

Abstract
Family, adoption and twin data each support substantial heritability for addictions. Most of this heritable influence is not substance-specific. The overlapping genetic vulnerability for developing dependence on a variety of addictive substances suggests large roles for “higher order” pharamacogenomics in addiction molecular genetics. We and others have now completed genome-wide association (GWA) studies of DNAs from individuals with dependence on a variety of addictive substances vs appropriate controls. Recently reported replicated GWA observations identify a number of genes based on comparisons between controls and European-American and African-American polysubstance abusers. Here we review the convergence between these results and data that compares control samples and a) alcohol dependent European Americans, b) methamphetamine dependent Asians and c) nicotine dependent samples from European backgrounds. We also compare these human data to quantitative trait locus (QTL) results from studies of addiction-related phenotypes in mice that focus on alcohol, methamphetamine and barbiturates. These comparisons support a genetic architecture built from largely-polygenic contributions of common allelic variants to dependence on a variety of legal and illegal substances. Many of the gene variants identified in this way are likely to alter specification and maintenance of neuronal connections.
Keywords: Association genome scanning, substance dependence, microarray, pooled, neuronal connections
I. Complex genetic influences on human addiction vulnerability are supported by classical genetic studies in humans and murine QTL studies
A. Studies of human addiction vulnerabilities
Vulnerability to addictions is a complex trait with strong genetic influences that are largely shared by abusers of different legal and illegal addictive substances [1–4]. This conclusion comes from family, adoption and twin studies. Family studies clearly document that first degree relative (eg sibs) of addicts display greater risk for developing substance dependence than more distant relatives [1, 5]. Adoption studies consistently find greater similarities between substance abuse phenotypes with biological relatives than with adoptive family members [1]. In twin studies, differences in concordance between genetically identical and fraternal twins also support heritability for vulnerability to addictions [3, 6–12]. Twin data allows quantitation of the amount, about 50%, of addiction vulnerability that is heritable. Twin data also supports the idea that the environmental influences on addiction vulnerability that are not shared among members of twin pairs are much larger than those that are shared by members of twin pairs. Many environmental influences on human addiction vulnerability are thus likely to come from outside of the family environment.
We can also seek differences between identical vs fraternal twin pairs in the degree to which one twin’s dependence on one substance enhances the chance that his or her co-twin will become dependent on a substance of a different class. Such twin data allows identification of the extent to which genetic influences on addiction vulnerability are common to dependence on multiple different substances vs the extent to which they are substance-specific. Interestingly, much of the genetic influence on addiction comes from genetic factors that are common to most addictive substances [2, 9, 10].
There are few studies of the ways in which most human addiction vulnerabilities move through families (eg segregation analyses). No such study indicates a “major” gene effect on addiction vulnerability in most current populations. There is one exception: the “flushing syndrome” variants at the aldehyde (ALDH) and alcohol (ADH) dehydrogenase loci in Asian individuals do provide genes of major effect in this population. Individuals with these gene variants are at much lower risk for becoming dependent on alcohol than individuals with other genotypes [13] in Chinese [14, 15], Korean [16], Japanese [17–22] and other populations [23, 24]. Homozygous ALDH2*2 individuals are strongly protected from alcohol dependence [17, 18].
Linkage-based analyses for addiction vulnerabilities would be expected to reproducibly identify any gene whose variants exerted a major influence on human addiction vulnerability (see below). Linkage-based genome scans ask how markers and genes move together through families. Existing linkage data for human dependence on alcohol, nicotine and a number of other substances reveals no evidence to support any major gene locus. That is, no locus individually appears to contribute a substantial fraction of the vulnerability to any addictive substance. There is a caveat: these data come from subjects that largely have European ethnic/racial backgrounds [25–41]. Nevertheless, as with many complex human disorders in which initial hopes for a tractable underlying genetic architecture supported use of linkage approaches, linkage peaks may be more likely to arise when polygenes that each contribute modest amounts to addiction vulnerability happen to lie near each other on human chromosomes rather than when there is a single gene variant of major effect for addiction vulnerability [42].
Our best current assessment for the genetic architecture of human dependence on legal and illegal addictive substances, then, is that each is influenced roughly 50% by polygenic genetic influences, that is by variants in individual genes that contribute modest amounts to this overall genetic vulnerability. The best current genetic architectural model posits that many of these influences are shared by individuals who are dependent on addictive substances from several different chemical classes, but that some of the genetic influences are specific to drugs of one class. While the best documented genetic heterogeneity for addictions comes from the chromosome 4 major gene effects found in poorly-alcohol-metabolizing (“flushing”) Asian individuals [17–19, 23], there are likely to be other examples of genetic heterogeneity that have yet to be elucidated.
B. Studies of murine strain differences in drug related phenotypes
Inbred mouse strains have members who are virtually genetically identical to each other but differ from members of other inbred strains. Comparing phenotypic differences among “unrelated” members of different strains thus provides one way in which individual differences in “unrelated” humans can be modeled. There is substantial conservation between the nature and function of most murine vs human genes. The large evolutionary distance from common ancestors to mice and men makes it unlikely that the same gene variants are maintained in members of both species through their separate evolutionary paths. However, common human variants and common murine strain differences both need to withstand pressures from natural selection processes including many that may overlap between these two species. There is thus some rationale for seeking addiction-associated variants in murine genes that have survived selection pressures on mice as potential models for common addiction-associated human variants that have also survived evolutionary selection pressures on humans. If there are a limited number of genes that can contain variants likely to influence vulnerability to addiction, not provide lethality and not provide strong negative selection, some of these genes from such a group may be more likely than a random group of genes to contain both murine strain differences and common human allelic variants that influence addiction phenotypes.
Inbred mouse strains do differ in a number of drug related phenotypes [43–48]. Many of these phenotypes have now been studied in “quantitative trait locus” paradigms. In these approaches, recombinant inbred (RI) mice are frequently studied [49, 50]. Each strain of these RI mice contains a different complement of reassorted chromosomal segments derived from two progenitor inbred strains. By comparing the ways in which the RI strain - to - RI strain variation in the phenotype co-varies with the RI strain - to - RI strain differences in the complement of chromosomal segments derived from each of the progenitor strains, chromosomal regions that contribute to variance in each trait can be mapped. The chromosomal regions identified in the most common RI strains are large. Thus, mapping of several of the most robust “QTLs” related to drug actions has been improved by developing congenic mouse lines that follow the drug response differences as the genomic intervals that derive from one of the progenitor strains are narrowed. Nevertheless, the intervals that are identified with many of the drug associated QTLs are still large, contain many genes, and thus often provide regions that are homologous with (syntenic with) segments of several different human chromosomes
Databases that contain many drug response QTLs and congenic intervals from fine mapping studies are available (eg http://www.ohsu.edu/parc/data/qtl/by_phen.shtml). These data provide opportunities to seek correlations between these QTLs and human addiction vulnerability genes. To do this, however, we must first relate the murine chromosomal regions that contain these QTLs to the corresponding syntenic human chromosomal regions (see below).
II. Human data supports “higher order” pharmacogenomics for much of the genetic influence on human addiction
There are several possible levels of analysis for pharmacogenomics and pharmacogenetics. It is useful to consider the classical genetic data noted above when considering which level to apply to the genetics of individual differences in human addiction vulnerability. We use “primary” pharmacogenomics to describe the genetics of individual differences in the adsorption, distribution, metabolism and/or excretion of a drug. Asian flushing-related gene variants provide a strong example of primary pharmacogenomics. We use “secondary” pharmacogenomics and pharmacogenetics to describe individual differences in drug targets, such as the G-protein coupled receptors, transporters and ligand gated ion channels that are the primary targets of opiates, psychostimulants and barbiturates, respectively. However, it is “higher order” pharmacogenomics and pharmacogenetics that appear most likely to influence vulnerability to addictive substances from several different chemical classes. This “higher order” pharmacogenomics and pharmacogenetics provides individual differences in post-receptor drug responses. These post-receptor drug responses are thus less likely to be specific to individual classes of addictive substances and more likely to be common to actions of abused substances of several different chemical classes. This description fits well with the description, noted above, of the nature of most of the genetic influence on human individual differences in vulnerability to substance dependence. We thus postulate that most of the human genetics of addition vulnerability is likely to represent “higher order” pharmacogenetics and pharmacogenomics as we define it here.
III. Linkage vs association for identifying gene variants that underlie addiction-related phenotypes
A. Linkage approaches
Most of the available literature that attempts to “positionally clone” gene variants for human addictions utilizes linkage-based methods. Linkage asks how addiction phenotypes and genetic markers (typically genotyped approximately every 1/400th of the genome) move together through pedigrees of related individuals. Many loci with nominally-significant linkage to addiction phenotypes have been identified for most common addictions. Several of these loci do overlap. However, the large numbers of linkage-based studies that are now reported yield large numbers of nominally-positive results. Virtually all chromosomes are covered [27, 30, 31, 51–60]. Thus, the overall convergence between these datasets may not differ from chance when all data are lumped together. Convergence between data for linkage to smoking related phenotypes that we have assembled in Figure 1, for example, is present at only chance levels.
