

Abstract
A long-standing problem of conventional cancer chemotherapy is the lack of tumor specificity. Tumor-targeting drug-delivery systems have been explored to overcome this problem. These systems combine a powerful cytotoxic anticancer agent with a tumor-targeting molecule via a ‘smart’ linker to form highly efficacious drug conjugates. These drug conjugates can deliver potent cytotoxic drugs specifically to tumors and tumor cells with minimal systemic toxicity. This review describes our groups’ research on the molecular approaches to the design and development of a novel drug-delivery system bearing highly potent new-generation taxoids for tumor-targeting chemotherapy in our laboratory.
Cancer is both the leading cause of death in developed countries and the second leading cause in developing countries [1]. Currently, one out of every four deaths in the USA is caused by cancer. It has been estimated that there will be approximately 1.6 million new cancer cases and over half a million deaths from cancer in the USA in 2011 [2]. Despite the significant advances in cancer detection, prevention, surgical oncology, chemotherapy and radiation therapy, there is still no common cure for this disease. Traditional chemotherapy relies on the premise that rapidly proliferating tumor cells are more likely to be destroyed than normal cells by cytotoxic agents. However, in reality, these cytotoxic agents have little or no specificity, leading to severe and dose-limiting side effects such as neutropenia, anemia, hair loss, damage to liver, kidney and bone marrow. Therefore, extensive efforts have been made to develop highly efficacious tumor-targeting drug conjugates to overcome the shortcomings of conventional chemotherapy in the last few decades [3,4]. These drug conjugates recognize and take advantage of intrinsic morphological and physiological differences between normal and cancerous cells/tissues. For example, rapidly growing cancer cells overexpress cancer-specific receptors to enhance the uptake of nutrients and vitamins. These receptors can be used as targets to deliver cytotoxic drugs specifically to cancer cells through receptor-mediated endocytosis (RME). A variety of ligands to those cancer-specific receptors, such as monoclonal antibodies (mAbs), polyunsaturated fatty acids (PUFAs), folic acid, transferrin, oligopeptides, hyaluronic acid and aptamers, have been exploited as the tumor-targeting modules (TTMs) to construct tumor-targeting drug conjugates, and a numbers of those drug conjugates are currently in clinical and preclinical development [3–6]. Tumor- targeting drug conjugates typically consist of a TTM connected to an anticancer agent directly or through a suitable ‘smart’ linker. These drug conjugates should be nontoxic and stable in blood circulation to minimize systemic toxicity, but should be effectively internalized inside the tumor cells and efficiently release the anticancer agent without loss of potency [3,7–9].
We describe an account of our research on the design and development of novel tumor-targeting drug-delivery systems for new-generation taxoid anticancer agents.
New-generation taxoids to be delivered specifically to tumors
Paclitaxel and docetaxel have had a significant impact on current cancer chemotherapy, but seriously suffer from the lack of tumor specificity and multidrug resistance (MDR). Paclitaxel and docetaxel are effective against solid tumors, such as breast, ovary and lung cancers, but do not show efficacy against colon, pancreatic, melanoma and renal cancers. For example, human colon carcinoma is inherently multidrug resistant due to the overexpression of P-gp, which is an effective ATP-binding cassette transporter. P-gp effluxes hydrophobic anticancer agents, including paclitaxel and docetaxel, out of cancer cells [10]. On the basis of our extensive structure– activity relationship study of taxoids, we have developed a series of highly potent new-generation taxoids [11–19], including ‘ortataxel’ which has advanced to Phase II human clinical trials [20]. Most of these new-generation taxoids exhibited two-to-three orders of magnitude higher potency than those of paclitaxel and docetaxel against drug-resistant cell lines expressing MDR phenotypes. Accordingly, we have used these highly potent taxoids as the cytotoxic agents for tumor-targeting drug-delivery systems. Selected new-generation taxoids are listed in Table 1 and their potencies in Table 2.
