

Abstract
Background
Studies of copy number variation (CNV) have successfully characterized loci and molecular pathways involved in a range of neuropsychiatric conditions. We conducted an analysis of rare CNVs in Tourette Syndrome (TS) to identify novel risk regions and relevant molecular pathways, evaluate the burden of structural variation in cases versus controls, and to assess the overlap of identified variations with those implicated in other neuropsychiatric syndromes.
Methods
We conducted a case-control study of 460 individuals with TS, including 148 parent-child trios and 1131 controls. CNV analysis was undertaken using 370K to 1M probe arrays, and genome-wide genotyping data was used to match cases and controls for ancestry. Transmitted and de novo CNVs present in < 1% of the population were evaluated.
Results
While there was no significant increase in the number of de novo or transmitted rare CNVs in cases versus controls, pathway analysis using multiple algorithms showed enrichment of genes within histamine receptor (H1R and H2R) signaling pathways (p=5.8×10-4-1.6×10-2) as well as “axon guidance”, “cell adhesion”, “nervous system development” and “synaptic structure and function” processes. Genes mapping within rare CNVs in TS showed significant overlap with those previously identified in autism spectrum disorders (ASD), but not intellectual disability or schizophrenia. Three large, likely-pathogenic, de novo events were identified, including one disrupting multiple gamma-Aminobutyric acid (GABA) receptor genes.
Conclusions
We identify further evidence supporting recent findings regarding the involvement of histaminergic and GABAergic mechanisms in the etiology of TS and show an overlap of rare CNVs in TS and ASD.
Keywords: Tourette syndrome, copy number variation, CNV, histamine, GABA, autism
INTRODUCTION
Tourette Syndrome (TS) is a developmental neuropsychiatric disorder which affects between 3 in 1000 individuals and 1 in 100 individuals (1-3). It is characterized by the presence of both motor and vocal tics and follows a waxing and waning course, often with improvement or remission in adulthood (4-7). While cellular and molecular mechanisms underlying TS pathophysiology remain uncertain, multiple lines of evidence point to the involvement of dopaminergic (DA) neurotransmission and abnormalities in the corticostriatal- thalamic-cortical (CTSC) circuits (8, 9). More recent post mortem data highlights abnormalities in striatal gamma-Aminobutyric acid (GABA)ergic interneurons (10, 11).
Three decades of research led to widespread agreement that genes play a significant role. TS and related conditions, including chronic tics, aggregate within families and show considerably higher concordance in monozygotic versus dizygotic twins (12-14). While early segregation analyses suggested single-gene autosomal dominant inheritance, contemporary data has pointed to a highly heterogeneous genetic architecture (15-18).
While the major emphasis over the last decade has been on the contribution of common genetic variations (>5% of the population), several recent findings highlight the importance of studying rare, highly penetrant variants, including the identification of mutations in the gene Slit and trk like family member 1 (SLITRK1) (19) and mapping of rare chromosomal abnormalities disrupting the Contactin-associated protein-like 2 (CNTNAP2) (20) and Neuroligin 4 (NLGN4) (21) genes, with these latter two also strongly implicated in autism spectrum disorders (ASD). Most recently, we characterized a highly penetrant mutation in the gene L-histidine Decarboxylase (HDC) in a dense TS pedigree, implicating histaminergic (HA) neurotransmission in the genesis or modulation of tics (22).
The emergence of microarray technologies that can detect sub-microscopic structural variation revealed extensive copy number variation (CNV) across the human genome (23-26) and provided new opportunities for genome-wide assessment of rare variation. Studies in schizophrenia (SCZ) (27-33) and ASD (34-37) demonstrated an over-representation of rare CNVs, particularly genic de novo variants (30, 34, 35, 38), and highlighted molecular mechanisms that likely play a role in these conditions. Moreover, recent replicated findings show that more than one developmental neuropsychiatric disorder may share the same rare variant as a risk factor. For example, evidence implicating structural variants at the regions 16p11.2 (33, 39), 22q11.2 (40, 41), 1q21 (27, 32, 42-44), and the genes Neurexin 1 (NRXN1) (34, 36, 37, 45, 46) and SH3 and multiple ankyrin repeat domains 3 (SHANK3) (47) in both SCZ and ASD support the hypothesis of shared biological pathways in these conditions. Similarly, the single genome-wide copy number variation study published to date in TS identified rare variants at the NRXN1 and catenin, alpha 3 (CTNNA3) loci, leading the authors to hypothesize an overlap of risk with both ASD and SCZ (48).
We evaluated 460 unrelated affected individuals (including 148 trios) and 1131 control individuals (including 436 trios) using Illumina genome-wide SNP microarrays, paying particular attention to guarding against known confounds in association studies, including population stratification and batch effects (49). Moreover, the study design includes an evaluation of both transmitted and de novo CNVs, providing important opportunities to advance the understanding of the contribution of rare structural variation to this disorder.
METHODS AND MATERIALS
Study Subjects
Patients who met Diagnostic and Statistical Manual of Mental Disorders, fourth edition, text revision (DSM-IV-TR) criteria for Tourette’s Disorder (50) and their parents, if available, were included. Two cohorts of Caucasian TS patients (n=645, including 248 trios) from independent studies in the United States and Netherlands were ascertained (see Methods in Supplement 1). Control subjects were comprised of unrelated children (n=546) and parents (n=1098) of European ancestry from the Simons Simplex Collection (SSC) who were extensively phenotyped (https://sfari.org/ssc-instruments) and showed no evidence of ASD (51), as well as a group of unrelated healthy subjects collected as part of a separate genetic study of intracranial aneurysms (YNIA, n=786) (Figure 1, Table S1 in Supplement 1).
Figure 1. Copy number variant (CNV) discovery, quality control, and annotation workflow.
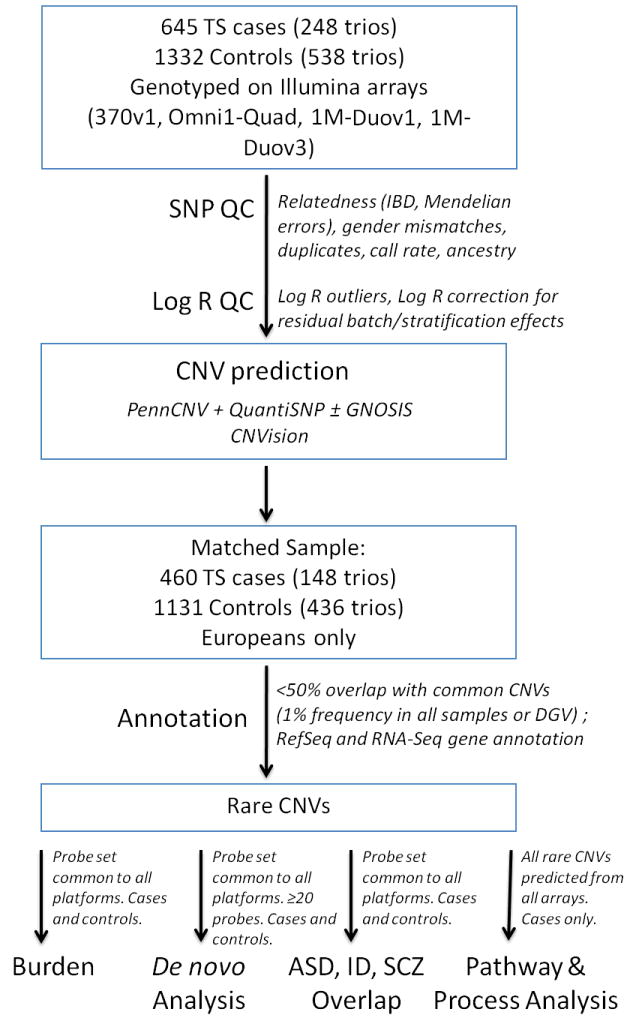
Tourette syndrome (TS) and control samples were genotyped on Illumina single nucleotide polymorphism (SNP) microarrays. Quality control was performed simultaneously in both cases and controls using SNP, LogR values, and CNV algorithm output. CNVs in remaining samples were annotated to determine rarity. Only CNVs meeting the definition of rare (occurring in <1% of all study samples and Database of Genomic Variants) were carried into further analysis for global burden, de novo burden, overlap with other neurodevelopmental disorders, and pathway analysis.
