

Abstract
Osteoarthritis (OA) is a multifactorial disease subject to the effects of many genes and environmental factors. Alterations in the normal pattern of chondrocyte gene control in cartilage facilitate the onset and progression of OA. Stable changes in patterns of gene expression, not associated with alterations in DNA sequences, occur through epigenetic changes, including DNA methylation, histone modifications, and alterations in chromatin structure, as well as by microRNA (miRNA)-mediated mechanisms. Moreover, the ability of the host to repair damaged cartilage is reflected in alterations in gene control circuits, suggestive of an epigenetic and miRNA-dependent tug-of-war between tissue homeostasis and OA disease pathogenesis. Herein, we summarize epigenetic and miRNA-mediated mechanisms impacting on OA progression and in this context offer potential therapeutic strategies for OA treatment.
What is OA disease and how does it develop?
Idiopathic osteoarthritis (OA) is a late-onset, complex disease of the joint, characterized by progressive failure of the extracellular matrix (ECM) of cartilage (Figure 1 and Box 1), with changes in and contributions by the synovium, subchondral bone, and other joint tissues. Risk factors for OA development can be divided into two fundamental mechanisms related to the adverse effects of trauma on an otherwise normal joint, to normal loading on a maligned joint, or a range of scenarios in between, all resulting in abnormal biomechanics that impact on biological responses. Although tremendous advances have been made in defining OA susceptibility genes, these are merely predictive of risk for disease development, whereas it is the interaction with the environment that determines whether individuals develop the disease.
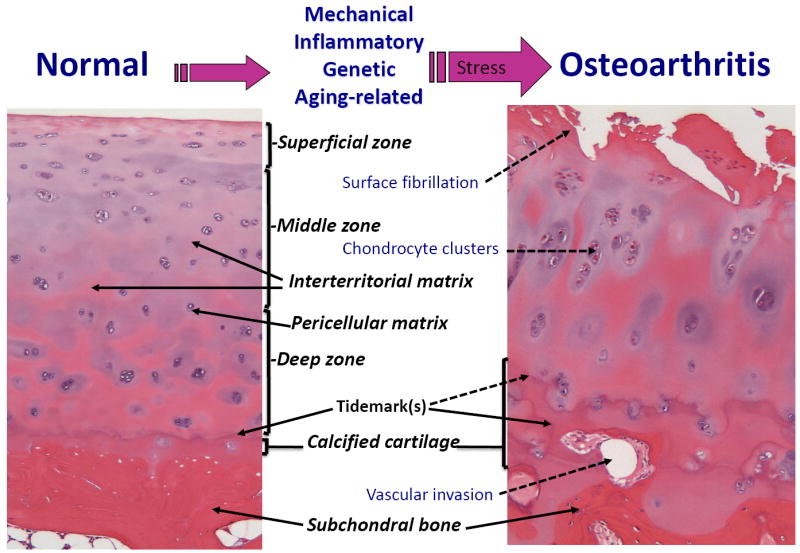
Figure 1. Major phenotypic changes by which normal cartilage tissue becomes compromised in osteoarthritis (OA).
Normal articular cartilage subjected to stresses such as inflammation, mechanical stress, genetic aberrations, and age-related degeneration can slowly develop phenotypic characteristics of OA tissue. These include increased stress and inflammatory signaling, proliferation of chondrocytes resulting in chondrocyte clusters, and degradation of the extracellular matrix (ECM). The role of the subchondral bone in osteoarthritis and its interactions with cartilage have been reviewed by Goldring and Goldring [88].
Text Box 1 ECM and chondrocytes in normal and OA cartilage.
The composition and cellular organization of human adult articular cartilage is complex, with qualitative and quantitative differences in matrix constituents ranging from the superficial through deep zones and between the interterritorial and territorial, or pericellular, regions. Chondrocytes, the unique cellular component of articular cartilage, maintain the matrix components under normal, low turnover conditions in which glycosaminoglycans on proteoglycans and other non-collagen molecules can be replaced. In adult articular cartilage, the collagen network, which consists of type II, IX, and XI collagens, and aggrecan are responsible, respectively, for the tensile strength and compressive resistance to mechanical loading. Normally, chondrocytes maintain a low turnover of these cartilage matrix proteins, but cells in the surface zone of OA cartilage undergo a phenotypic change in which catabolic genes that are not normally expressed by articular chondrocytes, such as matrix-degrading proteases, are switched on [38]. Moreover, recent large-scale expression profiling studies, using full-thickness cartilage, showed that many anabolic genes, including COL2A1, are upregulated in late-stage OA [50, 51]. These findings apply predominantly to chondrocytes in the middle and deep zones, as found by laser capture microdissection, whereas expression of type II collagen within chondrocytes of the surface zone is considerably reduced, suggesting that phenotypic change also involves repression of anabolic genes [89]. Mutations in ECM genes account for several human chondrodysplasias, in which early onset OA occurs [90], and GWAS studies have indicated that polymorphisms in genes involved in cartilage and joint development, such as GDF5 and SMAD3, are OA risk alleles [91, 92]. The dramatic phenotypic differences between normal and OA disease cartilage, which may reflect genetic and epigenetic changes, can be appreciated in Figure 1.
Much has been learned about factors involved in cartilage degeneration and other joint pathologies from animal models of post-traumatic OA, including transgenic and knockout mice subjected to surgically induced OA disease [1]. Genetic models with abnormal composition and structure of articular cartilage or other joint tissues may develop spontaneous or accelerated OA due to altered biomechanics. Nevertheless, there are common mediators across these models that determine initiation and progression of cartilage damage. Work during the past few years has discovered roles for a number of mediators that impact on the disease process and may inform us about new directions for targeted therapies [2]. The activation of stress- and inflammation-induced signaling, as well as transcriptional and post-transcriptional events, may cause phenotypic shift, apoptosis, and aberrant expression of both catabolic and anabolic genes in cartilage and other joint tissues [3].
The importance of the fine ECM protein network and its stability in joint mechanics and cartilage health over time is well documented in three mouse models that each present age-dependent cartilage degeneration similar to OA patients: Col11a1 haplo-insufficient mice (heterozygous chondrodysplasia, cho/+), type IX collagen-deficient mice (Col9a1-/-), and Timp3-/- mice (see Glossary). A requirement for aggrecan depletion in the initiation of cartilage erosion was revealed in Adamts5 knockout mice (see Glossary), which are protected against OA progression in a surgical inductive OA model [4, 5]. However, aggrecan depletion alone does not drive cartilage erosion, as demonstrated in recent studies with Mmp13 knockout mice in which matrix metalloprotease (MMP)-13 deficiency inhibited OA progression despite continued aggrecan depletion [6]. Moreover, the pericellular matrix that encapsulates the chondrocytes, distinct from the interterritorial matrix, consists of matrilin 3, fibronectin, biglycan, fibromodulin, COMP, and collagen VI, but little type II collagen (see Box 1 for information on ECM and cartilage structure). This pericellular matrix has recently been shown to protect the chondrocyte from activation [7]. Importantly, the latter work uncovered a critical role of the serine protease, high temperature requirement A1 (HTRA1), which is increased in both human OA cartilage and the articular cartilage of mouse models of OA [7]. Once HTRA1 activity disrupts the pericellular matrix, which may occur early before overt symptoms of OA develop, chondrocyte receptors such as DDR2 may be exposed to activation by type II collagen in fibrillar form, leading to preferential up-regulation of MMP-13 and further degradation of the interterritorial matrix [8]. This review focuses on epigenetic changes and specific miRNAs linked to cartilage homeostasis and its breakdown in OA disease.
