

Abstract
Context
The CACNA1C gene (alpha 1C subunit of the L-type voltage-gated calcium channel) has been identified as a risk gene for both bipolar disorder and schizophrenia but the mechanism of association has not been explored.
Objective
To identify the neural system mechanism that explains the genetic association between the CACNA1C gene and psychiatric illness, using neuroimaging and human brain expression.
Design
We used BOLD fMRI to measure brain activation in circuitries related to bipolar disorder and schizophrenia by comparing CACNA1C genotype groups in healthy subjects. We tested the effect of genotype on mRNA levels of CACNA1C in post-mortem human brain. A case-control analysis was used to determine the association of CACNA1C genotype and schizophrenia.
Setting
National Institutes of Health Clinical Center
Patients
Healthy Caucasian men and women participated in the fMRI study. Post-mortem samples from normal human brains were used for the brain expression study. Patients with schizophrenia and healthy subjects were used in the case-control analysis.
Main Outcome Measures
BOLD fMRI, mRNA levels in post-mortem brain samples, and genetic association with schizophrenia
Results
The risk associated single nucleotide polymorphism (SNP rs1006737) in CACNA1C predicted increased hippocampal activity during emotional processing (puncorr=0.001, pFDR=0.052, Z=3.20) and increased prefrontal activity during executive cognition (puncorr=2.8e-05, pFDR=0.011, Z=4.03). The risk SNP also predicted increased expression of CACNA1C mRNA in human brain (p=0.0017). CACNA1C was associated with schizophrenia in our case-control sample (OR 1.77, p=0.026).
Conclusions
The risk associated SNP in CACNA1C maps to circuitries implicated in genetic risk for both bipolar disorder and schizophrenia. Its effects in human brain expression implicate a molecular and neural systems mechanism for the clinical genetic association.
Introduction
Several research groups have performed independent genome-wide association studies of bipolar disorder, with little agreement among the most associated loci.1–3 However, a comparison of the Wellcome Trust Case Control Consortium (WTCCC) and STEP-UCL studies identified CACNA1C (alpha 1C subunit of the L-type voltage-gated calcium channel) as showing the strongest consistent signal.3 The best single SNP in this region (rs1006737) across the two studies also showed association in a separate dataset, the so-called ED-DUB-STEP2, as well as a combined analysis of all three datasets (rs1006737, p = 7.0 × 10−8),4 thus providing further evidence that CACNA1C is a credible susceptibility locus for bipolar disorder.
Because statistical association with clinical diagnosis does not establish biological significance nor identify a mechanism of risk, it is important to extend the statistical evidence with biological data. One approach that has become increasingly informative in translating clinical associations with psychiatric disorders into potential neural mechanisms of risk has been the use of neuroimaging to map gene effects in brain.5–7 We thus used functional MRI to test the effects of risk associated variation in this gene on specific patterns of brain activity that have been associated with mental illness and with increased genetic risk for mental illness. Patients with bipolar disorder have previously been shown to exhibit elevated amygdala8 and hippocampal activity9 in response to emotional stimuli. There also is evidence that individuals at increased genetic risk for bipolar disorder show similar patterns of brain activity, suggesting that this may reflect a neural mechanism of genetic risk.10, 11 As a risk gene for bipolar disorder, we thus hypothesized that healthy subjects who are carriers for the risk associated allele (A; minor allele) of CACNA1C would have increased amygdala and hippocampal activity in response to emotional stimuli compared to the common allele (G). Because this gene has also recently been associated with risk for schizophrenia, though with less statistical power,12 we further hypothesized that individuals with this risk associated allele would also show inefficient prefrontal activity during a working memory task, which has been identified as a potential biologic intermediate phenotype related to genetic risk for schizophrenia.13
Methods
Imaging subjects
Healthy adults participated in a functional MRI study in the Clinical Brain Disorders Branch (CBDB) Sibling Study at the National Institute of Mental Health (NIMH), NIH.14 The study was approved by the NIMH Intramural Program Institutional Review Board. All participants were assessed using the Structured Clinical Interview for DSM-IV. All subjects were physically and psychiatrically healthy; specific exclusion criteria have been previously reported.6 Only Caucasians of self-identified European descent were included in this data set in order to minimize population stratification artifacts. Subject demographics for the imaging study are listed in Table 1.
Table 1.
