

Abstract
Unregulated cellular proliferation, caused by mutation or dysregulation of growth-promoting proteins, is an underlying cause of cancer. Many such growth-promoting proteins exhibit an increased dependence on the activity of the chaperone heat-shock protein 90 (Hsp90) for correct folding and maturation in the cell. One can therefore envision that inhibition of Hsp90 would be an effective and broadly applicable strategy for the development of anticancer agents. Hsp90 functions in multichaperone complexes driven by the binding and hydrolysis of ATP. Encouraging results have been obtained by inhibiting Hsp90 with 17-AAG, an active-site binding ATP analog. Here we present the results of a different approach to inhibiting Hsp90 by disrupting its interaction with a cochaperone named Hsp organizing protein (HOP). We have used an AlphaScreen technology based high-throughput in vitro screen to identify compounds that inhibit this interaction. In addition, we demonstrate that these compounds are active in vivo. Treatment of human breast cancer cell lines BT474 and SKBR3 with these compounds decreases the levels of the Hsp90-dependent client protein HER2, with associated cell death.
Dysregulation of signal transduction pathways, for example, by the overexpression of growth factor receptors, is a well-known contributor to the growth and progression of many tumors. For instance, approximately one-third of all breast cancer cells overproduce the cell surface receptor HER2 (designated HER2 positive) (1). The more HER2 a cell produces, the more aggressive its growth and the poorer the patient prognosis (2, 3). One of the most effective developments in the treatment of breast cancer in the past 20 years is a humanized monoclonal antibody (Herceptin (trastuzumab)) that binds to the extracellular domain of HER2 on the surface of breast cancer cells. Herceptin has been proposed to work via several different mechanisms, among which are blockade of downstream HER2 signaling pathways (4) and targeting of the cancer cells for destruction by the immune system (5).
An alternative and more general approach to anticancer therapeutics would be to prevent HER2 and other oncogenic proteins from ever folding and maturing in the cell. Certain proteins are dependent on the activity of other proteins, known as chaperones, for correct folding and maturation. About half of all proteins whose folding depends on the activity of the chaperone Hsp90 are proteins whose mutation or overproduction promotes cancer. Examples include but are not limited to pp60v-src, Bcr-Abl, p53, Akt, Flt3, HIF-1α, B-Raf, EGFR, and HER2. Inhibition of Hsp90 function therefore presents an effective route by which to disrupt multiple growth-promoting signaling pathways simultaneously (6).
Hsp90 activity is ATP-dependent, and inhibitors that bind at the ATP active site, for example, geldanamycin (GA) and its derivative 17-AAG, have already shown promise as anticancer agents (7, 8). 17-AAG has anti-tumor activity in several human xenograft models. Moreover, 17-AAG is currently being tested in clinical trials for use alone or in combination with other anticancer agents against melanoma, breast, prostate, and thyroid cancer (9).
Although 17-AAG illustrates the potential of inhibiting Hsp90 as a route to novel anticancer therapeutics, it is not a “magic bullet”. The most common dose-limiting side effects of 17-AAG in cancer patients are anorexia, nausea, diarrhea, hepatoxicity, fatigue, thrombocytopenia, and anemia. 17-AAG has low solubility in aqueous solution, which is also limiting for its clinical applications (10, 11). Although the precedent of 17-AAG is encouraging, there is clearly a need for novel Hsp90 inhibitors with higher solubility and fewer side effects.
Here we present an entirely new approach to inhibiting Hsp90 activity. Hsp90 does not function in isolation but rather is part of a multiprotein complex (12). The current consensus model for Hsp90-dependent protein folding assumes that a newly synthesized unfolded polypeptide first binds to Hsp40 and is then passed from Hsp40 to Hsp70. Hsp70 and Hsp90 are brought into close proximity by their C-terminal peptide interactions with two independent tetratricopeptide repeat (TPR) domains of HOP, TPR1, and TPR2A, respectively. Substrates are transferred from Hsp70 to Hsp90 to complete the final stage of maturation. The interaction of Hsp90 with HOP, via its TPR2A domain, is absolutely essential for Hsp90 activity (13), with a dissociation constant (Kd) of 11 μM (14). We seek to inhibit this interaction as a novel route by which to inhibit Hsp90 activity (Figure 1).
Figure 1.

Schematic illustration of Hsp90-mediated protein folding and proposed mechanism of identified compounds. a) For Hsp90-dependent proteins, the newly synthesized polypeptide chain (black line) is chaperoned through the initial steps of folding by Hsp70 (yellow rectangle). Hsp70 and Hsp90 (orange dimer) are brought together in a complex, via their interactions with the TPR1 (blue) and TPR2A (purple) domains of HOP, respectively. Partially folded polypeptide is passed from Hsp70 to Hsp90, where the final steps of folding occur. If Hsp90 activity is inhibited, for example, by 17-AAG, the Hsp90-dependent protein, e.g., HER2, dissociates from the multichaperone complexes, does not complete its folding, and is targeted for degradation by the proteasome. b) Proposed mode of action of molecules that inhibit the Hsp90–HOP interaction. The first, Hsp70-dependent step of folding will initiate as normal. The inhibitory molecule will prevent the interaction of Hsp90 with HOP. Thus Hsp90 cannot form the HOP-mediated complex with Hsp70 and consequently cannot receive the partially folded substrate. The incompletely folded protein is targeted for degradation by the proteasome.