Impact of epigenetic and miRNA-mediated gene control in normal and OA chondrocytes
In normal adult chondrocytes, as in all somatic cells, epigenetic mechanisms involving DNA methylation at CpG motifs, modification of histone tails, and changes in chromatin structure mediated by enzymatic activities and non-coding RNAs, together generate stable, heritable phenotypes. Growing evidence points to interactions with the environment yielding epigenetic alterations in gene expression that lead to transgenerational phenotypes and disease etiologies transmitted to daughter cells throughout many doublings [9]. Of particular interest in this regard are paramutational-like effects described in mice whereby specific miRNAs control gene expression by a germ line transmissible, epigenetic mechanism [10], which can result in pathophysiologies such as heritable cardiac hypertrophy [11]. OA disease develops in the context of chondrocytes responding to a stress-associated, pro-inflammatory environment. Thus, it is also of particular importance to explore how alterations in the regulation of miRNAs that maintain a long-term normal chondrocyte phenotype or control inflammatory response mediators and their associated signaling pathways (reviewed in [12], [13]) directly or indirectly affect OA disease progression. Other recent evidence also points to the critical importance of miRNA-mediated regulation of gene control circuitry that impacts on the epigenetic-like, heritable conversion of normal cells into cancerous ones [14].
Regulation of cartilage gene expression by chromatin modification
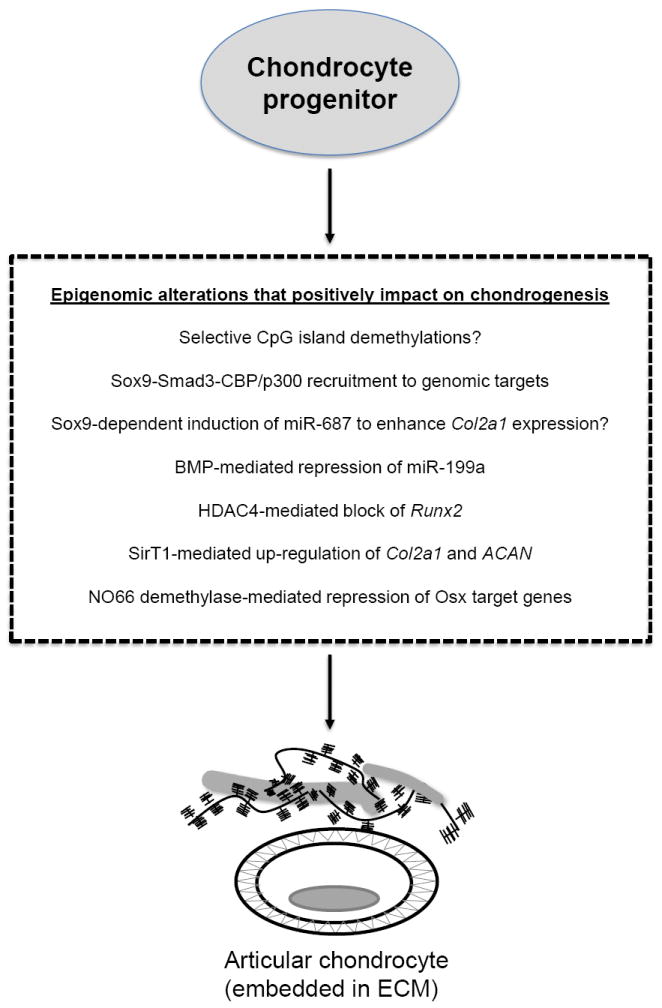
Histone modifications involving acetylation and deacetylation strongly impact the anabolic and catabolic phenotypes of chondrocytes. Histone acetylation is a critical step for regulatory factors gaining access to the transcriptional machinery and the subsequent initiation of gene expression, whereas deacetylation primarily plays a role in the termination or repression of gene expression. The regulation of anabolic genes such as COL2A1 and CD-RAP in chondrocytes requires an open chromatin network maintained by Sox9 and its physical interaction with the transcriptional co-activator and CBP/p300 [15, 16]. Sox9 forms a protein complex with the Smad3 transcription factors, which enhances CBP/p300 recruitment and subsequent acetylation-mediated Sox9 stabilization [17]. Figure 2 illustrates some of the major epigenetic events associated with chondrogenic programming and the genesis and maintenance of articular cartilage.
Figure 2. Major epigenetic players and microRNAs (miRNAs) that help to drive the genesis of articular chondrocytes from osteo-chondro progenitors.
The ECM and physiological status of chondrocyte before and after progression to OA are shown. Note that the pericellular matrix surrounding normal chondrocytes is absent in OA cartilage. Statements with question marks indicate regulatory mechanisms based on in vitro evidence that require further validation in vivo.
The impact of histone acetylations on chondrocyte phenotype has been explored at the level of specific histone deacetylases (HDAC). HDACs consist of two families: the classical HDAC family and the NAD+-dependent SIR2 family (also known as class III HDACs). Based on phylogeny, the classical HDACs can be grouped into 3 classes: Class I (HDAC1, 2, 3, and 8), related to yeast RPD3; class II (HDAC4, 5, 6, 7, 9, and 10), more closely related to yeast HDA1; and class IV (HDAC11). HDACs were found not only to inhibit the expression of MMPs induced by inflammatory cytokines and other mediators [18-21] but also to suppress expression of cartilage-specific genes, including COL2A1 and aggrecan (ACAN) [22].
Specific HDACs seem to be involved in different processes and to target different chondrocyte-specific genes [22, 23]. A correlation between MMP13 and HDAC7 gene expression has been reported in human knee osteoarthritis [24], and HDAC4 plays a role in cartilage development by interacting with Runx2 and blocking its activity, preventing premature chondrocyte hypertrophy in prehypertrophic chondrocytes [25]. The expression of HDACs 1 and 2 are elevated in OA compared to normal human cartilage [22]. In addition, evidence was reported suggesting that HDAC1- and HDAC2-mediated gene repression of specific target genes, including COL2A1, at least in part involves their recruitment by the Snail transcription factor, which interacts with their carboxy-terminal domains [22]. In spite of the latter observation and the fact that HDAC inhibitors were reported to suppress the expression of specific MMPs, implementing HDAC inhibitors as a potential treatment for OA disease has been questioned by other findings showing that HDAC blockers also suppress COL2A1 expression by an indirect mechanism involving activation of Wnt5a, a repressor of type II collagen expression [26]. The impact of specific HDACs on cartilage development and OA disease progression are summarized in Figures 2 and 3, respectively.