Figure 1.
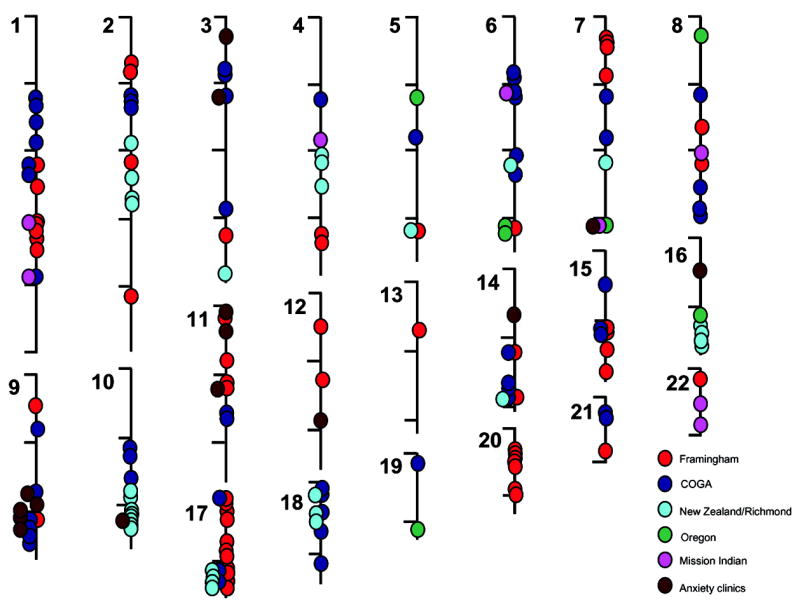
Diagram of the chromosomal locations of linkage peaks with nominally significant LOD scores (as defined by the papers authors) for smoking related phenotypes in individual or repeated analyses of data from Framingham study participants, participants in the Collaborative Study on the Genetics of Alcoholism (COGA), smokers recruited in New Zealand and in Richmond, Virginia, nicotine family study participants in Oregon, Mission Indian samples studied for alcohol and smoking phenotype and participants in anxiety clinics [27, 30, 31, 51–60]. Convergence between these results does not differ from chance in Monte Carlo simulations.
These sorts of results could be viewed as discouraging for attempts to identify the genes and gene variants that underlie human addictions. On the other hand, these data could also indicate that most of the genetic architecture for human addiction vulnerability is likely to be polygenic in most populations. A genetic architecture with modest individual contributions from variants at a number of loci that are likely to exert influences in each addicted individual supports use of association based approaches. Genome wide association (see below) seems to be the best way to aid in defining which reported linkage results are more or less likely to be “real”, eg to represent reproducible observations (reviewed in [61–67])
B. Association approaches
There is now an increasing consensus that genome-wide association approaches are likely to yield positive results in complex disorders, such as addictions. Association asks how addiction phenotypes and genetic markers (genotyped approximately every 1/600,000th of the genome in current datasets) are found together in nominally-unrelated individuals (though we are all distantly related to each other, of course). We and others have developed these methods, relying on the increasing density of single nucleotide polymorphism (SNP) markers that can be assessed with decreasing costs/SNP using “SNP chip” microarrays of increasing sophistication.
These association approaches, now termed genome-wide association (GWA) or whole genome association, can help elucidate chromosomal regions and genes that contain allelic variants that predispose to substance abuse without requiring family member participation. GWA gains power as densities of genomic markers increase. Association identifies smaller chromosomal regions than linkage-based approaches. GWA fosters pooling strategies that preserve confidentiality and reduce costs, such as the microaray-based approaches that we and others have developed and extensively validated [68–78]. GWA provides ample genomic controls that can minimize the chances of unintended ethnic mismatches between disease and control samples.
The large number of comparisons that are key components of GWA do raise concerns, however. These considerations mandate statistical approaches, such as the Monte Carlo methods that we use here, that correct for many multiple comparisons. Considerations based on this multiple testing problem also impel us to focus on results that can be replicated in several independent samples. We thus especially focus here on data that provides convergence between observations made in several independent samples. To seek results that are the most likely to be generalizable, we focus on results that replicate in 1) studies of individuals with dependences on several different classes of addictive substances and 2) in studies of individuals who come from European-American, African-American (largely admixed West African and European heritages) and Asian genetic backgrounds. Reproducible observations made in individuals of each of these groups are more likely to provide a solid set of observations that will be reproducible elsewhere. Seeking such convergence will exclude population-specific variants and variants that predispose to addictions to only specific substance classes, however.
IV. Human genome wide association and murine quantitative trait locus data provide opportunities for identifying convergent information about specific genes with increasing power and selectivity
A. GWA studies of human addiction vulnerabilities
1) Initial data from African- and European-American polysubstance abusers
To elucidate genes likely to harbor addiction-associated variants [72, 79, 80], we have recently reported GWA studies that assess allele frequencies for 639,401 autosomal SNPs using four Affymetrix microarray types [68] in each of two samples.
Samples
We have studied two samples, one African- and the other European-American, of polysubstance abusers who reported dependence on at least one illegal substance. We have compared these data to data from ethnically- matched controls who reported no significant lifetime use of any addictive substance. DNAs for these studies came from research volunteers, largely for nontherapeutic studies, who came to the NIDA-IRP in Baltimore Maryland. Each subject provided an informed consent, self-reported his or her ethnicity, provided drug use histories and provided DSMIII-R or IV diagnoses of substance use disorders [72, 79, 80]. “Abusers” displayed heavy lifetime use of illegal substances [77] and dependence on at least one illegal substance. “Controls” displayed neither abuse nor dependence on any addictive substance and report no significant lifetime histories of use of any addictive substance. Control individuals thus combined individuals with no lifetime experience with any addictive substance with individuals who had modest exposures to legal addictive substances.
Genotyping
We assessed allele frequencies in about 73 DNA pools, each containing equal amounts of DNA from 20 individuals of the same racial and phenotype group. Each DNA pool was assessed on four sets of four arrays, two from “100k” and two from “500k” Affymetrix microarray sets. Importantly, the methods used for these samples revealed r = 0.95 correlations between pooled and individual genotyping in extensive validation studies that are reported in detail in ref [68]. Data for each SNP thus provided a score that reflected a continuous percentage of its two alleles in hetero- and homozygotes in each pool. For SNPs that displayed nominally-significant differences between addicted and control individuals, there was a r = 0.9 correlation between the magnitude of the differences between dependent vs nondependent data that derived from pooled vs the magnitude of the differences derived from individual genotyping approaches. Variance from array-to-array that assessed the same DNA pool and variance from pool-to-pool were both modest, ca 3%, supporting good validity of this pooling data.
2) Convergence with human substance dependence datasets
a: European-American alcohol dependence [77]
Sample 1
Unrelated individuals sampled from pedigrees collected by the Collaborative Study on the Genetics of Alcoholism provided an interesting sample for this approach for several reasons. Dependence on alcohol and other substances has been carefully characterized in these individuals using validated instruments. Unrelated control individuals free from substance abuse or dependence diagnoses, largely individuals who marry into these pedigrees, were also available. We thus identified 120 unrelated alcohol dependent individuals and 160 unrelated unaffected controls with self-reported European-American ethnicities.
Genotyping
We assessed allele frequencies in 14 DNA pools, each containing equal amounts of DNA from 20 individuals of the same phenotype group, on four sets of four arrays, two from “100k” and two from “500k” Affymetrix SNP arrays, using approaches that were extensively validated as described above.
b) Asian methamphetamine-dependence
Sample 2
Unrelated subjects were recruited in Taipei included 140 methamphetamine-dependent individuals diagnosed using DSM IV criteria [81] [82–86]. 240 matching Han Chinese normal controls denied any history of use of illegal drugs and denied any histories of psychotic symptoms.
Sample 3
Subjects who were born and resided in Japan included one hundred methamphetamine dependent subjects were in- or outpatients of psychiatric hospitals that participated in the Japanese Genetics Initiative for Drug Abuse (JGIDA) and met ICD-10-DCR criteria for methamphetamine dependence in diagnoses made by two trained psychiatrists. Controls were one hundred age-, gender-, and geographically-matched staff recruited at the same institutions who were free from exposure to any illegal substance, abuse or dependence of any legal substance, comorbidity with any psychotic psychiatric illness and family histories of substance dependence or psychotic psychiatric illness based on detailed psychiatric interviews.
Genotyping
We assessed allele frequencies in these 29 DNA pools, each containing equal amounts of DNA from 20 individuals of the same phenotype group, on four sets of arrays. For sample 3, two arrays from “100k” and two from “500k” Affymetrix SNP arrays were used. For sample 2, two array sets from “500k” Affymetrix SNP arrays were used. Approaches that were extensively validated as described above were used for this genotyping.
c) Australian and US dependent vs nondependent smokers of European ancestry [87]
Samples
Participants of European ancestry from the Collaborative Genetic Study of Nicotine Dependence or the Nicotine Addiction Genetics study in the US and Australia, respectively were sampled. Smokers with dependence diagnoses by the Fagerström Test for Nicotine Dependence were compared to smokers who had no FTND dependence.