In previous studies we have demonstrated the potential of the proposed approach. We have shown that a designed TPR domain, which binds to the C-terminus of Hsp90 more tightly and more specifically than TPR2A, can compete for binding to Hsp90 after being transfected into breast cancer cells and thereby prevent formation of the functional Hsp90–HOP–Hsp70 complex. Hsp90 is thus inhibited, the levels of HER2 are reduced, and the cells die (15). Here we present the identification, via a high-throughput screen (HTS), of small molecules that inhibit the Hsp90–TPR2A interaction. Moreover, we show that such compounds are effective in vivo, causing a decrease in HER2 levels in human breast cancer cells (BT474 and SKBR3), with associated cell death. Because our compounds act by a mechanism completely different from those of 17-AAG, Herceptin, and traditional nonspecific chemotherapeutics, they have high potential to be combined synergistically with these and other anticancer agents.
RESULTS AND DISCUSSION
Hit Identification Using the High-Throughput AlphaScreen Assay
Small molecule inhibitors of the Hsp90–HOP interaction, rather than peptides, are required for the development of this approach as a therapeutic strategy. We therefore developed an AlphaScreen technology based HTS to identify novel inhibitors of the Hsp90–TPR2A interaction. A schematic of the AlphaScreen is shown in Figure 2. We demonstrated the suitability of this assay using free Hsp90 peptide. Free Hsp90 peptide effectively competes with biotinylated Hsp90 peptide for its binding to TPR2A, resulting in decreased fluorescence signals. Measuring the concentration-dependent inhibition of free Hsp90 peptide gives an IC50 of about 5 μM, which is consistent with the known Kd of the Hsp90-TPR2A interaction (14).
Figure 2.

Schematic illustration of the AlphaScreen. The donor bead (D, red) is coated with streptavidin (dark blue), which binds N-terminally biotinylated (gray square) Hsp90 peptide (gray oval). The acceptor bead (A, orange) is coated with Ni-NTA (black Y), which binds the C-terminal His6 tag (blue diamond) of TPR2A (green oval). a) In the absence of inhibitors, the Hsp90–TPR2A interaction brings the donor and acceptor beads together. Excitation of the donor beads at 680 nm produces excited singlet state oxygen species, which can diffuse up to 200 nm and react with a chemiluminescor in the acceptor bead, further activating flurophores in the acceptor bead, which emit light at 520–620nm. b) In the presence of small molecule inhibitors (black circles) that inhibit the Hsp90–TPR2A interaction, the donor and acceptor beads are no longer brought to within 200 nm of each other, and no acceptor bead fluorescence is observed upon excitation of the donor bead.
Using the AlphaScreen in a 384-well format, we screened the MicroSource Discovery Systems GenPlus library (960 compounds) and the Maybridge diversity library (20,000 compounds) at ~10 μM concentration of each compound. The NCGC compound library (76,174 compounds) was screened in a 1536-well assay format at a titration series of seven concentrations with 0.1% BSA and 0.01% Tween-20 included in the assay buffer to decrease the likelihood of compounds acting by aggregate formation for promiscuous inhibition (16). The average Z′ factor value for all assays in both formats is about 0.7, indicating the robustness. Although we identified early “hits” in each of the libraries, the compounds shown to have the desired properties by follow-up assays were all from the NCGC compound library. For clarity, therefore, we present the results only from this library.
For each of the compounds, a concentration-dependent inhibition curve was determined, and 149 compounds with IC50 values of 10 μM or less were identified as primary hits. Figure 3 is a flowchart showing the characterization of the hit compounds using secondary in vitro and in vivo assays. Following structure clustering analysis, 41 representatives were selected for further competition confirmation and subjected to a counter screen for false-positive identification. In the counter-screen, compounds were tested for their inhibition in a version of the AlphaScreen in which the donor and acceptor beads are brought together by a covalent biotin-His6 linker. Compounds that inhibit fluorescence, mimic either biotin or NTA, or inhibit by any nonspecific mechanism will be eliminated by the counter-screen.
Figure 3.

Flowchart of the steps in hit identification and verification.
Only three compounds remained as true positive hits after these steps. An examination of the structures of these three compounds revealed that they have a common core. We therefore purchased and tested five additional structurally related compounds that were not present in the original library. Only three of the five compounds tested positive in the AlphaScreen (Figure 4). All of the concentration–response competition curves for the six active compounds had Hill slopes close to –1, indicating no cooperative binding of multiple molecules or compound aggregation. Also it is reassuring to see that none of this class of compounds was identified as “promiscuous inhibitors” in an aggregate profiling screen against the same NCGC compounds by monitoring, with or without 0.01% Triton X-100, their inhibition of the activity of AmpC β-lactamase, a protein target completely unrelated to our study (see Pub-Chem AID = 584, 585). At this point we continued with further characterization of the six positive hit compounds and one of the inactive compounds, to serve as a negative control.