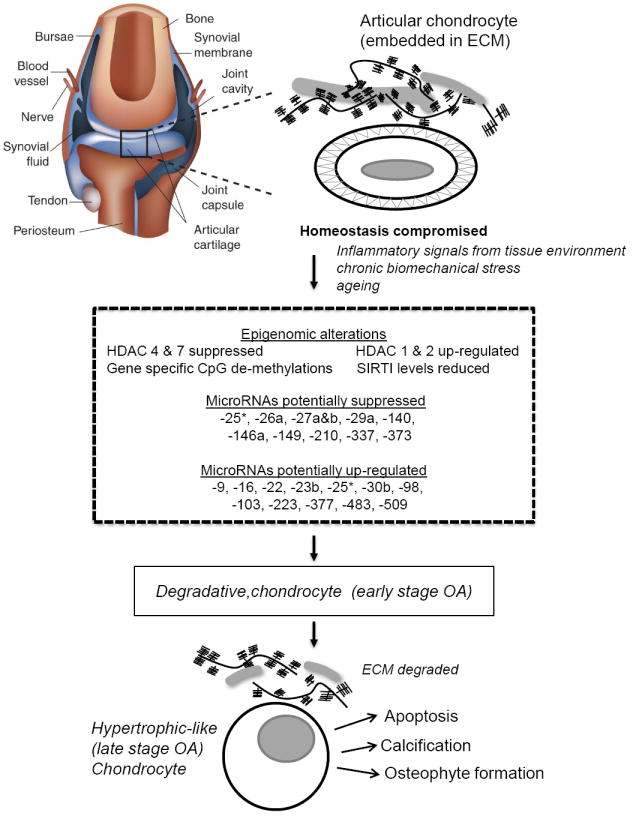
Figure 3. The path towards osteoarthritis (OA).
Summary of the epigenetic markers and associated regulatory alterations impacting on the physiology of growth- and differentiation-arrested normal articular chondrocytes leading them down a path towards OA disease. Again as in Figure 2, the presence of a pericellular matrix surrounding normal chondrocytes is absent in OA cartilage. *miR-25 was found to be up-modulated by Jones et al. [58] and suppressed by Iliopoulos et al. [56] in OA cartilage.
Of the SIR2 family, SIRT1, a lysine deacetylase that plays an important role in age-related diseases, is of interest because it promotes cartilage-specific ECM gene expression, including COL2A1, by enhancing Sox9-mediated transcription via the recruitment of histone acetyltransferases (HATs) to the promoter and enhancer sites [27]. SIRT1 also blocks apoptosis in chondrocytes by multiple mechanisms [28, 29], and findings in OA cartilage compared with normal cartilage suggest that reduced SIRT1 expression and activity in chondrocytes may correlate with increased apoptosis and the altered pattern of gene expression (reduced COL2A1 and ACAN and increased COL10A1 and ADAMTS5) evident at certain stages of OA [27, 28, 30]. Although not as yet shown in OA disease states, SIRT1 also has a broad anti-inflammatory function in a variety of tissues [31], in which it initiates a gene-specific transcriptional repression program in part by deacetylating the p65 subunit of nuclear factor κB (NF-κB), which blocks NF-κB DNA binding [32] and can lead to the recruitment of RelB-dependent transcriptional repressor complexes to terminate acute inflammatory responses [33]. Interestingly, a recent study showed that treating chondrocytes with tumor necrosis factor (TNF)-α induced cathepsin B-mediated cleavage of SIRT1, thereby reducing its enzymatic activity in association with reduced Sox9 binding to the COL2A1 enhancer [34].
OA is an inflammatory disease and thus it is also important to note that under some circumstances, histone demethylation can induce a pro-inflammatory gene expression program within a cell. For example, in activated immune effector cells canonical NF-κB signaling has been shown to inhibit the formation of the polycomb repressor complexes that are dependent on the triply methylated lysine 27 of histone 3 (H3K27me3) by inducing the expression of Jmjd3, a Jumonji C (JmjC) catalytic domain-containing protein, that functions as an H3K27me3 demethylase [35]. The canonical NF-κB signaling pathway driven by the Iκ kinase β (IKKβ) has been reported to drive MMP-13-dependent ECM remodeling in differentiating cultures of primary human OA articular chondrocytes [36]; thus, it will be interesting to know if IKKβ-mediated NF-κB signaling involves alterations in chromatin structure in OA disease development in vivo. Recent evidence indicates that expression of NFAT1 (NFATc2/NFATp; nuclear factor of activated T cells 1), an essential transcriptional regulator of articular cartilage homeostasis, is associated with increased H3K4me2 (and knock-down of lysine-specific demethylase-1), whereas decreased NFAT1 expression, which causes OA-like changes in adult mice) is correlated with increased H3K9me2 (and knock-down of JmjC-containing histone demethylase-2a) [37].
Epigenetic mechanisms involving DNA methylation
Methylation of genomic DNA represents a significant mechanism for regulating tissue-specific gene expression. Pathological loss of DNA methylation may result in aberrant gene induction, whereas increased methylation may silence normally expressed genes. Methylation is affected by dietary and other environmental factors, and the DNA methylation pattern for a specific gene varies between cell types. In general, active promoters that drive transcription of genes are hypomethylated in cells, whereas hypermethylation of promoters silences genes. DNA methylation status is heritable such that the cell type-specific gene expression repertoire is stably transmitted to daughter cells. Thus aberrant methylation patterns will also be transmitted to daughter cells, which may result in either continued abnormal expression of silenced genes or suppression of normally expressed genes.
Previous studies have demonstrated epigenetic de-repression associated with DNA methylation loss on chondrocyte genes including MMP3, MMP9, MMP13, ADAMTS4, IL-1β, and LEP, which encode enzymes contributing to the degradative phenotype of OA cartilage [38-43]. An inhibitor of NF-κB, partly through modulation of DNA methyltransferase-1 expression, can prevent cytokine-induced demethylation of a specific site in the IL-1β promoter, which is associated with decreased IL-1β expression [44]. A recent study demonstrated that whereas two genes encoding suppressors of cytokine signaling (SOCS), SOCS2 and CIS-1, but not SOCS1 and SOCS3, are suppressed in OA chondrocytes, the CpG methylation status of the SOCS2 promoter is unchanged [45]. Interestingly, loss of expression of osteogenic protein-1 (OP-1, or bone morphogenetic protein-7) in aging chondrocytes is associated with hypermethylation of the OP-1 promoter. Exposing chondrocytes to OP-1 can downregulate stress and inflammatory response genes [46].