Genotyping
Allele frequency data was assessed in 16 DNA pools, each containing equal amounts of DNA from 60 individuals on a single set of 49 arrays that assessed 2,427,354 SNPs. Methods used for these samples revealed r = 0.85 correlations between pooled and individual genotyping. There was a modest, r = 0.58 correlation between dependent vs nondependent data derived from pooled vs individual genotyping for the SNPs that displayed nominally significant differences. Individual genotyping was performed to follow up nominally-positive results for 39,213 SNPs in 1,050 dependent and 879 nondependent smokers. The convergence analyses presented below use the data from these 39,213 SNPs and from the subset of these SNPs that lie within exons of genes or within 0.1Mb 3′ and 5′ flanking regions of these exons.
B. QTL studies of mouse strain differences in drug-related phenotypes
Mouse quantitative trait locus (QTL) regions for traits related to addictive substances were assembled from data from QTL mapping and from the refined chromosomal regions defined by congenic mice for these phenotypes by Portland Alcohol Research Center investigators. These QTLs are found at http://www.ohsu.edu/parc/data/qtl/by_phen.shtml.). This site provides current information for alcohol, methamphetamine and pentobarbital QTLs. Traits mapped include: ethanol metabolism, ethanol-conditioned taste aversion and place preference, acute and chronic alcohol withdrawal, acute pentobarbital withdrawal, acute alcohol stimulated activity, alcohol hypothermia, alcohol-induced loss of righting reflex, alcohol preference drinking and methamphetamine stimulated locomotor activity (see Table II). QTLs with genome-wide “significant” at p < 0.01 or “suggestive” linkage at p < 0.05, as well as provisional loci “prov” without these levels of genome-wide significance when corrected for multiple comparisons, were selected for potential inclusion in the current analyses.
QTLs were mapped on mouse chromosomes to several degrees of resolution, since for some QTLs, only the peak marker location was available, while for others, more detailed mapping information included information from congenic mice. 1) Single point data with cM values, 2) single point data with Mb values, 3) chromosomal intervals with cM values and 4) chromosomal intervals with Mb values. For single point data with cM values, we used genes to bridge between mouse QTL regions and the human chromosomal regions that were syntenic with these murine QTLs. We followed the protocol “mouse QTL cM--> mouse genes in this cM area --> homologous human genes -->syntenic human regions”. We used the mapping files available at (http://www.informatics.jax.org/reports/homologymap/mouse_human.shtml). For single point data with Mb values, we transformed each specific position on mouse chromosome to human chromosome, using data from the UCSC website [88]. For broad intervals in either Mb or cM formats, we transformed murine data to murine chromosome bands, then to human chromosomal bands, then to human Mb regions. For example, we transformed “mouse 28-47cM” to human 2q33-q36. Since mouse/human synteny is discontinuous, we identified more human chromosomal regions, each smaller, than the initial 60 mouse chromosomal regions.
We focused on QTLs whose positions corresponded to human autosomes. We omitted QTL regions for which no synteny could be identified and two regions that were very large. For the 132 remaining human chromosomal regions that correspond to (eg are syntenic with) mouse regions identified with addictive substance QTLs, we assessed Monte Carlo p values for the overlap with the reproducibly-positive SNPs from [68] as noted above.
V. Remarkable convergence between human genome wide association studies of dependence on a variety of addictive substances
There is a remarkable degree of overall convergence between the clustered, reproducibly positive SNPs in the NIDA samples and those identified by comparisons of alcohol dependent and methamphetamine dependent individuals.
600,000+ SNP genome scans for the alcohol dependence vs control samples yielded 1098 clustered SNPs that displayed nominally-positive dependence vs control differences and also overlapped with the clustered nominally positive SNPs identified in each of two NIDA samples that lay within genes. Corresponding values for the two methamphetamine dependent Asian samples were 498 and 291 for 500,000 SNP GWA studies. Each of these overlaps was highly significant. Using Monte Carlo methods that correct for repeated comparisons; fewer than 1 in 10,000 simulation trials identified such overlaps by chance in each case. Confirmatory permutation analyses that also correct for repeated comparisons also identify fewer than 1 in 10,000 permutations that provide such results by chance alone in each case in which we have applied them.
There was more modest overlap between these data and data from comparisons of nondependent vs dependent smokers [87]. Several comparisons did reach statistical significance in Monte Carlo trials that provide correction for the repeated comparisons involved. When we focus on SNPs that distinguish dependent vs nondependent smokers that lie within genes, there is a significant, p = 0.03, overlap with 294 of the SNPs that display nominally-significant abuser vs control differences in each of the two NIDA polysubstance abuser vs control samples. The 64 NIDA SNPs that are clustered within genes display a p = 0.088 trend toward significant overlap with this data that does not reach significance.
VI. Additional support for genes identified in Liu et al [68] provided by convergence between human genome wide association studies of dependence
One approach to describing the convergence between these datasets relies on the overall convergence, as described above. Another approach relies on identifying the numbers of nominally-positive SNPs that lie in genes nominated by an initial dataset. We begin here with the set of genes identified in [68], which were initially identified on the basis of strong support from clustered, reproducibly-positive SNPs in European- and African American samples from NIDA with more modest levels of support from 100k SNP genome wide association datasets from JGIDA methamphetamine-dependence and COGA alcohol-dependence samples.
Current availability of data based on 520,000 – 630,000 autosomal SNPs from these samples, our ability to move these datasets to the more-complete build 36.1 of the NCBI human genome assembly and the availability of data from other samples listed above provides the data for Table I. To be able to display data from all of the samples in the same way, this table documents convergence of data from all nominally positive SNPs (not just those that cluster). Nevertheless, this data strongly supports the overall convergences documented in the simulations above for virtually all of the genes identified in Liu et al [68]. Altered marker locations in human genome build 36 data do move previously-reported positive association signals away from the PCDH9, DEFB1 and XKR5 genes that are included in build 35 data in Liu et al [68], however.
Table I.
Genes and classes of genes that contain clustered SNPs that provide nominally-significant abuser vs control differences in both European-American and African-American NIDA (N) samples [68]. The numbers of SNPs within each gene (exons +/− 100kb flank) that display nominally-significant differences in comparisons between N: polysubstance abusers and control individuals in both replicate NIDA samples [51] C: alcohol dependent vs control COGA samples [68, 77], J: methamphetamine dependent vs control samples from JGIDA (Drgon et al, manuscript in preparation), T: methamphetamine dependent vs control samples from Taiwan (Uhl et al, manuscript in preparation) and B: nicotine dependent smokers vs nondependent smokers from Australia and the US [87]. Genes are grouped by the class of the function to which they contribute: “CAM” cell adhesion, “CHA” channels, “DIS” disease associated, “ENZ” enzymes, “OTHER” other functions, “PROT” protein processing, “REC” receptors (combining single TM, seven TM and ligand-gated channel families), “STR” structural, “TF” transcriptional regulation, “TRANSP” transporters, and unknown functions.
Chromosome number and initial chromosomal position for the cluster (bp, NCBI Mapviewer Build 36.1) are listed. Several genes are identified by the same clusters of positive SNPs.