Table 1.
Selected new-generation taxoids.
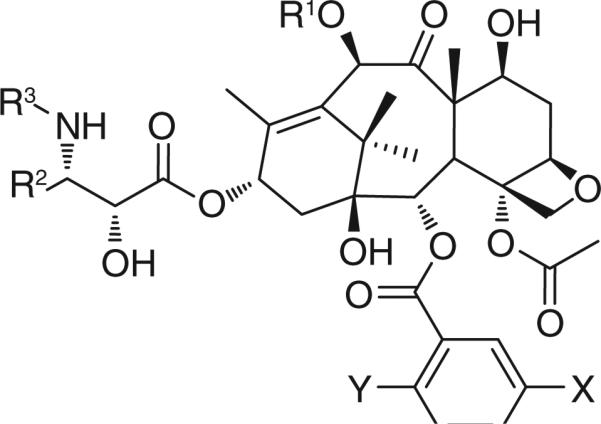 | |||||
|---|---|---|---|---|---|
| Taxane | R1 | R2 | R3 | X | Y |
| Paclitaxel | Ac | Ph | PhCO | H | H |
| Docetaxel | H | Ph | t-Boc | H | H |
| SB-T-1213 | EtCO | i-butenyl | t-Boc | H | H |
| SB-T-1214 | c-PrCO | i-butenyl | t-Boc | H | H |
| SB-T-1216 | Me2NCO | i-butenyl | t-Boc | H | H |
| SB-T-11033 | EtCO | i-Bu | t-Boc | MeO | H |
| SB-T-121303 | EtCO | i-butenyl | t-Boc | MeO | H |
| SB-T-121313 | EtCO | i-butenyl | t-Boc | MeO | MeO |
| SB-T-121602 | Me2NCO | i-butenyl | t-Boc | Me | H |
Table 2.
Cytotoxicity (IC50; nM) of new-generation taxoids against selected cancer cell lines.
| Taxane | MCF7† | NCI/ADR‡ | LCC6-MDR§ | CFPAC-1¶ | HT-29# | DLD-1†† |
|---|---|---|---|---|---|---|
| Paclitaxel | 1.7 | 550 | 346 | 68 | 12 | 300 |
| Docetaxel | 1.0 | 723 | 120 | - | - | - |
| SB-T-1213 | 0.18 | 4.0 | - | 4.6 | 0.37 | 3.9 |
| SB-T-1214 | 0.20 | 3.9 | - | 0.38 | 0.73 | 3.8 |
| SB-T-1216 | 0.13 | 7.4 | - | 0.66 | 0.052 | 5.4 |
| SB-T-11033 | 0.36 | 0.61 | 0.80 | - | - | - |
| SB-T-121303 | 0.36 | 0.79 | 0.90 | 0.89 | - | - |
| SB-T-121313 | 0.30 | - | - | 0.025 | 0.56 | - |
| SB-T-121602 | 0.08 | - | - | 0.31 | 0.003 | 0.46 |
Human mammary cancer cell line (P-gp-).
Human ovarian cancer cell line (P-gp+).
mdr1-fransduced human breast cancer cell line (P-gp+).
Human pancreatic cancer cell line.
Human colon cancer cell line (P-gp-).
Human colon cancer cell line (P-gp+).
MDR: Multidrug resistance.
Recently, it has been shown that the ineffectiveness of standard chemotherapeutic agents could be attributed to the existence of relatively rare, highly drug resistant, quiescent or slowly proliferating tumor-initiating cells (i.e., cancer stem cells [CSCs]) [21,22]. These cells have heightened expression of many stem cell-related genes, such as those in the Notch and Wnt signaling pathways, exhibit similar pluripotency, and are notoriously malignant. CSCs are exclusively endowed with tumor-initiating capacity in the majority of cancer types, and are responsible for sustaining tumor growth, meta stasis and relapse.
One of the new-generation taxoids, SB-T-1214, exhibited remarkable efficacy against highly drug-resistant (P-gp+) colon tumor xeno-graft in severe combined immune deficiency (SCID) mice, inducing complete regression in all surviving mice with tumor growth delay >187 days [18]. The observed total suppression of tumor recurrence by SB-T-1214 might indicate that this taxoid can kill or regulate CSCs by inhibiting stem cell-related gene expression of CSCs and/or promoting their differentiation. Thus, we examined the activity of SB-T-1214 against colon CSCs from HCT116, HT-29 and DLD-1 cell lines using cancer spheroids in 3D cultures [23]. Administration of 100 nM SB-T-1214 to the HCT116, HT-29 and DLD-1 spheroids for 48 h resulted in marked suppression of the growth of the secondary spheroids in all cells (Figure 1). Most importantly, viable cells that survived this treatment regimen significantly lost the ability to form secondary spheroids, which indicates that colon CSC population was critically affected. In fact, the cells that survived drug treatment displayed profound morphological abnormalities, indicating a clear sign of the mitotic catastrophe.
Figure 1. Effects of SB-T-1214 on colon cancer stem cells (HCT 116, HT29 and DLD-1).