Single Nucleotide Polymorphism (SNP) Genotyping
Genomic DNA was extracted from peripheral blood or cell line lymphoblasts using standard protocols. TS subjects and controls were genotyped using the Illumina HumanCNV 370v1-duo, HumanOmni1-Quad, Human1M-Duo v1, and Human 1M-Duo v3 BeadChips, according to the standard Illumina protocol (see Methods in Supplement 1).
SNP Quality Control
Genotypes were analyzed using Plink (52) and removed from the analyses if: 1) sample call rate was less than 97%; 2) genotypes were inconsistent with recorded gender; or 3) Mendelian inconsistencies or cryptic relatedness were detected (see Methods in Supplement 1).
Population Outlier Exclusions
After removing individuals with cryptic relatedness, Golden Helix SNP and Variation Suite v7.4.0 (SVS; Golden Helix, Inc.) was used to perform a genotype principal component analysis (PCA) among all cases and controls and to plot derivative Log Ratio spread and Log Ratio average by chromosome in order to remove outlying samples (see Methods in Supplement 1).
Residual Batch Effect Control
Correction of LogR values to control for residual batch effects was performed using Golden Helix SVS (see Methods in Supplement 1). PCA-corrected data was used for all CNV predictions included in our analyses.
CNV Predictions
Genotypic data from all samples, including both cases and controls, were processed by three CNV detection algorithms: PennCNV (Rev. 220) (53), QuantiSNP (v1.1) (54), and GNOSIS (38). The results of all three algorithms were analyzed and merged using the program CNVision ((38) and http://www.cnvision.org) (Figure 1) (see Methods in Supplement 1).
CNV Quality Control
Samples were discarded from final analysis if they failed any one of the quality control checks for the three individual CNV algorithms (Figure 1) (see Methods in Supplement 1).
CNV Confirmation
Confirmations of all predicted de novo CNVs in TS cases were performed using whole-blood derived DNA (18 ng), evaluated using real-time qPCR analysis (see Methods in Supplement 1).
CNV Annotation
A CNV was classified as rare, genic, exonic, and/or brain expressed based on pre-determined criteria (see Methods in Supplement 1).
CNV Burden Analysis
To ensure comparable CNV detection from different array types, CNV predictions from the shared set of probes common to all array platforms (n=213,819) was used for all case-control comparisons. For the overall burden analysis, we compared the proportion of subjects in each group harboring at least one predicted rare CNV.
Relative frequencies of RefSeq genes, annotated as mapping within the boundaries of genic CNVs in TS and controls, were calculated to determine whether specific genes were overrepresented in TS.
To assess whether genes and CNVs previously associated with ASD, intellectual disability (ID), and SCZ were enriched in TS cases versus controls, we compared previously defined gene lists (55, 56) to one generated from all rare genic CNVs in TS cases, predicted from the shared set of probes common to all array platforms (see Methods in Supplement 1).
Biological Pathway and Processes Enrichment Analysis
As these analyses do not involve a case-control comparison, our input included all genes corresponding to all high confidence CNVs predicted from cases using full probe sets from each array. This provided for maximal detection resolution from each array-type. We determined whether RefSeq genes mapping within rare exonic CNV intervals in TS subjects were over-represented in one or more biological processes or functionally defined pathways compared to all known genes, using MetaCore (MC), PANTHER (PA) and Ingenuity Pathway Analysis (IPA) (see Methods in Supplement 1).
RESULTS
After completing quality control and case-control matching, a total of 460 cases (148 trios) and 1131 controls (436 trios) were included in our analyses (Figure 1; see also Table S1, Figure S1-2 in Supplement 1). PCA of genotypic data yielded a genomic inflation factor (λ) of 1.14. We further corrected for residual differences in batch effects by adjusting LogR values for 9 principal components (Figure S3 in Supplement 1). From this matched sample, using the set of probes shared by all array platforms, we identified a total of 745 rare high confidence CNVs in cases and 1910 in controls (Table S2 in Supplement 2). Among these, we found 4 cases (2.7%) and 5 controls (1.1%) carrying at last one rare de novo CNV (Table S3 in Supplement 1). The rate of de novo CNVs in our control sample was highly consistent with that found in prior studies (38, 57).
Overall, we found no statistically significant differences in rare CNV burden in cases versus controls, even using a nominal uncorrected p-value of 0.05. This result did not vary when we examined de novo or transmitted CNVs or proportions of deletions or duplications separately (Figure S4, Table S3 in Supplement 1). We conducted an exploratory analysis evaluating multiple types of rare CNVs, including those that map to coding regions of the genome, those that overlap exons, and those that overlap genes expressed in human brain. No subgroup was significantly overrepresented in cases versus controls (Figure S4, Table S3 in Supplement 1). We did observe larger (mean size: 14Mb versus 662.9kb) and more gene-rich (mean gene number: 28.0 versus 6.4 genes per CNV) rare de novo CNVs in cases versus controls, a pattern seen in other neuropsychiatric disorders (38). However, these differences were not statistically significant (p=0.3 using two-tailed t-test for each comparison), likely due to the small number of observations in each group (n=5 in cases, n=4 in controls). Similarly, neither mean CNV size (220kb versus 150kb) nor mean gene content (1.8 versus 1.2 genes per CNV) was significantly different between cases and controls when all (de novo and transmitted) rare CNVs were considered together (p=0.3 using two-tailed t-test for each comparison).
We next evaluated whether the total group of genes mapping within rare genic CNV intervals in cases pointed to the involvement of particular biological pathways or processes. As described above, to maximize the sensitivity of this analysis, we considered all predicted high confidence rare exonic CNVs in cases derived from full array probe sets. We relied on three programs, MetaCore (MC), PANTHER (PA), and Ingenuity Pathway Analysis (IPA). A list of all 2,646 genes identified in TS cases was used as input for each algorithm. The five pathways showing greatest significance (smallest p-values), relative to all known genes in the genome, included pathways involving ubiquitin (MC, p=9.6 × 10-5), GABA receptor signaling (MC, p=1.9 × 10-4), sphingolipid metabolism (IPA, p=3.1 × 10-4), histamine receptor signaling (MC, p=5.8 × 10-4, H1R), and alpha-2 adrenergic receptor function (MC, p=7.7 × 10-4). Overlapping results among differing algorithms occurred for GABA (IPA, p=6.0 × 10-3; MC, p=1.9 × 10-4) and histamine (MC, p=5.8×10-4, H1R; PA, p=0.016A, H2R; p=0.058, H1R) signaling pathways only (Table 1; Figure S5 in Supplement 1).
Table 1.
Pathway analysis of 2,646 genes within exonic rare copy number variants (CNV) in Tourette syndrome (TS).
