

Abstract
Natural inhibitors occupy an important place in the potential to neutralize the toxic effects caused by snake venom proteins and enzymes. It has been well recognized for several years that animal sera, some of the plant and marine extracts are the most potent in neutralizing snake venom phospholipase A2 (svPLA2). The implication of this review to update the latest research work which has been accomplished with svPLA2 inhibitors from various natural sources like animal, marine organisms presents a compilation of research in this field over the past decade and revisiting the previous research report including those found in plants. In addition to that the bioactive compounds/inhibitor molecules from diverse sources like aristolochic alkaloid, flavonoids and neoflavonoids from plants, hydrocarbones 2, 4 dimethyl hexane, 2 methylnonane, and 2, 6 dimethyl heptane obtained from traditional medicinal plants Tragia involucrata (Euphorbiaceae) member of natural products involved for the inhibitory potential of phospholipase A2 (PLA2) enzymes in vitro and also decrease both oedema induced by snake venom as well as human synovial fluid PLA2. Besides marine natural products that inhibit PLA2 are manoalide and its derivatives such as scalaradial and related compounds, pseudopterosins and vidalols, tetracylne from synthetic chemicals etc. There is an overview of the role of PLA2 in inflammation that provides a rationale for seeking inhibitors of PLA2 as anti-inflammatory agents. However, more studies should be considered to evaluate antivenom efficiency of sera and other agents against a variety of snake venoms found in various parts of the world. The implications of these new groups of svPLA2 toxin inhibitors in the context of our current understanding of snake biology as well as in the development of new novel antivenoms therapeutics agents in the efficient treatment of snake envenomations are discussed.
Keywords: Inhibitors, Phospholipase A2, Snake venom phospholipase A2, Envenomation, Inflammation
Background
Snake envenomation is a serious medical problem, especially in the farms where snakes are abundant [1]. The common species such as Crotalus rhodostoma (38%), Trimeresurus albolabris (27%), Daboia russeli siamensis (14%), Naja atra (10%), and others (11%) are responsible for snakebites in Southeast Asia [2]. Snake venoms constitute a rich source of phospholipase A2 (PLA2) enzymes, which show remarkable functional diversity. Snake venom phospholipase A2 (svPLA2) can induce several additional effects such as cardiotoxicity, myotoxicity, pre or postsynaptic neurotoxicity, edema, hemolysis, hypotension, convulsion, platelet aggregation inhibition and anticoagulation [3–5]. svPLA2 catalyze the hydrolysis of 2-acyl ester bonds of 3- sn-phospholipids producing fatty acids and lysophospholipids [6]. The Ca2+ ion, an essential cofactor, and an Asp residue at position 49 are required for catalysis on artificial substrates [6]. Their catalytic activity upon cell membranes of specific tissues suggests an important role of these enzymes in venoms toxicity.
PLA2s were recently divided into 15 groups based on biochemical and structural criteria, considering their molecular weight, disulfide bonds profile, phospholipid substrates, amino acid sequence, and sensibility to Ca2+ ions, catalytic activity and genic structure [7–9]. Snake venoms are especially rich in group I and II PLA2s, found in the Elapidae or Viperidae families, respectively. Group II PLA2s can be further subdivided into two main types commonly referred to as Asp-49 and Lys- 49 isoforms [10]. Since then, the isoforms of basic myotoxic PLA2s were isolated from Bothrops snakes and classified into group II, where the Asp-49 residue was replaced by Lys-49, resulting thus two classes of this enzyme: (a) Asp-49 myotoxins showing moderate catalytic activity and (b) Lys-49 myotoxins with low or no enzymatic activity upon artificial substrates [9–10]. Previously, myonecrotic venom components were extensively studied. Several myotoxic Bothrops PLA2s were characterized, such as B. asper PLA2s, B. moojeni myotoxins I and II, and B. jararacussu bothropstoxins I (Lys-49) and II (Asp-49), responsible for several biological effects including myonecrosis, edema, irreversible neuromuscular blockage and in vitro cell lysis [11–14] etc. Many acidic PLA2s have been isolated from Agkistrodon halys, Pseudechis papuanus, Bothrops neuwiedi, Bothrops lanceolatus, Trimeresurus jerdonii, Heloderma horridum, Ophiophagus hannah, Lachesis muta, B. jararaca, B. moojeni and B. jararacussu [15–19]. Toxicity and pharmacological effects differ in acidic isoforms. For example, the acidic PLA2 isolated from Lachesis muta venom is myonecrotic, proteolytic, anticoagulant and platelet aggregation inhibitor. Another myotoxic PLA2 from the same venom did not show anticoagulant or lethal activity.
It has been suggested that PLA2s represent a class of versatile enzymes and, as multifunctional proteins; they are extremely relevant as mediators of several inflammatory diseases and promising agents for use in biotechnological areas [20–21]. An increasing search for use of these enzymes is therefore not surprising, including their general anesthetic action, treatment of rheumatoid arthritis, bactericidal action, novel class of antiparasitary agents, HIV inhibitors and others [22–26]. In this review, we broadly discuss the implications of the PLA2 inhibitor groups of plants, marine organisms serve as sources of compounds on current understanding of snake biology, as well as in the development of new therapeutic drugs for treatment of snake envenomations.
Venom neutralization by bio-active compounds from plant
a) Neutralization potential of crude mixture of plants:
Many plants are used in traditional medicine as active agents against various effects induced by snakebite [27]. Viperidae snakes (Daboia russelli russelli) are well known for sever local effects and these effects commonly treated with plant extracts without any scientific validation in triabl areas, India [28]. There are few survey reports that reveal the practice of herbal medicine by either folk or indigenous communities [29–31]. However, in most cases the efficacy of this traditional treatment regimen is unproven. Thus, the study of herbal antidotes against snake venom is of great importance in the management of snakebite. Recently, various research groups actively enganged for antivenom research by using diversed crude extracts and elucidated active compounds from various plants sources [27,32–34]. In addition, their natural PLA2 inhibitors also play an important role in some traditional medicines used for immune support, such as the ashwaganda plant (Withania somnifera) used in Ayurvedic medicine. Aqueous extracts of this plant have been reported to neutralize venom of the Indian speckled cobra -Naja naja [35] another report, ethanolic seed extract of Strychnas nux-vomica also showed antisnake venom activity [36]. Ethanol leaf extract of Acalypha indica potentially neutralized Viper russelli russelli (Russell's viper) venom [37]. Morus alba leaf extract abolished the in vitro proteolytic and hyaluronolytic activities of Indian vipera ‘D. russelli russelli’ venom. Edema, hemorrhage and myonecrotic activities were also neutralized effectively [28]. Aqueous root extract of Mimosa pudica inhibited the hyaluronidase and protease activities of Indian snake (Naja naja, Vipera russelli and Echis carinatus) venoms by dose dependant manner [38].
The ethanol leaf extract and essential oil of Nectandra angustifolia was the most active and inhibited both venom activities (hemolytic and coagulant), while the oil was only active on the coagulant activity [39]. Methanol extract of fresh leaves (Camellina sinensis L.) showed potential to inhibit PLA2 enzymes with hydrolytic activity in Naja naja kaouthia Lesson and Calloselasma rhodostoma Kuhl venoms [40]. Another, methanolic extract from Cordia verbenacea ‘baleeira’, ‘whaler’ (Cv) significantly inhibited paw edema induced by Bothrops jararacussu snake venom and by its main basic phospholipase A2 homologs, namely bothropstoxins I and II (BthTXs). Alam et al, [41] also reported that the isolation, purification and partial characterization of viper venom inhibiting factor from the root extract of the Indian medicinal plant sarsaparilla (Hemidesmus indicus R.Br). Viper venom-induced inflammation and inhibition of free radical formation by pure compound (2- hydroxy-4-methoxy benzoic acid) isolated and purified from anantamul (Hemidesmus indicus R.BR) root extract [42].
In our previous study, the herbal practitioners in the study area were interviewed, and information on medicinal plants was collected from the traditional healers called “Vaidyars”. This survey covers 72 medicinal plants belonging to 53 families that are used for the treatment of snakebite in a traditional way. Traditional approach was evaluated scientifically with some selected plant extracts (7.2 mg/kg, bodoy weight) and partially purified fractions (2.4 mg/kg, b.w) were orally administered to mice experimentally envenomed with rattlesnake venom s.c. injection (2.5 - 15 mg/kg, b.w). Tested fractions (Aristolochia indica, Hemidesmus indicus, Gloriosa superba, Strychnos nuxvomica, Eclipta prostrata, and Andrographis paniculata) showed potent neutralizing effect against the venom. Compared to the extracts, administration of purified fractions was more effective in increasing the body weight. Control mice injected with the venom alone showed weight loss and severe toxicity at 15 mg/kg, b.w. The purified fractions (2.4 mg/kg, b.w) produced significant protection against venom induced changes in serum SOD and LPx levels. The isolated fractions effectively inhibited the toxic effect of snake venoms in vitro than in vivo. The above observations confirmed the protective activity of plants - Aristolochia indica, Hemidesmus indicus, Gloriosa superba, Strychnos nux-vomica, Eclipta prostrata, and Andrographis paniculata against the lethal action of snake venom [33].