To evaluate possible alterations in sphere-forming capacity, control and drug-treated (with 100 nM SB-T-1214) spheroids/cells were dissociated to a single-cell suspension and an equal number of viable cells (400 cells) were plated on ultra-low attachment six-well plates in MSCB medium containing 10% Matrigel. The number of secondary spheroids was counted 10 days after plating. Untreated cells (dark purple) and SB-T-1214-treated cells (light purple).
Adapted with permission from [23].
Also, it was found that the treatment of HCT-116, DLD-1 and HT-29 CSCs with SB-T-1214 led to the downregulation of a number of stem cell-related genes and significant inhibition of genes involved in retaining pluripotency [23]. Relatively low concentrations of SB-T-1214 (100 nM for 24 or 48 h) inhibited the majority of stem cell-related genes in all colon CSCs examined. It is worthy of note that many of these genes are related to self-renewal, regulation of symmetric/asymmetric division and pluripotency.
These results provided additional strong support for the use of new-generation taxoids as the highly potent cytotoxic anti-tumor agents for novel tumor-targeting drug-delivery systems.
PUFA as a TTM of drug-delivery system
Omega-3 PUFA such as docosahexaenoic acid (DHA), eicosapentaenoic acid and linolenic acid are naturally occurring compounds found in vegetable oils, cold-water fish and meat. In a single-arterial perfusion study, it was shown that some PUFAs are taken up more rapidly by tumor tissue than normal cells [24]. In addition, some omega-3 PUFAs have shown anticancer and drug-sensitizing activity against various cancers in both clinical and preclinical trials [25,26]. It has also been shown that PUFAs are readily incorporated into the lipid bilayer of tumor cells, which disrupts the morphology of the cell and presumably influences the susceptibility of the tumor cells to anticancer agents [27,28].
PUFAs are utilized as cellular membrane components, energy resources and signaling molecules. It has also been shown that PUFAs and their metabolites affect a number of cellular processes involved in carcinogenesis including inflammation, apoptosis and cell division. Furthermore, PUFAs are readily taken up by malignant cells, thus making them ideal TTMs for drug conjugation. The conjugation of cytotoxic drugs to PUFAs has been found to substantially alter their pharmacokinetics (PK), resulting in the tumor-specific accumulation of the cytotoxic agent [29].
The PUFA component of the PUFA–drug conjugate readily binds to human serum albumin (HSA) to form a complex, which helps improve solubility in blood plasma [30]. There is evidence to suggest that gp60 and secreted protein, which is acidic and rich in cysteine, play a crucial role in the transport of HSA-bound complexes from blood vessels to the tumor interstitium leading to an accumulation of the HSA-bound complexes in the tumor tissue (Figure 2) [31–33].
Figure 2. gp60-mediated transcytosis.

(1) HSA (orange) bound to drug conjugate (green) travels through blood stream and binds to gp60 (purple) on the tumor epithelial cell surface. (2) Ligand binding initiates transcytosis. (3) Vesicles containing drug conjugate–protein complex are transported across the cell. (4) Fusion of the vesicle to the interstitial cell wall occurs. (5) Drug conjugate–HSA complex is released into the tumor interstitium. (6) Binding of SPARC to HSA causes release of a free drug conjugate, which permeates into tumor cells.
HSA: Human serum albumin; PUFA: Polyunsaturated fatty acid; SPARC: Secreted protein acidic and rich in cysteine.
Reproduced with permission from [33].
It is very likely that PUFA–drug conjugates are accumulated into the tumor tissue, followed by uptake to tumor cells, wherein the PUFA– drug linkage is cleaved, releasing the PUFA and the free drug. It is possible that once internalized, certain PUFAs and their metabolites may result in synergism with cytotoxic drugs due to their ability to modulate several key intracellular processes [34]. Thus, conjugation of drugs to PUFAs results in tumor-specific drug accumulation, lower volume of distribution to benign tissues and enhanced efficacy.
DHA–paclitaxel (Figure 3), which has advanced to Phase III clinical evaluation against several cancer types [101], has been shown to have better PK properties and increased efficacy than its parent compound paclitaxel [29]. DHA–paclitaxel exhibits less systemic toxicity than the parent compound by one or two orders of magnitude, until the ester linkage is cleaved. In addition, the high levels of serum protein binding significantly reduces clearance rate and volume of distribution, resulting in less systemic toxicity, hence reduced side effects [35]. Another advantage of DHA–paclitaxel over paclitaxel is that the DHA–drug conjugate exhibits a wider therapeutic window, and therefore allows the delivery of higher concentrations of paclitaxel to the tumor. It is unlikely, however, that DHA– paclitaxel would be effective against MDR tumors that overexpress P-gp, as paclitaxel is a well-documented P-gp substrate.
Figure 3.