| GeneGo (Metacore) Pathway Maps | p-value | Ratio | RefSeq Genes | # TS subjects |
|---|---|---|---|---|
| Proteolysis_Role of Parkin in the Ubiquitin-Proteasomal Pathway | 9.6 × 10-5 | 6/24 | CUL1, PSMD13, PARK2, SEPT5, SIAH1, TUBB4Q, TUBB8, UBE2J1 | 10 |
| Neurophysiological process_GABA-A receptor life cycle | 1.9 × 10-4 | 6/27 | CLTB, GABRA1, GABRA6, GABRB1, GABRB2, GABRD, GABRG1, GABRG2, TUBB4Q, TUBB8 | 9 |
| Cell adhesion_Histamine H1 receptor signaling in the interruption of cell barrier | 5.8 × 10-4 | 7/45 | CTNNA3, GNB4, GNG7, ITPR1, MYH11, MYLK3, OCLN, PPP1R12A, TLN1 | 17 |
| Transport_Alpha-2 adrenergic receptor regulation of ion channels | 7.7 × 10-4 | 7/47 | CACNA1C, GNB4, GNG7, ITPR1, PRKACB, PRKCZ | 11 |
| Regulation of metabolism_Bile acids regulation of glucose and lipid metabolism via FXR | 0.0012 | 6/37 | ACACA, ACACB, APOC4, FOXO1, ADD1 | 5 |
| Muscle contraction_ GPCRs in the regulation of smooth muscle tone | 0.0015 | 9/83 | ADRB2, CACNA1C, CAMK1, ITPR1, MYH11, MYLK3, OXT, OXTR, PPP1R12A, PRKACB | 10 |
| Cytoskeleton remodeling_Role of PKA in cytoskeleton reorganization | 0.0018 | 6/40 | ADD1, GNB4, GNG7, ITPR1, MYLK3, PPP1R12A, PRKACB | 12 |
| Regulation of lipid metabolism_Regulation of acetyl-CoA carboxylase 1 activity in lipogenic tissue | 0.0019 | 4/17 | ACACA, ACACB, PRKAB2, ADD1 | 4 |
| Muscle contraction_S1P2 receptor-mediated smooth muscle contraction | 0.0026 | 5/30 | GNB4, GNG7, ITPR1, MYLK3, PPP1R12A, PRKCZ | 10 |
| Cell adhesion_Gap junctions | 0.0026 | 5/30 | GJA5, GJA8, OCLN, PRKCZ, TUBB4Q, TUBB8 | 8 |
| PANTHER Pathways | p-value | Ratio | RefSeq Genes | # TS |
| Beta2 adrenergic receptor signaling | 0.0047 | 8/44 | PRKX, CACNA1C, GNB4, SNAP29, PRKACB, ADRB2, GNB1L, GNG7 | 12 |
| Coenzyme A biosynthesis | 0.0054 | 3/6 | PANK2, PANK3, PANK4 | 3 |
| 5HT4 type receptor mediated signaling | 0.010 | 6/31 | HTT, HTR4, GNB4, SNAP29, GNB1L, GNG7 | 9 |
| Muscarinic acetylcholine receptor 2 and 4 signaling | 0.011 | 9/62 | PRKX, SLC18A3, CACNA2D1, GNB4, SNAP29, PRKACB, CHAT, GNB1L, GNG7 | 12 |
| Beta1 adrenergic receptor signaling | 0.016 | 7/44 | PRKX, CACNA1C, GNB4, SNAP29, PRKACB, GNB1L, GNG7 | 11 |
| 5HT1 type receptor mediated signaling | 0.016 | 7/44 | PRKX, HTT, GNB4, SNAP29, PRKACB, GNB1L, GNG7 | 11 |
| Histamine H2 receptor mediated signaling | 0.016 | 5/25 | PRKX, GNB4, PRKACB, GNB1L, GNG7 | 10 |
| Heterotrimeric G-protein signaling pathway-rod outer segment phototransduction | 0.017 | 7/45 | PRKX, GRK4, GRK6, GNB4, PRKACB, GNB1L, GNG7 | 13 |
| Oxytocin receptor mediated signaling | 0.026 | 8/60 | PRKCZ, CACNA1C, OXTR, GNB4, SNAP29, PLCH2, GNB1L, GNG7 | 11 |
| Muscarinic acetylcholine receptor 1 and 3 signaling pathway | 0.028 | 8/61 | ITPR1, PRKCZ, SLC18A3, GNB4, SNAP29, CHAT, GNB1L, GNG7 | 10 |
| Heterotrimeric G-protein signaling pathway-Gi alpha and Gs alpha mediated pathway | 0.036 | 16/166 | PRKX, GRM7, ADRA18, CLTCL1, RGS14, GRK4, GRK6, HTR4, GNB4, CLTB, CKB, PRKACB, ADRB2, HTR7, GNB1L, GNG7 | 17 |
| 5HT2 type receptor mediated signaling | 0.051 | 8/69 | PRKCZ, CACNA1C, HTT, GNB4, SNAP29, PLCH2, GNB1L, GNG7 | 11 |
| Histamine H1 receptor mediated signaling | 0.058 | 6/47 | ITPR1, PRKCA, GNB4, PLCH2, GNB1L, GNG7 | 10 |
| IPA Pathways | p-value | Ratio | RefSeq Genes | # TS |
| Sphingolipid Metabolism | 3.1 × 10-4 | 14/82 | ARSB, ARSD, ARSE, ARSF, ARSH, ASAH2, GBA3, GLB1L2, GLB1L3, PPAP2A, SGMS1, SGPP1, SPHK1, SULF1 | 14 |
| Glycosaminoglycan Degradation | 0.0010 | 8/36 | ARSB, FGFRL1, GBA3, GLB1L2, GLB1L3, HEXB, IDUA, SULF1 | 11 |
| GABA Receptor Signaling | 0.0060 | 8/47 | AP1B1, GABRA1, GABRA6, GABRB1, GABRB2, GABRD, GABRG1, GABRG2 | 6 |
| N-Glycan Degradation | 0.027 | 5/29 | EDEM1, GBA3, GLB1L2, GLB1L3, HEXB | 5 |
| Neuropathic Pain Signaling in Dorsal Horn Nucleus | 0.033 | 11/102 | CAMK1, GRIA1, GRINA, GRM7, ITPR1, PIK3C3, PLCH2, PRKACB, PRKCZ, TAC1, TACR1 | 14 |
The assessment of relative representation is compared to the entire genome, using the programs MetaCore (GeneGo, Inc), PANTHER, and Ingenuity Pathway Analysis (IPA). Ratios indicate the proportion of genes (PANTHER and IPA) or network objects (GeneGo Metacore) in known biological pathways that coincide with rare TS gene CNVs. The top five pathways with lowest p-values are shaded grey. Pathways with overlap between algorithms are highlighted in bold. “#TS” is number of unique TS subjects contributing at least one gene to the pathway result.
Recognizing that results from pathway analysis tools can be biased by the clustering of gene families within structural variants (58), we next removed CNVs containing large numbers of genes, defined by exceeding three standard deviations from the mean of all genic TS CNVs (mean = 2.3, SD = 13.3). Two duplications, one involving 447 RefSeq genes on chromosome 5q and the other involving 56 genes on chromosome 22q11, met this criterion. An analysis of the remaining list of 2,143 genes resulted in the elimination of the GABA receptor signaling findings and ranked the histamine receptor signaling pathway as having the smallest relative p-value (Table S4 in Supplement 1). We found no evidence that enrichment of histamine receptor signaling could have been the result of similar clustering. The histamine pathways identified by both PA and MC were composed of between 4 and 9 genes from rare CNVs found in 10 (PA) or 17 (MC) TS subjects (Table 1; Table S4 in Supplement 1). No single CNV contributed more than one gene to the histamine pathways results.
Using this list of 2,143 genes, we also examined the enrichment of all TS rare CNVs in biological processes relative to the genome using MC and PA. Significant p-values (<0.05) with agreement between algorithms was observed for “axon guidance”, “cell adhesion”, “nervous system development” and “synaptic structure and function” (Table S5 in Supplement 1).
As multiple recent studies suggested overlapping risks among divergent neuropsychiatric disorders, including ASD, SCZ, and ID (40, 44, 55), we next adopted previously defined gene lists from recent large scale studies of these disorders to evaluate genic overlap with TS (38, 55, 56). We found a significant enrichment of genes classified as “ASD implicated” (2% vs. 0.4%, p=0.006) and “ASD implicated + ASD candidates” (10.2% vs. 5.7%, p=0.002) among individuals with TS versus matched controls (Table 2, Figure 2). This enrichment withstands correction for multiple comparisons. In contrast, no significant overlap was observed with either ID (p=0.9) or SCZ (p=0.2) gene lists (Figure 2).
Table 2.