It remains unclear if either hypo- or hyper-methylation of DNA alters the regulation of cartilage matrix genes in OA. As observed by Poschl and colleagues [47], DNA methylation is not a key component in the downregulation of ACAN, whose promoter has 33 CpG sites within a 340 bp CpG island that are unmethylated in both normal and OA cartilage samples. Although p21WAF1/CIP1, an inhibitor of cell proliferation, is regulated by epigenetic modulation in other contexts and is also expressed in normal chondrocytes, DNA methylation of its promoter appears to be responsible for its down-regulation in OA chondrocytes [48]. Other studies demonstrated that the methylation status of a CpG island near to the transcription start site of the COL2A1 promoter is demethylated in human articular chondrocytes, mesenchymal stem cells (MSCs), and MSC-derived chondrocytes independent of COL2A1 expression [49]. However, the methylation status of the CpG sites in the 309 bp enhancer region of the COL2A1 gene, which is required for its Sox9-dependent transcription, remains to be examined. By contrast, induction of type X collagen during MSC chondrogenesis is correlated with demethylation of two CpG sites in the promoter region of the COL10A1 gene. Conceivably, the increased expression of COL2A1 in OA samples in the absence of differential methylation could be explained by the efforts of the OA chondrocytes to regenerate the ECM as an anabolic-like response within the degradative environment, consistent with findings in microarray analyses of human cartilage [50, 51]. The Human Epigenome Project has shown that genes such as COL2A1 with large numbers of CpG islands are generally not methylated in normal cells, irrespective of expression with exceptions being tumor suppressor genes in cancer [52, 53]. For example, despite decreased expression in OA chondrocytes, as mentioned above, the methylation status of SOCS2 promoter CpG sites is unaltered [45], whereas in tumor cells the CpG sites in the promoter regions of SOCS genes are hypermethylated in association with decreased expression [54, 55].
In summary, epigenetic changes in OA involving hypomethylation and consequent activation of aberrant genes are more prevalent than those involving hypermethylation and silencing of anabolic genes. No doubt other such genes will be identified in the future. Although DNA methylation will not always explain permanently altered gene expression in OA, increased understanding of the methylation patterns in OA tissues offers significant therapeutic potential for new approaches to prevent or reverse epigenetic changes.
Impact of microRNAs (miRNAs) on chondrocyte physiology and OA disease
Over the past few years, the influence of specific microRNAs in cartilage homeostasis and OA disease has received greater attention [56-65]. Dicer, an enzyme required for miRNA biogenesis, is essential for murine skeletal development, with Dicer-null growth plates presenting a pronounced lack of chondrocyte proliferation in conjunction with enhanced differentiation to postmitotic hypertrophic chondrocytes [57]. The latter results may be explained by Dicer loss having distinct functional effects at different stages of chondrocyte development [57]. The miRNA expression profile of mesenchymal stem cells is significantly altered once they differentiate into chondrocytes [66, 67]. MiR-1 represses ACAN gene expression in the human chondrosarcoma cell line, HCS-2/8; and MiR-1 levels diminish at the late hypertrophic stage of chondrocyte differentiation [68]. MiR-199a expression is decreased during BMP-2 induced chondrogenesis, indicating that it may function as a suppressor of early steps in the chondrogenic program [69]. Indeed, enforced miR-199a expression in murine C3H10T1/2 stem cells or in the prechondrogenic cell line ATDC5 suppresses multiple markers of early chondrogenesis, including type II collagen and cartilage oligomeric matrix protein (COMP), while anti-miR-199a has an opposite, stimulatory effect [69]. Consistent with these observations, Smad1, a positive downstream mediator of BMP-2 signaling and a regulator of bone and cartilage development, was shown to be a direct miR-199a target [69]. Thus, BMP-2-mediated repression of miR-199a would prevent its post-transcriptional repression of Smad1. In addition, a number of miRNAs have also been identified as regulators of osteoblastogenesis, including miR-29, miR-141, miR-200a, miR-206, miR-210 and miR-2861 [70]. Osteoblast differentiation from osteo-chondroprogenitors also involves the activities of novel transcription factors, including Osterix (Osx) [71]. From an epigenetic perspective, Osx target genes are repressed in osteo-chondroprogenitors due to the activity of NO66, a JmjC-domain containing histone demethylase, which removes activating H3K4me and H3K36me chromatin marks from the promoters of osteoblast specific genes [71]. Taken together, these data suggest that cartilage tissue engineering approaches might be enhanced by manipulating the programming of osteochondro-progenitors with different combinations of miRNAs and/or the activities of chromatin modifying factors such as NO66 (see Figure 2 for specific miRs and chromatin modifications associated with chondrogenesis and cartilage homeostasis).
Unbiased genomic expression profiling approaches have uncovered a number of miRNAs whose expression are either significantly upregulated or suppressed in human OA chondrocytes, in comparison with their normal articular counterparts. One screen identified 17 miRNAs whose expression varied by 4-fold or more [58] in normal versus late stage OA cartilage. An independent study revealed a signature of 16 miRNA that distinguishes normal from OA cartilage tissue, with 9 miRNAs significantly up-regulated and 7 miRNAs down-regulated in OA tissue compared with normal controls [56] (see Figure 3). In addition, over-expression experiments with specific miRNAs or their targeted LNA (Locked Nucleic Acid) inhibitors implicated miR-9 in the regulation of MMP13 as well as miR-9, miR-98 and miR-146 in the control of TNFα [58]. Although sufficient direct functional target analysis is lacking for most of these differentially expressed miRNAs, integrating some of these results with proteomics analysis of normal vs. OA cartilage revealed several miRNA-gene target pairs potentially involved in cartilage homeostasis and structure (miR-140-ADAMTS5, miR-483-ACAN, miR-509-Sox9, miR-223-GDF5), in biomechanics (miR-25-ITGA5), in apoptosis (miR-373-CASP6, miR-210-CASP10), as well as lipid metabolism (miR-22-PPAR, miR-22-BMP7, miR-29a-LEP) [56]. Functional experiments on selected miR-gene pairs verified that miR-22 regulates BMP-7 and PPARA at the RNA and protein levels, respectively [56]. Moreover, enforced miR-22 expression or siRNA-mediated suppression of either PPARA or BMP7 resulted in increases in IL-1β and MMP-13 protein levels, whereas inhibiting endogenous miR-22 in OA chondrocytes elevated PPARA and BMP7, while simultaneously inhibiting IL-1β and MMP-13, with a concomitant cartilage-protective increase in aggrecan protein [56]. Alterations in miRNA signatures associated with OA disease development and progression are highlighted in Figure 3.