Several plants are used in traditional medicine as active agents against various effects induced by snakebite [27, 32]. Folk medicine and its isolated constituents recommended as snake venom antidotes for the protection against Jararaca venom [43] for i.e. haemorrhagic effect of Bothrops atrox venom and their strong lethal enzymatic effect was neutralized by plants [44–45]. Furthermore, previoulsy we reported [33] hydrocarbones such as 2, 4 dimethyl hexane, 2 methylnonane, 2, 6 dimethyl heptane obtained from traditional medicinal plants Tragia involucrata (Euphorbiaceae) member showed effective venom and PLA2 neutralisation in mice (Figure 1A). PLA2 inhibitors have also been isolated from plants such as Horsfieldia amygdaline [46].
Figure 1.
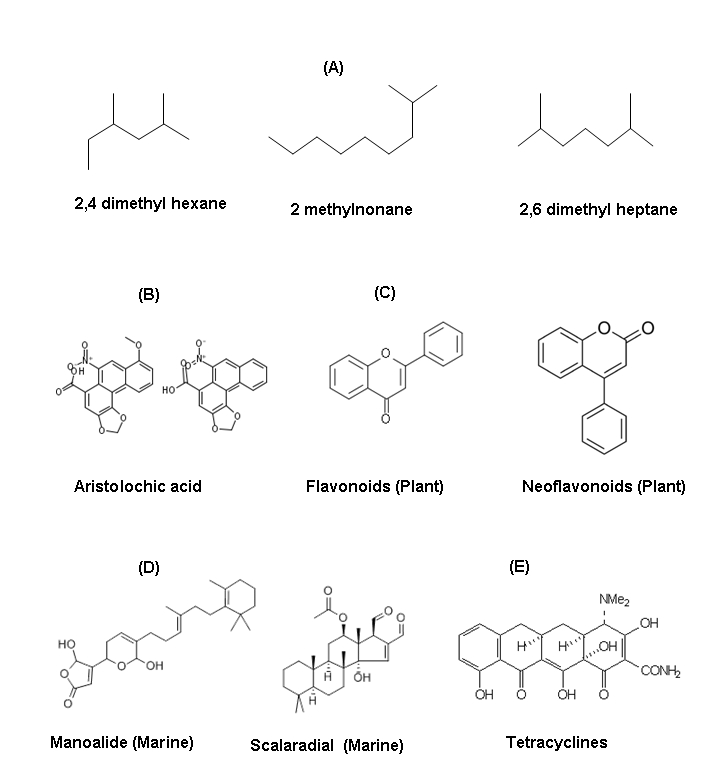
Structure of various bioactive molucules involved for the inhibitory potential of phospholipase A2 (PLA2) enzymes in vitro and also decrease both oedema induced by snake venom as well as human synovial fluid PLA2.(A) Hydrocarbons like 2, 4 dimethyl hexane, 2 methylonane, 2,6 dimethyl heptane obtained from traditional medicine plants Tragia involucrata (Euphorbiaceae) member (Samy et al, J Ethnopharmacol. 2006,107(1):99-106), (B) Aristolochic alkaloid, (C) Flavonoids and neoflavonoids from plants (D) Manolide and scalaradial from marine sources (E) tetracylne (antibiotics)
The active component was isolated and identified as rosmarinic acid (RA). RA is an ester of caffeic acid and 3, 4- dihydroxyphenyllactic acid 2-O-cafeoil-3-(3, 4-di-hydroxyphenyl)- R-lactic [32]. RA in the species C. verbenacea and of its anti-inflammatory and antimyotoxic properties against snake venoms and isolated toxins reported for the first-time. RA inhibited the edema and myotoxic activity induced by the basic PLA2s BthTX-I and BthTX-II. The pure compound potentiated the ability of the commercial equine polyvalent antivenom in neutralizing lethal and myotoxic effects of the crude venom as well as isolated PLA2s in experimental models. Circular dichorism data also suggested that after binding, no significant conformation changes were occurred either in the Cv-RA or in the target PLA2. A possible model for the interaction of RA with Lys49-PLA2 BthTX-I is proposed [32]. A compound AIPLAI purified from the methanolic leaf extract of Azadirachta indica (Neem) inhibits the Cobra and Russell's viper venoms (RVVs) PLA2 enzymes in a dose-dependent manner and the AIPLAI holds good promise for the development of novel anti-snake venom drug in future [34].
The secondary metabolites (chemical constituents) are responsible for the neutralizing effect of plants that are in popular use against the action of snake venoms [47–48]. An animal study has also been well documented in the case of immunity of the opossum (Didelphis marsupials) towards the venom of the Jararaca (Bothrops jararaca) snake [49]. Plants are reputed to be capable of neutralizing the action of snake venom, and their antivenom activity has been related to certain chemical compounds identified in those plants [43]. Several chemical constituents like alkaloids, flavonoids, sitosterol or glucoside, lupeol, gymnemagenin, phenolics, pentacyclic triterpenes like oleanoic acid, ursolic, tannins, taraxasterol, α and β amyrin are found to be present in varying proportions in plants. They have also been previously reported for antisnake venom activity. All these classes of chemical compounds are capable of interacting with macromolecular targets with enzymes or receptors and it can effectively inhibit the toxic effect of snake venoms in vitro than in vivo [50]. Besides, several other phytochemicals of plant origin showed the most potent inhibition of human synovial fluid phospholipase (HSF- PLA2) was observed with compounds such as scalaradial and ellagic acid, which both have IC50 values of 0.02 µM (using autoclaved [3H]-arachidonic-acid (AA)-labelled Escherichia coli membranes as substrate).
b) Alkaloids:
Recently, alkaoid has been reported that aristolochic acid inhibits PLA2 in vitro and also decrease both oedema induced by snake venom as well as human synovial fluid PLA2 (Figure 1B). Several authors studied the anti-inflammatory activity of aristolochic acid and to investigate the effect of the alkaloid on the enzymes involved in the release of eicosanoids. Results demonstrated that aristolochic acid was inhibited the inflammation induced by immunological, immune complexes, and non-immunological agents such as carrageenan or croton oil [51].
c) Flavonoids:
The inhibitory effect of the plant flavonoid and rutin were investigated against group I PLA2 from porcine pancreas and Naja naja as well as group II PLA2s from Vipera russelli and Crotalus atrox etc (Figure 1C). Rutin efficiently inhibited group II PLA2s obtained both V. russelli and C. atrox but it was shown only a weak inhibitor of PLA2-I from porcine pancreas and N. naja. The lack of strong inhibition of pancreatic PLA2-I was not due to contaminating proteins in the enzyme preparation, since the same weak inhibition was obtained against pancreatic PLA2 purified to homogeneity as well [52]. The phytotherapeutic agent like flavonoids selectively inhibited the group II PLA2 of venom origins. Previously, Alcaraz and Hoult [53] reported that the hypolaetin-8-glucoside and related flavonoids inhibited the snake venom phospholipase A2 effectively. Porcine pancreatic PLA2 hydrolyses phosphatidylcholine when in the lamellar state as well as in the micellar state. We have found that alphatocopherol, the most active form of vitamin E, is able to inhibit PLA2 activity only toward lamellar fluid membranes, thus protecting phospholipids toward this lytic enzyme. This compound decreases both the initial rate and the extent of hydrolysis. The inhibition is of the non-competitive type and the evidence strongly suggests that it is mainly due to an effect of alpha-tocopherol on the substrate, i.e. the membrane and not, on the enzyme [54].
d) Lipids:
Membrane lipid-derived second messengers are generated by PLA2 during synaptic activity. Overstimulation of this enzyme during neurotrauma results in the accumulation of bioactive metabolites such as arachidonic acid, oxygenated derivatives of arachidonic acid, and platelet-activating factor (PAF). Several of these bioactive lipids participate in cell damage, cell death, or repair-regenerative neural plasticity. Neurotransmitters may activate PLA2 directly when linked to receptors coupled to G proteins and/or indirectly as calcium influx or mobilization from intracellular [55]. Chronic inflammatory diseases are often accompanied by intense angiogenesis. A model of inflammatory angiogenesis is the murine air pouch granuloma which has a hyperangiogenic component. Proinflammatory lipid mediator generation is also a hallmark of chronic inflammation and the role of endogenous production of these mediators in angiogenesis is not known. The 14 kDa PLA2 deacylates phospholipid, liberating arachidonic acids are used for leukotriene and lysophospholipid, production of plateletactivating factor (PAF) [56]. Another report, 1-alkyl ether lipids were studied for their ability to inhibit PLA2 and antagonize PAF responses. Studies with synthetic micellar substrate (1- stearyl-2-arachidonyl phosphocholine), at concentrations ranging from 0.02 to 1000 µM, demonstrate that CL 118326 inhibits porcine pancreatic PLA2 in vitro. As the substrate concentration increases, there is a dose-dependent increase in the IC50 value (IC50 ranges: 2.6-137 µM). CL 118326 inhibits mammalian pancreatic PLA2, but not snake or bee venom PLA2. CL 118326 inhibits thrombin effectively at IC50 = 7.9 µM dose [57].