Tourette syndrome (TS) rare copy number variants (CNVs) in genes and regions previously associated with autism spectrum disorders (ASD). Underlined genes showed recurrence and relative enrichment in TS versus controls (see Table 3).
| Gene/Regiona | Location (NCBI 36/hg18) | All [exonic] (de novo)b | Deletions [exonic] (de novo) | Duplications [exonic] (de novo) | |||
|---|---|---|---|---|---|---|---|
| TS | Controls | TS | Controls | TS | Controls | ||
| GPR89A | chr1:144,475,952-144,538,460 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| DISC1 | chr1:229,829,184-230,243,641 | 1 [1] | 0 | 0 | 0 | 1 [1] | 0 |
|
| |||||||
| GALNT13 | chr2:154,436,672-155,018,735 | 1 [1] | 1 [1] | 1 [1] | 0 | 0 | 1 [1] |
|
| |||||||
| NRXN1 | chr2:50,000,992-51,113,178 | 3 [2] | 2 [1] | 3 [2] | 2 [1] | 0 | 0 |
|
| |||||||
| CNTN4 | chr3:2,117,247-3,074,645 | 1 [1] | 4 [2] | 0 | 2 [0] | 1 [1] | 2 [2] |
|
| |||||||
| FHIT | chr3:59,710,076-61,212,173 | 12 [1] | 5 [1] | 12c [1] | 4 [0] | 0 | 1 [1] |
|
| |||||||
| OXTR | chr3:8,767,095-8,786,300 | 1 [1] | 0 | 0 | 0 | 1 [1] | 0 |
|
| |||||||
| GABRG1 | chr4:45,732,544-45,820,839 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| GABRA4 | chr4:46,615,674-46,690,337 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| GABRB1 | chr4:46,728,336-47,123,202 | 1 [1] | 1 [1] | 0 | 0 | 1 [1] | 1 [1] |
|
| |||||||
| CTNND2 | chr5:11,024,952-11,957,110 | 1 [1] | 0 | 1 [1] | 0 | 0 | 0 |
|
| |||||||
| KLHL3 | chr5:136,981,088-137,099,678 | 1 [1] (1) | 0 | 0 | 0 | 1 [1] (1) | 0 |
|
| |||||||
| NSD1 | chr5:176,493,439-176,659,820 | 1 [1] (1) | 0 | 0 | 0 | 1 [1] (1) | 0 |
|
| |||||||
| CDH18 | chr5:19,508,912-20,017,044 | 1 [1] | 1 [1] | 1 [1] | 1 [1] | 0 | 0 |
|
| |||||||
| CDH10 | chr5:24,522,967-24,680,668 | 0 | 1 [1] | 0 | 1 [1] | 0 | 0 |
|
| |||||||
| SEMA5A | chr5:9,088,138-9,599,233 | 1 [1] | 0 | 1 [1] | 0 | 0 | 0 |
|
| |||||||
| PARK2 | chr6:161,688,580-163,068,824 | 5 [1] | 18 [12] | 4 [0] | 8 [4] | 1 [1] | 10 [8] |
|
| |||||||
| GRM8 | chr7:125,865,888-126,670,805 | 0 | 1 [1] | 0 | 1 [1] | 0 | 0 |
|
| |||||||
| CNTNAP2 | chr7:145,444,386-147,749,019 | 2 [0] | 0 | 2 [0] | 0 | 0 | 0 |
|
| |||||||
| TMEM195 | chr7:15,206,468-15,568,165 | 0 | 1 [1] | 0 | 1 [1] | 0 | 0 |
|
| |||||||
| DPP6 | chr7:153,380,710-154,316,928 | 1 [1] | 5 [5] | 0 | 0 | 1 [1] | 5 [5] |
|
| |||||||
| AUTS2 | chr7:68,701,841-69,895,821 | 2 [1] | 1 [1] | 2 [1] | 1 [1] | 0 | 0 |
|
| |||||||
| RB1CC1 | chr8:53,697,571-53,789,579 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| ASTN2 | chr9:118,227,328-119,217,138 | 2 [1] | 0 | 1 [1] | 0 | 1 [0] | 0 |
|
| |||||||
| KCNMA1 | chr10:78,299,368-79,067,583 | 1 [0] | 0 | 1 [0] | 0 | 0 | 0 |
|
| |||||||
| CACNA1C | chr12:2,032,677-2,677,376 | 1 [1] | 0 | 1 [1] | 0 | 0 | 0 |
|
| |||||||
| DUOXA1 | chr15:43,196,978-43,209,349 | 1 [1] | 0 | 0 | 0 | 1 [1] | 0 |
|
| |||||||
| A2BP1 | chr16:7,322,752-7,702,500 | 2 [0] | 5 [1] | 1 [0] | 2 [1] | 1 [0] | 3 [0] |
|
| |||||||
| CDH13 | chr16:81,218,079-82,387,700 | 1 [1] | 1 [0] | 1 [1] | 1 [0] | 0 | 0 |
|
| |||||||
| NF1 | chr17:26,446,121-26,728,821 | 1 [1] | 0 | 1 [1] | 0 | 0 | 0 |
|
| |||||||
| MACROD2 | chr20:13,924,146-15,981,841 | 9 [1] | 13 [3] | 9 [1] | 13 [3] | 0 | 0 |
|
| |||||||
| PAK7 | chr20:9,466,037-9,767,687 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| PDE9A | chr21:42,946,931-43,068,687 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| WDR4 | chr21:43,136,273-43,172,747 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| TBX1 | chr22:18,124,226-18,134,855 | 1 [1] (1) | 1 [1] (0) | 0 | 0 | 1 [1] (1) | 1 [1] (0) |
|
| |||||||
| ADSL | chr22:39,072,450-39,092,521 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| 16p11.2 | chr16:29,550,000-30,200,000 | 0 | 1 [1] | 0 | 0 | 0 | 1 [1] |
|
| |||||||
| 22q11.21 | chr22:16,926,349-20,666,469 | 1 [1] (1) | 1 [1] (0) | 0 | 0 | 1 [1] (1) | 1 [1] (0) |
CNVs are included if there is any overlap with an ASD gene or with ≥50% of an ASD region
De novo CNV count in parentheses
Includes one homozygous deletion
Figure 2. Rare copy number variant (CNV) burden in known autism spectrum disorder (ASD), intellectual disability (ID), and schizophrenia (SCZ) genes/regions.
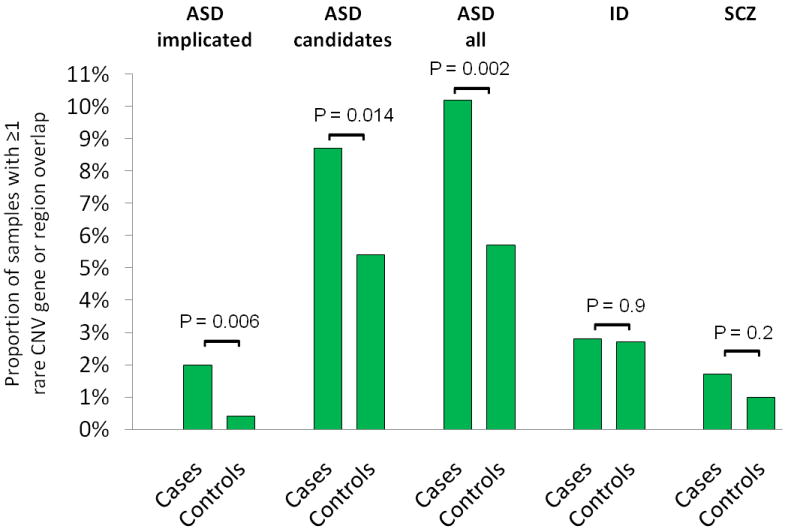
P-values were calculated using two-tailed Fisher exact test and are shown for each comparison between cases and controls. Comparisons were made for proportion of samples with CNV overlap with ASD implicated genes, ASD candidate genes, all (implicated + candidate) ASD genes, ID genes, and SCZ genes. Threshold for significance using a standard Bonferroni approach is p<0.013. TS subjects show significantly more overlap for ASD genes compared to controls, but no significant difference for ID or SCZ genes.
We next evaluated both CNV intervals and RefSeq transcripts mapping within rare CNVs that were recurrently observed as restricted to or over-represented in cases (Table 3). To avoid biasing our interpretation, we also evaluated a randomly selected subset of controls of equal size (N=460) and identified genes that were either restricted to or overrepresented in unaffected individuals (n=41 genes, data not shown). While these could potentially include protective alleles, we reasoned that the majority would have no association with disease and consequently, we added this number to the total found in cases (N=26) as the basis for a correction for multiple comparisons in evaluating association (N=67 total events). To ensure comparable CNV detection from different array types, CNVs in both cases and controls were detected using probes shared among the arrays (n=213,819).
Table 3.