| Nominally-positive SNPS | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene [51] | Class | gene description | ch | bp | N | C | J | T | B |
| CSMD2 | CAM | CUB and Sushi multiple domains 2 | 1 | 33752196 | 8 | 27 | 6 | 11 | |
| LRP1B | CAM | low density lipoprotein-related protein 1B | 2 | 140705466 | 15 | 73 | 38 | 16 | 4 |
| CNTN6 | CAM | contactin 6 | 3 | 1109629 | 4 | 17 | 6 | 13 | 6 |
| CNTN4 | CAM | contactin 4 | 3 | 2117247 | 6 | 32 | 14 | 10 | |
| LRRN1 | CAM | leucine rich repeat neuronal 1 | 3 | 3816554 | 3 | 13 | 9 | 1 | |
| LPHN3 | CAM | latrophilin 3 | 4 | 62045434 | 3 | 7 | 9 | 3 | |
| CTNND2 | CAM | catenin delta 2 | 5 | 11024952 | 5 | 53 | 17 | 8 | |
| TRIO | CAM | triple functional domain/PTPRF interacting | 5 | 14196829 | 6 | 11 | 3 | 7 | |
| BAI3 | CAM | brain-specific angiogenesis inhibitor 3 | 6 | 69404158 | 6 | 24 | 7 | 1 | |
| SEMA3C | CAM | semaphorin 3C | 7 | 80209790 | 4 | 10 | 7 | 0 | |
| CSMD1 | CAM | CUB and Sushi multiple domains 1 | 8 | 2782789 | 19 | 115 | 48 | 36 | 12 |
| SGCZ | CAM | sarcoglycan zeta | 8 | 13991744 | 11 | 61 | 35 | 17 | |
| PTPRD | CAM | receptor type protein tyrosine phosphatase D | 9 | 8307268 | 4 | 40 | 13 | 7 | 2 |
| LRRN6C | CAM | leucine rich repeat neuronal 6C | 9 | 27938528 | 10 | 44 | 23 | 21 | 18 |
| CTNNA3 | CAM | catenin alpha 3 | 10 | 67349937 | 11 | 33 | 18 | 7 | 21 |
| CNTN5 | CAM | contactin 5 | 11 | 98397081 | 7 | 42 | 18 | 9 | 7 |
| ANKS1B | CAM | ankyrin repeat and sterile alpha motif domain containing 1B | 12 | 97653202 | 5 | 17 | 13 | 5 | |
| POSTN | CAM | periostin | 13 | 37034779 | 6 | 10 | 11 | 4 | |
| CDH13 | CAM | cadherin 13 | 16 | 81218079 | 9 | 107 | 41 | 39 | 2 |
| DSCAM | CAM | Down syndrome cell adhesion molecule | 21 | 40306213 | 4 | 24 | 31 | 16 | |
| GJA5 | CHA | connexin 40 | 1 | 145695517 | 3 | 9 | 4 | 0 | |
| KCNQ3 | CHA | KQT-like voltage-gated potassium channel member 3 | 8 | 133210438 | 6 | 16 | 7 | 4 | 2 |
| TRPC4 | CHA | transient receptor potential cation channel C,4 | 13 | 37108795 | 4 | 11 | 5 | 4 | |
| KCNH5 | CHA | voltage-gated potassium channel H 5 | 14 | 62243698 | 6 | 13 | 6 | 3 | |
| RYR3 | CHA | ryanodine receptor 3 | 15 | 31390469 | 7 | 37 | 17 | 10 | 3 |
| ALS2CR19 | DIS | amyotrophic lateral sclerosis 2 chromosome region candidate 19 | 2 | 205118761 | 4 | 25 | 11 | 8 | 3 |
| PTHB1 | DIS | parathyroid hormone-responsive B1 | 7 | 33135677 | 4 | 9 | 8 | 3 | |
| AAA1 | DIS | AAA1 protein | 7 | 34356559 | 5 | 38 | 3 | 11 | 5 |
| ATXN10 | DIS | ataxin 10 | 22 | 44446351 | 4 | 9 | 5 | 1 | |
| TTLL7 | ENZ | tubulin tyrosine ligase-like family member 7 | 1 | 84107645 | 3 | 11 | 3 | 1 | |
| ACP6 | ENZ | lysophposphatidic acid phosphatase 6 | 1 | 145585792 | 4 | 8 | 4 | 0 | 1 |
| PTGS2 | ENZ | prostaglandin-endoperoxide synthase 2 | 1 | 184907592 | 3 | 7 | 2 | 0 | |
| DAF | ENZ | decay accelerating factor for complement | 1 | 205561488 | 5 | 3 | 21 | 8 | |
| SIPA1L2 | ENZ | signal-induced proliferation-associated 1 like 2 | 1 | 230600337 | 5 | 15 | 10 | 4 | |
| CAPN13 | ENZ | calpain 13 | 2 | 30799141 | 3 | 6 | 9 | 1 | 2 |
| CPS1 | ENZ | carbamoyl-phosphate synthetase 1 | 2 | 211129583 | 3 | 11 | 5 | 1 | |
| FHIT | ENZ | fragile histidine triad gene | 3 | 59710076 | 9 | 65 | 35 | 18 | 2 |
| PDE4D | ENZ | phosphodiesterase 4D | 5 | 58302468 | 4 | 11 | 12 | 9 | |
| UST | ENZ | uronyl-2-sulfotransferase | 6 | 149110157 | 5 | 8 | 4 | 5 | 4 |
| DGKB | ENZ | diacylglycerol kinase beta | 7 | 14153770 | 6 | 29 | 9 | 3 | |
| CHN2 | ENZ | chimerin 2 | 7 | 29200646 | 7 | 25 | 8 | 7 | |
| PDE1C | ENZ | phosphodiesterase 1C | 7 | 31795772 | 3 | 8 | 7 | 0 | |
| CAMK1D | ENZ | calcium/calmodulin-dependent protein kinase ID | 10 | 12431589 | 3 | 16 | 10 | 9 | |
| PRKG1a | ENZ | cGMP-dependent protein kinase I | 10 | 52504299 | 14 | 36 | 19 | 24 | |
| HPSE2 | ENZ | heparanase 2 | 10 | 100208867 | 5 | 29 | 14 | 8 | 3 |
| PZP | ENZ | pregnancy-zone protein | 12 | 9192703 | 3 | 3 | 6 | 2 | |
| SERPINA1 | ENZ | serpin peptidase inhibitor A 1 | 14 | 93914451 | 4 | 4 | 4 | 1 | |
| SERPINA2 | ENZ | serpin peptidase inhibitor A 2 | 14 | 93900404 | 4 | 3 | 4 | 1 | |
| USP31 | ENZ | ubiquitin specific peptidase 31 | 16 | 22986868 | 3 | 6 | 3 | 3 | |
| CHST9 | ENZ | carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase 9 | 18 | 22749593 | 3 | 9 | 6 | 1 | |
| RBMS3 | OTHER | RNA binding motif, single stranded interacting protein | 3 | 29297947 | 5 | 21 | 11 | 2 | 8 |
| RPA3 | OTHER | replication protein A3 | 7 | 7643100 | 5 | 11 | 2 | 0 | |
| HHLA1 | OTHER | HERV-H LTR-associating 1 | 8 | 133145212 | 4 | 7 | 5 | 0 | |
| RAB2-like | OTHER | FLJ45872 protein | 8 | 138890869 | 3 | 19 | 10 | 5 | |
| FGF14 | OTHER | fibroblast growth factor 14 | 13 | 101173036 | 4 | 29 | 9 | 6 | 5 |
| A2BP1 | OTHER | ataxin 2-binding protein 1 | 16 | 6009133 | 11 | 102 | 37 | 21 | 13 |
| OSBPL1A | OTHER | oxysterol binding protein-like 1A | 18 | 19996007 | 4 | 10 | 4 | 1 | |
| EPB41L2 | PROT | erythrocyte membrane protein band 4.1-like 2 | 6 | 131202180 | 3 | 17 | 2 | 1 | |
| ELMO1 | PROT | engulfment and cell motility 1 | 7 | 36860486 | 5 | 24 | 8 | 12 | |
| SORCS1 | PROT | sortilin-related VPS10 domain containing receptor 1 | 10 | 108323411 | 4 | 17 | 10 | 7 | 3 |
| MICALCL | PROT | MICAL C-terminal like | 11 | 12271946 | 3 | 10 | 9 | 3 | |
| MCTP2 | PROT | multiple C2 domains, transmembrane 2 | 15 | 92642499 | 3 | 5 | 5 | 4 | |
| IMPACT | PROT | Impact homolog | 18 | 20260680 | 4 | 8 | 3 | 1 | |
| DOK6 | PROT | docking protein 6 | 18 | 65219271 | 6 | 15 | 1 | 2 | |
| NAPB | PROT | neuritis with brachial prediliction | 20 | 23303165 | 3 | 7 | 1 | 7 | |
| CRIM1 | REC | cysteine rich transmembrane BMP regulator 1 | 2 | 36436901 | 5 | 16 | 13 | 12 | |
| GPR39 | REC | G protein-coupled receptor 39 | 2 | 132891086 | 4 | 8 | 3 | 4 | |
| bFGFR-like | REC | hypothetical protein FLJ32955 | 2 | 150332266 | 3 | 10 | 5 | 2 | |
| GRM7 | REC | glutamate receptor, metabotropic 7 | 3 | 6877927 | 5 | 59 | 24 | 14 | |
| GABRG1 | REC | GABA A receptor gamma 1 | 4 | 45732543 | 3 | 7 | 2 | 0 | |
| NPSR1 | REC | neuropeptide S receptor 1 | 7 | 34664422 | 5 | 32 | 3 | 8 | 3 |
| HRH4 | REC | histamine receptor H4 | 18 | 20294591 | 3 | 7 | 1 | 0 | |
| ACTN2 | STR | alpha 2 actinin | 1 | 234916422 | 3 | 5 | 3 | 2 | |
| OC90 | STR | similar to Otoconin 90 precursor | 8 | 133105910 | 4 | 5 | 5 | 0 | |
| AKAP13 | STR | A kinase anchor protein 13 | 15 | 83724875 | 3 | 12 | 7 | 4 | |
| FERD3L | TF | Fer3-like | 7 | 19150930 | 3 | 2 | 2 | 0 | |
| TWIST1 | TF | twist homolog 1 | 7 | 19121616 | 3 | 2 | 2 | 0 | |
| CNDB1 | TF | cyclic nucleotide binding domain containing 1 | 8 | 87947789 | 5 | 4 | 5 | 0 | |
| NFIB | TF | nuclear factor I/B | 9 | 14071847 | 3 | 13 | 3 | 3 | |
| E2F7 | TF | E2F transcription factor 7 | 12 | 75939159 | 4 | 2 | 2 | 1 | |
| NPAS3 | TF | neuronal PAS domain protein 3 | 14 | 32478200 | 8 | 43 | 24 | 5 | |
| ZNF407 | TF | zinc finger protein 407 | 18 | 70474282 | 5 | 20 | 2 | 0 | |
| NRIP1 | TF | nuclear receptor interacting protein 1 | 21 | 15255427 | 4 | 5 | 2 | 1 | |
| SLC9A9 | TRANSP | sodium/hydrogen exchanger 9 | 3 | 144466754 | 6 | 17 | 6 | 5 | 5 |
| XKR4 | TRANSP | XK family 4 | 8 | 56177571 | 8 | 20 | 4 | 11 | |
| ABCC4 | TRANSP | ATP-binding cassette C 4 | 13 | 94470090 | 3 | 11 | 10 | 4 | |
| CCDC93 | unknown | coiled-coil domain containing 93 | 2 | 118389617 | 5 | 6 | 1 | 1 | |