Phospholipase A2 (PLA2) inhibitors from marine organisms
Phospholipase A2 (PLA2) inhibitor from marine organisms that are provides a rationale for seeking inhibitors of PLA2 as antiinflammatory agents [58]. Marine natural products that inhibit PLA2 are manoalide [59–63] and its derivatives such as scalaradial [64] and related compounds like pseudopterosins [65], vidalols, and a group of terpenoids that contain masked 1,4-dicarbonyl moieties respectively.
a) Manoalide:
Several marine terpenoids that contain at least one reactive aldehyde group, such as manoalide (MLD) and its congeners, possess interesting anti-inflammatory activities that are mediated by the covalent inactivation of secretory phospholipase A2 [63]. Manoalide is a potent analgesic and antiinflammatory sesterterpene isolated in 1980 from a marine sponge. The anti-inflammatory activity of manoalide is due to inhibition of PLA2, through irreversible binding to several lysine residues (Figure 1D). This binding is realized by means of the two masked aldehyde functions present in the polar part of manoalide. Of the two aldehyde groups, only that present in the g-hydroxybutenolide ring seems to be essential, since cacospongionolides, naturally occurring analogues lacking the second masked aldehyde group, were also shown to be irreversible PLA2 inhibitors. It appears that the minimum structural requirement for exhibiting manoalide-like PLA2 inhibition would be the presence in the inhibitor of functional groups able to seize the amino groups of PLA2 lysine residues with formation of stable covalent bonds. Many manoalide analogues have been isolated from marine sponges, most of them sharing PLA2 inhibitory properties [62].
MLD inactivated bee venom phospholipase A2s, localization of the manoalide binding site on bee venom PLA2 [59–60]. MLD drug inhibited purified human synovial fluid phospholipase A2 (HSF-PLA2). Inhibition of HSF-PLA2 by MLD was concentration and time dependent with IC50 values of 0.2 and 0.02 µM for dipalmitoylphosphatidylcholine (DPPC) as the substrate (38.0 µM and 18.8 µmol) [3H]arachidonic acid released/min/mg protein with Escherichia coli as a natural substrate respectively [61].
b) Scalaradial:
Scalaradial (SLD), a 1, 4-dialdehyde marine terpenoid that was isolated from the sponge (Cacospongia mollior) possesses antiinflammatory properties in vivo and in vitro. SLD is a potent inactivator of bee venom PLA2 with an IC50 value of 0.07 µM. Inactivation of bee venom PLA2 occurred in a time-dependent manner. The rate of inactivation was reduced markedly in the presence of excess phosphatidylcholine, suggesting that modification of the enzyme occurs at or near the substrate binding site [64]. Due to its peculiar dialdehyde structural feature, it has been proposed that scalaradial exerts its enzymatic inactivation by means of an irreversible covalent modification of its target [63].
c) Petrosaspongiolide:
Petrosaspongiolide M (PM) is an anti-inflammatory marine metabolite that displays a potent inhibitory activity toward group II and III secretory phospholipase A2 (PLA2) enzymes. The details of the mechanism, which leads to a covalent adduct between PLA2 and gamma-hydroxybutenolide-containing molecules such as PM. The model provides an interesting insight into the PM-PLA2 inhibition process and may prove useful in the design of new anti-inflammatory agents that target PLA2 secretory enzymes [65].
d) Vidalols:
Chemical studies of the Caribbean red alga Vidalia obtusaloba have resulted in the isolation of two new bromophenolic metabolites, vidalols A and B (1, 2). The new compounds were discovered as part of an organized effort to isolate new naturally-occurring anti-inflammatory agents with a focus upon those which may function through the inhibition of PLA2 [66]. A chemically synthesized novel phenolic metabolite, vidalenolone, as well as two previously described and structurally simple phenols were isolated as SH2-inactive substances. Their structures were determined by spectroscopic methods such as 2D NMR [67]. The cytotoxic activities of all the synthetic compounds were determined against DLD-1 cells using the MTT assay. Compounds with IC50 < 20 µmol were also tested against HCT-116 cells. Compound 3c (2, 5-dibromo- 3, 4-dihydroxybenzyl n-propyl ether) was the most active compound against both cell lines (IC50 = 1.72 and 0.80 µmol, respectively), and its effect on the cell cycle was studied using flow cytometry [68].
Other lead molecules as PLA2 inhibitors
Analogues of lead molecules were generated by substituting different types of functional groups in order to obtain a molecule with optimal PLA2 inhibition. The lead molecules characterized as PLA2 inhibitors are acenaphthenes, azetidinones, indoles, piperazines, isoxazolidines, isoxazolines, diazepinones, and several substrate analogues [69]. Among these analogue piperazine derivatives on optimization with respect to hydrophobicity and electronegativity showed inhibition at nanomolar levels. Structural analysis of many lead molecules indicated that a PLA2 inhibitor should have both hydrophobic moiety and polar functional groups. However, the docking studies clearly showed that the binding mechanisms of plant (acalyphin, chlorogenic acid, stigmasterol, curcumin and tectoridin) and marine compounds (gracilin A and aplysulphurin A), performed with Russell's viper and bovine pancreatic PLA2 as target molecules. These compounds interacted with the amino acid (AA) residues at the active site of both PLA2s, and proved the efficacy as anti-inlfammaotry and as well antidotes properties [70].
Antibiotic as a PLA2 inhibitor
a) Tetracyclines as PLA2 inhibitor:
Tetracyclines are primarily bacteriostatic broad-spectrum antimicrobial agents; they are thought to exert their effect by the inhibiting protein synthesis. Tetracyclines at low doses were found to interfere with the production of prostaglandins and leukotrienes [71–72], scavenge oxygen radicals [73], interfere with the expression of nitrous oxide synthase [74], and enhance natural inhibitors of matrix metalloproteinases [75–76] after this novel discovery non-antimicrobial chemically modified tetracyclines (CMT) were prepared and widely investigated in different type of diseases (Figure 1E).
Tetracyclines have been recognized as useful agents for therapy of inflammatory arthritis [77], non-infected corneal ulcers, serious (sometimes life-threatening) skin-blistering diseases, rheumatoid arthritis and osteoarthritis, systemically as well as locally induced bone loss, suppress connective tissue breakdown during periodontal disease and perhaps even tumor-induced angiogenesis [78–80]. However, prolonged use of tetracyclines is limited by their detrimental antimicrobial properties. Recently, a group of chemically modified tetracyclines CMT, CMT-3 and CMT-5 [81] devoid of antimicrobial properties has been synthesized. However, the CMT-3 (COL-3) is produced by deletion of the dimethylamino group from carbon 4 in the A ring of tetracycline, which abolishes the antibiotic activity but not the anti-MMP activity of the molecule. Further modification by replacement of the carbon 11 carbonyl oxygen and the carbon 12 hydroxyl groups with nitrogen, abolishes the anti-MMP activity, giving rise to CMT-5 (COL-5), which is neither antibiotic nor anti-MMP [79]. Some CMT were found to inhibit various matrixmetalloprotinase (MMPs) [79, 82, 83]. Nearly 10 synthetic CMT were tested for inhibition of sPLA2 human porcine PLA2, and Naja naja PLA2. PLA2 activity was assessed by radio labeled Escherichia coli assay using standard and high calcium concentrations. Of which 6 CMT inhibited sPLA2 activity at 50 µg/ml or lower concentrations [77]. However, the other antibiotics such as tetracyclines, macrolides, chloramphenicol and carbenicillin inhibited the activity of Crotalus adamanteus PLA2 towards phospholipids of egg-yolk emulsions [84]. For example, an assessment of metalloproteinase inhibitors clodronate and doxycyline showed effective neutralization of hemorrhage and coagulopathy induced by Bothrops asper snake venom [85].