Specific miRNA signatures in synovial fluids may serve as novel biomarkers of cartilage disease. The concentrations of miR-16, miR-132, miR-146a, and miR-223 are reduced in the synovial fluid of individuals suffering from OA, compared with healthy controls [60], whereas miR-16, miR-132, miR-223, miR-146a and miR-155 [72] are expressed at higher levels in the synovial fluid of rheumatoid arthritis (RA) patients. However, the differences in signatures in OA cartilage compared to synovial fluids, including miR-16 and miR-223, which are upregulated in OA cartilage [56], may represent tissue-specific differences in disease activities. Evidence that specific miRNAs may impact on stress-related articular cartilage mechanotransduction has also been reported [59]. MiRNA-34a has been reported to modulate chondrocyte apoptosis [73]. MiR-222, which targets the p27Kip1 cyclin-dependent kinase inhibitor [74], was shown to be upregulated in the anterior weight-bearing cartilage of the knee joint as opposed to its posterior non-weight-bearing counterpart, suggesting that miR-222 may contribute to enhanced chondrocyte proliferation in the cartilage superficial zone of mechanically stressed joints [59].
MiRNA-140 is encoded by the miR140 gene, located in an intron of the E3 ubiquitin protein ligase gene Wwp2, is evolutionarily conserved amongst vertebrates and is abundantly and specifically expressed by mouse embryo chondrocytes during bone development [61, 75-77]. Importantly, miR-140 expression is greatly reduced in OA cartilage [63] and is suppressed by IL-1β signaling [63]. MiR-140 was first shown to target HDAC4 [76], a known co-repressor of Runx2 and MEF2C transcription factors essential for chondrocyte hypertrophy and bone development. MiR-140 also targets CXCL12 (stromal cell-derived factor 1) [78] and SMAD3 [79], both of which are implicated in chondrocyte differentiation. More recently, miR-140 was shown to target ADAMTS5 [64]. Finally, miR-140 also suppresses Dnpep, an aspartyl aminopeptidase, which has been suggested to antagonize BMP signaling downstream of Smad activation [80]. Importantly, miR140 knockout mice are predisposed to age-related OA-like changes, and over-expression of miR140 in chondrocytes is protective against surgically induced OA [61, 64].
Other recent findings indicate that miR-27b, miR27a, and miR-146a suppress ADAMTS and/or MMP expression in chondrocytes [65, 81, 82]. MiR-27b targets MMP-13 mRNA, and MiR-27b expression is suppressed by mitogen-activated protein kinase (MAPK) and NF-κB signaling [81]. Another report provided evidence that miR-27a may indirectly regulate the levels of MMP-13 and the proanabolic insulin-like growth factor binding protein (IGFBP)-5 by targeting an upstream positive effector, or effectors, of both genes [82]. Although miR-146a is highly expressed in low grade OA cartilage miR-146a levels plummet in late stage OA [62]; and it is strongly up-regulated by IL-1β signaling [62] with its induced transcription dependent on canonical NF-κB activation [83]. MiR-146a was previously found to directly target mRNAs encoding TRAF6 and IRAK, thereby attenuating proinflammatory signaling pathways [83]. MiR-146a has also been suggested to function as a negative feedback regulator of MMP-13 [62] and as a suppressor of autoimmunity and myeloproliferation [84]. Furthermore, miR-146a has recently been implicated in the control of knee joint homeostasis and OA-associated algesia by balancing inflammatory responses in cartilage and synovium with pain-related factors in glial cells. As such, it may be useful for the treatment of both cartilage regeneration and pain symptoms caused by OA [65].
The potential importance of miRNAs in maintaining cartilage integrity and homeostasis is supported by a recent study demonstrating that the major chondrogenic transcription factor Sox9 positively regulates the COL2A1 gene in human chondrocytes by a miR-675-dependent mechanism [85]. MiRNA-675 is derived from a primary transcript encoded by the H19 gene, and expression of H19 and miR-675 RNAs are Sox9-dependent. Importantly, enforced miR-675 expression rescues COL2A1 mRNA levels in either Sox9- or H19-depleted primary human articular chondrocytes. Although the OA expression status of miR-675 and its target remain unknown, these data indicate that miR-675 may modulate cartilage homeostasis by suppressing a COL2A1 transcriptional repressor [85]. Profiling studies of primary adult articular chondrocytes show that miRNAs are differentially expressed in cells with a range of differentiated, chondroblast-like, and dedifferentiated phenotypes, suggesting that manipulating miRNA profiles may provide new strategies for improving cartilage tissue engineering [86, 87].
Future perspectives
OA in humans develops slowly with time and may not be symptomatic until significant joint damage occurs. Thus, it has been extremely difficult to develop disease-modifying drugs and prove their effectiveness in clinical trials owing to the lack of biomarkers and sensitive techniques for identifying and assessing patients with early changes. Because the cytotoxicity and off-target effects of HDAC inhibitors may preclude their use for treatment of a relatively benign disease, compared to cancer, more specific inhibitors that target OA disease-specific processes, such as inflammation and mechanotransduction, or targeting the inhibitors to the joint tissues themselves must be considered. Inhibitors of signaling pathways that regulate DNA methylation might be another approach. Profiling unique patterns of epigenetic changes represent a novel strategy to distinguish diverse chondrocyte phenotypes, including chondrogenic programming, articular cartilage homeostasis, and OA disease progression to identify novel biomarkers of early OA. Correlating DNA methylation, chromatin marks, and miRNA signatures in human OA disease with those found in well-defined OA animal models should allow us to define the regulatory requirements for stress-related pro-inflammatory signaling and the acquired hypertrophic-like phenotypes of OA chondrocytes. To design effective miRNA-based treatment modalities without potentially deleterious off-target effects, future work must be directed toward identifying key miRNA targets that functionally impact on early OA onset and disease progression. Overall we are increasingly hopeful that rational molecular therapies may be on the horizon to combat the pro-inflammatory tissue destructive phases of OA disease and to simultaneously enhance cartilage integrity and maintain joint flexibility with progressing age.
Text Box 2 Epigenetic regulation of gene expression.
Gene expression is regulated by both epigenetic and non-epigenetic mechanisms. Non-epigenetic gene regulation governs genes that are part of the expression repertoire of a specific somatic cell type and is dependent upon the presence and activities of specific transcription factors and other DNA binding proteins that directly impact on the transcriptional output of a cell. In contrast, one major level of epigenetic regulation of gene expression involves the long-term silencing of genes that are not normally expressed by a given cell type. This silencing is important for genomic stability and cellular homeostasis, and alterations in epigenetic status can be associated with disease. Epigenetic modification consists of three distinct processes: DNA methylation of carbon 5 of cytosine residues in CpG nucleotides; some histone modifications, in particular acetylation and methylation on specific lysines [93] and some epigenetic changes are also subject to regulation by non-coding RNAs [9] and a complex network of miRNAs [94].