Recurrent rare copy number variant (CNV) gene overlap with enrichment in Tourette syndrome (TS) versus controls. Underlined genes have been associated with autism spectrum disorders (ASD) (see Table 2).
| Gene | Location (NCBI 36/hg18) | Count in TS | Freq in TS (N=460) | Del/Dup | Count in Controls | Freq in Controls (N=1131) | Del/Dup | Overall Enrichme nt in TSa | p-valueb |
|---|---|---|---|---|---|---|---|---|---|
| FHIT | chr3:59,710,076-61,212,173 | 12 | 2.61% | 12 c/0 | 5 | 0.44% | 4/1 | 5.90 | 4.2×10-4 |
| LEPREL1 | chr3:191,157,316-191,321,412 | 5 | 1.09% | 5/0 | 1 | 0.09% | 1/0 | 12.29 | 0.0091 |
| DAP | chr5:10,732,342-10,814,387 | 3 | 0.65% | 1/2 | 0 | 0.00% | 0/0 | specific | 0.024 |
| HGSNAT | chr8:43,114,749-43,177,127 | 3 | 0.65% | 0/3 | 0 | 0.00% | 0/0 | specific | 0.024 |
| POTEA | chr8:43,266,742-43,337,485 | 3 | 0.65% | 0/3 | 0 | 0.00% | 0/0 | specific | 0.024 |
| LOC285692 | chr5:9,694,427-9,956,936 | 12 | 2.61% | 12/0 | 13 | 1.15% | 13/0 | 2.27 | 0.040 |
| AGBL4 | chr1:48,771,114-50,262,213 | 2 | 0.43% | 2/0 | 0 | 0.00% | 0/0 | specific | 0.083 |
| BHLHE40 | chr3:4,996,208-5,001,861 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| ARL8B | chr3:5,138,930-5,197,601 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| KCNIP4 | chr4:20,339,337-21,308,416 | 2 | 0.43% | 2/0 | 0 | 0.00% | 0/0 | specific | 0.083 |
| MYO10 | chr5:16,715,016-16,989,385 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
| PCDHB12 | chr5:140,568,475-140,571,882 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
| PCDHB13 | chr5:140,573,693-140,577,177 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
| PPP2R2B | chr5:145,949,261-146,415,893 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
| CNTNAP2 | chr7:145,444,386-147,749,019 | 2 | 0.43% | 2/0 | 0 | 0.00% | 0/0 | specific | 0.083 |
| PTPRN2 | chr7:157,024,516-158,073,179 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| PPP1R3B | chr8:9,031,177-9,045,630 | 2 | 0.43% | 2/0 | 0 | 0.00% | 0/0 | specific | 0.083 |
| SGCZ | chr8:13,991,744-15,140,163 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
| TEK | chr9:27,099,147-27,220,172 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
| ASTN2 | chr9:118,227,328-119,217,138 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
| COL5A1 | chr9:136,673,473-136,876,509 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| PIWIL1 | chr12:129,388,567-129,422,826 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| RIMBP2 | chr12:129,446,634-129,568,415 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| EPSTI1 | chr13:42,360,122-42,464,377 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| DNAJC15 | chr13:42,495,362-42,581,306 | 2 | 0.43% | 0/2 | 0 | 0.00% | 0/0 | specific | 0.083 |
| PIWIL3 | chr22:23,445,001-23,500,683 | 2 | 0.43% | 1/1 | 0 | 0.00% | 0/0 | specific | 0.083 |
CNVs in TS and controls were detected using probes shared among all array platforms (n=213,819).
Del = deletions, Dup = duplications
Relative frequency = frequency in TS / frequency in controls
Fisher exact test, two-tailed; p<7.5×10-4 is Bonferroni-adjusted significance level for 67 comparisons
One CNV is a homozygous deletion, confirmed by qPCR
One CNV is a de novo duplication
Only a single gene, fragile histidine triad (FHIT), reached statistical significance based on this corrected p-value threshold of 7.5 × 10-4 (Table 3; Figure S6 in Supplement 1). However, a detailed inspection of this locus showed that the majority of CNVs in this interval had highly similar start and stop coordinates, despite the absence of flanking segmental duplications. In addition, one affected individual was homozygous for what appeared to be the identical CNV (Figure S6 in Supplement 1). These observations raised the prospect of residual population stratification. Consequently, we further evaluated ancestral clustering in cases and controls (59, 60) (see Methods in Supplement 1) and found that all carriers of the identical FHIT CNV (N=17), showed significant ancestral clustering, compared to a randomly chosen control sample of equal size (p=0.028) (Figure S7 in Supplement 1). Interestingly, when FHIT CNV carriers were grouped based on affected status, the TS carriers (N=12) were more similar in ancestry than a random sample of non-carriers (p=0.002) while control carriers (N=5) were not (p=0.58). While this last observation deserves further scrutiny, overall these results suggest that the FHIT association with TS identified in our cohort is most likely the result of population stratification.
Finally, we reasoned that while the overall number of de novo structural variations identified might not differ between cases and controls, specific CNVs might nonetheless be conferring disease risk. Consequently, we focused on large rare de novo exonic variants, given both the trend toward larger and more gene-rich de novo CNVs in cases versus controls in this study and strong evidence from studies in ASD, demonstrating that these CNVs carry significant risks (27, 38, 61). To ensure that we did not overlook such an event due to reliance on the reduced “consensus” probe sets, we re-analyzed cases for de novo CNVs using predictions derived from the full array probe sets. Our results were identical. We detected four de novo CNVs which were subsequently all confirmed using qPCR. The largest of these is a heterozygous duplication that spans 51.8Mb and 447 RefSeq genes (Figure 3A) on chromosome 5q (chr5: 127,500,000-179,295,570). Three other de novo variants were detected: a 316 kb duplication at 6p25.3 (chr6: 744,618-1,060,897; 1 RefSeq gene), a 1.2 Mb deletion at 20p13 (chr20: 2,758,098-3,942,609; 27 RefSeq genes) (Figure 3B), and a 2.5 Mb duplication at 22q11.21 (chr22:17,269,490-19,792,353; 56 RefSeq genes) (Figure 3C). Though the number of events evaluated here is small, the distribution of CNV size and gene content in TS cases as well as controls in this study is similar to the pattern we observed in a recent study of ASD (Figure 4), in which large highly genic de novo events were essentially restricted to affected individuals (38).
Figure 3. Large de novo copy number variants (CNV) in Tourette syndrome (TS) subjects.
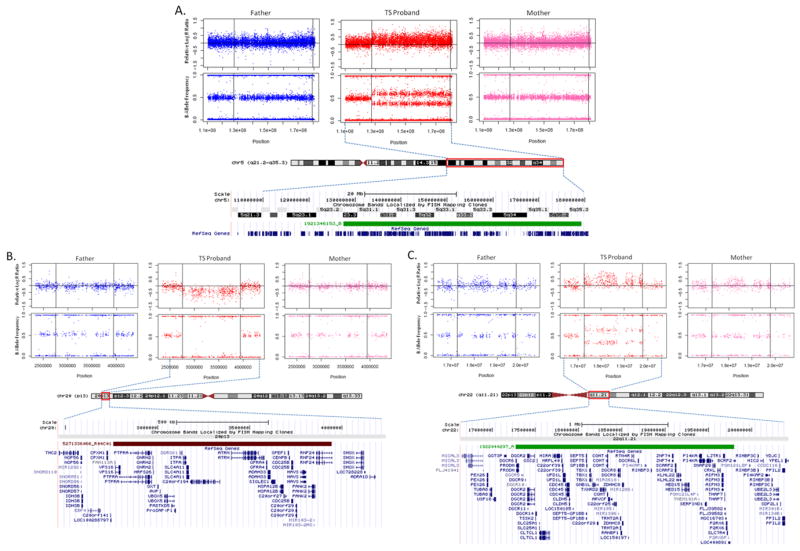
Plots of relative microarray probe Log R Ratios (LRR) and B Allele Frequencies (BAF) in unaffected father, TS proband, and unaffected mother. CNV regions are bounded by horizontal lines in LRR and BAF plots, outlined in red on chromosome ideograms, and represented by green (duplication) or red (deletion) below chromosome bands. Data is shown for (A) a 51.8 Mb heterozygous duplication on chromosome 5 (chr5: 127,500,000-179,295,570), containing 447 RefSeq transcripts; (B) a 1.2 Mb region on chromosome 20p13 (chr20: 2,758,098-3,942,609), containing 27 RefSeq transcripts; and (C) a 2.5 Mb region on chromosome 22q11.21 (chr22:17,269,490-19,792,353), containing 56 RefSeq transcripts. All de novo deletions were confirmed by qPCR.
Figure 4. Distribution of number of RefSeq genes overlapped by rare de novo copy number variants (CNV) in Tourette syndrome (TS) probands, autism probands, and sibling controls.