Subject demographics for imaging studies.
| 1a. Emotional Memory | ||||
|---|---|---|---|---|
| GG (n=57) | GA (n=43) | AA (n=16) | All genotypes (n=116) | |
| Age | 29.37 (8.938) | 29.47(9.570) | 27.88 (8.016) | 29.20 (9.001) |
| Sex | 30 F (53%) | 22 F (51%) | 8 F (50%) | 60 (52%) |
| IQ | 107.8 (7.851) | 107.7 (9.561) | 107.2 (7.774) | 107.6 (8.429) |
| 1b. Emotional Faces | ||||
|---|---|---|---|---|
| GG (n=64) | GA (n=53) | AA (n=14) | All genotypes (n=131) | |
| Age | 29.20 (8.778) | 28.98 (8.835) | 27.07 (7.976) | 28.89 (8.679) |
| Sex | 34 F (54%) | 30 F (57%) | 7 F (50%) | 71 F (54%) |
| sIQ | 107.9 (8.421) | 108.5 (7.630) | 106.3 (9.401) | 107.9 (8.181) |
| 1c. Working Memory (N-back) | ||||
|---|---|---|---|---|
| GG (n=146) | GA (n=141) | AA (n=29) | All genotypes (n=316) | |
| Age | 30.92 (9.260) | 31.48 (9.548) | 30.07 (9.787) | 31.09 (9.417) |
| Sex | 80 F (55%) | 74 F (52%) | 16 F (55%) | 170 F (54%) |
| IQ | 108.4 (8.875) | 109.5 (9.482) | 107.6 (9.248) | 108.8 (9.178) |
fMRI tasks
Emotional memory task
The emotional memory task involved the encoding and retrieval of aversive scenes,15 which has been shown to reliably engage the hippocampus in healthy volunteers.16–18 The scenes, selected from the International Affective Picture System,19 were presented in a block designed with two blocks of aversive/neutral scenes alternating with blocks of resting state, for both encoding and retrieval blocks. During experimental blocks, six scenes of similar valence (neutral or aversive) were presented serially to subjects for 3 seconds each. During resting blocks, participants were asked to attend to a fixation cross presented in the center of the screen for 18 seconds. These fixations blocks were treated as a baseline in the fMRI analyses. During the encoding blocks, subjects were instructed to choose whether the scene presented depicted an “indoor” or “outdoor” scene. During the retrieval blocks, subjects were instructed to select the scenes seen during the encoding session (i.e. “old”) or the scenes not seen during the encoding session (i.e. “new”). In each retrieval block, half of the scenes were old (i.e. presented during the encoding session). Each session (encoding or retrieval) consisted of 17 blocks (four aversive, four neutral, and nine rest conditions). Subjects completed the entire encoding session before beginning the retrieval session after a brief delay (about 2 min). For the encoding session, the presentation of “indoor” and “outdoor” scenes, and for the retrieval session, the presentation of “old” and “new” scenes, was counterbalanced within each block. In addition, the presentation order of aversive and neutral blocks was counterbalanced across subjects. The total scan time was 5 min 40 sec for this task. For this study, only the aversive encoding and aversive retrieval tasks were analyzed. There were no significant differences between genotype groups during the aversive retrieval task. BOLD fMRI was performed on a General Electric 3-Tesla Signa scanner (Milwaukee, WI) using a gradient-echo, echo-planar imaging sequence for all fMRI tasks. Specific parameters for the emotional memory task are the following: axial slices=24, slice thickness=4 mm, gap=1 mm, TR=2000 msec, TE=28 msec, FOV=24 cm, matrix =64*64.
Emotional faces task
The face matching task is a simple perceptual task, which has previously been shown to robustly engage the amygdala.5, 20, 21 The block fMRI paradigm consists of two experimental conditions: an emotional face matching condition and a sensorimotor control task. The face matching task consisted of five 30-second duration blocks. Blocks 1, 3, and 5 were sensorimotor blocks, and blocks 2 and 4 were emotion blocks. Each sensorimotor and emotion block consisted of six 5-second duration trials. Each trial involved the presentation of two images in the lower panel and one image in the upper panel. In the six trials of each sensorimotor block, the two lower images were shapes, and the upper panel image was identical to one of the shapes in the lower panel. Subjects responded using button presses (left or right) to indicate which image in the lower panel matches the upper panel image. In the six trials of each emotion block, the lower panel consisted of two faces, one angry and one afraid, derived from a standard set of pictures of facial affect.22 The upper panel consists of one of the two faces shown in the lower panel. Subjects respond using button presses (left or right) to indicate which lower panel face matches the face in the upper panel. BOLD fMRI parameters for the emotional faces task are the following: axial slices=24, thickness=4 mm, gap=1 mm, TR=2000 msec, TE=28 msec, FOV=24 cm, matrix=64*64.
N-back
Participants also performed a N-back working memory task administered using a block design, with the 2-back working memory condition alternating with a no-back control condition as previously described.13 During the 0-back control task block, the subject simply responded with the current digit presented (1–4 in a diamond shaped box). This alternated with the 2-back block in which the subject serially responded with numbers presented 2 previous (“n” = 2). BOLD fMRI parameters for the N-back task are the following: axial slices=24, thickness=6 mm, TR=2000 msec, TE=28 msec, FOV=24 cm, matrix=64*64.