Relaxed nucleosomal chromatin indicates transcriptionally accessible or active regions and is generally associated with hypomethylated DNA, acetylation of histones H3 and H4, as well as methylation of histone H3 at lysine 4 (H3-K4). Compact heterochromatin is transcriptionally silent, consisting of non-acetylated histones and hypermethylation of H3 at lysines 9 and 27 (K9 and K27) [95, 96]. DNA methylation occurs by the addition of methyl groups to 5'-cytosines next to guanines (CpG sites) by DNA methyltransferases (DNMTs) [97]. These methyl groups primarily originate from serine. Methylated CpG sites attract methyl-binding proteins, which can recruit histone deacetylases and histone methyl transferases. Thus, there is a reciprocal relationship between DNA methylation and histone modifications. DNA methylation is secondary to histone modifications in establishing silencing but is important for maintaining silencing in the long term because DNA methylation is generally stable in somatic cells throughout adult life [98, 99]. During DNA replication, the methylation pattern is rapidly reproduced on the nascent strand by the methyl transferase DNMT1, which associates with proliferating-cell nuclear antigen and tracks with DNA polymerase [100]. The histone code can then be re-established after cell division by various mechanisms involving methyl binding domains (MBDs) and DNMTs that interact with histone methyltransferases and histone deacetylases.
Acknowledgments
Work related to the subject of this review is supported by National Institutes of Health grant RC4-AR060546. We thank Dr. Cecilia Dragomir for providing the histological sections in Figure 1.
Glossary
- ADAMTS
a disintegrin and metalloprotease with thrombospondin domains
- Aggrecan
the major large aggregating proteoglycan of articular cartilage that consists of glycosaminoglycan side-chains on the aggrecan core protein that attaches to the hyaluronic acid polymers through link protein.
- Articular cartilage
The specialized tissue that covers bone in articulating joints such as the knee.
- Cartilage-specific collagens
Type II collagen is the major component of the collagen network in cartilage, which also contains type IX collagen and type XI collagen that are important for determining the shape and size of collagen fibrils
- Chondrocytes
cells producing cartilage matrix. Cartilage matrix is rich in type II collagen encoded by COL2A1 and a highly sulphated cartilage-specific proteoglycan, aggrecan, encoded by ACAN.
- Chondrodysplasias
Genetic abnormalities of cartilage formation that generally result from mutations in genes encoding cartilage matrix genes, growth and differentiation factors, or their receptors as well as other genes involved in skeletal development.
- Hypertrophic chondrocyte
large chondrocytes producing type X collagen, encoded by COL10A1, found at the growth plate adjacent to the junction between cartilage and bone.
- DNA methylation
methylation of cytosine at CpG dinucleotide pairs. This methylation modification alters the transcriptional state of target genes.
- Chromatin
DNA and the associated nuclear proteins
- Chromatin remodeling
bending or sliding of nucleosomes along DNA to facilitate transcription
- COMP
cartilage oligomeric matrix protein
- Glycosaminoglycans
glycosylated and sulfated carbohydrate side-chains on aggrecan and other proteoglycan core proteins synthesized by glycosyl transferases and sulfotransferases
- Histones
proteins around which DNA is wrapped in the nucleus. The core histones are H2, H3, and H4
- Histone modifications
acetylation, methylation and other modifications to histones give rise to the “histone code”. This code is “read” by specific binding proteins in the nucleus and is important for regulating transcription.
- Nucleosome
repeating subunits of ~156bp DNA wrapped around a histone octamer.
- Osteoblasts
cells in bone responsible for deposition of the mineralized bone matrix consisting of type I collagen and bone-specific non-collagenous proteins such as osteocalcin.
- Osterix (OSX)
C2H2-type zinc finger transcription factor highly specific to osteoblasts.
- Paramutation
epigenetic modifications yielding mitotically stable and sexually transmissible changes in gene expression by non-Mendelian patterns.
- RUNX2
Runt family transcription factor essential for chondrocyte terminal differentiation and endochondral ossification
- SOX 9 (Sry-type HMG box)
a transcription factor belonging to a subfamily of DNA-binding proteins with greater than 50% homology with the testis determination factor Sry and containing a high-mobility-group domain.
- Subchondral bone
The bone beneath the calcified base of the articular cartilage that modifies mechanical forces through the articular cartilage.
- Synovium
A thin membrane (without a basement membrane) that lines the joint cavity and is the site of synovial fluid production.
- TIMP
tissue inhibitor of metalloproteinases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Little CB, Fosang AJ. Is cartilage matrix breakdown an appropriate therapeutic target in osteoarthritis--insights from studies of aggrecan and collagen proteolysis? Curr Drug Targets. 2010;11:561–575. doi: 10.2174/138945010791011956. [DOI] [PubMed] [Google Scholar]
- 2.van den Berg WB. Osteoarthritis year 2010 in review: pathomechanisms. Osteoarthritis Cartilage. 2011;19:338–341. doi: 10.1016/j.joca.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Goldring MB, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cell Mater. 2011;21:202–220. doi: 10.22203/ecm.v021a16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glasson SS, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- 5.Stanton H, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- 6.Little CB, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60:3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polur I, et al. Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol Histopathol. 2010;25:599–608. doi: 10.14670/hh-25.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L, et al. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. 2010;62:2736–2744. doi: 10.1002/art.27582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skinner MK, et al. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends in Endocrinology & Metabolism. 2010;21:214–222. doi: 10.1016/j.tem.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuzin Fo, et al. Inherited variation at the epigenetic level: paramutation from the plant to the mouse. Current Opinion in Genetics and Development. 2008;18:193–196. doi: 10.1016/j.gde.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Wagner KD, et al. RNA Induction and Inheritance of Epigenetic Cardiac Hypertrophy in the Mouse. Developmental Cell. 2008;14:962–969. doi: 10.1016/j.devcel.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 12.McCall CE, et al. Epigenetics, bioenergetics, and microRNA coordinate gene-specific reprogramming during acute systemic inflammation. Journal of Leukocyte Biology. 2011;90:439–446. doi: 10.1189/jlb.0211075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcu K, et al. NF-kappaB Signaling: Multiple Angles to Target OA. Curr Drug Targets. 2010;11:599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zahnow CA, Baylin SB. Epigenetic Networks and miRNAs in Stem Cells and Cancer. Molecular Cell. 2010;39:661–663. doi: 10.1016/j.molcel.2010.08.036. [DOI] [PubMed] [Google Scholar]
- 15.Furumatsu T, et al. Sox9 and p300 cooperatively regulate chromatin-mediated transcription. J Biol Chem. 2005;280:35203–35208. doi: 10.1074/jbc.M502409200. [DOI] [PubMed] [Google Scholar]
- 16.Imamura T, et al. A novel tumor necrosis factor alpha-responsive CCAAT/enhancer binding protein site regulates expression of the cartilage-derived retinoic acid-sensitive protein gene in cartilage. Arthritis Rheum. 2008;58:1366–1376. doi: 10.1002/art.23438. [DOI] [PubMed] [Google Scholar]