Tetracyclines also inhibit cell proliferation, cell migration, and synthesis of the extracellular matrix in a variety of cell types studied in culture [86]. However, the earlier studies have shown that CMT-3 and CMT-8 inhibit tumor metastasis and arthritisaffected synoviocyte invasion in animal models [87]. In addition, doxycycline, CMT-1, and CMT-8 exerted a direct dose-dependent inhibition of porcine periodontal ligament epithelial cell medium MMP activity as assayed by gelatin enzymography. Both the 92-kDa (MMP-9) and 72-kDa (MMP-2) gelatinases were inhibited by the tetracyclines added to the conditioned medium. Culturing the cells in the presence of the tetracyclines required considerably smaller concentrations to reduce the secreted MMP activity. The drugs were not toxic to the epithelial cells at 4 to 250 µg/ml concentrations up to 24 h of culture. Doxycycline and the chemically modified tetracyclines, in addition to inhibiting the MMP activity may also reduce the enzyme expression at the transcriptional level [88]. Doxycycline and 4-de-dimethylaminotetracycline (CMT-1) have been tested in vitro for their ability to inhibit human neutrophil and fibroblast interstitial collagenases and collagenase in human gingival crevicular fluid (GCF). Human fibroblast collagenase and GCF collagenase from LJP patients were both relatively resistant to tetracycline inhibition; the IC50 for doxycycline and CMT-1 for these 2 sources of collagenase were 280 and 500 µM, respectively [89–90]. The presence of 25 µM CMT-3, the 48/80- induced histamine release from rat serosal mast cells was inhibited significantly, to 43.0 ± 7.3% of control. Similarly, the activation-induced secretion of TNF-α and IL-8 by HMC-1 cells were decreased in the presence of 25 µM CMT-3 to 13.5 ± 4.1% and 9.7 ± 1.1% of control, respectively [91]. Adding CMT-3 at final concentrations of 5 - 20 µM inhibited MT1-MMP gelatinolytic and caseinolytic activity, blocked MT1-MMP activation of pro-MMP-2, and decreased invasiveness (using the Matrigel system) of HT-1080 fibrosarcoma cells [92]. Six different CMT to inhibit human and chicken MMP were studied in vitro. The ability of tetracycline and its analogs to inhibit MMP appears to depend on the Ca++/Zn++ binding site at C11 (carbonyl oxygen) and C12 (OH group) of the molecule, which is lacking in CMT-5, the pyrazole derivative of tetracycline [93].
b) Lipophilic tetracyclines such as minocycline and doxycycline:
Previous study reported that the doxycycline, minocycline [74], and chemically modified tetracyclines (CMTs) [94] inhibit inducible NO synthase (iNOS) expression in murine macrophages. Tilley and colleagues [95], reported that the tetracyclines, particularly minocycline and doxycycline, are potent inhibitors of collagenases and gelatinases, two matrix metalloproteinases [96, 97] showed a significant reduction of the collagenolytic activity of synovial tissue obtained from six patients with rheumatoid arthritis who received minocycline before total joint arthroplasties compared with the collagenolytic activity of synovium obtained from the same patients during a previous arthroplasty when they had not received minocycline. Minocycline and doxycycline also inhibit PLA2 in vitro [77, 98], another mediator of inflammation [99]. Tetracycline has also been reported to decrease leukotaxis and to inhibit phagocytosis by neutrophils, lymphocyte proliferative responses, and anti-CD3-induced synovial T-cell proliferation in a dose-dependent manner [79, 100]. Some of these antiinflammatory properties have been shown in a chemically modified tetracycline analog that has no antibacterial effects [79, 101, 102]. Recently, several in vitro and fewer in vivo studies have shown that tetracyclines, non-antibacterial, non-chelating hydroxypyrazoline derivative of minocycline, 12 S-hydroxy-1, 12-pyrazolinominocycline (PMIN), also induced antinociceptive and anti-inflammatory effects in rats [103].
Cazalis et al, [104] investigated that the effects of tetracycline (TC), doxycycline (doxy), and chemically modified tetracycline- 3 (CMT-3) on the production of pro-inflammatory mediators and MMPs in an ex vivo human whole blood (WB) model stimulated with Porphyromonas gingivalis lipopolysaccharide (LPS). Results show that TC, doxy, and CMT-3 can reduce the production of pro-inflammatory mediators (interleukin-1beta (IL-1beta), interleukin-6 (IL-6), interleukin-8 (IL-8), MMP-8, and MMP-9. However, the secretion of IL-1beta and -8 and TNFalpha by macrophages decreased significantly (P <0.05) when they were pretreated with 2 µM doxycycline, whereas a concentration of 10 µM was required to significantly reduce IL-6 secretion [105]. Pretreatment of macrophages with 10 µM doxycycline prior to A. actinomycetemcomitans LPS stimulation resulted in a marked decrease in the phosphorylation of extracellular signal-regulated kinase 1/2 (- 76%).
A previous study describes the interaction of α-Lipoic acid (ALA) and dihydrolipoic acid (DHALA) with pro-inflammatory secretory PLA2 enzymes from inflammatory fluids and snake venoms. In vitro enzymatic inhibition of sPLA2 from V. russellii, N. naja and partially purified sPLA2 enzymes from human ascitic fluid (HAF), human pleural fluid (HPF) and normal human serum (HS) by ALA and DHLA was studied [106]. Since in experimental models lipophilic tetracyclines such as minocycline and doxycycline are anti-inflammatory, they examined their effects on PLA2 activity using two assay systems in vitro. They found that minocycline and to a lesser degree doxycycline were markedly inhibitory to both pancreatic and non-pancreatic PLA2. Using [14C] oleic acid labeled Escherichia coli membrane phospholipids as substrate, the IC50 values for minocycline and doxycycline were 3.6 × 10-5 M (18 µg/ml) and 0.98 × 10-4 M (47 µg/ml), respectively [77]. In addition, minocycline has its greatest effect on lipid types, which result from degradation (lipase) reactions, suggesting a lipase inhibition effect and/or direct effect on ocular flora [107]. Currently, patients were randomized to receive 20 mg of subantimicrobial-dose (SD) doxycycline or placebo twice daily for 6 months. Doxycycline treatment significantly reduced the number of inflammatory and non-inflammatory lesions in patients with moderate facial acne, was well tolerated, had no detectable antimicrobial effect on the skin flora, and did not result in any increase in the number or severity of resistant organisms [108].
Snake venom PLA2 inhibitors and therapeutic potential
Certain molecules like endogenous antitoxic serum proteins with neutralizing capacity against snake venoms have been previously reported [109, 110]. Snake blood-derived inhibitors have been grouped into three major classes (α, β and γ), based on common structural motifs found in other proteins with diverse physiological properties [111–114]. PLA2 inhibitors (PLIs) from snake blood Agkistrodon, Trimeresurus, Bothrops, Crotalus, Naja, Laticaudata and Elaphe. Several reviews dealing with inhibitors of snake venom hemorrhagins [115–120] myotoxins and neurotoxins [121] have been published along the past two decades. Lizano et al. [114] reviewed the current knowledge regarding PLA2s inhibitor proteins derived from both snake and mammalian blood, with particular emphasis on the classification, molecular and functional characterization of myotoxic PLA2 inhibitors.
The search for new strategies for treatment of snake envenomations has prompted the discovery of proteins which neutralize the major toxic components of these venoms. These proteins have been isolated from both venomous and nonvenomous snakes, mammals, and most recently from medicinal plant extract [114]. Phospholipases A2s (PLA2s) are commonly found in snake venoms from Viperidae, Hydrophidae and Elaphidae families have been extensively studied due to their pharmacological and physiopathological effects in living organisms [122]. Several substances have been evaluated regarding their effects against snake venoms and isolated toxins, including plant extracts and compounds from marine animals, mammals and snakes serum plasma, in addition to poly or monoclonal antibodies and several synthetic molecules. The enzyme first attracted the interest of investigators seeking cures for snake-bite, since it is a component of toxic venoms from a number snakes. These venoms produce neurotoxicity, myotoxicity, and inflammation. Interestingly, some nonvenomous snakes secrete PLA2 inhibitors. Venomous snakes produce a natural PLA2 inhibitor that has also been isolated. Neuroprotective polypeptide (diffusible survival evasion peptide or DSEP) was used to treat rats with traumatic brain injuries. The peptide, which inhibits the enzymatic activity of sPLA2, blocked the inflammatory response to injury and prevented neural degeneration [123]. Furthermore, the biotechnological potential of PLA2 inhibitors may provide therapeutic molecular models with antiophidian activity to supplement the conventional serum therapy against these multifunctional enzymes.