| KIAA1679 | unknown | KIAA1679 protein | 2 | 137464932 | 3 | 23 | 17 | 4 | |
| FLJ37543 | unknown | hypothetical protein FLJ37543 | 5 | 60969393 | 3 | 4 | 7 | 4 | |
| FLJ20323 | unknown | hypothetical protein FLJ20323 | 7 | 7573149 | 5 | 7 | 4 | 0 | |
| KIAA0143 | unknown | KIAA0143 protein | 8 | 133021330 | 3 | 5 | 8 | 0 | |
| KIAA1713 | unknown | KIAA1713 | 18 | 29412569 | 5 | 7 | 9 | 1 | |
| CCDC102B | unknown | coiled-coil domain containing 102B | 18 | 64655575 | 3 | 11 | 5 | 0 | |
The genes that we identify in this way are involved in cell adhesion, enzymatic activities, protein translation, trafficking and degradation; transcriptional regulation, receptor, ion channel and transport processes, disease processes, cell structures and other functions (Table I). Most are expressed in the brain. The genes whose products are involved in cell adhesion processes provide especially interesting results. Cell adhesion mechanisms are central for properly establishing and regulating neuronal connections during development and can play major roles in mnemonic processes in adults [89, 90]. Almost all of these cell adhesion-related genes are expressed in brain regions involved in memory, including hippocampus and cerebral cortex. By contrast, substantial expression in mesolimbic/mesocortical dopamine “reward system” neurons is not documented for most. These genes represent glycophosphoinositol (GPI)-anchored, single-transmembrane, and seven transmembrane cell adhesion molecules as well as genes whose products bind to cell adhesion-related protein complexes including CTNND2, TRIO, CTNNA3, ANKS1B and POSTN. These cell adhesion related genes thus provide attractive candidates for future studies of their possible relationships with addiction phenotypes.
Our identification of SNP markers whose allelic frequencies distinguish addicts of several different ethnicities from matched controls supports “common disease/common allele” genetic architecture [68] for addiction vulnerability. The convergent data derived from studies of individuals with addictions to substances in several different pharmacological classes supports the idea that “higher order pharmacogenomic/pharmacogenetic” variations enhance vulnerability to many addictions. These results do not exclude additional contributions to addiction vulnerability from genomic variants that influence vulnerability to specific substances or variants that are found only in specific populations. Nevertheless, the findings presented here provide promise for enhancing understanding of features that are common to human addictions in ways that could facilitate efforts to personalize prevention and treatment strategies for debilitating addictive disorders.
VII. Convergence between human genome wide association studies of dependence on a variety of addictive substances and murine QTLs for drug response phenotypes
There was also a statistically significant overlap between the data from the SNPs that provided reproducibly nominally positive results in the NIDA data and the human regions with synteny to the murine QTL regions. 38 QTL regions received such support from reproducibly-positive SNPs from the human NIDA samples, with overall p = 0.049 (Table II). While this p value is modest in comparison to data from the human convergences noted above, the fact that there is any significant convergence across these distinct species and phenotypes is remarkable.
VIII. Genes identified in Liu et al [68] and by murine QTLs for drug response phenotypes
Table II lists the best-supported QTLs and the gene or genes that are identified by convergent human data [68, 89–92] for each QTL. The genes identified by both of these approaches again can be related to cell adhesion processes, such as PTPRD, LRP2 and alpha 6 integrin, transcriptional regulation by NFI/B, NPAS2 and enzymes such as NIMA-related kinase 7.
It is interesting that PTPRD is identified in both datasets. Congenic mice that are made to narrow the chromosomal segment that contains this QTL, however, have suggested that murine variants in an adjacent gene, MPDZ confers the behavioral phenotype rather than PTPRD [93].
It is also interesting that formin 1 and formin 2 are both identified by multiple nominally-positive human SNPs in these convergent datasets. These genes are each highly expressed in brain where they are likely to play substantial roles in regulation of actin polymerization events. Controlled actin polymerization, in turn, is crucial to regulated formation and stabilization of neuronal processes, which in turn provide major elements for neuronal connections.
IX. Summary and conclusions
It is an exciting time to be able to summarize and review the rapidly-emerging data on the complex genetics of human addiction vulnerability. Recent availability of data from genome wide association studies provide results for dependence on several different classes of addictive substances that converge with each other in striking fashion that is highly unlikely to be due to chance. Present availability of data from genome wide association studies of dependence phenotypes in European-American, African-American and Asian samples all support the idea that many of the allelic variants that predispose to these common disorders are so evolutionarily old that they are present in members of each major current human population. These data, combined with the varying results from linkage-based studies, do not support any single gene of major effect in most populations. An underlying genetic architecture with polygenic contributions from currently common allelic variants thus fits with the data that we assemble here from molecular genetic studies. Such a genetic architecture is quite consistent with data from classical genetics, eg family, adoption and twin approaches.
The genes identified in this work should not displace consideration of the important roles that reward processes play in events that lead to substance dependence. However, identification of these genes that harbor variants that influence vulnerability to addictions does contribute to a growing body of data that implicates cell adhesion and related memory-like processes in addiction. Studies that alter reconsolidation and other memory-related processes using knockout mice, protein synthesis inhibitors and/or pharmacologic treatments can provide powerful influences on addictions [94–96]. This empirical evidence enriches theoretical work that increasingly recognizes memory-like features for addiction [97] and work that implicates memory-associated brain regions in relapse to addiction [98, 99]. Our current data also complement clinical observations which document that addicts’ enhanced vulnerabilities to substance abuse relapse can persist for decades after their last prior use of addictive substances.
As we elaborate and extend these molecular genetic approaches, it seems likely that the number of genes with variants that contribute to individual differences in human addiction vulnerability will continue to grow, and that many of the genes listed here will receive additional support. With complementary genetic studies of other clinically important phenomena, such as the ability to successfully quit use of an addictive substance [100], we will have more and more tools to be able to both better understand, better prevent and better treat these common and debilitating conditions.