Figure 4.

Competition curves for selected compounds in the AlphaScreen assay. a) Competition curves for different compounds using the AlphaScreen with the donor and acceptor beads brought together by Hsp90 peptide and TPR2A protein. Compounds 8–13 all show typical dose-dependent inhibition of the acceptor fluorescence. The inactive structural analogue compound does not. b) AlphaScreen in counter-screen format. A covalent linker peptide (biotin-His6 peptide) directly brings the donor and acceptor beads together. Any compounds that inhibit in this assay are considered to be false positives. Percent of control binding (no inhibition – 100%) is plotted against log[compound]. Error values were calculated from multiple experiments. c) Core structure representative of the hit compounds.
Confirmation of Compound Binding Using a Fluorescence Polarization Assay
We tested these six hit compounds using an independent in vitro assay, to rule out any false positives that may have been missed in the AlphaScreen counter-screen. We therefore developed a fluorescence polarization (FP) assay by which to monitor the fluorescein-labeled C-terminal Hsp90 peptide–TPR2A interaction and its inhibition. Compounds were screened for their ability to inhibit the Hsp90–TPR2A interaction, as monitored by decreases in the polarization of the bound fluorescein-labeled Hsp90 peptide. The intensities from both the perpendicular and horizontal orientations were checked to make sure that the FP value (ratio between these two) decreases were not due to abnormal intensity changes that might come from artificial compound interference. No abnormal intensity changes were observed. Also, we measured the intrinsic fluorescence of compounds and observed very low background signal in the detecton range for both FP and AlphaScreen assays. Thus the data supports the hypothesis that the changes in FP observed when the active compounds are added are because of differences in the tumbling of the fluorescent Hsp90 peptide when free as compared to when bound to TPR2A.
All six compounds tested positive in the FP assay, with Kd values in the micromolar range, consistent with those measured from the AlphaScreen competition assay. Moreover, the compound that was inactive in the AlphaScreen, which we brought forward as a negative control, was also inactive in the FP assay (Figure 5, panel a).
Figure 5.

Characterization of the binding between compounds and TPR2A protein using secondary in vitro binding assays. a) Examples of inhibition curves for selected compounds in the fluorescence polarization assay. A peptide corresponding to the C-terminus of Hsp90 (10-mer peptide, N-terminally labeled with fluorescein) and His6-tagged TPR2A protein were used in these experiments. Percent of control binding (peptide in complex with TPR2A, FP is high = 100%) is plotted versus log[compound]. Data are reported as mean and standard deviation of two experiments. b) Binding constant and stoichiometry for the interaction of TPR2A with compound 8 were determined by ITC measurements. Top: Incremental heat effect upon titration of compound 8 (0.5 mM) into TPR2A (40 μM). Bottom: integrated heat effects normalized to the amount of injected compound 8 and fitted curve based on a 1:1 binding model. Constants determined were Kd = 16 μM, stoichiometry N = 1.07.
Demonstration of Direct Binding between Compound 8 and TPR2A
The data presented to date show that we have identified compounds that compete for the TPR2A–Hsp90 interaction. To demonstrate direct binding between compounds and protein or peptide, we performed isothermal titration calorimetry (ITC) measurements with the representative compound 8 and either the TPR2A protein or Hsp90 peptide. As we might have anticipated, because it is the larger molecule and has a “binding cleft”, ITC showed a direct interaction between TPR2A and compound 8, with a Kd of 16 μM and a 1:1 binding stoichiometry (Figure 5, panel b). No interaction between the compound and Hsp90 peptide was detected (data not shown).
In Vivo Activity of Selected Compounds
Activity in vitro does not necessarily mean that the compounds will be active in vivo. Not least of the requirements for in vivo activity is that the compounds must be able to enter cells and function in a cellular context. We therefore tested the activity of the 6 hit compounds and the one inactive compound in cell based assays to assess the overall capacity of the compounds to inhibit cancer cell growth. For comparison, we also tested the effects of 17-AAG as a positive control.
Figure 6, panel a shows examples of concentration-dependent killing of BT474 cells by the compounds as measured by a WST-1 cell proliferation assay. Only the compounds that showed activity in vitro showed cell killing activity. Although we do not know the exact concentrations in the cells of the compounds and their differential partition into each cellular compartment, the observation that the range of concentrations over which the compounds are active coincides with the Kd values measured in vitro suggests that these compounds permeate the cell membrane well and are likely inhibiting the targeted interaction in cells. Because all six hit compounds behaved similarly in the in vitro binding assays and the BT474 cell proliferation assay, we chose just one representative (compound 8) for more detailed follow-up assays.
Figure 6.

Effects of compounds on the growth of various breast cells. a) Effects of compounds on the growth of BT474 breast cancer cells. b) Effect of 17AAG on the growth of BT474, SKBR3, and MCF-12F cells. c) Effect of compound 8 on the growth of BT474, SKBR3, and MCF-12F cells. Data are reported as mean and standard deviation of six replicates.
