
- 10.Johnson JL, Rajagopalan KV. Purification and properties of sulfite oxidase from human liver. J Clin Invest. 1976;58:543–550. doi: 10.1172/JCI108499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kisker C, Schindelin H, Pacheco A, Wehbi WA, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC. Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell. 1997;91:973–983. doi: 10.1016/s0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- 12.Qiu JA, Wilson HL, Pushie MJ, Kisker C, George GN, Rajagopalan KV. The structures of the C185S and C185A mutants of sulfite oxidase reveal rearrangement of the active site. Biochemistry. 2010;49:3989–4000. doi: 10.1021/bi1001954. [DOI] [PubMed] [Google Scholar]
- 13.Feng C, Wilson HL, Hurley JK, Hazzard JT, Tollin G, Rajagopalan KV, Enemark JH. Essential role of conserved arginine 160 in intramolecular electron transfer in human sulfite oxidase. Biochemistry. 2003;42:12235–12242. doi: 10.1021/bi0350194. [DOI] [PubMed] [Google Scholar]
- 14.Doonan CJ, Wilson HL, Rajagopalan KV, Garrett RM, Bennett B, Prince RC, George GN. Modified Active Site Coordination in a Clinical Mutant of Sulfite Oxidase. J Am Chem Soc. 2007 doi: 10.1021/ja071402a. [DOI] [PubMed] [Google Scholar]
- 15.George GN, Kipke CA, Prince RC, Sunde RA, Enemark JH, Cramer SP. Structure of the active site of sulfite oxidase. X-ray absorption spectroscopy of the Mo(IV), Mo(V), and Mo(VI) oxidation states. Biochemistry. 1989;28:5075–5080. doi: 10.1021/bi00438a026. [DOI] [PubMed] [Google Scholar]
- 16.Pacheco A, Hazzard JT, Tollin G, Enemark JH. The pH dependence of intramolecular electron transfer rates in sulfite oxidase at high and low anion concentrations. J Biol Inorg Chem. 1999;4:390–401. doi: 10.1007/s007750050325. [DOI] [PubMed] [Google Scholar]
- 17.Wilson HL, Rajagopalan KV. The role of tyrosine 343 in substrate binding and catalysis by human sulfite oxidase. J Biol Chem. 2004;279:15105–15113. doi: 10.1074/jbc.M314288200. [DOI] [PubMed] [Google Scholar]
- 18.Feng C, Wilson HL, Hurley JK, Hazzard JT, Tollin G, Rajagopalan KV, Enemark JH. Role of conserved tyrosine 343 in intramolecular electron transfer in human sulfite oxidase. J Biol Chem. 2003;278:2913–2920. doi: 10.1074/jbc.M210374200. [DOI] [PubMed] [Google Scholar]
- 19.Schrader N, Fischer K, Theis K, Mendel RR, Schwarz G, Kisker C. The crystal structure of plant sulfite oxidase provides insights into sulfite oxidation in plants and animals. Structure. 2003;11:1251–1263. doi: 10.1016/j.str.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Karakas E, Wilson HL, Graf TN, Xiang S, Jaramillo-Busquets S, Rajagopalan KV, Kisker C. Structural insights into sulfite oxidase deficiency. J Biol Chem. 2005;280:33506–33515. doi: 10.1074/jbc.M505035200. [DOI] [PubMed] [Google Scholar]
- 21.Banks GR, Shelton PA, Kanuga N, Holden DW, Spanos A. The Ustilago maydis nar1 gene encoding nitrate reductase activity: sequence and transcriptional regulation. Gene. 1993;131:69–78. doi: 10.1016/0378-1119(93)90670-x. [DOI] [PubMed] [Google Scholar]
- 22.Johnstone IL, McCabe PC, Greaves P, Gurr SJ, Cole GE, Brow MA, Unkles SE, Clutterbuck AJ, Kinghorn JR, Innis MA. Isolation and characterisation of the crnA-niiA-niaD gene cluster for nitrate assimilation in Aspergillus nidulans. Gene. 1990;90:181–192. doi: 10.1016/0378-1119(90)90178-t. [DOI] [PubMed] [Google Scholar]
- 23.Crawford NM, Smith M, Bellissimo D, Davis RW. Sequence and nitrate regulation of the Arabidopsis thaliana mRNA encoding nitrate reductase, a metalloflavoprotein with three functional domains. Proc Natl Acad Sci U S A. 1988;85:5006–5010. doi: 10.1073/pnas.85.14.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skipper L, Campbell WH, Mertens JA, Lowe DJ. Pre-steady-state kinetic analysis of recombinant Arabidopsis NADH:nitrate reductase: rate-limiting processes in catalysis. J Biol Chem. 2001;276:26995–27002. doi: 10.1074/jbc.M100356200. [DOI] [PubMed] [Google Scholar]