Docosahexaenoic acid– paclitaxel (Taxoprexin®).
As mentioned above, a number of new-generation taxoids exhibit two to three orders of magnitude better activity compared with paclitaxel against MDR cancer cell lines [11,12,18]. Thus, PUFA conjugates that bear a new-generation taxoid should be more efficacious than DHA–paclitaxel against MDR cell lines. Accordingly, novel PUFA–taxoid conjugates were synthesized and assayed in vivo for their efficacy against different drug-resistant and drug-sensitive human tumor xenografts in SCID mice (Figure 4)[36]. Several of these conjugates led to a complete remission in all surviving mice with minimal systemic toxicity. Remarkably, DHA-SB-T-1214 led to a complete regression of the highly drug-resistant DLD-1 colon tumor xenograft in five out of five mice without recurrence of tumor growth for more than 190 days after treatment. Again, little systemic toxicity was observed, implying the tumor specificity of these conjugates (Figure 4). DHA-SB-T-1214 is currently undergoing the last stage of preclinical evaluations, and an Investigational New Drug application to the US FDA will be filed in the near future.
Figure 4. Effect of docosahexaenoic acid–taxoid conjugates on human colon tumor xenograft DLD-1.

Adapted with permission from [36].
mAb as a TTM of drug-delivery system
Cancer cells overexpress certain antigens on the cell surface, and these (tumor-specific) antigens can be used as a biomarker to differentiate tumor tissues from normal tissues [37]. mAbs with high binding specificity to tumor-specific antigens can be developed, which can be used as drug-delivery vehicles to carry a payload of cytotoxic agents specifically to the tumor site. Upon binding to the cell surface antigen, an mAb– drug conjugate is internalized via RME and anticancer agents would be released inside the tumor cell. For example, brentuximab vedotin is a mAb–drug conjugate targeting CD30 and recently approved by the FDA for the treatment of Hodgkin's lymphoma and several other mAb– drug conjugates are currently in human clinical trials. Brentuximab vedotin consists of an anti-CD30 mAb and monomethyl auristatin E, a potent antimitotic agent, which are conjugated through a citruline–valine dipeptide linker (Figure 5) [38,39]. This linker, containing unnatural amino acid citruline, was chosen to confer added stability to the mAb–drug conjugate in the blood circulation, while maintaining the ability to release monomethyl auristatin E by the action of cathepsin B upon internalization into CD30-expressing tumor cells.
Figure 5.

Brentuximab vebotin.
The efficacy of the mAb–drug conjugate depends not only on the specificity of the mAb and potency of the cytotoxic drug, but also on the linker. For the synthesis of a mAb–taxoid conjugate, we chose a cleavable disulfide linker due to the observation that the concentration of glutathione (GSH) in cancer tissues is three orders of magnitude greater than that in the blood circulation [40]. In this first-generation mAb–taxoid conjugate (Figure 6), modification of a taxoid structure was necessary to form a disulfide linkage to the mAb via a thiol-disulfide exchange reaction [41]. We successfully conjugated highly cytotoxic C-10 methyldisulfanylpropanoyl taxoids to different IgG class mAbs recognizing the EGFR (Figure 6) [41]. These immunoconjugates showed exceptional target specificity in vitro. For example, KS78-SB-T-12136 showed high potency (IC50 = 1.5 nM) against the A431 cell line, while a negative control, mN901-SB T-12136, exhibited no cytotoxicity against the A431 cell line at least up to 100 mM. Moreover, KS61-SB-T-12136 showed excellent anti-tumor activity in vivo against A431 human epidermoid carcinoma xenografts in SCID mice, resulting in complete inhibition of tumor growth in all treated mice with no noticeable toxicity to the animals (Figure 7) [41]. Necropsy on day 75, followed by histopathological examination, showed residual calcified material at the tumor site but no evidence of tumor cells. It is noteworthy that treatment with free taxoid SB-12136 at the equi- molar dose to the immunoconjugate showed no therapeutic effect at all (Figure 7). The results unambiguously indicated that targeted delivery of the taxoid using a tumor-specific mAb was essential for the activity. The results also indicated the aliphatic disulfide linker was stable in blood circulation long enough for delivery to the tumor, but efficiently cleaved by intracellular reducing substances in cancer cells.
Figure 6. Synthesis of first-generation monoclonal antibody–taxoid conjugates.