Cell-Line Specificity
The molecular basis for the cell-line specificity of different anticancer agents is not well understood and cannot be predicted a priori. To determine whether our hit compounds can selectively kill breast cancer over normal breast cells, we compared the cell-killing activity of 17-AAG and compound 8 with that of an inactive structural homologue in two different HER2 positive human breast cancer cell lines (BT474, SKBR3) alongside MCF-12F (a non-tumorigenic mammary epithelial cell line). Figure 6, panels b and c shows the concentration-dependent cell killing of these three cell lines by 17-AAG and by compound 8. 17-AAG was significantly more potent in killing BT474 cells than SKBR3 and normal cells. Compound 8 also showed some selectivity in preferentially killing BT474 cells, but this effect was less pronounced. The inactive structural homologue was inactive in all three cell lines (data not shown). Many factors can contribute to the differences in cell-line specificity of a given compound, including cellular uptake and metabolism or “off target” interactions with other cellular components. Optimization of compound 8 to improve its tumor cell specificity will be the topic of further studies.
Effect of Active Compounds on the Levels of HER2 and Hsp70
The cell-killing effects suggest that the compounds act in the hypothesized fashion. However, to more explicitly explore their mechanism of action, it was important to study their effects on Hsp90 client proteins. BT474 and SKBR3 cells are both HER2 positive and have been extensively used in HER2 and Hsp90 research. We therefore investigated the concentration-and time-dependence of the compounds’ effect on HER2 levels in these cell lines. Again, we compared the activity of our compounds with that of 17-AAG. We performed Western Blots on total cell extracts, using appropriate antibodies. Examples of the data obtained in these experiments are shown in Figure 7. Shown on the left is a time-dependent decrease in HER2 levels in BT474 cells after treatment with compound 8 or 17-AAG. Also compared are the levels of the chaperone Hsp70 and the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which serves as a non-Hsp90-dependent control. On the right are the same experiments performed in SKBR3 cells.
Figure 7.

Effect of compounds on the levels of HER2 and Hsp70 in BT474, SKBR3 breast cancer cells. Cells treated with compounds at different concentrations or untreated were harvested at indicated time points and lysates were prepared and immunoblotted as described in Methods.
We observed a very reproducible behavior in BT474 cells: HER2 levels rapidly decrease after compound treatment for 6 h and then increase at 12 h. Similar results have been reported by others for cells treated with 17-AAG. Although compound cell entry, metabolism, or unknown features of HER2 regulation could all contribute to this effect, the underlying mechanism has not yet been elucidated and will be a topic of future studies.
It has long been observed that treatment of cells, by GA or other ATPase inhibitors of Hsp90, induces the expression of the chaperone Hsp70 (17, 18). Induction of Hsp70 is undesirable, because Hsp70 has potent anti-apoptotic activity (19, 20), which to some extent can counteract the desired cell killing. In our experiments, we also observed a significant increase in Hsp70 levels when cells were treated with 17-AAG (Figure 7). By contrast, none of our compounds induced Hsp70 overproduction, as can be clearly seen in the Western blots with compound 8 as an example. This result is consistent with our hypothesis that these compounds act via a different mechanism from that of 17-AAG and suggests a potential advantage of these compounds over the ATPase inhibitors of Hsp90.
Conclusion
We have described the identification of a class of small molecules that inhibit the interaction of the C-terminal peptide of Hsp90 and the TPR2A domain of HOP. We have demonstrated their inhibitory activity in vitro, using three different assays, the Alpha-Screen, a fluorescence polarization assay, and isothermal titration calorimetry. We have also demonstrated that the compounds are active in vivo, in reducing the levels of the cancer-promoting, Hsp90-dependent client protein HER2 and in cell killing. Not only do these compounds inhibit Hsp90 by a unique mechanism, but they also have the desirable property of not inducing overexpression of Hsp70. The compounds show modest selectivity for cancer versus noncancer cell lines.
All six active compounds have in common a 7-azapteridine ring system (pyrimido[5,4-e] [1,2,4]triazine-5,7-dione) (Table 1). In the in vitro and in vivo assays performed to date, the six related compounds show similar activities, with no substitution causing a dramatic change in activity. We speculate that the relatively flat structure–activity relationship (SAR) is because most of the compounds were different by substitutions at a single position, presumably not a key position. It is interesting to note, however, that the active compound 8 and the inactive analogue differ by only a single methyl group at position 1. Although this subtle difference might account for the lack of activity of the inactive analogue in cell based assays, as a result of its increased polarity and consequent decreased cell membrane permeability, the observation that this compound is also inactive in the in vitro binding assays prompts us to speculate that this methyl group could be involved in a key hydrophobic interaction in the TPR binding pocket. Clearly, however, future extensive SAR studies, combined with structural characterizations of the TPR2A–compound complexes, will provide a more detailed picture. More extensive characterization of the in vivo effects of the compounds, e.g., exploring cell type specificity and the range of Hsp90 client proteins against which the compounds are effective, is underway. It is also of our interest to test the synergistic effects of the compounds with 17-AAG, Herceptin, and nonspecific chemotherapeutic agents such as Paclitaxol.