a) PLA2 inhibitors peptides:
A basic nonapeptide P2 (antiflammin-2, HDMNKVLDL) which is identical to a portion of the amino acid sequence (residues 246-254) of lipocortin I, has been described to have antiinflammatory activity in a rat paw edema model [124]. P2 (0.05 µM) was also reported to inhibit porcine pancreatic PLA2. The effect of synthetic P2 (98% pure) on PLA2 was evaluated in two assay systems. Using porcine pancreatic PLA2 and phosphatidylcholine/deoxycholate mixed micellar substrate, P2 (0.005-50 µM) had no effect on PLA2 activity, even in the presence of 2 [125]. Two anti-inflammatory peptides, named antiflammins (AFs), corresponding to a region with high amino acid similarity between lipocortin-1 and uteroglobin were tested for their ability to inhibit transglutaminase (TG) and lowmolecular- mass PLA2. Porcine pancreatic PLA2 activity and guinea pig hepatic TG activity were determined by arachidonyl release from arachidonyl-phosphatidylcholine and by the incorporation of putrescine into succinylated casein, respectively. AFs inhibited TG activity but did not affect PLA2 activity. Moreover, porcine pancreatic PLA2 was activated by TG [126]. This antiflammins showed significant effects on PLA2 activation by the enzyme called transglutaminase. However, the peptides (antiflammins) failed to show their inhibit effect against purified human synovial fluid PLA2 (HSF-PLA2), even at such high concentrations of peptides as 50 µM. In vivo antiinflammatory activity of these peptides was evaluated in several experimental models of inflammation induced by carrageenan, croton-oil [127].
b) Co-crystallization studies PLIs:
In 1984, the first venom PLA2 with a lysine substituting for the highly conserved aspartate 49 was discovered, in the North American crotalid snake Agkistrodon piscivorus piscivorus [128]. Ten years later, the first mapping of a ‘toxic region’ on a Lys-49 PLA2 was reported, in B. asper myotoxin II [129]. After a further decade of research on the Lys-49 PLA2s, a better understanding of their structural determinants of toxicity and mode of action is rapidly emerging, with myotoxic effector sites identified at the C-terminal region in at least four proteins: B. asper myotoxin II, A. p. piscivorus (K49 PLA2), A. contortrix laticinctus (ACL myotoxin), and B. jararacussu (bothropstoxin I) [130].
A new myotoxic Lys49-PLA2 isolated from B. moojeni snake venom has been crystallized. Suggest a new quaternary structure for this Lys49-PLA2 in contrast to the dimeric and monomeric structures solved so far for this class of proteins [131, 132]. BnSP-7 and BnSP-6, Lys49-PLA2 isolated from B. neuwiedi pauloensis snake venom, were co-crystallized with alpha-tocopherol and X-ray diffraction data were collected for both complexes (2.2 and 2.6 A). A new “alternative” quaternary conformation for these two complexes compared with all other dimeric Lys49-PLA2 has been observed [133]. Large single crystals of piratoxin I a Lys49-PLA2 homologue with low enzymatic activity have been obtained. The crystals belong to the orthorhombic system space group P2 (1)2(1)2(1), and diffract X-rays to a resolution of 2.8 A. Preliminary analysis reveals the presence of two molecules in the crystallographic asymmetric unit [134]. A myotoxic PLA2, bothropstoxin II, which exhibits low hydrolytic activity, was crystallized and Xray diffraction data were collected to a resolution of 2.2 A. Preliminary analyses reveals the presence of three molecules in the asymmetric unit.
c) Structure based functional studies of PLIs:
Several crystal structures of the tetrahedral mimic inhibitors with an sn-2-phosphate, substituent, L-1-(O-octyl-2-(heptyl phosphonyl)-sn-glycero-3-phosphoethanolamine, complexed with secreted PLA2 from Chinese cobra venom [135, 136], PLA2 and peptide inhibitor from Russells viper venom [137], bee venom [138, 139], and inflammatory exudates [140, 141] have been reported. Also the crystal structure of a sn-2-acyl-amino analogue of phospholipids complexed with the engineered (without the residues of the surface 62-66) porcine PLA2 [142, 143] has been determined. The crystal structures of the complexes of bovine pancreatic PLA2 with an inhibitor ndodecyl- phosphorylcholine [144–147] and human synovial PLA2 with an acyl amino analogue of phospholipids [148] are also of some interest. Recently, a series of indole inhibitors of human secretory PLA2 were developed [149–151].
Despite differences in their primary sequences (~30% homology only), crystal structures of PLA2 from bovine pancreas (type I) and human synovial fluid (type II) are almost superimposable and, not surprisingly, the active sites from both types of enzymes are virtually identical [152]. Asp-99 and His-48 form an essential catalytic dyad in the fashion of serine proteases [153]. Tyr-52 and Tyr-73 appear to be associated with these two residues via a hydrogen bonding network, but there is evidence [154] that Tyr-52 is not essential for the catalytic reaction. From structure-function studies of type I and II enzymes, the first eight residues at the NH2 terminus together with Tyr-69, based on the numbering system of Renetseder et al, [155], are thought to play a functional role in interfacial binding of the enzyme to aggregated substrate [156]. Both chemical modification [157] and site-directed mutagenesis [158–160] studies have shown that the NH2-terminal residue is crucial for activity on micellar substrates. Further studies with type I PLA2 [160, 161] have shown that significant alterations of the invariant hydrophobic face of the NH2-terminal amphipathic helix (residues 2, 5, or 9) are detrimental to enzyme activity. Residue 10 is necessary for interfacial binding, and changes in other residues have a modest or no effect on catalytic activity. Production of the fully active enzyme depends on conformational changes of the NH2- terminal helical region that occur firstly on interfacial binding to the phospholipid micelle and secondly on substrate binding at the active site [162].
Nuclear magnetic resonance (NMR) spectroscopic studies of porcine pancreatic PLA2 indicated that the first few residues of the NH2 terminus are not alpha-helical in solution but become helical and rigid on formation of a ternary complex with micelles or a substrate analogue [163–165]. X-ray crystallographic studies showed that the NH2-terminal and β- loop regions (residues 62-73) are held by hydrogen bonding in the active enzyme conformation and form the catalytic network involving His-48, Tyr-52, Tyr-73, and Asp-99 [166–169]. Recent studies have shown that there is a strong but non-covalent association between residues of the NH2-terminal amphipathic helix and the β-loop region, suggesting that a novel approach to enzyme inhibition may be to provide one or another of these domains as free peptide. Peptides from the β-loop region, corresponding to residues 70-74 in three different type II PLA2 molecules, were made and added to mixed micelle assays for the PLA2. Significant enzyme inhibition occurred for each of the three peptides, and inhibition was specific to the PLA2 from which the peptide was derived.
Mechanism action of PLA2 inhibitors
Natural inhibitor inhibits p38 MAPK phosphorylation and slows down the activation of transcription factor which control the transcription of sPLA2, MMPs and inflammatory cytokines. MAPKK –kinase, sPLA2-IIA-secretory phospholipase A2 Group IIA, PGE2-prostaglandin E2.
Secretory phospholipase A2 (sPLA2-IIA) is an inflammatory protein known to play a role in the pathogenesis of envenomation. Over the last 30 years, interest in PLA2 has grown beyond its enzymatic capacity to cleave phospholipids. PLA2 cleave membrane phospholipids to release arachidonic acid, the precursor to a large family of pro-inflammatory eicosanoids including prostaglandins and leukotrienes. Current therapies include non-steriodal anti-inflammatory drugs (NSAIDs') that inhibit cyclooxygenases (COX-1, COX-2) but have no effect on the production of leukotrienes or platelet activating factor (PAF) [170, 171]. In addition, existing treatment with non-steroidal drugs is the only choice for the treatment of anti-inflammatory. However, the available treatments (such as targeting tumour necrosis factor and TNF signaling) do not cure the disease in the clinical settings, and ultimately leads to adverse side effects [172]. A simple mechanism of action of inhibitors on interleukin (IL) induced expression of sPLA2 and matrix metalloproteinase proposed. Cytokines induced the expression of sPLA2-IIA and MMP via activation of mitogenactivated protein kinase (MAPK) etc. Natural inhibitor inhibits p38 MAPK phosphorylation and slows down the activation of transcription factor which control the transcription of sPLA2, matrix metalloproteinase (MMP) such as MMP-1, MMP-2, MMP-3 and MMP-9 and inflammatory cytokine like IL-1, IL-6 and TNF -alpha etc. The effects of inhibitors inhibit the sPLA2 action and decrease the formation of arachidonic acid for prostaglandin production (Figure 2).
Figure 2.