- 17.Furumatsu T, et al. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J Biol Chem. 2005;280:8343–8350. doi: 10.1074/jbc.M413913200. [DOI] [PubMed] [Google Scholar]
- 18.Clark IM, et al. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol. 2008;40:1362–1378. doi: 10.1016/j.biocel.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Chabane N, et al. Histone deacetylase inhibitors suppress interleukin-1beta-induced nitric oxide and prostaglandin E2 production in human chondrocytes. Osteoarthritis Cartilage. 2008;16:1267–1274. doi: 10.1016/j.joca.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, et al. Inhibition of histone deacetylases antagonized FGF2 and IL-1beta effects on MMP expression in human articular chondrocytes. Growth Factors. 2009;27:40–49. doi: 10.1080/08977190802625179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young DA, et al. Histone deacetylase inhibitors modulate metalloproteinase gene expression in chondrocytes and block cartilage resorption. Arthritis Res Ther. 2005;7:R503–512. doi: 10.1186/ar1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong S, et al. A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J. 2009;23:3539–3552. doi: 10.1096/fj.09-133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haberland M, et al. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higashiyama R, et al. Correlation between MMP-13 and HDAC7 expression in human knee osteoarthritis. Mod Rheumatol. 2010;20:11–17. doi: 10.1007/s10165-009-0224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vega RB, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 26.Huh YH, et al. Regulation of Type II Collagen Expression by Histone Deacetylase in Articular Chondrocytes. Journal of Biological Chemistry. 2007;282:17123–17131. doi: 10.1074/jbc.M700599200. [DOI] [PubMed] [Google Scholar]
- 27.Dvir-Ginzberg M, et al. Regulation of cartilage-specific gene expression in human chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J Biol Chem. 2008;283:36300–36310. doi: 10.1074/jbc.M803196200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gagarina V, et al. SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010;62:1383–1392. doi: 10.1002/art.27369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takayama K, et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 2009;60:2731–2740. doi: 10.1002/art.24864. [DOI] [PubMed] [Google Scholar]
- 30.Fujita N, et al. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J Orthop Res. 2011;29:511–515. doi: 10.1002/jor.21284. [DOI] [PubMed] [Google Scholar]
- 31.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeung F, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu TF, et al. NAD+-dependent SIRT1 Deacetylase Participates in Epigenetic Reprogramming during Endotoxin Tolerance. Journal of Biological Chemistry. 2011;286:9856–9864. doi: 10.1074/jbc.M110.196790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dvir-Ginzberg M, et al. Tumor necrosis factor alpha-mediated cleavage and inactivation of sirT1 in human osteoarthritic chondrocytes. Arthritis Rheum. 2011;63:2363–2373. doi: 10.1002/art.30279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Santa F, et al. The Histone H3 Lysine-27 Demethylase Jmjd3 Links Inflammation to Inhibition of Polycomb-Mediated Gene Silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 36.Olivotto E, et al. Differential requirements for IKKalpha and IKKbeta in the differentiation of primary human osteoarthritic chondrocytes. Arthritis & Rheumatism. 2008;58:227–239. doi: 10.1002/art.23211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodova M, et al. Nfat1 regulates adult articular chondrocyte function through its age-dependent expression mediated by epigenetic histone methylation. J Bone Miner Res. 2011;26:1974–1986. doi: 10.1002/jbmr.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roach HI, et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52:3110–3124. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- 39.Roach HI, Aigner T. DNA methylation in osteoarthritic chondrocytes: a new molecular target. Osteoarthritis Cartilage. 2007;15:128–137. doi: 10.1016/j.joca.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Hashimoto K, et al. DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 2009;60:3303–3313. doi: 10.1002/art.24882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.da Silva MA, et al. Cellular and epigenetic features of a young healthy and a young osteoarthritic cartilage compared with aged control and OA cartilage. J Orthop Res. 2009;27:593–601. doi: 10.1002/jor.20799. [DOI] [PubMed] [Google Scholar]
- 42.Cheung KS, et al. Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol Int. 2009;29:525–534. doi: 10.1007/s00296-008-0744-z. [DOI] [PubMed] [Google Scholar]
- 43.Iliopoulos D, et al. Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: possible molecular target for osteoarthritis therapeutic intervention. Ann Rheum Dis. 2007;66:1616–1621. doi: 10.1136/ard.2007.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imagawa K, et al. The epigenetic effect of glucosamine and a nuclear factor-kappa B (NF-kB) inhibitor on primary human chondrocytes--implications for osteoarthritis. Biochem Biophys Res Commun. 2011;405:362–367. doi: 10.1016/j.bbrc.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Andres MC, et al. Suppressors of cytokine signalling (SOCS) are reduced in osteoarthritis. Biochem Biophys Res Commun. 2011;407:54–59. doi: 10.1016/j.bbrc.2011.02.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chubinskaya S, et al. Regulation of chondrocyte gene expression by osteogenic protein-1. Arthritis Res Ther. 2011;13:R55. doi: 10.1186/ar3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poschl E, et al. DNA methylation is not likely to be responsible for aggrecan down regulation in aged or osteoarthritic cartilage. Ann Rheum Dis. 2005;64:477–480. doi: 10.1136/ard.2004.022509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sesselmann S, et al. DNA methylation is not responsible for p21WAF1/CIP1 down-regulation in osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2009;17:507–512. doi: 10.1016/j.joca.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 49.Zimmermann P, et al. Correlation of COL10A1 induction during chondrogenesis of mesenchymal stem cells with demethylation of two CpG sites in the COL10A1 promoter. Arthritis Rheum. 2008;58:2743–2753. doi: 10.1002/art.23736. [DOI] [PubMed] [Google Scholar]
- 50.Aigner T, et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54:3533–3544. doi: 10.1002/art.22174. [DOI] [PubMed] [Google Scholar]
- 51.Ijiri K, et al. Differential expression of GADD45beta in normal and osteoarthritic cartilage: potential role in homeostasis of articular chondrocytes. Arthritis Rheum. 2008;58:2075–2087. doi: 10.1002/art.23504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brena RM, et al. Toward a human epigenome. Nat Genet. 2006;38:1359–1360. doi: 10.1038/ng1206-1359. [DOI] [PubMed] [Google Scholar]
- 53.Eckhardt F, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marini A, et al. Epigenetic inactivation of tumor suppressor genes in serum of patients with cutaneous melanoma. J Invest Dermatol. 2006;126:422–431. doi: 10.1038/sj.jid.5700073. [DOI] [PubMed] [Google Scholar]
- 55.Sutherland KD, et al. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. 2004;23:7726–7733. doi: 10.1038/sj.onc.1207787. [DOI] [PubMed] [Google Scholar]