Acknowledgments
This research was supported financially by the NIH Intramural Research Program, NIDA, DHSS and by grants AA10760 AA011114 and DA05228and support from the Department of Veterans Affairs to JC and KB. We are grateful for dedicated help with clinical characterization of NIDA subjects from Dan Lipstein, Fely Carillo, Carlo Contoreggi, Judith Hess and other Johns Hopkins-Bayview support staff and passionate discussions of statistical issues with Dr Daniel Naiman. We are also grateful to collaborators in the work cited here including COGA investigators, investigators in the Japanese Initiative for the Genetics of Drug Abuse (JGIDA), the Taiwan Methamphetamine study group. We thank the NicSNP group for providing access to the data from individual genotypes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Uhl GR, Elmer GI, Labuda MC, Pickens RW. Genetic influences in drug abuse. In: Gloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. New York: Raven Press; 1995. pp. 1793–2783. [Google Scholar]
- 2.Tsuang MT, Lyons MJ, Meyer JM, Doyle T, Eisen SA, Goldberg J, et al. Co-occurrence of abuse of different drugs in men: the role of drug-specific and shared vulnerabilities. Arch Gen Psychiatry. 1998;55:967–72. doi: 10.1001/archpsyc.55.11.967. [DOI] [PubMed] [Google Scholar]
- 3.Karkowski LM, Prescott CA, Kendler KS. Multivariate assessment of factors influencing illicit substance use in twins from female-female pairs. Am J Med Genet. 2000;96:665–70. [PubMed] [Google Scholar]
- 4.True WR, Heath AC, Scherrer JF, Xian H, Lin N, Eisen SA, et al. Interrelationship of genetic and environmental influences on conduct disorder and alcohol and marijuana dependence symptoms. Am J Med Genet. 1999;88:391–7. doi: 10.1002/(sici)1096-8628(19990820)88:4<391::aid-ajmg17>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 5.Merikangas KR, Stolar M, Stevens DE, Goulet J, Preisig MA, Fenton B, et al. Familial transmission of substance use disorders. Arch Gen Psychiatry. 1998;55:973–9. doi: 10.1001/archpsyc.55.11.973. [DOI] [PubMed] [Google Scholar]
- 6.Woodward CE, Maes HH, Silberg JL, Meyer JM, Eaves LJ. Tobacco, alcohol and drug use in 8-16 year old twins. NIDA Res Monograph. 1996;162:309. [Google Scholar]
- 7.Tsuang MT, Lyons MJ, Eisen SA, Goldberg J, True W, Lin N, et al. Genetic influences on DSM-III-R drug abuse and dependence: a study of 3,372 twin pairs. Am J Med Genet. 1996;67:473–7. doi: 10.1002/(SICI)1096-8628(19960920)67:5<473::AID-AJMG6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 8.Kendler KS, Prescott CA. Cocaine use, abuse and dependence in a population-based sample of female twins. Br J Psychiatry. 1998;173:345–50. doi: 10.1192/bjp.173.4.345. [DOI] [PubMed] [Google Scholar]
- 9.Kendler KS, Aggen SH, Tambs K, Reichborn-Kjennerud T. Illicit psychoactive substance use, abuse and dependence in a population-based sample of Norwegian twins. Psychol Med. 2006;36:955–62. doi: 10.1017/S0033291706007720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agrawal A, Neale MC, Prescott CA, Kendler KS. A twin study of early cannabis use and subsequent use and abuse/dependence of other illicit drugs. Psychol Med. 2004;34:1227–37. doi: 10.1017/s0033291704002545. [DOI] [PubMed] [Google Scholar]
- 11.Grove WM, Eckert ED, Heston L, Bouchard TJ, Jr, Segal N, Lykken DT. Heritability of substance abuse and antisocial behavior: a study of monozygotic twins reared apart. Biol Psychiatry. 1990;27:1293–304. doi: 10.1016/0006-3223(90)90500-2. [DOI] [PubMed] [Google Scholar]
- 12.Gynther LM, Carey G, Gottesman II, Vogler GP. A twin study of non-alcohol substance abuse. Psychiatry Res. 1995;56:213–20. doi: 10.1016/0165-1781(94)02609-m. [DOI] [PubMed] [Google Scholar]
- 13.Chen CC, Lu RB, Chen YC, Wang MF, Chang YC, Li TK, et al. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am J Hum Genet. 1999;65:795–807. doi: 10.1086/302540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomasson HR, Edenberg HJ, Crabb DW, Mai XL, Jerome RE, Li TK, et al. Alcohol and aldehyde dehydrogenase genotypes and alcoholism in Chinese men. Am J Hum Genet. 1991;48:677–81. [PMC free article] [PubMed] [Google Scholar]
- 15.Chen WJ, Loh EW, Hsu YP, Chen CC, Yu JM, Cheng AT. Alcohol-metabolising genes and alcoholism among Taiwanese Han men: independent effect of ADH2, ADH3 and ALDH2. Br J Psychiatry. 1996;168:762–7. doi: 10.1192/bjp.168.6.762. [DOI] [PubMed] [Google Scholar]
- 16.Shen YC, Fan JH, Edenberg HJ, Li TK, Cui YH, Wang YF, et al. Polymorphism of ADH and ALDH genes among four ethnic groups in China and effects upon the risk for alcoholism. Alcohol Clin Exp Res. 1997;21:1272–7. [PubMed] [Google Scholar]
- 17.Higuchi S. Polymorphisms of ethanol metabolizing enzyme genes and alcoholism. Alcohol Alcohol Suppl. 1994;2:29–34. [PubMed] [Google Scholar]
- 18.Higuchi S, Matsushita S, Imazeki H, Kinoshita T, Takagi S, Kono H. Aldehyde dehydrogenase genotypes in Japanese alcoholics. Lancet. 1994;343:741–2. doi: 10.1016/s0140-6736(94)91629-2. [DOI] [PubMed] [Google Scholar]
- 19.Higuchi S, Matsushita S, Murayama M, Takagi S, Hayashida M. Alcohol and aldehyde dehydrogenase polymorphisms and the risk for alcoholism. Am J Psychiatry. 1995;152:1219–21. doi: 10.1176/ajp.152.8.1219. [DOI] [PubMed] [Google Scholar]
- 20.Maezawa Y, Yamauchi M, Toda G, Suzuki H, Sakurai S. Alcohol-metabolizing enzyme polymorphisms and alcoholism in Japan. Alcohol Clin Exp Res. 1995;19:951–4. doi: 10.1111/j.1530-0277.1995.tb00972.x. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura K, Iwahashi K, Matsuo Y, Miyatake R, Ichikawa Y, Suwaki H. Characteristics of Japanese alcoholics with the atypical aldehyde dehydrogenase 2*2. I. A comparison of the genotypes of ALDH2, ADH2, ADH3, and cytochrome P-4502E1 between alcoholics and nonalcoholics. Alcohol Clin Exp Res. 1996;20:52–5. doi: 10.1111/j.1530-0277.1996.tb01043.x. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka F, Shiratori Y, Yokosuka O, Imazeki F, Tsukada Y, Omata M. Polymorphism of alcohol-metabolizing genes affects drinking behavior and alcoholic liver disease in Japanese men. Alcohol Clin Exp Res. 1997;21:596–601. [PubMed] [Google Scholar]
- 23.Luczak SE, Elvine-Kreis B, Shea SH, Carr LG, Wall TL. Genetic risk for alcoholism relates to level of response to alcohol in Asian-American men and women. J Stud Alcohol. 2002;63:74–82. [PubMed] [Google Scholar]
- 24.Schuckit MA, Duby J. Alcohol-related flushing and the risk for alcoholism in sons of alcoholics. J Clin Psychiatry. 1982;43:415–8. [PubMed] [Google Scholar]
- 25.Porjesz B, Begleiter H, Reich T, Van Eerdewegh P, Edenberg HJ, Foroud T, et al. Amplitude of visual P3 event-related potential as a phenotypic marker for a predisposition to alcoholism: preliminary results from the COGA Project. Collaborative Study on the Genetics of Alcoholism. Alcohol Clin Exp Res. 1998;22:1317–23. [PubMed] [Google Scholar]
- 26.Dick DM, Edenberg HJ, Xuei X, Goate A, Kuperman S, Schuckit M, et al. Association of GABRG3 with alcohol dependence. Alcohol Clin Exp Res. 2004;28:4–9. doi: 10.1097/01.ALC.0000108645.54345.98. [DOI] [PubMed] [Google Scholar]
- 27.Gelernter J, Liu X, Hesselbrock V, Page GP, Goddard A, Zhang H. Results of a genomewide linkage scan: support for chromosomes 9 and 11 loci increasing risk for cigarette smoking. Am J Med Genet B Neuropsychiatr Genet. 2004;128:94–101. doi: 10.1002/ajmg.b.30019. [DOI] [PubMed] [Google Scholar]
- 28.Saccone NL, Kwon JM, Corbett J, Goate A, Rochberg N, Edenberg HJ, et al. A genome screen of maximum number of drinks as an alcoholism phenotype. Am J Med Genet. 2000;96:632–7. doi: 10.1002/1096-8628(20001009)96:5<632::aid-ajmg8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 29.Schuckit MA, Edenberg HJ, Kalmijn J, Flury L, Smith TL, Reich T, et al. A genome-wide search for genes that relate to a low level of response to alcohol. Alcohol Clin Exp Res. 2001;25:323–9. [PubMed] [Google Scholar]
- 30.Bergen AW, Yang XR, Bai Y, Beerman MB, Goldstein AM, Goldin LR. Genomic regions linked to alcohol consumption in the Framingham Heart Study. BMC Genet. 2003;4(Suppl 1):S101. doi: 10.1186/1471-2156-4-S1-S101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bierut LJ, Rice JP, Goate A, Hinrichs AL, Saccone NL, Foroud T, et al. A genomic scan for habitual smoking in families of alcoholics: common and specific genetic factors in substance dependence. Am J Med Genet A. 2004;124:19–27. doi: 10.1002/ajmg.a.20329. [DOI] [PubMed] [Google Scholar]