TABLE 1.
Summary of hits information
| Hit | Structure | Source | Kd (M) (ALPHA) | Kd(M) (FP) | IC50 (M) (WST-1) |
|---|---|---|---|---|---|
| 8 |
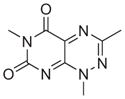
|
NCGC00046775 | 4.92e-7 | 3.65e-7 | 9.46e-7 |
| 9 |
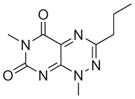
|
Commercial | 4.14e-7 | 2.02e-7 | 9.17e-7 |
| 10 |
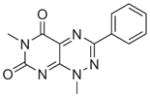
|
Commercial | 5.85e-7 | 1.04e-6 | 6.90e-7 |
| 11 |
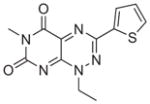
|
NCGC00034880 | 3.65e-7 | 3.08e-7 | 1.21e-6 |
| 12 |
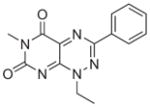
|
NCGC00034018 | 3.60e-7 | 9.28e-7 | 1.72e-6 |
| 13 |
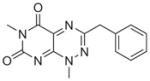
|
Commercial | 1.82e-7 | 5.66e-7 | 4.27e-6 |
| Inactive |
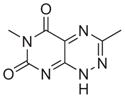
|
Commercial | N/A | N/A | N/A |
Although some have considered it a challenge to disrupt protein–protein interactions using small molecules, recent experimental and statistical approaches have shown that not all residues at a protein–protein interface contribute equally to the binding free energy (22). Recent reports of small molecule inhibitors of protein–protein interactions attest to the idea that inhibitors of protein–protein interactions do not have to be large, they just have to inhibit key interactions. In the context of the TPR2A–Hsp90 peptide interaction, amidation of the free carboxy-terminus of the peptide MEEVD-COO- is sufficient to abolish detectable binding, a “hot spot” at the interaction interface. We speculate that the small molecules we have identified inhibit the HOP–HSP90 interaction by binding at a key position on the TPR2A interaction interface. Future structural characterizations will reveal the details of this interaction and may suggest how the compounds can be chemically modified to achieve higher potency and specificity. Relatively straightforward methods for the total synthesis of the structural class of compounds identified in this study have been described (23, 24), which would make it feasible to test a greater diversity of analogues and to fine-tune their properties.
The compounds identified in this study serve as the first case of a novel class of small molecule inhibitors of Hsp90 by disrupting its cochaperone interaction and provide a strong rationale for pursuing the development of small molecule compounds that specifically interrupt the interaction between Hsp90 and cochaperones. The current generation of compounds show moderate (micromolar) activity in various in vitro and in vivo assays. Future generations of compounds with high affinity and specificity for different cochaperone proteins can also be used as chemical probes to dissect the complex Hsp90-cochaperone interactions, with the potential to be developed into novel anticancer drugs.
METHODS
Materials
AlphaScreen Histag fusion detection 10k assay point kit and Opti-384 plates used in the screening were purchased from PerkinElmer (PerkinElmer). His6-tagged TPR2A was produced using a bacteria expression system and purified using Ni-NTA superflow resin from Qiagen. Hsp90 C-terminal peptide with or without an N-terminal biotin group was synthesized using automated solid-phase synthesis by the Yale Keck facility (TEEMPPLEGDDDTSRMEEVD). 17-Allylamino-17-demethoxygeldanamycin (17-AAG) was purchased from Assay Designs. The 17-AAG stock was dissolved in 100% DMSO at a stock concentration of 1.7 mM and kept at −20 °C protected from light. It was further diluted in sterile PBS buffer for the various experiments in cell culture. The cell proliferation agent WST-1 salt was purchased from Roche Diagnostics Corp. The six toxoflavin structural class compounds identified through the screening and two inactive structural homologues were provided by the National Chemical Genomical Center (NCGC) as 20 mM stock solution in 100% DMSO. They were further diluted in sterile PBS for the various experiments in cell culture. HEPES, NaCl, BSA, PBS, DMSO, and DTT were purchased from Sigma.