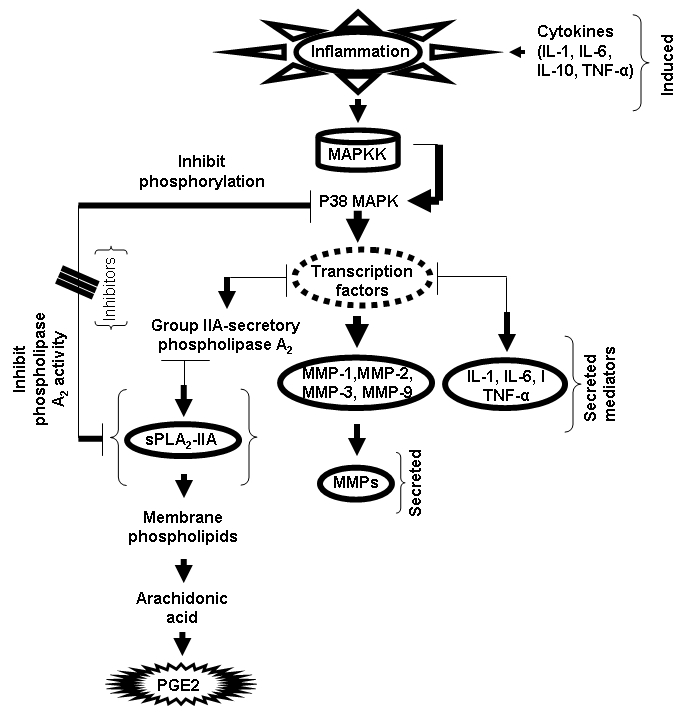
Flow chart represents the simple mechanism of action of inhibitors on interleukin (IL) induced expression of secretory phospholipase A2 (sPLA2) and matrix metalloproteinase. Cytokines induced the expression of sPLA2-IIA and MMP via activation of mitogen-activated protein kinase (MAPK) etc.
Conclusions and future impacts
Snakebite envenomations and their severity remain is not clear yet. However, this poorly neutralized by conventional antivenom serotherapy. In severe cases, these local effects may lead to permanent tissue loss, disability or amputation. A renewed interest in the phenomenon of inter- and intra-species resistance towards the toxicity of snake venoms, coupled with the search for new strategies for treatment of snake envenomations, has prompted the discovery of proteins which neutralize the major toxic components of these venoms. Naturally available inhibitors occupy an important place in the potential to neutralize the toxic effects caused by snake venom proteins. It has been well recognized for several years that animal sera, some plant and marine extracts, antibiotics from synthetic chemicals are the most potent in neutralizing snake venoms. The implications of these new groups of PLA2 toxin inhibitors in the context of our current understanding of snake biology as well as in the development of novel therapeutic reagents in the treatment of snake envenomations world-wide are discussed. Furthermore, the biotechnological potential of PLA2 inhibitors may provide therapeutic molecular models with antiophidian activity to supplement the conventional serum therapy against these multifunctional enzymes. In addition to that the clinically importance of these natural inhibitors may lead to the development of new novel antivenoms therapeutics for the efficient treatment of the envenomation.
Acknowledgments
The authors are grateful to the National University of Singapore for the financial support (Grant No: R-181-000 078 122) provided under the ARF.
Footnotes
Citation:Samy et al, Bioinformation 8(1): 048-057 (2012)
References
- 1.P Pithayanukul, et al. J Ethnopharmacol. 2004;90:347. doi: 10.1016/j.jep.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 2.C Viravan, et al. Trans R Soc Trop Med Hyg. 1992;86:100. doi: 10.1016/0035-9203(92)90463-m. [DOI] [PubMed] [Google Scholar]
- 3.JM Gutiérrez, B Lomonte. Toxicon. 1995;33:1405. doi: 10.1016/0041-0101(95)00085-z. [DOI] [PubMed] [Google Scholar]
- 4.CL Ownby, et al. J Toxicol Toxins Rev. 1998;17:213. [Google Scholar]
- 5.C Teixeira, et al. Toxicon. 2009;4:67. [Google Scholar]
- 6.BZ Yu, et al. Biochemistry. 1998;37:12576. doi: 10.1021/bi9728607. [DOI] [PubMed] [Google Scholar]
- 7.J Balsinde, et al. J Biol Chem. 1999;274:25967. doi: 10.1074/jbc.274.37.25967. [DOI] [PubMed] [Google Scholar]
- 8.G Lambeau, M Lazdunski. Trends Pharmacol Sci. 1999;20:162. doi: 10.1016/s0165-6147(99)01300-0. [DOI] [PubMed] [Google Scholar]
- 9.DA Six, EA Dennis. Biochim Biophys Acta. 2000;1488:1. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 10.CL Ownby, et al. Toxicon. 1999;37:411. doi: 10.1016/s0041-0101(98)00188-3. [DOI] [PubMed] [Google Scholar]
- 11.JM Gutiérrez, et al. Toxicon. 1984;22:115. [Google Scholar]
- 12.AM Soares, et al. Toxicon. 1998;36:503. [Google Scholar]
- 13.Y Angulo, et al. Toxicon. 2005;46:291. [Google Scholar]
- 14.SH Andrião-Escarso, et al. Biochimie. 2000;82:755. doi: 10.1016/s0300-9084(00)01150-0. [DOI] [PubMed] [Google Scholar]
- 15.L Faria de, et al. Toxicon. 2001;39:825. [Google Scholar]
- 16.S Xu. Acta Cryst. 2002;58:1836. [Google Scholar]
- 17.R Ramasamy, et al. Clinical and Experimental Pharmacology and Physiology. 2005;32:7. doi: 10.1111/j.1440-1681.2005.04236.x. [DOI] [PubMed] [Google Scholar]
- 18.Y Wang. Toxicon. 2008;51:1131. doi: 10.1016/j.toxicon.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 19.J Fernández. Biochime. 2010;92:273. [Google Scholar]
- 20.CFP Teixeira, et al. Toxicon. 2003;42:947. [Google Scholar]
- 21.V Iansante, et al. Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry. 2009;8:51. doi: 10.2174/187152309789151977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.KA Brogden, et al. Nature Reviews Microbiology. 2005;3:238. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 23.WH Schwarz, VV Zverlov. Mol Microbiol. 2006;60:1323. doi: 10.1111/j.1365-2958.2006.05181.x. [DOI] [PubMed] [Google Scholar]
- 24.MM Thwin, et al. J Med Chem. 2007;50:5938. doi: 10.1021/jm070385x. [DOI] [PubMed] [Google Scholar]
- 25.M Silvana, et al. Current Topics in Medicinal Chemistry. 2007;7:743. doi: 10.2174/156802607780487614. [DOI] [PubMed] [Google Scholar]
- 26.V Arce-Estrada. Vaccine. 2009;27:460. [Google Scholar]
- 27.AM Soares, et al. Current Medicinal Chemistry. 2005;12:2625. doi: 10.2174/092986705774370655. [DOI] [PubMed] [Google Scholar]
- 28.KT Chandrashekara, et al. Phytother Res. 2009;23:1082. doi: 10.1002/ptr.2735. [DOI] [PubMed] [Google Scholar]
- 29.VH Harsha, et al. Fitoterapia. 2002;73:281. doi: 10.1016/s0367-326x(02)00078-3. [DOI] [PubMed] [Google Scholar]
- 30.VH Harsha, et al. J Ethnopharmacol. 2003;84:37. [Google Scholar]
- 31.M Parinitha, et al. J Ethnopharmacol. 2005;98:307. doi: 10.1016/j.jep.2005.01.035. [DOI] [PubMed] [Google Scholar]
- 32.FK Ticli, et al. Toxicon. 2005;46:318. [Google Scholar]
- 33.R Perumal Samy, et al. J Ethnopharmacol. 2008;115:302. doi: 10.1016/j.jep.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 34.AK Mukherjee, et al. Toxicon. 2008;51:1548. [Google Scholar]
- 35.S Lizano, et al. Toxicon. 2003;42:963. [Google Scholar]
- 36.I Chatterjee, et al. Indian J Experimental Biol. 2004;42:468. [Google Scholar]
- 37.A Shirwaikar, et al. J Ethnopharmacol. 2004;94:267. doi: 10.1016/j.jep.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 38.H Grish, et al. Fitoterapia. 2004;75:378. [Google Scholar]
- 39.AM Torres, et al. Nat Prod Coummun. 2011;6:1393. [Google Scholar]
- 40.P Pithayanukul, et al. Phytother Res. 2010;24:S56. doi: 10.1002/ptr.2903. [DOI] [PubMed] [Google Scholar]
- 41.MI Alam, et al. Toxicon. 1994;32:1551. doi: 10.1016/0041-0101(94)90314-x. [DOI] [PubMed] [Google Scholar]
- 42.MI Alam, A Gomes. Toxicon. 1998;b36:207. [Google Scholar]