Image analysis
Images were processed as described previously18 using SPM5 (http://www.fil.ion.ucl.ac.uk/spm). Briefly, images were realigned to the first image of the scan run, spatially normalized into a standard stereotactic space (MNI template) using an affine and nonlinear (4×5×4 basis-functions) transformation, smoothed with a FWHM Gaussian filter (8-mm FWHM for emotion tasks and 10-mm for N-back) and ratio normalized to the whole-brain global mean. In the first level analyses, linear contrasts were computed producing t-statistical parameter maps at each voxel for emotional tasks. Similarly, t-statistical parameter maps were produced for the 2-back WM condition using the 0-back condition as a baseline. These statistical images were entered in a second-level model to identify significant activations within and between genotype groups, thresholded at p<0.01 uncorrected for the ROI using SPM5. Initially, each of the imaging studies was evaluated using an additive genetic model, with three levels of genotype. This was not significant for any of the studies. Therefore, a secondary model, a recessive risk allele model, was tested comparing homozygotes for the risk allele against other genotypes using a two-sample t-test (AA vs. GA + GG). Results are presented based on this analysis. For both emotional tasks, the ROI was defined as the amygdala, hippocampus, and parahippocampus using the WFU Pickatlas.23 For the N-back task, the ROI was defined as BA 9, 10, and 46 using the WFU Pickatlas.23 Of note, there were no behavioral differences between the three genotype groups in the accuracy and reaction times for any of the tasks.
Genetic Association Cohort
The cohort used in the genetic association study was the CBDB/NIMH Sibling Study sample, which consists of subjects collected as part of an ongoing investigation into neurobiological traints related to genetic risk for schizophrenia.14 Participants were between the ages of 18 and 60. Only Caucasians of self-identified European descent were analyzed to reduce genetic heterogeneity. Collection details, screening, diagnostic procedures, and exclusion criteria has been previously described.14 Sample demographics for genetic association study are listed in Table 2.
Table 2.
Subject demographics for genetic cohort study.
| Cases (n=282) | Controls (n=440) | |
|---|---|---|
| Sex | 66 F (23%) | 236 F (54%) |
| Age | 36.44 y (10.54) | 33.09 y (10.09) |
| Age of onset | 21.65 y (5.457) | N/A |
Genotyping
The CACNA1C single nucleotide polymorphism rs1006737 was determined in the clinical samples by standard allelic discrimination Taqman assay that uses the 5′ nuclease activity of Taq DNA polymerase to detect a fluorescent reporter signal generated after PCR amplification. The assay cocktail (Assays on Demand) for rs1006737 was obtained from Applied Biosystems (Foster City, CA). Genotype reproducibility was routinely assessed by regenotyping all samples for the selected SNP and was generally >99%. Genotyping completion rate was >95%. Genotypes in all groups were in Hardy Weinberg equilibrium determined by an exact test (all p>0.1).
Human brain expression study
Human brain tissue was collected as part of the CBDB/NIMH Brain Collection. Details about the collection, screening, and dissection processes for human brain tissues have been described.24 Expression of mRNA was measured in human dorsolateral prefrontal cortex from prenatal samples (gestational week 14 through 20, and from day of birth through old age) using Illumina custom microarrays. Sample demographics for human brain tissue are listed in Table 3. Microarray chips were generated in the NHGRI Microarray Core Facility from 44,544 70mer probes obtained from the Illumina Oligoset HEEBO (http://www.microarray.org/sfgf/heebo.do). RNA samples from the CBDB brain series across the lifespan collection (500 ng) were amplified and labeled using fluorescent dye (Cy5). Samples were each hybridized to microarrays simultaneously with a reference standard (labeled with Cy3) consisting of a pool of RNA from many brain tissue samples. The data were normalized using the loess method from Limma R package25 and data outside 3 standard deviations were treated as outliers. Human fetal tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland. Genotyping of these samples was performed using Illumina 1M Infinium SNP chips. rs1006737 is not found on this SNP platform, so a proxy SNP was selected for genetic analysis based on LD in the HAPMAP EU sample. Subjects were genotyped for CACNA1C, rs2159100, which is in allelic identity with rs1006737 (R2=1.0).26 Because the postmortem dataset contained a larger percentage of minor allele carriers than the clinical samples, only an additive genetic model was tested using a linear regression to determine the effect of genotype on CACNA1C expression based on three genotype groups.
Table 3.
Sample demographics for human brain expression study.
| Total (n=261) | GG (n=108) | GA (n=116) | AA (n=37) |
|---|---|---|---|
| Age: mean (sd) | 27.35y (22.06) | 26.11y (23.39) | 31.59 y (19.70) |
| Sex | 36F (33%) | 42F (36%) | 13 F (35%) |
| Race | 46 AA (43%) 62CAUC (57%) |
72 AA (62%) 44CAUC (38%) |
30 AA (81%) 7 CAUC (19%) |
African American (AA), Caucasian (CAUC)
Results
For each imaging task, subjects in each genotype group were matched for age, sex, IQ, and task performance, thus isolating the effect of genotype on brain information processing not confounded by general brain function parameters or by task performance. The first two tasks focused on the engagement of medial temporal lobe structures implicated in emotion processing and associated with mood disorders and increased genetic risk for mood disorders.