- 32.Dick DM, Jones K, Saccone N, Hinrichs A, Wang JC, Goate A, et al. Endophenotypes successfully lead to gene identification: results from the collaborative study on the genetics of alcoholism. Behav Genet. 2006;36:112–26. doi: 10.1007/s10519-005-9001-3. [DOI] [PubMed] [Google Scholar]
- 33.Dick DM, Nurnberger J, Jr, Edenberg HJ, Goate A, Crowe R, Rice J, et al. Suggestive linkage on chromosome 1 for a quantitative alcohol-related phenotype. Alcohol Clin Exp Res. 2002;26:1453–60. doi: 10.1097/01.ALC.0000034037.10333.FD. [DOI] [PubMed] [Google Scholar]
- 34.Dick DM, Plunkett J, Wetherill LF, Xuei X, Goate A, Hesselbrock V, et al. Association between GABRA1 and drinking behaviors in the collaborative study on the genetics of alcoholism sample. Alcohol Clin Exp Res. 2006;30:1101–10. doi: 10.1111/j.1530-0277.2006.00136.x. [DOI] [PubMed] [Google Scholar]
- 35.Edenberg HJ, Foroud T. The genetics of alcoholism: identifying specific genes through family studies. Addict Biol. 2006;11:386–96. doi: 10.1111/j.1369-1600.2006.00035.x. [DOI] [PubMed] [Google Scholar]
- 36.Guerrini I, Cook CC, Kest W, Devitgh A, McQuillin A, Curtis D, et al. Genetic linkage analysis supports the presence of two susceptibility loci for alcoholism and heavy drinking on chromosome 1p22.1-11.2 and 1q21.3-24.2. BMC Genet. 2005;6:11. doi: 10.1186/1471-2156-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinnaduwage D, Briollais L. Comparison of genotype- and haplotype-based approaches for fine-mapping of alcohol dependence using COGA data. BMC Genet. 2005;6(Suppl 1):S65. doi: 10.1186/1471-2156-6-S1-S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porjesz B, Begleiter H, Wang K, Almasy L, Chorlian DB, Stimus AT, et al. Linkage and linkage disequilibrium mapping of ERP and EEG phenotypes. Biol Psychol. 2002;61:229–48. doi: 10.1016/s0301-0511(02)00060-1. [DOI] [PubMed] [Google Scholar]
- 39.Reck BH, Mukhopadhyay N, Tsai HJ, Weeks DE. Analysis of alcohol dependence phenotype in the COGA families using covariates to detect linkage. BMC Genet. 2005;6(Suppl 1):S143. doi: 10.1186/1471-2156-6-S1-S143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang HC, Chang CC, Lin CY, Chen CL, Lin CY, Fann CS. A genome-wide scanning and fine mapping study of COGA data. BMC Genet. 2005;6(Suppl 1):S30. doi: 10.1186/1471-2156-6-S1-S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong X, Zhang H. Linkage analysis and association analysis in the presence of linkage using age at onset of COGA alcoholism data. BMC Genet. 2005;6(Suppl 1):S31. doi: 10.1186/1471-2156-6-S1-S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–7. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 43.Orsini C, Bonito-Oliva A, Conversi D, Cabib S. Susceptibility to conditioned place preference induced by addictive drugs in mice of the C57BL/6 and DBA/2 inbred strains. Psychopharmacology (Berl) 2005;181:327–36. doi: 10.1007/s00213-005-2259-6. [DOI] [PubMed] [Google Scholar]
- 44.Orsini C, Buchini F, Piazza PV, Puglisi-Allegra S, Cabib S. Susceptibility to amphetamine-induced place preference is predicted by locomotor response to novelty and amphetamine in the mouse. Psychopharmacology (Berl) 2004;172:264–70. doi: 10.1007/s00213-003-1647-z. [DOI] [PubMed] [Google Scholar]
- 45.Rodgers RJ, Boullier E, Chatzimichalaki P, Cooper GD, Shorten A. Contrasting phenotypes of C57BL/6JOlaHsd, 129S2/SvHsd and 129/SvEv mice in two exploration-based tests of anxiety-related behaviour. Physiol Behav. 2002;77:301–10. doi: 10.1016/s0031-9384(02)00856-9. [DOI] [PubMed] [Google Scholar]
- 46.Rodgers RJ, Davies B, Shore R. Absence of anxiolytic response to chlordiazepoxide in two common background strains exposed to the elevated plus-maze: importance and implications of behavioural baseline. Genes Brain Behav. 2002;1:242–51. doi: 10.1034/j.1601-183x.2002.10406.x. [DOI] [PubMed] [Google Scholar]
- 47.Krasnova IN, Ladenheim B, Jayanthi S, Oyler J, Moran TH, Huestis MA, et al. Amphetamine-induced toxicity in dopamine terminals in CD-1 and C57BL/6J mice: complex roles for oxygen-based species and temperature regulation. Neuroscience. 2001;107:265–74. doi: 10.1016/s0306-4522(01)00351-7. [DOI] [PubMed] [Google Scholar]
- 48.Golden GT, Ferraro TN, Smith GG, Snyder RL, Jones NL, Berrettini WH. Acute cocaine-induced seizures: differential sensitivity of six inbred mouse strains. Neuropsychopharmacology. 2001;24:291–9. doi: 10.1016/S0893-133X(00)00204-9. [DOI] [PubMed] [Google Scholar]
- 49.Ferraro TN, Golden GT, Smith GG, Martin JF, Schwebel CL, Doyle GA, et al. Confirmation of a major QTL influencing oral morphine intake in C57 and DBA mice using reciprocal congenic strains. Neuropsychopharmacology. 2005;30:742–6. doi: 10.1038/sj.npp.1300592. [DOI] [PubMed] [Google Scholar]
- 50.Berrettini WH, Ferraro TN, Alexander RC, Buchberg AM, Vogel WH. Quantitative trait loci mapping of three loci controlling morphine preference using inbred mouse strains. Nat Genet. 1994;7:54–8. doi: 10.1038/ng0594-54. [DOI] [PubMed] [Google Scholar]
- 51.Bergen AW, Korczak JF, Weissbecker KA, Goldstein AM. A genome-wide search for loci contributing to smoking and alcoholism. Genet Epidemiol. 1999;17(Suppl 1):S55–60. doi: 10.1002/gepi.1370170710. [DOI] [PubMed] [Google Scholar]
- 52.Duggirala R, Almasy L, Blangero J. Smoking behavior is under the influence of a major quantitative trait locus on human chromosome 5q. Genet Epidemiol. 1999;17(Suppl 1):S139–44. doi: 10.1002/gepi.1370170724. [DOI] [PubMed] [Google Scholar]
- 53.Ehlers CL, Wilhelmsen KC. Genomic screen for loci associated with tobacco usage in Mission Indians. BMC Med Genet. 2006;7:9. doi: 10.1186/1471-2350-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goode EL, Badzioch MD, Kim H, Gagnon F, Rozek LS, Edwards KL, et al. Multiple genome-wide analyses of smoking behavior in the Framingham Heart Study. BMC Genet. 2003;4(Suppl 1):S102. doi: 10.1186/1471-2156-4-S1-S102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li MD, Ma JZ, Cheng R, Dupont RT, Williams NJ, Crews KM, et al. A genome-wide scan to identify loci for smoking rate in the Framingham Heart Study population. BMC Genet. 2003;4(Suppl 1):S103. doi: 10.1186/1471-2156-4-S1-S103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saccone NL, Neuman RJ, Saccone SF, Rice JP. Genetic analysis of maximum cigarette-use phenotypes. BMC Genet. 2003;4(Suppl 1):S105. doi: 10.1186/1471-2156-4-S1-S105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Straub RE, Sullivan PF, Ma Y, Myakishev MV, Harris-Kerr C, Wormley B, et al. Susceptibility genes for nicotine dependence: a genome scan and followup in an independent sample suggest that regions on chromosomes 2, 4, 10, 16, 17 and 18 merit further study. Mol Psychiatry. 1999;4:129–44. doi: 10.1038/sj.mp.4000518. [DOI] [PubMed] [Google Scholar]
- 58.Sullivan PF, Neale BM, van den Oord E, Miles MF, Neale MC, Bulik CM, et al. Candidate genes for nicotine dependence via linkage, epistasis, and bioinformatics. Am J Med Genet B Neuropsychiatr Genet. 2004;126:23–36. doi: 10.1002/ajmg.b.20138. [DOI] [PubMed] [Google Scholar]
- 59.Swan GE, Hops H, Wilhelmsen KC, Lessov-Schlaggar CN, Cheng LS, Hudmon KS, et al. A genome-wide screen for nicotine dependence susceptibility loci. Am J Med Genet B Neuropsychiatr Genet. 2006;141:354–60. doi: 10.1002/ajmg.b.30315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang D, Ma JZ, Li MD. Mapping and verification of susceptibility loci for smoking quantity using permutation linkage analysis. Pharmacogenomics J. 2005;5:166–72. doi: 10.1038/sj.tpj.6500304. [DOI] [PubMed] [Google Scholar]
- 61.Evans DM, Cardon LR. Genome-wide association: a promising start to a long race. Trends Genet. 2006;22:350–4. doi: 10.1016/j.tig.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 62.Newton-Cheh C, Hirschhorn JN. Genetic association studies of complex traits: design and analysis issues. Mutat Res. 2005;573:54–69. doi: 10.1016/j.mrfmmm.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 63.Farrall M, Morris AP. Gearing up for genome-wide gene-association studies. Hum Mol Genet. 2005;14(Spec No 12):R157–62. doi: 10.1093/hmg/ddi273. [DOI] [PubMed] [Google Scholar]
- 64.Craig DW, Stephan DA. Applications of whole-genome high-density SNP genotyping. Expert Rev Mol Diagn. 2005;5:159–70. doi: 10.1586/14737159.5.2.159. [DOI] [PubMed] [Google Scholar]