Cells and Antibodies
BT474 (ATCC HTB-20) and SBKR3 (ATCC HTB-30) breast cancer cells and MCF-12F (ATCC CRL-10783) non-carcinogenic epithelial breast cells were purchased from American Type Culture Collection (ATCC). BT474 and SKBR3 are human breast cancer cells that overexpress HER2. BT474 cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 2 mM glutamine, 10 μg mL−1 insulin, penicillin, and streptamycin. SKBR3 cells were cultured in McCoy’s media with 10% heat-inactiveated fetal bovine serum, 2 mM glutamine, penicillin and streptomycin. MCF-12F normal breast cells were grown in a medium containing a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium and Ham’s F12 medium, with 20 ng mL−1 epidermal growth factor (EGF), 0.01 mg mL−1 insulin, 500 ng mL−1 hydrocortisone, and 5% horse serum. All cells were grown in a humidified atmosphere of 5% CO2 at 37 °C. Western Blot antibody for rabbit polyclonal anti-HER2 (sc-284) was purchased from Santa Cruz Biotechnology. Mouse anti-Hsp70 monoclonal (SPA-810) antibody was purchased from Stressgen/Assay Designs. Secondary HRP conjugated antimouse and antirabbit antibodies were from Santa Cruz Biotechnology, Inc. and Amersham, respectively. Horseradish-peroxidase conjugated anti-GAPDH (ab9482) was from abCam.
Compounds Libraries
Three libraries with a total of 97,134 compounds have been screened using this assay. A library of 960 compounds with known biological functions was purchased from Microsource Discovery Inc. A library of 20,000 compounds with chemically diverse structures was purchased from Maybridge (ACROS Janssen Pharmaceuticalaan 3A). These compounds were stored at a stock concentration of 10 mM in DMSO in 384-well plates. A third set of 76,174 compounds from different sources were screened at NCGC at a series of diluted compound concentrations ranging from 0.3 to 40 μM in the assay buffer of 25 mM HEPES, 100 mM KCl, 2 mM MgCl2, 5 mM DTT, and 0.01% Tween-20 with 0.1% bovine serum albumin (BSA).
Primary Hits Identification using AlphaScreen Technology Based HTS and Counter Screen
We have screened the above-mentioned three libraries for compounds that interrupt the interaction between TPR2A domain and Hsp90 peptide. A counter screen using a covalently linked biotin-His6 peptide was performed to identify false positives that interfere with the assay by mechanisms unrelated to the interaction, e.g., singlet oxygen quenchers, biotin mimetics, fluorescence quenchers, and light scatterers. The ratio between the IC50 values for the competition assay and counter-screen was used to define specific inhibitors. Any compounds having similar activities in both the competition and counter screen assays (IC50 ratio <10) were considered nonspecific (e.g., inactive structural analogue). Compounds 8–13, with IC50 ratios of ~500, were thus confirmed as specific inhibitors and subjected to further secondary assay characterization. All of the detailed technical information regarding the assay development, optimization and implementation is described elsewhere (Yi, F.; Zhu, P.J.; Southall, N.; Ingelse, J.; Zheng, W.; Regan, L. Unpublished results).
Fluorescence Polarization Assay
A fluorescence polarization assay was performed to confirm the binding between the hit compounds identified from the primary AlphaScreen. Purified His6-tagged TPR2A protein and fluorescein-labeled 10 mer C-terminal Hsp90 peptide (DDTSRMEEVD) were used in this assay. Assays were performed in a total volume of 25 μL in the buffer of 20 mM Tris, pH 8.0, 50 mM NaCl, 1 mM DTT. For competition studies, 20 μL of premixed 20 nM fluorescein-labeled Hsp90 peptide and 5 μM His6-tagged TPR2A protein were added into each well of a 384-well black assay plate with a nonbinding surface coating (Corning). These concentrations were chosen to allow for approximately 50% peptide bound in the presence of a protein concentration close to the Kd (5 μM) of the interaction of interest, to also yield a reasonable polarization shift upon protein binding by the labeled peptide. Approximately 250 nL of each compound was transferred from a stock (10 mM in DMSO) into two wells of the assay plate using an automatic liquid handler (Tecan EVO) outfitted with a pin tool (V&P Scientific). Fixed tips on the liquid handler performed 12 three-fold serial dilutions, resulting in final compound concentrations ranging from 100 μM to 0.56 nM, and then transferred 5 μL of the diluted compounds into the wells with assay mixture in the assay plate. Results from duplicate dilution series were averaged for each compound. Each plate included 48 wells of free fluorescein-labeled Hsp90 peptide and the same peptide in the presence of TPR2A protein. Plates were incubated at RT for 2 h prior to measurement to ensure binding equilibrium.
Measurements were performed on a PerkinElmer Envision plate reader using a 480 nm (30 nm bandwidth) excitation filter and two polarized 535 nm emission filters (40 nm bandwidths) integrating signals over 100 flashes. Upon excitation, fluorescence emission intensities from both the parallel (S) and perpendicular (P) orientation were measured, and polarization values were expressed in milliploarization units (mP), calculated using the formula mP =1000(Is − GIp)/(Is + GIp), where Is and Ip are the parallel and perpendicular emission intensity for the sample respectively, G is the G factor (= Is/Ip, measured as 0.94 for this study). The fraction of specific binding is defined as the contribution to bound peptide signal to the total signal, calculated using the equation b = (mP − mPf)/(mPb − mPf), where b is the fraction of bound peptide, mP is the total mP value recorded, mPb is the mP value of the bound peptide control, and mPf is the mP value for the free fluorescein-Hsp90 peptide. The assay window was defined as mPb−mPf.