- 25.Fischer K, Barbier GG, Hecht HJ, Mendel RR, Campbell WH, Schwarz G. Structural basis of eukaryotic nitrate reduction: crystal structures of the nitrate reductase active site. The Plant cell. 2005;17:1167–1179. doi: 10.1105/tpc.104.029694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fersht AR, Winter GP. Redesigning enzymes by site-directed mutagenesis. Ciba F Symp. 1985;111:204–218. doi: 10.1002/9780470720929.ch14. [DOI] [PubMed] [Google Scholar]
- 27.Temple CA, Graf TN, Rajagopalan KV. Optimization of expression of human sulfite oxidase and its molybdenum domain. Arch Biochem Biophy. 2000;383:281–287. doi: 10.1006/abbi.2000.2089. [DOI] [PubMed] [Google Scholar]
- 28.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 29.Johnson JL. Molybdenum. Method Enzymol. 1988;158:371–382. doi: 10.1016/0076-6879(88)58069-2. [DOI] [PubMed] [Google Scholar]
- 30.Yu L, Wolin MJ. Hydrogenase measurement with photochemically reduced methyl viologen. J Bacteriol. 1969;98:51–55. doi: 10.1128/jb.98.1.51-55.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borek D, Minor W, Otwinowski Z. Measurement errors and their consequences in protein crystallography. Acta Crystallogr D Biol Crystallogr. 2003;59:2031–2038. doi: 10.1107/s0907444903020924. [DOI] [PubMed] [Google Scholar]
- 32.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Word JM, Bateman RC, Jr, Presley BK, Lovell SC, Richardson DC. Exploring steric constraints on protein mutations using MAGE/PROBE. Protein Sci. 2000;9:2251–2259. doi: 10.1110/ps.9.11.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 38.Barbier GG, Joshi RC, Campbell ER, Campbell WH. Purification and biochemical characterization of simplified eukaryotic nitrate reductase expressed in Pichia pastoris. Protein Expr Purif. 2004;37:61–71. doi: 10.1016/j.pep.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 39.Pollock VV, Conover RC, Johnson MK, Barber MJ. Bacterial expression of the molybdenum domain of assimilatory nitrate reductase: production of both the functional molybdenum-containing domain and the nonfunctional tungsten analog. Arch Biochem Biophys. 2002;403:237–248. doi: 10.1016/s0003-9861(02)00215-1. [DOI] [PubMed] [Google Scholar]
- 40.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 41.Cramer SP, Wahl R, Rajagopalan KV. Molybdenum sites of sulfite oxidase and xanthine dehydrogenase. A comparison by EXAFS. J Am Chem Soc. 1981;103:7721–7727. [Google Scholar]
- 42.Harris HH, George GN, Rajagopalan KV. High-resolution EXAFS of the active site of human sulfite oxidase: comparison with density functional theory and X-ray crystallographic results. Inorg Chem. 2006;45:493–495. doi: 10.1021/ic0512274. [DOI] [PubMed] [Google Scholar]
- 43.Kappler U, Friedrich CG, Truper HG, Dahl C. Evidence for two pathways of thiosulfate oxidation in Starkeya novella (formerly Thiobacillus novellus) Arch Microbiol. 2001;175:102–111. doi: 10.1007/s002030000241. [DOI] [PubMed] [Google Scholar]
- 44.Feng C, Kedia RV, Hazzard JT, Hurley JK, Tollin G, Enemark JH. Effect of solution viscosity on intramolecular electron transfer in sulfite oxidase. Biochemistry. 2002;41:5816–5821. doi: 10.1021/bi016059f. [DOI] [PubMed] [Google Scholar]
- 45.Feng C, Kappler U, Tollin G, Enemark JH. Intramolecular electron transfer in a bacterial sulfite dehydrogenase. J Am Chem Soc. 2003;125:14696–14697. doi: 10.1021/ja038197t. [DOI] [PubMed] [Google Scholar]
- 46.Kappler U, Bailey S. Molecular basis of intramolecular electron transfer in sulfite-oxidizing enzymes is revealed by high resolution structure of a heterodimeric complex of the catalytic molybdopterin subunit and a c-type cytochrome subunit. J Biol Chem. 2005;280:24999–25007. doi: 10.1074/jbc.M503237200. [DOI] [PubMed] [Google Scholar]