Emotional imaging tasks
The first task involves the encoding and retrieval of aversive stimuli. For this emotional memory task, scans from 116 subjects (57 GG genotype, 43 GA, and 16 AA) were used in an ROI analysis of the amygdala and hippocampus. Initial testing of an additive genetic model yielded non-significant results. Testing of the recessive genetic model showed an effect of genotype on the engagement of the hippocampus. Homozygotes for the risk associated allele (AA) had greater bilateral hippocampal activity during the encoding of aversive images compared to carriers of the common allele (right hippocampus: puncorr=0.001, pFDR=0.052, Z=3.20; left hippocampus: puncorr=0.003, pFDR=0.052, Z=2.77), figure 1. While the uncorrected P values are still significant after correcting for the two different genetic models tested, the FDR corrected results remain significant only at the trend level.
Figure 1.
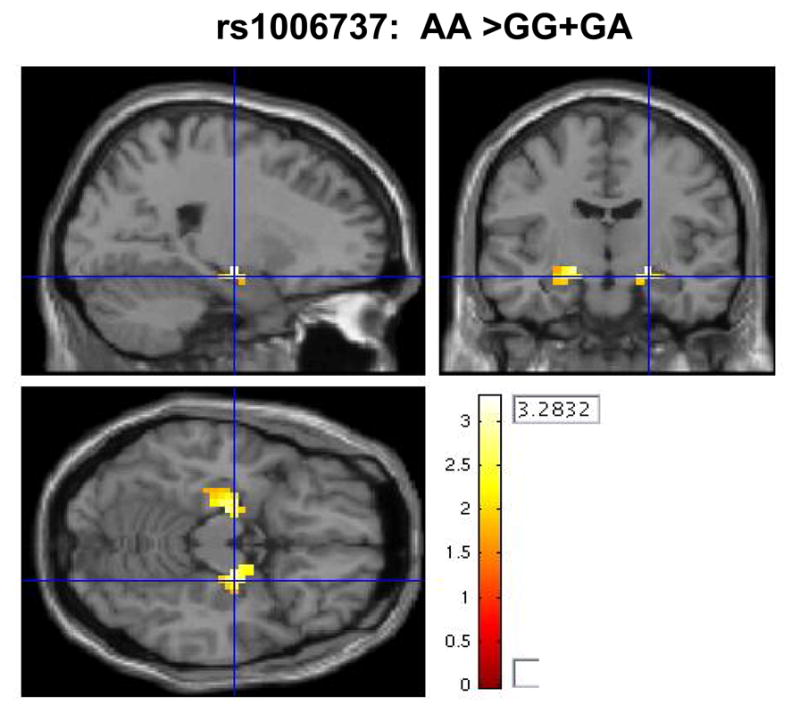
Risk-associated allele homozygotes (AA) have a trend for greater hippocampal activity during encoding of aversive images (19 voxels [peak voxel 22, -11, -11] for the right hippocampus; 28 voxels [peak voxel -19, -15, -15] for the left hippocampus).
The second task involved matching of emotional faces (i.e. angry or fearful), which has been previously shown to robustly activate the amygdala and the hippocampus.5 For this emotional faces task, scans from 131 subjects (64 GG genotype, 53 GA, and 14 AA) were used in a region-of-interest (ROI) analysis of the amygdala and hippocampus. There were no regions in the amygdala or hippocampus that survived a threshold of p<0.01 uncorrected, however when thresholded at p<0.05 risk allele homozygotes (AA) had slightly greater right amygdala activity compared to carriers of the common allele (puncorr=0.015, pFDR=0.213, Z=2.17). Although this activation was not significant by correction for multiple voxels within the region, it is similar to the pattern of hippocampal activation during encoding of aversive scenes.