- 56.Iliopoulos D, et al. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE. 2008;3:e3740. doi: 10.1371/journal.pone.0003740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi T, et al. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proceedings of the National Academy of Sciences. 2008;105:1949–1954. doi: 10.1073/pnas.0707900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones SW, et al. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-[alpha] and MMP13. Osteoarthritis and Cartilage. 2009;17:464–472. doi: 10.1016/j.joca.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 59.Dunn W, et al. Profiling microRNA expression in bovine articular cartilage and implications for mechanotransduction. Arthritis & Rheumatism. 2009;60:2333–2339. doi: 10.1002/art.24678. [DOI] [PubMed] [Google Scholar]
- 60.Murata K, et al. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2010;12:R86. doi: 10.1186/ar3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Araldi E, Schipani E. MicroRNA-140 and the silencing of osteoarthritis. Genes Dev. 2010;24:1075–1080. doi: 10.1101/gad.1939310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamasaki K, et al. Expression of MicroRNA-146a in osteoarthritis cartilage. Arthritis & Rheumatism. 2009;60:1035–1041. doi: 10.1002/art.24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miyaki S, et al. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis & Rheumatism. 2009;60:2723–2730. doi: 10.1002/art.24745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miyaki S, et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010;24:1173–1185. doi: 10.1101/gad.1915510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X, et al. MicroRNA-146a is linked to pain-related pathophysiology of osteoarthritis. Gene. 2011;480:34–41. doi: 10.1016/j.gene.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan C, et al. MicroRNA regulation associated chondrogenesis of mouse MSCs grown on polyhydroxyalkanoates. Biomaterials. 2011;32:6435–6444. doi: 10.1016/j.biomaterials.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 67.Sorrentino A, et al. Isolation and characterization of CD146+ multipotent mesenchymal stromal cells. Exp Hematol. 2008;36:1035–1046. doi: 10.1016/j.exphem.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 68.Sumiyoshi K, et al. Identification of miR-1 as a micro RNA that supports late-stage differentiation of growth cartilage cells. Biochem Biophys Res Commun. 2010;402:286–290. doi: 10.1016/j.bbrc.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 69.Lin EA, et al. miR-199a*, a Bone Morphogenic Protein 2-responsive MicroRNA, Regulates Chondrogenesis via Direct Targeting to Smad1. J Biol Chem. 2009;284:11326–11335. doi: 10.1074/jbc.M807709200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kapinas K, Delany AM. MicroRNA biogenesis and regulation of bone remodeling. Arthritis Res Ther. 2011;13:220. doi: 10.1186/ar3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sinha KM, et al. Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. EMBO J. 2010;29:68–79. doi: 10.1038/emboj.2009.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stanczyk J, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis & Rheumatism. 2008;58:1001–1009. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- 73.Abouheif MM, et al. Silencing microRNA-34a inhibits chondrocyte apoptosis in a rat osteoarthritis model in vitro. Rheumatology (Oxford) 2010;49:2054–2060. doi: 10.1093/rheumatology/keq247. [DOI] [PubMed] [Google Scholar]
- 74.le Sage C, et al. Regulation of the p27Kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wienholds E, et al. MicroRNA Expression in Zebrafish Embryonic Development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 76.Tuddenham L, et al. The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Letters. 2006;580:4214–4217. doi: 10.1016/j.febslet.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 77.Nicolas FE, et al. mRNA expression profiling reveals conserved and non-conserved miR-140 targets. RNA Biol. 2011;8 doi: 10.4161/rna.8.4.15390. [DOI] [PubMed] [Google Scholar]
- 78.Nicolas FE, et al. Experimental identification of microRNA-140 targets by silencing and overexpressing miR-140. RNA. 2008;14:2513–2520. doi: 10.1261/rna.1221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pais H, et al. Analyzing mRNA expression identifies Smad3 as a microRNA-140 target regulated only at protein level. RNA. 2010;16:489–494. doi: 10.1261/rna.1701210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nakamura Y, et al. Chondrocyte-Specific MicroRNA-140 Regulates Endochondral Bone Development and Targets Dnpep To Modulate Bone Morphogenetic Protein Signaling. Mol Cell Biol. 2011;31:3019–3028. doi: 10.1128/MCB.05178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akhtar N, et al. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010;62:1361–1371. doi: 10.1002/art.27329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tardif G, et al. Regulation of the IGFBP-5 and MMP-13 genes by the microRNAs miR-140 and miR-27a in human osteoarthritic chondrocytes. BMC Musculoskeletal Disorders. 2009;10:148. doi: 10.1186/1471-2474-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taganov KD, et al. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boldin MP, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. The Journal of Experimental Medicine. 2011;208:1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dudek KA, et al. Type II collagen expression is regulated by tissue-specific miR-675 in human articular chondrocytes. J Biol Chem. 2010;285:24381–24387. doi: 10.1074/jbc.M110.111328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karlsen TA, et al. Human primary articular chondrocytes, chondroblasts-like cells, and dedifferentiated chondrocytes: differences in gene, microRNA, and protein expression and phenotype. Tissue Eng Part C Methods. 2011;17:219–227. doi: 10.1089/ten.TEC.2010.0200. [DOI] [PubMed] [Google Scholar]
- 87.Lin L, et al. Assessment of the profiling MicroRNA expression of differentiated and dedifferentiated human adult articular chondrocytes. J Orthop Res. 2011;29:1578–1584. doi: 10.1002/jor.21423. [DOI] [PubMed] [Google Scholar]
- 88.Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci. 2010;1192:230–237. doi: 10.1111/j.1749-6632.2009.05240.x. [DOI] [PubMed] [Google Scholar]
- 89.Fukui N, et al. Regional differences in chondrocyte metabolism in osteoarthritis: a detailed analysis by laser capture microdissection. Arthritis Rheum. 2008;58:154–163. doi: 10.1002/art.23175. [DOI] [PubMed] [Google Scholar]
- 90.Kannu P, et al. Employing molecular genetics of chondrodysplasias to inform the study of osteoarthritis. Arthritis Rheum. 2009;60:325–334. doi: 10.1002/art.24251. [DOI] [PubMed] [Google Scholar]
- 91.Valdes AM, et al. The GDF5 rs143383 polymorphism is associated with osteoarthritis of the knee with genome-wide statistical significance. Ann Rheum Dis. 2011;70:873–875. doi: 10.1136/ard.2010.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Valdes AM, et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum. 2010;62:2347–2352. doi: 10.1002/art.27530. [DOI] [PubMed] [Google Scholar]
- 93.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iorio MV, et al. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta. 2010;1799:694–701. doi: 10.1016/j.bbagrm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 95.Gan Q, et al. Concise review: epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem Cells. 2007;25:2–9. doi: 10.1634/stemcells.2006-0383. [DOI] [PubMed] [Google Scholar]
- 96.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 97.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 98.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 99.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 100.Attwood JT, et al. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci. 2002;59:241–257. doi: 10.1007/s00018-002-8420-z. [DOI] [PMC free article] [PubMed] [Google Scholar]