- 43.PN Pereira, et al. Planta Medica. 1994;60:99. [Google Scholar]
- 44.R Otero, et al. J Ethnopharmacol. 2000;73:233. [Google Scholar]
- 45.R Otero, et al. J Ethnopharmacol. 2000;71:505. doi: 10.1016/s0378-8741(99)00197-x. [DOI] [PubMed] [Google Scholar]
- 46.A Miyake, et al. J Pharmacol Exp Ther. 1992;263:130. [PubMed] [Google Scholar]
- 47.WB Mors, et al. Toxicon. 1989;27:1003. doi: 10.1016/0041-0101(89)90151-7. [DOI] [PubMed] [Google Scholar]
- 48.WB Mors, et al. Phytochemistry. 2000;55:627. [Google Scholar]
- 49.GB Domont, et al. Toxicon. 1991;29:1183. doi: 10.1016/0041-0101(91)90191-s. [DOI] [PubMed] [Google Scholar]
- 50.MH Borges, et al. J Ethnopharmacol. 2005;98:21. [Google Scholar]
- 51.JJ Moreno, et al. Immunopharmacology. 1993;26:1. [Google Scholar]
- 52.M Lindahl, C Tagesson. Inflammation. 1997;21:347. doi: 10.1023/a:1027306118026. [DOI] [PubMed] [Google Scholar]
- 53.MJ Alcaraz, JRS Hoult. International Pharmacodynamics. 1985;278:4. [Google Scholar]
- 54.A Grau, A Ortiz. Chem Phys Lipids. 1998;91:109. doi: 10.1016/s0009-3084(97)00101-1. [DOI] [PubMed] [Google Scholar]
- 55.NG Bazan, et al. J Neurotrauma. 1995;12:791. doi: 10.1089/neu.1995.12.791. [DOI] [PubMed] [Google Scholar]
- 56.JR Jackson, et al. Biochim Biophys Acta. 1998;1392:145. doi: 10.1016/s0005-2760(98)00012-5. [DOI] [PubMed] [Google Scholar]
- 57.C Kohler, et al. Inflammation. 1993;17:245. [Google Scholar]
- 58.BC Potts, et al. J Natural Products. 1992;55:1701. doi: 10.1021/np50090a001. [DOI] [PubMed] [Google Scholar]
- 59.KB Glaser, RS Jacobs. Biochem Pharmacol. 1986;35:449. doi: 10.1016/0006-2952(86)90218-2. [DOI] [PubMed] [Google Scholar]
- 60.KB Glaser, et al. Biochem. Pharmacol. 1988;37:3639. doi: 10.1016/0006-2952(88)90396-6. [DOI] [PubMed] [Google Scholar]
- 61.PB Jacobson, et al. Biochemical Pharmacol. 1990;39:1557. doi: 10.1016/0006-2952(90)90521-l. [DOI] [PubMed] [Google Scholar]
- 62.A Scoriente, et al. Curr Med. Chem. 1999;6:415. [PubMed] [Google Scholar]
- 63.MC Monti, et al. Chembiochem. 2007;8:1585. [Google Scholar]
- 64.MS Carvalho de, RS Jacobs. Biochem Pharmacol. 1991;42:1321. [Google Scholar]
- 65.F Dal Piaz, et al. Chembiochem. 2002;29:687. [Google Scholar]
- 66.DE Wiemer. Experientia. 1991;47:851. doi: 10.1007/BF01922471. [DOI] [PubMed] [Google Scholar]
- 67.H Yoo, et al. J Nat Prod. 2002;65:51. [Google Scholar]
- 68.NA Shoeib, et al. J Nat Prod. 2004;67:1445. doi: 10.1021/np0305268. [DOI] [PubMed] [Google Scholar]
- 69.JN Narendra Sharath Chandra, et al. Curr Top Med Chem. 2007;7:787. doi: 10.2174/156802607780487678. [DOI] [PubMed] [Google Scholar]
- 70.N Nirmal, et al. Indian J Biochem Biophys. 2008;45:256. [PubMed] [Google Scholar]
- 71.TM ElAttar, et al. J Periodontal Res. 1988;23:285. doi: 10.1111/j.1600-0765.1988.tb01418.x. [DOI] [PubMed] [Google Scholar]
- 72.W Pruzanski, et al. Biochem Pharmacol. 1992;44:1165. doi: 10.1016/0006-2952(92)90381-r. [DOI] [PubMed] [Google Scholar]
- 73.HM Van Barr, et al. (Letter) Br J Dermatol. 1987;117:131. [Google Scholar]
- 74.AR Amin, et al. Proc Natl Acad Sci. USA. 1996;93:14014. [Google Scholar]
- 75.LM Golub, et al. J Periodontal Res. 1984;19:651. doi: 10.1111/j.1600-0765.1984.tb01334.x. [DOI] [PubMed] [Google Scholar]
- 76.RL Smith, et al. J Bone Joint Surg [Am] 1987;69:1063. [Google Scholar]
- 77.W Pruzanski, et al. J. Rheumatol. 1998;25:1807. [PubMed] [Google Scholar]
- 78.LM Golub, et al. Crit Rev Oral Biol Med. 1991;2:297. doi: 10.1177/10454411910020030201. [DOI] [PubMed] [Google Scholar]
- 79.LM Golub, et al. Curr Opin Dent. 1992;2:80. [PubMed] [Google Scholar]
- 80.S Steinsvoll, et al. Microbial Ecology in Health and Disease. 2004;16:1. [Google Scholar]
- 81.MM Islam, et al. American J Pathol. 2003;163:1557. doi: 10.1016/S0002-9440(10)63512-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.ME Ryan, et al. Curr Med Chem. 2001;8:305. doi: 10.2174/0929867013373598. [DOI] [PubMed] [Google Scholar]
- 83.JF Fisher, S Mobashery. Cancer and Metastasis Reviews. 2006;25:115. doi: 10.1007/s10555-006-7894-9. [DOI] [PubMed] [Google Scholar]
- 84.J Sugatani, et al. J Antibiot (Tokyo) 1979;32:734. [Google Scholar]
- 85.A Rucavado, et al. Toxicon. 2008;52:754. [Google Scholar]
- 86.Q Meng, et al. Clin Exp Metastasis. 2000;18:139. doi: 10.1023/a:1006732424102. [DOI] [PubMed] [Google Scholar]
- 87.RE Seftor, et al. Clin Exp Metastasis. 1998;16:217. doi: 10.1023/a:1006588708131. [DOI] [PubMed] [Google Scholar]
- 88.VJ Uitto, et al. Ann NY Acad Sci. 1994;6:140. [Google Scholar]
- 89.LM Golub, et al. J. Den. Res. 1987;66:1310. [Google Scholar]
- 90.T Ingman, et al. J Periodontol. 1993;64:82. [Google Scholar]
- 91.C Sandler, et al. Inflammation Res. 2005;54:304. [Google Scholar]
- 92.HM Lee, et al. Curr Med Chem. 2001;8:257. doi: 10.2174/0929867013373660. [DOI] [PubMed] [Google Scholar]
- 93.T Sorsa, et al. J Rheumatol. 1998;25:975. [PubMed] [Google Scholar]
- 94.AR Amin, et al. FEBS Lett. 1997;410:259. doi: 10.1016/s0014-5793(97)00605-4. [DOI] [PubMed] [Google Scholar]
- 95.BC Tilley, et al. Ann Intern Med. 1995;122:81. doi: 10.7326/0003-4819-122-2-199501150-00001. [DOI] [PubMed] [Google Scholar]
- 96.LH Nip, et al. J Periodontal Res. 1993;28:379. doi: 10.1111/j.1600-0765.1993.tb01082.x. [DOI] [PubMed] [Google Scholar]
- 97.RA Greenwald, et al. J Rheumatol. 1987;14:28. [PubMed] [Google Scholar]
- 98.HE Paulus. Annals of Internal Medicine. 1995;122:147. doi: 10.7326/0003-4819-122-2-199501150-00012. [DOI] [PubMed] [Google Scholar]
- 99.P Vadas, et al. Am J Pathol. 1989;134:807. [PMC free article] [PubMed] [Google Scholar]
- 100.M Kloppenburg, et al. Br J Rheumatol. 1992;31:S41. [Google Scholar]
- 101.PD Agostino, et al. International J Immunopharmacol. 2001;1:1765. [Google Scholar]
- 102.G Webster, JQ Del Rosso. Dermatologic Clinics. 2007;25:133. doi: 10.1016/j.det.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 103.LFS Bastos, et al. British J Pharmacol. 2009;55:714. [Google Scholar]
- 104.J Cazalis. Inflammation. 2009;32:130. [Google Scholar]
- 105.J Cazalis, et al. J Periodontol. 2008;79 [Google Scholar]
- 106.NM Jameel, et al. Life Sciences. 2006;80:146. doi: 10.1016/j.lfs.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 107.WE Shine, et al. Experimental Eye Research. 2003;76:417. doi: 10.1016/s0014-4835(03)00005-8. [DOI] [PubMed] [Google Scholar]