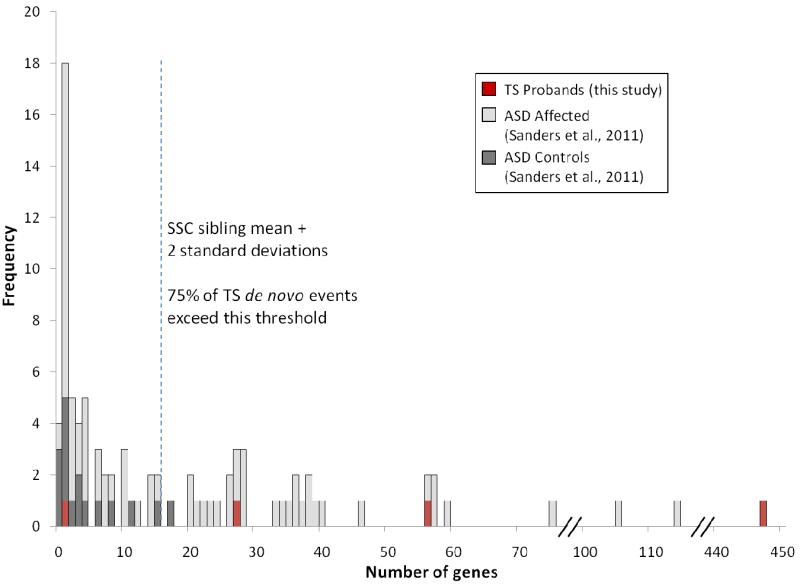
Dashed line represents two standard deviations from the mean number of genes overlapped by de novo CNVs in a large study of unaffected sibling control subjects (dark grey). Number of genes for TS cases (red) and autism probands (light grey) (38) are also plotted. Three of four TS de novo CNVs (this study) and 46% of autism proband de novo CNVs are beyond this threshold.
DISCUSSION
Our data supports recent findings implicating histaminergic neurotransmission in the etiology or modulation of tics and highlights the potential involvement of GABAergic mechanisms as well. In addition, the results reinforce the notion of shared genetic risks among clinically-distinct syndromes, in this case ASD and tic disorders, and identify three novel, large, rare, genic, de novo CNVs that are likely carrying risk in the individuals in which they were identified, based on their de novo status and high gene content relative to controls.
Analysis of genes mapping within rare genic TS CNVs show significant enrichment for a variety of pathways involved in CNS development and function (Table 1; Figure S6 in Supplement 1). However, after correcting for the impact of gene family clusters mapping within large CNVs, only histaminergic signaling was identified by multiple algorithms. The result is a particularly intriguing, given our recent discovery of a dense TS pedigree segregating a rare loss-of-function mutation in the HDC gene (22), the rate-limiting enzyme in histamine biosynthesis. HA signaling in the central nervous system is mediated by four G protein-coupled receptors, located both presynaptically (predominantly H3 as well as H4) and postsynaptically (H1-H3). Presynaptic HA receptors regulate not only the release of HA, but also a variety of other neurotransmitters, including dopamine. Several lines of evidence suggest that HA acts in a counter-regulatory fashion, with increased HA resulting in decreased DA signaling and vice versa (62, 63). H2 and H3 receptors are enriched in the striatum and cortex, regions of the brain implicated in TS (64), and studies of rodents with decreased brain HA show increased sensitivity to stereotypies when administered DA agonists (65). As H3R antagonists and inverse agonists are in late-stage clinical development and being considered for other neuropsychiatric indications (66, 67), there may well be near-term opportunities to translate a deeper understanding of the relationship of HA and tics into novel treatment approaches.
As noted, our initial pathway analysis also identified enrichment of GABA-A receptor life cycle and GABA receptor signaling genes; however, the finding was accounted for by a single large multigenic de novo CNV (Figure 3A). Consequently, the implications are not clear: on the one hand, the clustering of gene families in genomic segments, as well as the relatively large target sizes of neuronal genes, has the potential to bias pathway analyses (58); on the other hand, there is strong evidence from prior studies of neurodevelopmental disorders, including ASD (38), that large de novo genic events are very likely to be pathogenic. As 447 genes map within the chromosome 5 de novo event, conclusions regarding which gene or genes may be contributing to TS in this instance are not possible. However, in light of two recent post mortem studies highlighting the GABA system in TS (10, 11), further study of genes within this interval and attention to the GABA system in general will be of particular interest in large-scale sequencing and follow-up CNV studies.
Both the ubiquination pathway (p=9.6 × 10-5) (Table 1) and the cell adhesion process (Table S5 in Supplement 1) were significantly enriched in our cohort, and both have been identified by similar pathway analyses of ASD cohorts (37). Our findings with regard to axonal guidance, neuronal development, and synaptic structure and function processes are likewise consistent with prior studies of ASD (34, 37, 55).
In a similar vein, our comparison of all genes mapping within rare TS CNVs to genes previously implicated in ID, ASD, and SCZ support the hypothesis of shared genetic risks, but only for ASD and TS (Table 2, Figure 2). Consistent with this, several case reports and cohort studies point to an increased rate of comorbidity between ASD and TS or tic disorders (68-72). A prior genome-wide CNV study of TS (48) supported this finding and also suggested a convergence of risks among TS and SCZ as well. We did not find evidence for the latter. However, it is important to note that the previous study did not have a sufficient number of observations to conduct a meaningful statistical analysis and, conversely, our study did indeed find genes within TS CNVs that have previously been identified as putative SCZ risks, including CNTNAP2 and ASTN2 (Tables 2 and 3). As we did not have comprehensive phenotypic data regarding social disability syndromes or psychosis in our TS cohort, it was not possible for us to assess phenotypic overlap in individuals with putatively overlapping genetic risks. However, recent findings in ASD, SCZ, ID all suggest that the diversity of phenotypic outcomes from apparently identical mutations is unlikely to be explained entirely by overt “co-morbidity” (73).
After carefully matching cases and controls using genome-wide genotyping data and removing outliers, we calculated a genomic inflation factor (λ) of 1.14. While there is no consensus regarding a maximum value for CNV studies - in fact, few CNV studies have addressed this issue – our value is slightly above that generally accepted for genome-wide association studies (74). To protect against potential liability for Type I error, we looked for residual population structure in any putative positive associations arising from our case-control analyses and identified that this likely played a role in the observed FHIT finding (Figure S7 in Supplement 1).
Interestingly, we did not find an overall excess of rare CNVs in TS cases versus controls, as has been observed for ASD and SCZ (33, 55). However, it is also worth noting that in our analysis of more than 1000 simplex autism families, the significant increase in the burden of all rare CNVs was entirely accounted for by the contribution of a proportionally small number of large multigenic de novo events. In the present study, we observed a similar trend, with the mean size of rare de novo events in cases approximately 20x larger than those in controls and encompassing about four times as many genes. However, in the current study these differences were not statistically significant, a plausible consequence of our small sample size. A larger family cohort will be required to more definitively test the hypothesis that de novo CNVs, and particularly large multi-genic events, carry risk for TS.
Also of interest is a 2.5 Mb rare de novo duplication on chromosome 22q11.2 which corresponds to the recurrent heterozygous deletion that results in velocardiofacial syndrome (Table 2, Figure 3C). Details regarding the clinical features of this individual were described in an earlier case report (75). There is convincing evidence for an increased rate of psychosis and ID in individuals carrying the 22q11.2 deletion (76-78). Moreover, data from multiple recent studies of de novo CNVs in this region in individuals with ASD has shown an association, but only when duplications and deletions are considered together (38). Overall, there is not sufficient data to determine whether this is justifiable, as it is not clear whether 22.q11.2 duplications are independently associated with a developmental phenotype (79-81) or represent an incidental finding. However, recent evidence with regard to the contribution of deletions and duplications at 16p11.2 to ASD and a range of other conditions suggests that duplications of the 22q11.2 locus certainly warrant further evaluation with regard to their role in the risk for a variety of developmental outcomes, including TS (38).