(A) Dithiothreitol (B) N-succinimidyl-4-(2-pyridyldithio) pentanoate (SPP, 10 equivalent in ethanol), potassium phosphate buffer (50 mM), pH 6.5, NaCl (50 mM), EDTA (2 mM), 90 min. (C) Potassium phosphate buffer (50 mM), pH 6.5; NaCl (50 mM); EDTA (2 mM); SB-T-12136-SH (1.7 equivalent per dithiopyridyl group, in EtOH), 24 h.
mAb: Monoclonal antibody.
Adapted with permission from [41].
Figure 7. Anti-tumor activity of anti-EGFR mAb–taxoid conjugates against A431 xenografts in severe combined immunodeficient mice.

Adapted with permission from [41].
Although the first-generation mAb–taxoid immunoconjugates exhibited impressive results, the potency of SB-T-1213, a parent taxoid, was compromised eightfold by the introduction of a sulfhydryl group at the C10 acyl position (i.e., SB-T-12136-SH) in order to conjugate to mAbs [41]. Therefore, novel self-immolative linkers were designed to efficiently release unmodified toxoids to fully exploit their exceptional potency [7,8].
Self-immolative disulfide linkers for a tumor-targeting drug-delivery system
The drug-release mechanism of the second-generation self-immolative bifunctional disulfide linkers involves a cascade process, featuring thiolactonization in the key step, which is triggered by the nucleophilic attack of an intracellular thiol (e.g., GSH) on the disulfide moiety. To promote the thiolactonization process, a phenyl moiety was strategically placed to direct the cleavage of the disulfide bond by an intracellular thiol, generating a thiophenolate or sulfhydrylphenyl species that attacks at the ester linkage to the drug molecule (Figure 8). This drug-release mechanism was validated in a model system using fluorine-labeling and monitoring by 19F NMR spectroscopy [7] as well as in a real system with cancer cells using fluorescence labeling and confocal fluorescence microscopy (CFM) [8,9,42].
Figure 8. Second-generation self- immolative disulfide linkers and their drug-release mechanism.

TTM: Tumor-targeting module.
Adapted with permission from [8].
Adaptive with permission from [8].
The self-immolative disulfide linker module can release a taxoid (or other anticancer agents) efficiently inside cancer cells by taking advantage of the 1000-times higher concentration of GSH in tumor as compared with that in blood plasma [40]. When the TTM navigates the drug conjugate to the target receptors on the tumor surface, the whole conjugate is internalized via RME (Figure 9). Then, an intracellular thiol-triggered cascade drug release takes place through thiolactonization and the released potent anticancer agent attacks its target protein, for example, microtubules for taxoids. These second-generation disulfide linkers have been successfully incorporated into various tumor-targeting drug conjugates and their efficacy evaluated in cancer cells [8,9,42].
Figure 9. Receptor-mediated endocytosis of a tumor-targeting drug conjugate, drug release and binding of the released drug to the target protein.