Working memory imaging task
We also examined the effect of CACNA1C genotype on a task not related to limbic processing of emotion and not previously associated with mood disorders, but linked with schizophrenia and prefrontal cognitive processing. If this gene is associated more strongly with mood disorders than with schizophrenia because of a primary effect on emotional circuitry, then a task targeting emotionally neutral cognitive processing related to prefrontal cortex should show a less robust effect. We chose the N-back, a working memory task that robustly engages prefrontal cortical circuitry and has been especially useful in characterizing neural mechanisms of genes associated with schizophrenia.6, 27 Scans from 316 subjects (146 GG, 141 GA, and 29 AA), comparing 2-back to 0-back, were analyzed using a ROI for BA 9, 10, and 46. Again, analysis under an additive genetic model was nonsignificant. Testing of the recessive genetic model, however, showed a significant effect of genotype on the efficiency of the prefrontal cortex. Homozygotes for the risk allele (AA) had greater activity in the prefrontal cortex (1st cluster: puncorr=2.8e-05, pFDR=0.011, Z=4.03; 2nd cluster: puncorr=5.67e-05, pFDR=0.011, Z=3.86), figure 2, a pattern of inefficient engagement previously found in association with several putative schizophrenia susceptibility genotypes.6, 27 Even using a whole brain analysis, these two clusters survived the threshold of p<0.001 and 10 contiguous voxels, which suggests that the effect of the CACNA1C gene on this working memory task is specific to the prefrontal cortex. These results also remain significant after correcting for the two different genetic models tested. While the significance level appears greater in the prefrontal cortex during this working memory task compared to the hippocampal response in the emotional memory task, this is likely due to a difference in sample size as the effect sizes are quite similar (0.76 for emotional memory and 0.78 for working memory).
Figure 2.
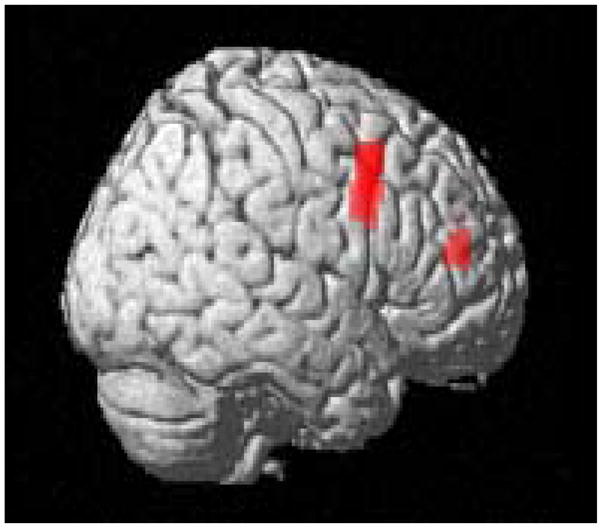
Risk allele homozygotes (AA) have prefrontal cortical inefficiency (greater activity) during the N-back working memory task (p<0.001 whole brain analysis; 88 voxels [peak voxel 54, 12, 39] for the first cluster; 20 voxels [peak voxel 33, 45, 15] for the second cluster. Red areas are significant voxels.
Clinical genetic association with schizophrenia
Our association with an intermediate brain phenotype linked with genetic risk for schizophrenia10 would suggest that CACNA1C might also show association with the clinical diagnosis of schizophrenia and indeed this has recently been reported, including a study with the same SNP as reported here (rs1006737; p=0.034).12 GWA results of the International Schizophrenia Consortium, also showed association with CACNA1C, though not to the same SNP (rs2238090, p = 7.7×10−6).28 We examined CACNA1C rs1006737 in a case-control analysis (282 cases and 440 controls) and found nominal association (p=0.026) with schizophrenia (odds ratio for risk allele homozygotes of 1.77 (CI: 1.07–2.91).
Association with expression of CACNA1C mRNA in human brain
CACNA1C is highly expressed in heart and in brain. In order to understand the molecular mechanism underlying the clinical association and the functional differences in brain circuitry, we tested the effect of genetic variation in CACNA1C on mRNA expression in a large cohort of human post-mortem brain samples. The proxy SNP (rs2159100) for rs1006737 (R2=1.0) showed a significant effect on CACNA1C expression; carriers of the risk genotype (AA) had highest expression, with heterozygotes (GA) having intermediate expression, and the common allele carriers (GG) having the lowest expression (linear regression, p=0.0017; figure 3). Because of the relatively small size of the postmortem sample, we also generated an empirical P-value by permutation analysis which involved randomizing the genotypes and level of expression and 100,000 repetitions of the regression analysis, yielding an empirical p-value of 0.00164. Expression of the probe did not significantly change across age, and genotype groups did not differ in mean age, therefore age was not used as a covariate. There was a difference in expression between Caucasians and African Americans, which is likely due to a difference in minor allele frequency (26% vs. 45% respectively). However there was not a race by genotype interaction, implicating genotype as an independent predictor of expression. An analysis using race as a covariate still showed a significant effect of genotype on expression (F= 3.275, p=0.039).
Figure 3.