Several limitations of our study deserve mention. First, the size of our patient cohort was relatively small, particularly with regard to the detection and analysis de novo CNVs. This issue was exacerbated by the removal of 185 probands during quality control steps, including matching for ancestry. On balance, though, the aforementioned findings with regard to the FHIT locus underscore the importance of rigorous control for population stratification. In addition, the reliance on differing arrays resulted in a smaller sample and lower resolution of CNV detection in order to avoid the significant confound of batch effects. In light of the fact that an increased burden of de novo CNVs in ASD and SCZ was initially identified using arrays with less than half the probe number present in the consensus set used in the current study (28, 35), it is unlikely that the failure to detect a statistically significant difference, if it is a false negative finding, was the result of insufficient resolution, but more likely the limitations imposed by sample size. Finally, our use of a subgroup of controls related to individuals with ASD could have resulted in the presence of affected individuals in our control sample. In this case, the liability would have been for Type II error, given the known increased burden of rare CNVs among individuals with ASD. Despite these issues, our data clearly demonstrate the value of pursuing rare variant and CNV analyses in TS, and highlight the pressing need for studies of larger cohorts to replicate, clarify, and extend these findings.
Supplementary Material
Acknowledgments
This study was funded by NIH grants: R01MH092289 (MWS), R01MH092293 (GAH and JAT), R01 MH092520 (DG) R01NS056276 (MWS), R01MH061940 (JFL), K05MH076273 (JFL), R25MH077823 (JFL), UL1RR024139 (SM), U24NS051869 (SM). Additional funding was provided by the Shepherd Foundation (MWS), the Overlook International Fund (MWS), the Yale Program on Neurogenetics (MG, MWS), and the New Jersey Center for Tourette Syndrome & Associated Disorders (through New Jersey Department of Health and Senior Services: 09-1839-FS-N-0) (GAH and JAT).
Footnotes
FINANCIAL DISCLOSURES Dr. Leckman has received support from the following: NIH (salary and research funding), Tourette Syndrome Association (research funding), Talecris Biotherapeutics (research funding), Klingenstein Third Generation Foundation (medical student fellowship program), C8Sciences (equity interest), John Wiley and Sons (book royalties), McGraw Hill (book royalties), Oxford University Press (book royalties). Dr. Gilbert has received research funding as a site investigator for pharmaceutical studies sponsored by Psyadon Pharmaceuticals and Otsuka Pharmaceuticals. He has received honoraria from the Tourette Syndrome Association/Centers for Disease Control and Prevention, the Movement Disorder Society, the American Academy of Neurology, and the American Academy of Pediatrics; serves on the medical advisory board for the Tourette Syndrome Association; writes board review questions for PREP SA (American Academy of Pediatrics); and has received research support from the NIH (NIMH R01 MH078160, NIMH R01 MH08185, and NINDS NS056276) and from the Cincinnati Children’s Hospital Research Foundation, the University of Cincinnati, and the Tourette Syndrome Association. Dr. Hoekstra has received research funding from National (the Netherlands Organization for Scientific Research and the Netherlands Organization for Health Research and Development) and European Science Foundations (European Union seventh Framework Programme) over the last two years. He also received honoraria from Desitin, Lilly, and Shire. Dr. State holds a patent with Yale University regarding the contribution of specific rare mutations in the gene CNTNAP2 and autism spectrum disorders.
Drs. Fernandez, Sanders, Ercan-Sencicek, Kim, Song, Yasuno, Ho, Bilguvar, Glessner, Chu, King, Heiman, Tischfield, Devlin, Hakkonarson, Mane, Gunel, Ms. Yurkiewicz and Raubeson, and Mr. Fishman have no biomedical financial interests or potential conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Centers for Disease Control and Prevention. Prevalence of Diagnosed Tourette Syndrome in Persons Aged 6–17 Years — United States. MMWR. 2007:581–585. [PubMed] [Google Scholar]
- 2.Robertson MM. The prevalence and epidemiology of Gilles de la Tourette syndrome. Part 1: the epidemiological and prevalence studies. J Psychosom Res. 2008;65:461–472. doi: 10.1016/j.jpsychores.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Robertson MM, Eapen V, Cavanna AE. The international prevalence, epidemiology, and clinical phenomenology of Tourette syndrome: a cross-cultural perspective. J Psychosom Res. 2009;67:475–483. doi: 10.1016/j.jpsychores.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Bloch M, Peterson B, Scahill L, Otka J, Katsovich L, Zhang H, et al. Adulthood outcome of tic and obsessive-compulsive symptom severity in children with Tourette syndrome. Arch Pediatr Adolesc Med. 2006;160:65–69. doi: 10.1001/archpedi.160.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin H, Yeh C, Peterson B, Scahill L, Grantz H, Findley D, et al. Assessment of symptom exacerbations in a longitudinal study of children with Tourette’s syndrome or obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2002;41:1070–1077. doi: 10.1097/00004583-200209000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Leckman J, Zhang H, Vitale A, Lahnin F, Lynch K, Bondi C, et al. Course of tic severity in Tourette syndrome: the first two decades. Pediatrics. 1998;102:14–19. doi: 10.1542/peds.102.1.14. [DOI] [PubMed] [Google Scholar]
- 7.Robertson M, Banerjee S, Kurlan R, Cohen D, Leckman J, McMahon W, et al. The Tourette syndrome diagnostic confidence index: development and clinical associations. Neurology. 1999;53:2108–2112. doi: 10.1212/wnl.53.9.2108. [DOI] [PubMed] [Google Scholar]
- 8.Harris K, Singer HS. Tic disorders: neural circuits, neurochemistry, and neuroimmunology. J Child Neurol. 2006;21:678–689. doi: 10.1177/08830738060210080901. [DOI] [PubMed] [Google Scholar]
- 9.Singer HS. Tourette’s syndrome: from behaviour to biology. Lancet Neurol. 2005;4:149–159. doi: 10.1016/S1474-4422(05)01012-4. [DOI] [PubMed] [Google Scholar]
- 10.Kataoka Y, Kalanithi PS, Grantz H, Schwartz ML, Saper C, Leckman JF, et al. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol. 2010;518:277–291. doi: 10.1002/cne.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalanithi PS, Zheng W, Kataoka Y, DiFiglia M, Grantz H, Saper CB, et al. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci U S A. 2005;102:13307–13312. doi: 10.1073/pnas.0502624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pauls D, Raymond C, Stevenson J, Leckman J. A family study of Gilles de la Tourette syndrome. Am J Hum Genet. 1991;48:154–163. [PMC free article] [PubMed] [Google Scholar]
- 13.Price R, Kidd K, Cohen D, Pauls D, Leckman J. A twin study of Tourette syndrome. Arch Gen Psychiatry. 1985;42:815–820. doi: 10.1001/archpsyc.1985.01790310077011. [DOI] [PubMed] [Google Scholar]
- 14.Walkup J, Leckman J, Price R, Hardin M, Ort S, Cohen D. The relationship between obsessive-compulsive disorder and Tourette’s syndrome: a twin study. Psychopharmacol Bull. 1988;24:375–379. [PubMed] [Google Scholar]
- 15.Eapen V, Pauls DL, Robertson MM. Evidence for autosomal dominant transmission in Tourette’s syndrome. United Kingdom cohort study. Br J Psychiatry. 1993;162:593–596. doi: 10.1192/bjp.162.5.593. [DOI] [PubMed] [Google Scholar]
- 16.Pauls DL, Leckman JF. The inheritance of Gilles de la Tourette’s syndrome and associated behaviors. Evidence for autosomal dominant transmission. N Engl J Med. 1986;315:993–997. doi: 10.1056/NEJM198610163151604. [DOI] [PubMed] [Google Scholar]
- 17.State MW. The genetics of child psychiatric disorders: focus on autism and Tourette syndrome. Neuron. 2010;68:254–269. doi: 10.1016/j.neuron.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.State MW. The genetics of Tourette disorder. Curr Opin Genet Dev. 2011;21:302–309. doi: 10.1016/j.gde.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- 20.Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA, et al. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics. 2003;82:1–9. doi: 10.1016/s0888-7543(03)00097-1. [DOI] [PubMed] [Google Scholar]
- 21.Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet. 2008;16:614–618. doi: 10.1038/sj.ejhg.5202006. [DOI] [PubMed] [Google Scholar]