Adapted with permission from [8].
Vitamins as TTMs for a drug-delivery system
Vitamins are required by all living cells for survival, but cancer cells particularly need certain vitamins and nutrients to sustain their rapid growth and proliferation; for example, vitamin B12, folic acid, biotin and riboflavin are essential for cell division. Therefore, the vitamin receptors are overexpressed on the cancer cell surface for the uptake of necessary vitamins. Accordingly, those vitamin receptors serve as useful targets for tumor-targeting drug delivery as well as biomarkers for identification and imaging of cancer cells. Among those vitamin receptors, folate and biotin receptors have been shown to be important and relevant targets.
Folic acid (vitamin B9), mostly in the form of folate, is essential for DNA synthesis, DNA repair, DNA methylation and RNA synthesis, and acts as a cofactor in certain biological processes [43]. Folate is not endogenously produced by animals. Thus, most mammalian cells obtain folate through low-affinity folate carriers, proton-coupled folate transporters or folate receptor on the cell surface [5,44]. The folate receptor was recognized as excellent target for tumor-targeting drug delivery, and considerable advances have been made [5,44–49]. It is worthy of note that folate–drug conjugates retain a high binding affinity to the folate receptor, but do not have affinity to the low-affinity folate carriers and proton-coupled folate transporters found on the majority of normal cells [5]. Biotin (vitamin B7) is essential for cell division, cell growth, fatty acid production, metabolism of fats and amino acids and plays a role in energy production [50], which makes it essential for rapidly dividing cancer cells. In contrast to folate receptors, biotin receptors had not been studied until it was reported in 2004 that those receptors were even more overexpressed than the folate and vitamin B12 receptors in a variety of cancer cells [51]. Thus, the biotin receptor has emerged as a new target for tumor-targeting drug delivery.
Vitamin–drug conjugates should be internalized into cancer cells via RME (Figure 9) [8,9]. The binding of the vitamin module to its receptor initiates a signaling cascade, which leads to the formation of a membrane invagination. The vitamin–receptor complex is encapsulated into a coated vesicle and individual vesicles fuse to form the early endosome. In the endosome, the vitamin–drug conjugate is released from the receptors and the receptors are recycled to the cell surface. In the endosome or lysosome, the vitamin–drug linkage can be cleaved by endogenous enzymes, lower pH or reducing substances, such as GSH, to release the drug in its active form. Then, the drug released from lysosome attacks its target, microtubules for taxoids.
We chose biotin as the TTM and conjugated it to a second-generation taxoid, SB-T-1214, with a self-immolative disulfide linker, successfully constructing biotin–linker–SB-T-1214 (1; Figure 10) [8,9]. The target specificity and potency of this drug conjugate 1 was evaluated using three cell lines, L1210FR (murine leukemia cell line, overexpressing folate and bioti receptors), L1210 (parent murine leukemia cell line) and WI-38 (normal human lung fibroblast cell line) (Table 3) [9]. Both L1210 and WI38 cell lines do not overexpress biotin receptors on their cell surfaces. As Table 3 shows, conjugate 1 exhibited high potency (IC50 = 8.8 nM) against L1210FR cell line. In sharp contrast, IC50 values against L1210 and WI38 cell lines were 59-times (522 nM) and 65-times (570 nM) larger, respectively. SB-T-1214 was also assayed against those three cell lines for comparison. As anticipated, the free taxoid was not able to distinguish these cell lines, resulting in very similar IC50 values [9]. The results clearly indicate that highly efficient vitamin–receptor-specific drug delivery to cancer cells has been achieved by conjugate 1.
Figure 10. Vitamin–linker–taxoid conjugate (1), fluorescence-labeled taxoid (2) and fluorescent probes (A–C).

Adapted and modified with permission from [9].
Table 3.
Cytotoxicities (IC50, nM) of taxoids and vitamin–linker–taxoid drug conjugates against leukemia and normal human lung fibroblast cells.
| Taxane/cojugate | L1210FR | L1210 | WI38 |
|---|---|---|---|
| Paclitaxel | 121 | 151 | 157 |
| SB-T-1214 | 9.50 | 9.72 | 10.7 |
| Biotin–linker–SB-T-1214 (1) | 8.80 | 522 | 570 |
| SB-T-1214–fluorescein (2) | 87.6 | 107 | 127 |
| Biotin–linker–SB-T-1214–fluoroscein (Probe C) | 81.7 | >5000 | >5000 |
In order to validate the mechanism of tumor-targeting drug delivery by conjugate 1, we monitored (i) RME, (ii) intracellular drug release, and (iii) drug-binding to the target protein (i.e., microtubules) using three fluorescently labeled probes (Figure 10) [8,9]. These probes are:
■ Biotin–fluorescein (Probe A) designed to observe RME;
■ Biotin–linker–coumarin conjugate (Probe B) designed to confirm internalization via RME and release of coumarin from the fluorogenic system via disulfide cleavage and thiolactonization;
■ Biotin–SB-T-1214–fluorescein conjugate (Probe C) designed to validate the internalization of the drug conjugate via RME and drug release followed by the binding of free fluorescent taxoid to microtubules.
The cellular uptake of these probes was monitored by CFM and fluorescence-activated cell sorting (FACS) flow cytometry [8,9]. Probe A (100 nM) was added to L1210FR cells at 37°C and incubated for 3 h. After thorough washing of the cells with phosphate-buffered saline (PBS), CFM analysis showed the efficient internalization of Probe A into the leukemia cells (Figure 11A). Endocytosis is an energy-dependent process and should be inhibited at low temperature (4°C) or in the presence of the metabolism inhibitor, typically NaN3 [52]. The CFM image and FACS of L1210FR cells incubated with Probe A at 4°C (100 nM, 3 h) showed dramatically diminished fluorescence in the cells (Figure 11b). In a similar manner, when L1210FR cells were incubated with Probe A (100 nM, 3 h) in the presence of 0.05% (w/v) NaN3 at 37°C, a significant decrease in fluorescence inside the cells was also observed (Figure 11C). The results unambiguously indicate that the Probe A was internalized through endocytosis. To further validate that the internalization of Probe A was exclusively through a RME process, excess free biotin (2 mM) was pre-incubated for 1 h to saturate the biotin receptors on the cancer cell surface, followed by the addition of Probe A (100 nM) at 37°C for 3 h. Both the CFM and FACS analysis showed drastic decrease in fluorescence (Figure 11D). The results confirm that Probe A was indeed internalized through RME.
Figure 11. Confocal fluorescence microscopy images and fluorescence- activated cell sorting analysis of L1210FR cells after incubation with Probe A under different conditions.