Isothermal Titration Calorimetry
Binding of TPR2A protein to a representative compound 8 was measured by ITC using a Microcal iTC200 calorimeter (Microcal Inc.). The syringe was loaded with 40 μL of compounds diluted using buffer from a 20 mM DMSO stock solution to 0.5 mM in a final 2.5% DMSO. About 200 μL of purified His6-tagged protein, at 40 μM in 20 mM Tris, 50 mM NaCl, 10 mM β-mercaptoethanol, pH 8.0, 2.5% DMSO was loaded into the cell. All experiments were performed with an initial injection of 0.5 μL, followed by 19 2-μL injections with 200 s spacing. The syringe was stirred at 1000 rpm with thermostatting at 25 °C. The binding isotherms were fit to a one-site binding model to obtain the thermodynamic parameters with the initial point discarded. Data analysis was conducted using Origin 7 software with the ITC200 add-on package.
WST-1 Colorimetric Cell Proliferation Assay
BT474, SBKR3, and MCF-12F cells were seeded in 96-well plates at a density of 2 × 104 cells (100 μL of culture)−1 well−1 except for the first and last columns to which only growth media was added. After a 24-h adhering period, the cells were either left untreated or treated with different concentrations of the hit compounds or 17-AAG or DMSO vehicle control. After a 4-day incubation period, the WST-1 tetrazolium salt colorimetric proliferation assay was performed by adding 10 μL of dissolved WST-1 solution into each well and incubating for 3 h at 37 °C in a 5% CO2 incubator. Absorbance at 440 nm was measured using a PerkinElmer Envision plate reader. The wells in the first and last rows for each column were discarded to avoid possible variation due to evaporation. Each data point represents an average of 6 replicate wells.
Western Blotting
BT474 and SKBR3 breast cancer cells were seeded at 2.5 × 106 cells (100-mm dish)−1 and allowed to adhere overnight. Cells were treated with 17-AAG (0.178 μM) or compound 8 (5 μM in SKBR3 cells, 1 μM in BT474 cells) or left untreated. BT474 cells were harvested at 6, 12, and 24 h after treatment with 1 μM compound 8 and 24 h after treatment with 0.178 μM 17AAG. SKBR3 cells were harvested at 3, 6, 12, and 24 h after treatment with 5 μM compound 8 and 24 h after treatment with 0.178 μM 17-AAG. Cells treated with compounds were washed using ice-cold PBS three times and then scraped into ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA and EGTA, 0.1% SDS, 1% NP-40, 1 mM Na3VO4, 25 mM NaF, 25 mM β-glycerol-phosphate), supplemented with Complete proteinase inhibitors (Roche Molecular Biochemicals). After incubating on ice for 30 min, the lysates were centrifuged at 14,000 rpm (4 °C) for 15 min to remove insoluble material. Total protein concentration was determined using the BCA assay (Pierce). Cell lysates of 25 μg of total protein were separated on a 4–12% gradient SDS-polyacrylamide gel (Biorad) followed by transfer to a 0.45 μm nitrocellulose PVDF membrane. Membrane was blocked in 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) with 1 mM Na3VO4 for 1 h at RT on shaker. Primary antibodies were diluted in 5% BSA at a concentration of 1:2000 (HER2, antirabbit) or 1:1000 (Hsp70, anti-mouse). Membrane was incubated in the primary antibodies at 4 °C overnight and washed five times at RT. Secondary HRP conjugated antibodies (antimouse antibody from Santa Cruz Biotechnology, Inc., antirabbit from Amersham) were diluted at 1:2000. Membrane was incubated in the secondary antibodies at RT for 1 h followed by washing 4 times in TBST and one last wash in TBS. Membrane was then processed using enhanced chemiluminescence (ECL, Amersham) development.
Data Analysis
Data were analyzed using Graphpad Prism 4 (GraphPad Software). A sigmoidal dose–response model with variable slope was used to fit the in vitro competition curves and growth inhibition curves. The IC50 values and the Hillslopes were generated from the curve fitting. All data were expressed as mean ± SD.
Assuming a one-site competition model, the IC50’s from the in vitro assays were converted to Kd’s using the Cheng and Prusoff equation (25):
where Kd(cpd) is the binding affinity for the compound-TPR2A interaction; [pep] is the Hsp90 peptide concentration used in the premixed complexes, which is 10 nM for the AlphaScreen assay and 20 nM for the FP assay; and Kd(pep) is the binding affinity for the Hsp90-TPR2A interaction, 10 μM is used in the calculation.
Acknowledgments
We thank Debbie Koay for her valuable help with the first cell proliferation assays; Dr. Elizabeth Rhoades for allowing us to use her cell culture facilities; Wei Zheng and Pingjun Zhu for their help with the NCGC screen; and Janie Merkel and Michael Salcius for help with the FP assay. We thank Aitziber Cortajarena, Clarence Chen, Tijana Grove, Meredith Jackrel, and Lenka Kundrat for helpful comments on the manuscript. We are grateful for the support of the Breast Cancer Alliance (L.R.) and for a Concept Award from the Department of Defense, Breast Cancer Research Program (W91ZSQ7167N696) and a NIH Molecular Libraries Screening Centers Network grant (XO1-MH077625-01) (F.Y.).