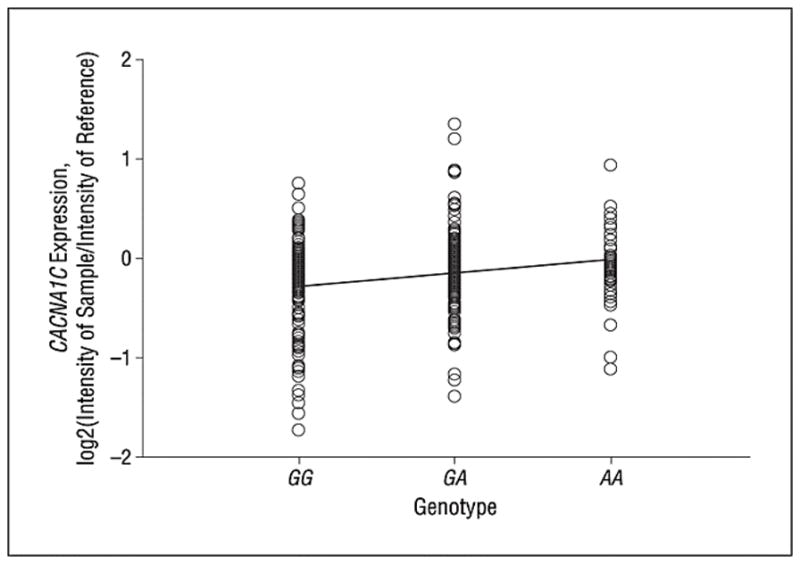
Risk-associated allele homozygotes (AA) have greater expression of CACNA1C than heterozygotes and common allele homozygotes (probe 28032) (rs2159100, P = .002 for linear regression analysis).
Comment
We have found that genetic variation previously implicated as a risk factor for bipolar disorder and for schizophrenia shows effects on brain functions related to medial temporal emotional processing and prefrontal cortical working memory processing that have been associated with risk for bipolar disorder and schizophrenia, respectively. Our results suggest that the pleotropic effects of the risk associated genotype on these diverse brain circuits parallels the diagnostic nonspecificity of the clinical associations and may reflect the underlying neural system mechanisms involved. Genetic variation in voltage-gated calcium channel genes has also been associated with several other complex multigenic neuropsychiatric disorders, including autism,29 epilepsy and migraine,30 as well as schizophrenia.28, 31 In addition, a missense mutation in CACNA1C results in Timothy syndrome, which is characterized by multiorgan dysfunction including cardiac arrhythmias and cognitive abnormalities.32
Data in this study suggest that calcium channel dysfunction may contribute in part to the genetic etiology of both bipolar disorder and schizophrenia through alterations in the functional activity of brain circuitries implicated in both conditions. This is analogous to other genes, e.g. COMT,6 GRM3,27 BDNF,18 and DISC133 that have been associated with both diagnoses and both patterns of neural circuitry effects. Studies of the N-back working memory task have shown that patients with schizophrenia and their healthy siblings have relatively increased prefrontal cortical activity for a given level of performance,13 suggesting that inefficiency in this circuitry is heritable and a good intermediate phenotype related to genetic risk for schizophrenia. Analogous studies have been done in patients with bipolar disorder using neuroimaging tasks that target mood circuitry in the temporal lobe.8, 9 Other studies have targeted serotonin signaling genes, and have found that healthy subjects who are carriers of the short allele of the serotonin transporter, the target of drugs that treat the mood symptoms of bipolar disorder, who have higher ratings of anxiety and depression34 have greater activation of the limbic circuitry,5, 20 similar to these data. While we found stronger statistical evidence of association with prefrontal processing, this was likely an artifact of the reduced power of the smaller sample in the emotional processing tasks, as the effect sizes were quite similar.
The molecular mechanism of genetic risk appears to relate at least in part to regulation of gene expression. Carriers of the risk allele had increased levels of CACNA1C mRNA. It may be that a specific transcript of CACNA1C that is measured by the oligonucelotide probe is involved in the function of the brain regions measured by functional MRI in this study. Calcium channels are involved in various aspects of neuronal development and in the establishment of maintenance of connectivity during development and throughout adulthood;35 therefore alterations in gene expression may also impact on brain structure, which could also affect brain function. A recent study reported that variation in CACNA1C results in alterations in cerebral gray matter. Total gray matter was highest in AA carriers of the risk SNP (rs1006737) compared to GA and GA carriers.36 It is not clear, however, whether and how this finding relates to the genetic association with psychiatric illness.