- 65.Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet. 2005;6:109–18. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- 66.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 67.Uhl GR. Molecular genetics of addiction vulnerability. NeuroRx. 2006;3:295–301. doi: 10.1016/j.nurx.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu QR, Drgon T, Johnson C, Walther D, Hess J, Uhl GR. Addiction molecular genetics: 639,401 SNP whole genome association identifies many “cell adhesion” genes. Am J Med Genet B Neuropsychiatr Genet. 2006;141:918–25. doi: 10.1002/ajmg.b.30436. [DOI] [PubMed] [Google Scholar]
- 69.Liu QR, Drgon T, Walther D, Johnson C, Poleskaya O, Hess J, et al. Pooled association genome scanning: validation and use to identify addiction vulnerability loci in two samples. Proc Natl Acad Sci U S A. 2005;102:11864–9. doi: 10.1073/pnas.0500329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meaburn E, Butcher LM, Liu L, Fernandes C, Hansen V, Al-Chalabi A, et al. Genotyping DNA pools on microarrays: tackling the QTL problem of large samples and large numbers of SNPs. BMC Genomics. 2005;6:52. doi: 10.1186/1471-2164-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meaburn E, Butcher LM, Schalkwyk LC, Plomin R. Genotyping pooled DNA using 100K SNP microarrays: a step towards genomewide association scans. Nucleic Acids Res. 2006;34:e27. doi: 10.1093/nar/gnj027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uhl GR, Liu QR, Walther D, Hess J, Naiman D. Polysubstance abuse-vulnerability genes: genome scans for association, using 1,004 subjects and 1,494 single-nucleotide polymorphisms. Am J Hum Genet. 2001;69:1290–300. doi: 10.1086/324467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Butcher LM, Meaburn E, Dale PS, Sham P, Schalkwyk LC, Craig IW, et al. Association analysis of mild mental impairment using DNA pooling to screen 432 brain-expressed single-nucleotide polymorphisms. Mol Psychiatry. 2005;10:384–92. doi: 10.1038/sj.mp.4001589. [DOI] [PubMed] [Google Scholar]
- 74.Butcher LM, Meaburn E, Knight J, Sham PC, Schalkwyk LC, Craig IW, et al. SNPs, microarrays and pooled DNA: identification of four loci associated with mild mental impairment in a sample of 6000 children. Hum Mol Genet. 2005;14:1315–25. doi: 10.1093/hmg/ddi142. [DOI] [PubMed] [Google Scholar]
- 75.Butcher LM, Meaburn E, Liu L, Fernandes C, Hill L, Al-Chalabi A, et al. Genotyping pooled DNA on microarrays: a systematic genome screen of thousands of SNPs in large samples to detect QTLs for complex traits. Behav Genet. 2004;34:549–55. doi: 10.1023/B:BEGE.0000038493.26202.d3. [DOI] [PubMed] [Google Scholar]
- 76.Sham P, Bader JS, Craig I, O’Donovan M, Owen M. DNA Pooling: a tool for large-scale association studies. Nat Rev Genet. 2002;3:862–71. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- 77.Johnson C, Drgon T, Liu QR, Walther D, Edenberg H, Rice J, et al. Pooled association genome scanning for alcohol dependence using 104,268 SNPs: validation and use to identify alcoholism vulnerability loci in unrelated individuals from the collaborative study on the genetics of alcoholism. Am J Med Genet B Neuropsychiatr Genet. 2006;141:844–53. doi: 10.1002/ajmg.b.30346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pearson JV, Huentelman MJ, Halperin RF, Tembe WD, Melquist S, Homer N, et al. Identification of the genetic basis for complex disorders by use of pooling-based genomewide single-nucleotide-polymorphism association studies. Am J Hum Genet. 2007;80:126–39. doi: 10.1086/510686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith SS, O’Hara BF, Persico AM, Gorelick DA, Newlin DB, Vlahov D, et al. Genetic vulnerability to drug abuse. The D2 dopamine receptor Taq I B1 restriction fragment length polymorphism appears more frequently in polysubstance abusers. Arch Gen Psychiatry. 1992;49:723–7. doi: 10.1001/archpsyc.1992.01820090051009. [DOI] [PubMed] [Google Scholar]
- 80.Persico AM, Bird G, Gabbay FH, Uhl GR. D2 dopamine receptor gene TaqI A1 and B1 restriction fragment length polymorphisms: enhanced frequencies in psychostimulant-preferring polysubstance abusers. Biol Psychiatry. 1996;40:776–84. doi: 10.1016/0006-3223(95)00483-1. [DOI] [PubMed] [Google Scholar]
- 81.Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI, Jr, et al. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol. 1994;55:149–58. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- 82.Itoh K, Hashimoto K, Shimizu E, Sekine Y, Ozaki N, Inada T, et al. Association study between brain-derived neurotrophic factor gene polymorphisms and methamphetamine abusers in Japan. Am J Med Genet B Neuropsychiatr Genet. 2005;132:70–3. doi: 10.1002/ajmg.b.30097. [DOI] [PubMed] [Google Scholar]
- 83.Iwata N, Inada T, Harano M, Komiyama T, Yamada M, Sekine Y, et al. No association is found between the candidate genes of t-PA/plasminogen system and Japanese methamphetamine-related disorder: a collaborative study by the Japanese Genetics Initiative for Drug Abuse. Ann N Y Acad Sci. 2004;1025:34–8. doi: 10.1196/annals.1316.004. [DOI] [PubMed] [Google Scholar]
- 84.Ujiie H, Inada T, Harano M, Komiyayama T, Yamada M, Sekine Y, et al. [Genetic studies on substance dependence] Seishin Shinkeigaku Zasshi. 2004;106:1598–603. [PubMed] [Google Scholar]
- 85.Morita Y, Ujike H, Tanaka Y, Uchida N, Nomura A, Ohtani K, et al. A nonsynonymous polymorphism in the human fatty acid amide hydrolase gene did not associate with either methamphetamine dependence or schizophrenia. Neurosci Lett. 2005;376:182–7. doi: 10.1016/j.neulet.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 86.Nishiyama T, Ikeda M, Iwata N, Suzuki T, Kitajima T, Yamanouchi Y, et al. Haplotype association between GABAA receptor gamma2 subunit gene (GABRG2) and methamphetamine use disorder. Pharmacogenomics J. 2005;5:89–95. doi: 10.1038/sj.tpj.6500292. [DOI] [PubMed] [Google Scholar]
- 87.Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, et al. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–7. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Welzl H, Stork O. Cell adhesion molecules: key players in memory consolidation? News Physiol Sci. 2003;18:147–50. doi: 10.1152/nips.01422.2002. [DOI] [PubMed] [Google Scholar]
- 90.Benson DL, Schnapp LM, Shapiro L, Huntley GW. Making memories stick: cell-adhesion molecules in synaptic plasticity. Trends Cell Biol. 2000;10:473–82. doi: 10.1016/s0962-8924(00)01838-9. [DOI] [PubMed] [Google Scholar]
- 91.Qiu S, Korwek KM, Weeber EJ. A fresh look at an ancient receptor family: emerging roles for low density lipoprotein receptors in synaptic plasticity and memory formation. Neurobiol Learn Mem. 2006;85:16–29. doi: 10.1016/j.nlm.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 92.Uhl GR. Molecular genetic underpinnings of human substance abuse vulnerability: likely contributions to understanding addiction as a mnemonic process. Neuropharmacology. 2004;47(Suppl 1):140–7. doi: 10.1016/j.neuropharm.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 93.Shirley RL, Walter NA, Reilly MT, Fehr C, Buck KJ. Mpdz is a quantitative trait gene for drug withdrawal seizures. Nat Neurosci. 2004;7:699–700. doi: 10.1038/nn1271. [DOI] [PubMed] [Google Scholar]
- 94.Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–22. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- 95.Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–79. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 96.Nestler EJ. Neurobiology Total recall-the memory of addiction. Science. 2001;292:2266–7. doi: 10.1126/science.1063024. [DOI] [PubMed] [Google Scholar]
- 97.Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–84. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 98.Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL. Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science. 2001;292:1175–8. doi: 10.1126/science.1058043. [DOI] [PubMed] [Google Scholar]
- 99.Volkow ND, Li TK. Drugs and alcohol: treating and preventing abuse, addiction and their medical consequences. Pharmacol Ther. 2005;108:3–17. doi: 10.1016/j.pharmthera.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 100.Uhl GR, Liu QR, Drgon T, Johnson C, Walther D, Rose JE. Molecular genetics of nicotine dependence and abstinence: whole genome association using 520,000 SNPs. BMC Genet. 2007;8:10. doi: 10.1186/1471-2156-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]