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Cooke T, Reeves J, Lanigan A, Stanton P. HER2 as a prognostic and predictive marker for breast cancer. Ann Oncol. 2001;1:S23–S28. doi: 10.1093/annonc/12.suppl_1.s23. [DOI] [PubMed] [Google Scholar]
- 3.Domingo-Domenech J, Fernandez PL, Filella X, Martinez-Fernandez A, Molina R, Fernandez E, Alcaraz A, Codony J, Gascon P, Mellado B. Serum HER2 extracellular domain predicts an aggressive clinical outcome and biological PSA response in hormone-independent prostate cancer patients treated with docetaxel. Ann Oncol. 2008;19:269–275. doi: 10.1093/annonc/mdm490. [DOI] [PubMed] [Google Scholar]
- 4.Yakes FM, Chinraianalab W, Ritter CA. Herceptin-induced inhibition of phophatidylinositol-3 kinase and Akt is required for antibody-medialed effects on p27 cyclin DL and antitumor action. Cancer Res. 2002;62:4132–4141. [PubMed] [Google Scholar]
- 5.Clynes RA, Towers TL, Presta LG. Inhibitory Fc receptore modulate in vivo cytoioxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 6.Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–530. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 7.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powers MV, Workman P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007;581:3758–3769. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 9.Stravopodis DJ, Margaritis LH, Voutsinas GE. Drug-mediated targeted disruption of multiple protein activities through functional inhibition of the Hsp90 chaperone complex. Curr Med Chem. 2007;14:3122–3138. doi: 10.2174/092986707782793925. [DOI] [PubMed] [Google Scholar]
- 10.Solit DB, Ivy SP, Kopil C, Sikorski R, Morris MJ, Slovin SF, Kelly W, DeLaCruz A, Curley T, Heller G, Larson S, Schwartz L, Egorin MJ, Rosen N, Scher HI. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin Cancer Res. 2007;13:1775–1782. doi: 10.1158/1078-0432.CCR-06-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramanathan RK, Egorin MJ, Eiseman JL, Ramalingam S, Friedland D, Agarwala SS, Ivy SP, Potter DM, Chatta G, Zuhowski EG, Stoller RG, Naret C, Guo J, Belani CP. Phase I and pharmacodynamic study of 17-(allylamino)-17-demethoxy geldanamycin in adult patients with refractory advanced cancers. Clin Cancer Res. 2007;13:1769–1774. doi: 10.1158/1078-0432.CCR-06-2233. [DOI] [PubMed] [Google Scholar]
- 12.Pratt WB, Dittmar KD. Studies with purified chaperones advance the understanding of the mechanism of glucocorticoid receptor-Hsp90 heterocomplex assembly. Trends Endocrinol Metab. 1998;9:244–252. doi: 10.1016/s1043-2760(98)00059-9. [DOI] [PubMed] [Google Scholar]
- 13.Chen S, Smith DF. Hop as an adaptor in the heat shock protein 70 (Hsp70) and Hsp90 chaperone machinery. J Biol Chem. 1998;273:35194–35200. doi: 10.1074/jbc.273.52.35194. [DOI] [PubMed] [Google Scholar]
- 14.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 15.Cortajarena AL, Yi F, Regan L. Designed TPR modules as novel anticancer agents. ACS Chem Biol. 2008;3:161–166. doi: 10.1021/cb700260z. [DOI] [PubMed] [Google Scholar]
- 16.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2002;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61:4003–4009. [PubMed] [Google Scholar]
- 18.Banerji U, O’Donnell A, Scurr M. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxy geldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23:4152–4161. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 19.Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–38739. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- 20.Garrido C, Schmitt E, Cande C, Vahsen N, Parcellier A, Kroemer G. HSP27 and HSP70: potentially oncogenic apoptosis inhibitors. Cell Cycle. 2003;2:579–584. [PubMed] [Google Scholar]
- 21.Onuoha SC, Coulstock ET, Grossmann JG, Jackson SE. Structural studies on the co-chaperone hop and its complexes with Hsp90. J Mol Biol. 2008;379:732–744. doi: 10.1016/j.jmb.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Thanos CD, DeLanon WL, Wells JA. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc Natl Acad Sci USA. 2006;103:15422–15427. doi: 10.1073/pnas.0607058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoneda F, Nagamatsu T. A convenient synthesis of toxoflavins, toxoflavin 4-oxides and 1-demethyltoxoflavins. Chem Pharm Bull (Tokyo) 1975;23:2001–2009. doi: 10.1248/cpb.23.2001. [DOI] [PubMed] [Google Scholar]
- 24.Wang S, Liu D, Zhang X, Li S, Sun Y, Li J, Zhou Y, Zhang L. Study on glycosylated prodrugs of toxoflavins for antibody-directed enzyme tumor therapy. Carbohydr Res. 2007;342:1254–1260. doi: 10.1016/j.carres.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of an inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]