Our data add to the growing literature that genes weakly associated with psychiatric diseases show stronger effects at the level of brain processing of emotional and cognitive information. This has been shown for several other genes that have been found to be positively but weakly associated with clinical diagnosis in large population studies only to show strong effects in imaging in much smaller samples. For example, a recent study of the gene ZNF804A, which showed significant genome wide significant association with schizophrenia (p<10−8) in a sample of over 23,000, also showed strong association (P=0.006) to an imaging phenotype in a much smaller sample (N=115) of normal subjects.37 We can explore this question of relative statistical power directly in our own data compared to the results of the large scale clinical study of CACN1AC that showed genome wide significant association (p<10-8) in a sample of 10,596 total subjects. Using the imaging data from our N-back study, which has a p-value of 2.8e-05 for a sample size of 316 subjects, if we increase the sample size to 10,000 to approximate the sample size in the previous GWA studies and hold the effect size constant, our p-value drops to 4.87e-109. By the same token, using our post-mortem expression data, which has a p-value of 0.0017 for a sample size of 261, if the sample size was increased to 10,000, the p-value would be 1.24e-70. We would suggest that it should come as no surprise that in principle our associations with quantitative biological measures related to clinical illness and to genetic risk for illness show greater effect sizes than association with clinical diagnosis for two reasons: 1) a quantitative trait phenotype has greater statistical power in general compared with a categorical variable, and 2) the measures we have studied in brain are likely closer to the neurobiology of the gene and its impact on illness, than is the clinical diagnosis. Analogous findings have been reported in other areas of complex medical genetics; for example, genes that show relatively weak association with complex syndromes such as hypertension or cardiovascular disease show much stronger statistical association with quantitative biological traits related to the biology of these syndromes, e.g. sodium homeostasis38 and lipid metabolism,39 respectively.
It is also worth noting that our associations of CACN1AC with brain-related quantitative phenotypes have potential clinical implications. Our demonstration of increased expression of the CACN1AC transcript suggests that if this translates into increase calcium channel activity, calcium channel inhibitors may have clinical value in treating these disorders. Indeed, there have been anecdotal reports and small clinical trials in the past suggesting benefit of these agents for some patients,40, 41 but the data have been inconsistent and limited. It may be worth considering genotype or brain imaging based phenotypes as individual predictors of response to these agents in future trials. While further studies are necessary to fully characterize the mechanism by which alterations in CACNA1C expression results in changes in brain function, this study identifies a potential mechanism of risk for bipolar disorder and/or schizophrenia.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health.
Footnotes
None of the authors have any competing interests.
References
- 1.Baum AE, Akula N, Cabanero M, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2007;13(2):197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sklar P, Smoller JW, Fan J, et al. Whole-genome association study of bipolar disorder. Mol Psychiatry. 2008;13(6):558–569. doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira MAR, O’Donovan MC, Meng YA, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40(9):1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hariri AR, Mattay VS, Tessitore A, et al. Serotonin transporter genetic variation and the response of the human amygdala.[see comment] Science. 2002 Jul 19;297(5580):400–403. doi: 10.1126/science.1071829. [DOI] [PubMed] [Google Scholar]
- 6.Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98(12):6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006;7(10):818–827. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]
- 8.Almeida JRC, Versace A, Hassel S, Kupfer DJ, Phillips ML. Elevated Amygdala Activity to Sad Facial Expressions: A State Marker of Bipolar but Not Unipolar Depression. Biological Psychiatry. doi: 10.1016/j.biopsych.2009.09.027. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whalley HC, McKirdy J, Romaniuk L, et al. Functional imaging of emotional memory in bipolar disorder and schizophrenia. Bipolar Disorders. 2009;11(8):840–856. doi: 10.1111/j.1399-5618.2009.00768.x. [DOI] [PubMed] [Google Scholar]
- 10.Kruger S, Alda M, Young LT, Goldapple K, Parikh S, Mayberg HS. Risk and Resilience Markers in Bipolar Disorder: Brain Responses to Emotional Challenge in Bipolar Patients and Their Healthy Siblings. Am J Psychiatry. 2006;163(2):257–264. doi: 10.1176/appi.ajp.163.2.257. [DOI] [PubMed] [Google Scholar]
- 11.Haldane M, Kempton M, Frangou S. Ventral prefrontal function mediates resileince to bipolar disorder: An fMRI study of BD patients and their unaffected siblings. European Psychiatry. 2009;24(Supplement 1):S916. [Google Scholar]
- 12.Green EK, Grozeva D, Jones I, et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry. 2009 doi: 10.1038/mp.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callicott JH, Egan MF, Mattay VS, et al. Abnormal fMRI response of the dorsolateral prefrontal cortex in cognitively intact siblings of patients with schizophrenia. American Journal of Psychiatry. 2003;160(4):709–719. doi: 10.1176/appi.ajp.160.4.709. [DOI] [PubMed] [Google Scholar]