- 108.R Skidmore, et al. Arch Dermatol. 2003;139:459. doi: 10.1001/archderm.139.4.459. [DOI] [PubMed] [Google Scholar]
- 109.MM Thwin, P Gopalakrishnakone. Toxicon. 1998;36:147. [Google Scholar]
- 110.MM Thwin, et al. Biochemistry. 2000;39:9604. doi: 10.1021/bi000395z. [DOI] [PubMed] [Google Scholar]
- 111.N Behrendt, et al. J Biol Chem. 1991;266:7842. [PubMed] [Google Scholar]
- 112.N Ohkura, et al. J Biochem. 1993;113:413. doi: 10.1093/oxfordjournals.jbchem.a124060. [DOI] [PubMed] [Google Scholar]
- 113.K Okumura, et al. J Biol Chem. 1998;273:19469. doi: 10.1074/jbc.273.31.19469. [DOI] [PubMed] [Google Scholar]
- 114.S Lizano, et al. Biochem J. 2000;346:631. [Google Scholar]
- 115.K Okumura, et al. IUBMB Life. 1999;48:99. doi: 10.1080/713803475. [DOI] [PubMed] [Google Scholar]
- 116.K Jeyaseelan, et al. Mol Biol Evol. 2000;17:1010. doi: 10.1093/oxfordjournals.molbev.a026382. [DOI] [PubMed] [Google Scholar]
- 117.CL Fortes-Dias, et al. Toxicon. 2003;41:909. [Google Scholar]
- 118.K Okumura, et al. IUBMB Life. 2003;55:539. doi: 10.1080/15216540310001620995. [DOI] [PubMed] [Google Scholar]
- 119.S Steve Quirós, et al. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology. 2006;46:60. [Google Scholar]
- 120.CZ Oliveira, et al. Biochimie. 2008;90:1506. doi: 10.1016/j.biochi.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 121.AL Fuly, et al. Toxicon. 2000;38:961. [Google Scholar]
- 122.S Marcussi, et al. Curr Top Med Chem. 2007;7:743. doi: 10.2174/156802607780487614. [DOI] [PubMed] [Google Scholar]
- 123.TJ Cunningham, et al. J Neurotrauma. 2004;21:1683. doi: 10.1089/neu.2004.21.1683. [DOI] [PubMed] [Google Scholar]
- 124.L Miele, et al. Nature. 1988;335:726. [Google Scholar]
- 125.WC Hope, et al. Agents Actions. 1991;34:77. doi: 10.1007/BF01993243. [DOI] [PubMed] [Google Scholar]
- 126.JJ Moreno, et al. In Immunopharmacol. 2006;6:300. [Google Scholar]
- 127.F Cabré, et al. Biochem Pharmacol. 1992;44:519. [Google Scholar]
- 128.JM Maraganore, et al. J Biol Chem. 1984;259:13839. [PubMed] [Google Scholar]
- 129.B Lomonte, et al. J Biol Chem. 1994;269:29867. [PubMed] [Google Scholar]
- 130.B Lomonte, et al. Toxicon. 2003;42:885. [Google Scholar]
- 131.DP Marchi-Salvador. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:600. doi: 10.1107/S174430910601801X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.DP Marchi-Salvador, et al. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:882–884. doi: 10.1107/S174430910502717X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.JI Santos dos, et al. Protein Pept Lett. 2005;2:819. doi: 10.2174/0929866054864283. [DOI] [PubMed] [Google Scholar]
- 134.F Canduri, et al. Toxicon. 1998;36:547. [Google Scholar]
- 135.SP White, et al. Science. 1990;250:1560. [Google Scholar]
- 136.YH Pan, et al. J Biol Chem. 2002;277:29086. doi: 10.1074/jbc.M202531200. [DOI] [PubMed] [Google Scholar]
- 137.V Chandra, et al. J Biol Chem. 2002;277:41079. doi: 10.1074/jbc.M206130200. [DOI] [PubMed] [Google Scholar]
- 138.DL Scott, et al. Science. 1990;250:1563. [Google Scholar]
- 139.D Fenard, et al. Molecular Pharmacol. 2001;60:341. doi: 10.1124/mol.60.2.341. [DOI] [PubMed] [Google Scholar]
- 140.DL Scott, et al. Science. 1991;254:1007. [Google Scholar]
- 141.AD Sedgwick, et al. Inflammtion. 2005;6:13. [Google Scholar]
- 142.MMGM Thunnissen, et al. Nature. 1990;347:689. doi: 10.1038/347689a0. [DOI] [PubMed] [Google Scholar]
- 143.JE Burke, EA Dennis. J Lipid Res. 2009;50:S237. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.K Tomoo, et al. Proteins: Struct Funct Genet. 1994;19:330. [Google Scholar]
- 145.K Sekar, et al. Biochemistry36. 1997;46:14186. [Google Scholar]
- 146.K Sekar. CurrTop Med Chem. 2007;7:757. [Google Scholar]
- 147.N Singh, et al. Curr Top Med Chem. 2007;7:757. doi: 10.2174/156802607780487669. [DOI] [PubMed] [Google Scholar]
- 148.SS Cha, et al. J Med Chem. 1996;39:3878. doi: 10.1021/jm960502g. [DOI] [PubMed] [Google Scholar]
- 149.RW Schevitz, et al. Nat Struct. Biol. 1995;2:458. doi: 10.1038/nsb0695-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.J Scott Sawyer, et al. J Med Chem. 2005;48:893. [Google Scholar]
- 151.L Zhou, et al. Nature. 1991;352:79. [Google Scholar]
- 152.JP Wery, et al. Nature. 1991;352:79. [Google Scholar]
- 153.BW Dijkstra, et al. Nature. 1981;289:604. doi: 10.1038/289604a0. [DOI] [PubMed] [Google Scholar]
- 154.C Dupureur, et al. Biochemistry. 1992;31:6402. doi: 10.1021/bi00143a007. [DOI] [PubMed] [Google Scholar]
- 155.R Renetseder, et al. J Biol Chem. 1985;260:11627. [PubMed] [Google Scholar]
- 156.A Randolph, et al. Toxicon. 1998;26:721. [Google Scholar]
- 157.John Wiley Yang C C, Sons . UK: 1997. p. 185. [Google Scholar]
- 158.S Di Marco, et al. J Biochem. 1992;112:350. doi: 10.1093/oxfordjournals.jbchem.a123904. [DOI] [PubMed] [Google Scholar]
- 159.F Marki, V Hanulak. J Biochem. 1993;113:734. doi: 10.1093/oxfordjournals.jbchem.a124112. [DOI] [PubMed] [Google Scholar]
- 160.B Maliwal, et al. Biochemistry. 1994;33:4509. doi: 10.1021/bi00181a010. [DOI] [PubMed] [Google Scholar]
- 161.X Liu, et al. Biochemistry. 1995;34:7322. doi: 10.1021/bi00022a005. [DOI] [PubMed] [Google Scholar]
- 162.PM Kilby, et al. Biochem J. 1995;305:935. doi: 10.1042/bj3050935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.N Dekker, et al. Eur J Biochem. 1991;199:601. doi: 10.1111/j.1432-1033.1991.tb16160.x. [DOI] [PubMed] [Google Scholar]
- 164.A Peters, et al. Biochemistry. 1992;31:10024. doi: 10.1021/bi00156a023. [DOI] [PubMed] [Google Scholar]
- 165.B van den Berg, et al. Struct Biol. 1995;2:402. [Google Scholar]
- 166.B Dijkstra, et al. J Mol Biol. 1981;147:97. [Google Scholar]
- 167.DL Scott, et al. Science. 1990;250:1541. [Google Scholar]
- 168.S Brunie, et al. J Biol Chem. 1985;260:9742. [PubMed] [Google Scholar]
- 169.BW Dijkstra, et al. J Mol Biol. 1982;147:97. [Google Scholar]
- 170.L Miele, et al. Curr Med Chem. 2005;12:3011. doi: 10.2174/092986705774462860. [DOI] [PubMed] [Google Scholar]
- 171.Z Zhang, et al. Mol Pharmacol. 2011 [Google Scholar]
- 172.JS Smolen, G Steiner. Nat Rev Drug Discov. 2003;2:473488. [Google Scholar]