(A) 100 nM, 37°C, 3 h. (B) 100 nM, 4°C, 3 h. (C) 100 nM, 37°C, 0.05% NaN3, 3 h. (D) 100 nM, 37°C, 3 h, pretreated with excess biotin (2 mM).
Adapted with permission from [8].
Next, Probe B (1 μm) was incubated with L1210FR at 37°C for 3 h. After washing the cells thoroughly with PBS, GSH ethyl ester (GSH-OEt, 2 mM) was added to the medium to assure cleavage of the disulfide bond, and cells were incubated for an additional 2 h at 37°C. As shown in the epifluorescence image of the treated cells (Figure 12a), coumarin molecules were released inside L1210FR cells, as indicated by the blue fluorescence. The result clearly indicates that the intracellular release of coumarin (a drug surrogate) via cleavage of the disulfide linkage by GSH and the subsequent thiolactonization proceeded as designed. In the absence of additional GSH-OEt, the observed blue fluorescence was markedly weaker (Figure 12B). The result indicates that the concentration of intracellular GSH in the L1210FR cell line is not sufficient for full cleavage of the disulfide linkage in 3 h in vitro, which are significantly different from those in vivo where the GSH supply in tumor tissues is more than adequate. Also, it should be noted that this experiment using additional GSH (as GSH-OEt) confirmed that the cleavage of the self-immolative disulfide linker and drug release was caused by GSH.
Figure 12. Confocal fluorescence microscopy analysis of L1210FR cells treated with Probe B.

(A) Confocal fluorescence microscopy image of L1210FR cells that were incubated with Probe B (nonfluorescent form), followed by treatment with glutathione-OEt to trigger the self-immolation of the linker. The coumarin released showed blue fluorescence. (B) Confocal fluorescence microscopy image of L1210FR cells after incubation with Probe B (nonfluorescent form) as control.
Adapted with permission from [8].
Finally, Probe C (20 μM) was incubated with L1210FR cells at 37°C for 2 h and analyzed by CFM. As Figure 13A shows, the whole conjugate was internalized in the same manner as that for Probe B. Then, GSH-OEt (2 mM) was added to the medium and the cells were incubated for another 1 h to ensure the drug release through disulfide cleavage and thiolactonization. As Figure 13B shows, the CFM image of the cells is dramatically different from that in Figure 13A. Figure 13b clearly indicates that the released fluorescent taxoid binds to the microtubules and highlights the microtubule bundles with green fluorescence. The result validates that the internalization and release of the taxoid proceeded through the designed mechanism, involving RME via biotin receptors and drug release via GSH-mediated self-immolative disulfide linker cleavage (Figure 9).
Figure 13. Confocal fluorescence microscopy analysis of L1210FR cells treated with Probe C.