- 22.Ercan-Sencicek A, Stillman A, Ghosh A, Bilguvar K, O’Roak B, Mason C, et al. L-histidine decarboxylase and Tourette’s syndrome. N Engl J Med. 2010;362:1901–1908. doi: 10.1056/NEJMoa0907006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 24.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nature genetics. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 25.Conrad D, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2009;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Redon R, Ishikawa S, Fitch K, Feuk L, Perry G, Andrews T, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walsh T, McClellan J, McCarthy S, Addington A, Pierce S, Cooper G, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 29.Wilson G, Flibotte S, Chopra V, Melnyk B, Honer W, Holt R. DNA copy-number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling. Hum Mol Genet. 2006;15:743–749. doi: 10.1093/hmg/ddi489. [DOI] [PubMed] [Google Scholar]
- 30.Xu B, Roos J, Levy S, van Rensburg E, Gogos J, Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 31.Mulle JG, Dodd AF, McGrath JA, Wolyniec PS, Mitchell AA, Shetty AC, et al. Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet. 2010;87:229–236. doi: 10.1016/j.ajhg.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Consortium IS. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCarthy S, Makarov V, Kirov G, Addington A, McClellan J, Yoon S, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshall C, Noor A, Vincent J, Lionel A, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szatmari P, Paterson A, Zwaigenbaum L, Roberts W, Brian J, Liu X, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glessner J, Wang K, Cai G, Korvatska O, Kim C, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo copy number variations (CNVs), including duplications of the 7q11.23 Williams-Buren syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss L, Shen Y, Korn J, Arking D, Miller D, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 40.Guilmatre A, Dubourg C, Mosca A, Legallic S, Goldenberg A, Drouin-Garraud V, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vassos E, Collier DA, Holden S, Patch C, Rujescu D, St Clair D, et al. Penetrance for copy number variants associated with schizophrenia. Hum Mol Genet. 2010;19:3477–3481. doi: 10.1093/hmg/ddq259. [DOI] [PubMed] [Google Scholar]
- 42.Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ikeda M, Aleksic B, Kirov G, Kinoshita Y, Yamanouchi Y, Kitajima T, et al. Copy number variation in schizophrenia in the Japanese population. Biol Psychiatry. 2010;67:283–286. doi: 10.1016/j.biopsych.2009.08.034. [DOI] [PubMed] [Google Scholar]
- 44.Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirov G, Gumus D, Chen W, Norton N, Georgieva L, Sari M, et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet. 2008;17:458–465. doi: 10.1093/hmg/ddm323. [DOI] [PubMed] [Google Scholar]
- 46.Vrijenhoek T, Buizer-Voskamp JE, van der Stelt I, Strengman E, Sabatti C, Geurts van Kessel A, et al. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am J Hum Genet. 2008;83:504–510. doi: 10.1016/j.ajhg.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauthier J, Champagne N, Lafrenière RG, Xiong L, Spiegelman D, Brustein E, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sundaram S, Huq A, Wilson B, Chugani H. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology. 2010;74:1583–1590. doi: 10.1212/WNL.0b013e3181e0f147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Consortium WTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Association AP. Diagnositc and statistical manual of mental disorders. 4. Washington, DC: 2000. text rev. [Google Scholar]
- 51.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 52.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant S, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colella S, Yau C, Taylor J, Mirza G, Butler H, Clouston P, et al. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jia P, Sun J, Guo AY, Zhao Z. SZGR: a comprehensive schizophrenia gene resource. Mol Psychiatry. 2010;15:453–462. doi: 10.1038/mp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, et al. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raychaudhuri S, Korn JM, McCarroll SA, Altshuler D, Sklar P, Purcell S, et al. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet. 2010;6:e1001097. doi: 10.1371/journal.pgen.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crossett A, Kent BP, Klei L, Ringquist S, Trucco M, Roeder K, et al. Using ancestry matching to combine family-based and unrelated samples for genome-wide association studies. Stat Med. 2010;29:2932–2945. doi: 10.1002/sim.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee C, Abdool A, Huang CH. PCA-based population structure inference with generic clustering algorithms. BMC Bioinformatics. 2009;10(Suppl 1):S73. doi: 10.1186/1471-2105-10-S1-S73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada K, Holmans P, et al. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferrada C, Ferré S, Casadó V, Cortés A, Justinova Z, Barnes C, et al. Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology. 2008;55:190–197. doi: 10.1016/j.neuropharm.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Munzar P, Tanda G, Justinova Z, Goldberg SR. Histamine h3 receptor antagonists potentiate methamphetamine self-administration and methamphetamine-induced accumbal dopamine release. Neuropsychopharmacology. 2004;29:705–717. doi: 10.1038/sj.npp.1300380. [DOI] [PubMed] [Google Scholar]
- 64.Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- 65.Kubota Y, Ito C, Sakurai E, Watanabe T, Ohtsu H. Increased methamphetamine-induced locomotor activity and behavioral sensitization in histamine-deficient mice. J Neurochem. 2002;83:837–845. doi: 10.1046/j.1471-4159.2002.01189.x. [DOI] [PubMed] [Google Scholar]
- 66.Lebois EP, Jones CK, Lindsley CW. The evolution of histamine H3 antagonists/inverse agonists. Curr Top Med Chem. 2011;11:648–660. doi: 10.2174/1568026611109060648. [DOI] [PubMed] [Google Scholar]
- 67.Brioni JD, Esbenshade TA, Garrison TR, Bitner SR, Cowart MD. Discovery of histamine H3 antagonists for the treatment of cognitive disorders and Alzheimer’s disease. J Pharmacol Exp Ther. 2011;336:38–46. doi: 10.1124/jpet.110.166876. [DOI] [PubMed] [Google Scholar]
- 68.Baron-Cohen S, Mortimore C, Moriarty J, Izaguirre J, Robertson M. The prevalence of Gilles de la Tourette’s syndrome in children and adolescents with autism. J Child Psychol Psychiatry. 1999;40:213–218. [PubMed] [Google Scholar]
- 69.Baron-Cohen S, Scahill VL, Izaguirre J, Hornsey H, Robertson MM. The prevalence of Gilles de la Tourette syndrome in children and adolescents with autism: a large scale study. Psychol Med. 1999;29:1151–1159. doi: 10.1017/s003329179900896x. [DOI] [PubMed] [Google Scholar]
- 70.Burd L, Li Q, Kerbeshian J, Klug MG, Freeman RD. Tourette syndrome and comorbid pervasive developmental disorders. J Child Neurol. 2009;24:170–175. doi: 10.1177/0883073808322666. [DOI] [PubMed] [Google Scholar]
- 71.Stern JS, Robertson MM. Tics associated with autistic and pervasive developmental disorders. Neurol Clin. 1997;15:345–355. doi: 10.1016/s0733-8619(05)70317-0. [DOI] [PubMed] [Google Scholar]
- 72.Canitano R, Vivanti G. Tics and Tourette syndrome in autism spectrum disorders. Autism. 2007;11:19–28. doi: 10.1177/1362361307070992. [DOI] [PubMed] [Google Scholar]
- 73.State MW, Levitt P. The Conundrums of Understanding Genetic Risks for Autism Spectrum Disorders. Nature Neuroscience. 2011 doi: 10.1038/nn.2924. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–812. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Clarke RA, Fang ZM, Diwan AD, Gilbert DL. Tourette syndrome and klippel-feil anomaly in a child with chromosome 22q11 duplication. Case Report Med. 2009;2009:361518. doi: 10.1155/2009/361518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- 77.Yan W, Jacobsen LK, Krasnewich DM, Guan XY, Lenane MC, Paul SP, et al. Chromosome 22q11.2 interstitial deletions among childhood-onset schizophrenics and “multidimensionally impaired”. Am J Med Genet. 1998;81:41–43. [PubMed] [Google Scholar]
- 78.Wang PP, Solot C, Moss EM, Gerdes M, McDonald-McGinn DM, Driscoll DA, et al. Developmental presentation of 22q11.2 deletion (DiGeorge/velocardiofacial syndrome) J Dev Behav Pediatr. 1998;19:342–345. doi: 10.1097/00004703-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 79.Ou Z, Berg JS, Yonath H, Enciso VB, Miller DT, Picker J, et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med. 2008;10:267–277. doi: 10.1097/GIM.0b013e31816b64c2. [DOI] [PubMed] [Google Scholar]
- 80.Wentzel C, Fernström M, Ohrner Y, Annerén G, Thuresson AC. Clinical variability of the 22q11.2 duplication syndrome. Eur J Med Genet. 2008;51:501–510. doi: 10.1016/j.ejmg.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 81.Portnoï MF. Microduplication 22q11.2: a new chromosomal syndrome. Eur J Med Genet. 2009;52:88–93. doi: 10.1016/j.ejmg.2009.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.