- 14.Egan MF, Goldberg TE, Gscheidle T, Weirich M, Bigelow LB, Weinberger DR. Relative risk of attention deficits in siblings of patients with schizophrenia. Am J Psychiatry. 2000;157(8):1309–1316. doi: 10.1176/appi.ajp.157.8.1309. [DOI] [PubMed] [Google Scholar]
- 15.Murty VP, Sambataro F, Das S, et al. Age-related alterations in simple declarative memory and the effect of negative stimulus valence. J Cogn Neurosci. doi: 10.1162/jocn.2009.21130. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertolino A, Rubino V, Sambataro F, et al. Prefrontal-hippocampal coupling during memory processing is modulated by COMT val158met genotype. Biological Psychiatry. 2006;60(11):1250–1258. doi: 10.1016/j.biopsych.2006.03.078. [DOI] [PubMed] [Google Scholar]
- 17.Meyer-Lindenberg A, Buckholtz JW, Kolachana B, et al. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc Natl Acad Sci U S A. 2006 April 18;103(16):6269–6274. doi: 10.1073/pnas.0511311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hariri AR, Goldberg TE, Mattay VS, et al. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. The Journal of Neuroscience. 2003;23(17):6690–6694. doi: 10.1523/JNEUROSCI.23-17-06690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang PJ, Bradley MM, Cuthbert BN. Technical Report A-6. Gainesville, FL: University of Florida; 2005. International affective picture system (IAPS): Affective ratings of pictures and instruction manual. [Google Scholar]
- 20.Hariri AR, Drabant EM, Munoz KE, et al. A susceptibility gene for affective disorders and the response of the human amygdala. Archives of General Psychiatry. 2005;62:146–152. doi: 10.1001/archpsyc.62.2.146. [DOI] [PubMed] [Google Scholar]
- 21.Rasetti R, Mattay VS, Wiedholz LM, et al. Evidence that altered amygdala activity in schizophrenia is related to clinical state and not genetic risk. American Journal of Psychiatry. 2009 February 1;166(2):216–225. doi: 10.1176/appi.ajp.2008.08020261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ekman P, Friesen WV. Pictures of Facial Affect. Palo Alto, CA: Consulting Psychologists Press; 1976. [Google Scholar]
- 23.Maldjian J, Laurienti PKR, Burdette J. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19(3):1233–1239. doi: 10.1016/s1053-8119(03)00169-1. [DOI] [PubMed] [Google Scholar]
- 24.Lipska BK, Peters T, Hyde TM, et al. Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Human Molecular Genetics. 2006 April 15;15(8):1245–1258. doi: 10.1093/hmg/ddl040. [DOI] [PubMed] [Google Scholar]
- 25.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3(1):Article 3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 26.Johnson AD, Handsaker RE, Pulit S, Nizzari MM, O’Donnell CJ, de Bakker PIW. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24(24):2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan H, Chen Q, Sust S, et al. Epistasis between catechol-O-methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc Natl Acad Sci U S A. 2007;104(30):12536–12541. doi: 10.1073/pnas.0610125104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459(7246):528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gargus JJ. Genetic calcium signaling abnormalities in the central nervous system: seizures, migraine, and autism. Ann N Y Acad Sci. 2009;1151:133–156. doi: 10.1111/j.1749-6632.2008.03572.x. [DOI] [PubMed] [Google Scholar]
- 31.Wei J, Hemmings GP. A further study of a possible locus for schizophrenia on the X chromosome. Biochem Biophys Res Commun. 2006;344(4):1241–1245. doi: 10.1016/j.bbrc.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 32.Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 33.Callicott JH, Straub RE, Pezawas L, et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci USA. 2005;102(24):8627–8632. doi: 10.1073/pnas.0500515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bigos KL, Hariri AR. Neuroimaging: technologies at the interface of genes, brain and behavior. Neuroimaging Clinics. 2007;17(4):459–467. doi: 10.1016/j.nic.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spitzer NC. Electrical activity in early neuronal development. Nature. 2006;444(7120):707–712. doi: 10.1038/nature05300. [DOI] [PubMed] [Google Scholar]
- 36.Kempton MJ, Ruberto G, Vassos E, et al. Effects of the CACNA1C Risk Allele for Bipolar Disorder on Cerebral Gray Matter Volume in Healthy Individuals. American Journal of Psychiatry. 2009;166(12):1413–1414. doi: 10.1176/appi.ajp.2009.09050680. [DOI] [PubMed] [Google Scholar]
- 37.Esslinger C, Walter H, Kirsch P, et al. Neural Mechanisms of a Genome-Wide Supported Psychosis Variant. Science. 2009;324(5927):605–605. doi: 10.1126/science.1167768. [DOI] [PubMed] [Google Scholar]
- 38.Newton-Cheh C, Larson MG, Vasan RS, et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet. 2009;41(3):348–353. doi: 10.1038/ng.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke R, Peden JF, Hopewell JC, et al. Genetic Variants Associated with Lp(a) Lipoprotein Level and Coronary Disease. New England Journal of Medicine. 2009;361(26):2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 40.Brunet G, Cerlich B, Robert P, Dumas S, Souetre E, Darcourt G. Open trial of a calcium antagonist, nimodipine, in acute mania. Clin Neuropharmacol. 1990;13(3):224–228. doi: 10.1097/00002826-199006000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Pazzaglia P, Post R, Ketter T, et al. Nimodipine monotherapy and carbamazepine augmentation in patients with refractory recurrent affective illness. J Clin Psychopharmacol. 1998;18(5):404–413. doi: 10.1097/00004714-199810000-00009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.