(A) Confocal fluorescence microscopy image of L1210FR cells after incubation with Probe C. (B) Confocal fluorescence microscopy image of L1210FR cells were incubated with Probe C, followed by treatment with glutathione-OEt to release the fluorescent taxoid 2. Then, the microtubule network in the cells was fluorescently labeled by 2 and visualized.
Adapted with permission from [8].
Since we synthesized SB-T-1214–fluorescein (2) and Probe C, we examined their tumor spec-ificity and potency as well. As shown in Table 3, the IC50 values of SB-T-1214, 2 and paclitaxel against L1210FR are 9.5, 87.6 and 122 nM, respectively. As mentioned above, SB-T-1214 exhibited comparable IC50 values against these three cell lines. Fluorescence-labeled taxoid 2 and paclitaxel showed slight to small differences in their IC50 values against the three cell lines. Thus, these three taxanes practically cannot distinguish targeted cancer cells from non-targeted cells (L1210) as well as normal cells (WI38), as anticipated.
We also examined the specificity and potency of the Probe C against the same three cell lines. As Table 3 shows, similar differences in IC50 values were observed to those for conjugate 1, albeit that the values for L1210 and WI38 were too high to determine. Thus, it is safe to conclude that the delivery of Probe C as well as the conjugate 1 to L1210FR is highly specific through vitamin–receptor recognition and RME based on both CFM/FACS and cytotoxicity analyses.
■ Taxoid conjugate with dual-targeting modules & that with dual anti-tumor agents
As an extension of the tumor-targeting drug conjugates bearing a vitamin as the TTM, a self-immolative disulfide linker and a taxoid described above, we have designed and successfully synthesized a novel drug conjugate bearing dual-targeting modules (3), such as biotin and folic acid, a trisubstituted 1,3,5-triazine splitter, a self-immolative disulfide linker and SB-T-1214, as well as another novel drug conjugate bearing dual anti-tumor agents (4); SB-T-1214 and topotecan, with self-immolative disulfide linkers, a biotin or folic acid as the TTM and a trisubstituted 1,3,5-triazine splitter (Figure 14) [53]. It is reasonable to hypothesize that the tumor-targeting efficacy would be enhanced if the drug conjugates possess different types of vitamins. Since tumors are heterogeneous (i.e., tumor cells are substantially different from each other), the relative levels of vitamin receptor overexpression differ from cell to cell in the tumor. Therefore, it would be beneficial if the tumor-targeting drug conjugates bear dual-vitamin–TTMs, which can target both vitamin receptors. Furthermore, it is also reasonable to assume that the anti-tumor efficacy would be substantially enhanced if two or more different anti-tumor agents were present in the same drug conjugate. This type of drug conjugate can deliver a combination of drugs in a single molecule, circumventing possible complications and PK issues associated with the standard combination drug therapy. The evaluation of the efficacy of these novel drug conjugates will be reported elsewhere in due course.
Figure 14. Tumor-targeting drug conjugates bearing dual-TTM or dual-cytotoxic drugs.

TTM: Tumor-targeting module.
Single-walled carbon nanotubes as a unique vehicle for tumor-targeting drug delivery
Single-walled carbon nanotubes (SWNTs) have been attracting extensive interest for their potential application in biomedical research as unique drug-delivery vehicles [54,55]. Functionalized SWNTs can incorporate multiple units of TTMs and drug molecule, which can penetrate the cell membrane, accumulate in the cytoplasm and release the drug molecules [54–56]. Building upon the successful construction of highly tumor-specific biotin–linker–taxoid conjugates (1 and Probe C) described above, we designed and devised a novel SWNT-based drug-delivery system [42] SWNTs can be functionalized with numerous units of TTM as well as the conjugate of a self-immolative linker and an anti-tumor agent to provide a novel tumor-targeting drug-delivery system (Figure 15), which can undergo mass delivery of cytotoxic payloads into cancer cells through tumor-specific RME.
Figure 15.

Fluorescently labeled single-walled carbon nanotubes conjugates 5, 6 and 7 as probes.
This SWNT-based drug-delivery system possesses the following advantages:
■ Multivalent ligand–receptor interactions on the tumor cell surface [57,58];
■ Increased drug-loading capacity;
■ Capability of controlled drug release inside tumor cells.
This unique drug-delivery system is multi-functional and multicomponent, consisting of TTMs (e.g., biotin) and cytotoxic payloads (e.g., second-generation taxoids) covalently linked to a SWNT scaffold to form a TTM–SWNT– linker–drug conjugate.
To evaluate the specificity and efficacy of the SWNT-based drug-delivery system, we designed and synthesized three fluorescently labeled SWNT-conjugates 5, 6 and 7, as molecular probes (Figure 14) [42]. Those SWNT conjugates were labeled by using either fluorescein isothiocyanate (FITC) (for conjugates 5 and 6) or fluorescein with a tether (for conjugate 7) and biotin was used as the TTM for conjugates 6 and 7. The fluorescent probes, SWNT– FITC (5) and biotin–SWNT–FITC (6), were designed to track the internalization of SWNT and biotin–SWNT, respectively, into the cancer cells. Biotin–SWNT–linker–taxoid–fluorescein (7) was designed to verify the internalization of the conjugate via RME and intracellular drug release in a manner similar to the conjugate 1 and Probe 3 described above (Figure 10).
We examined cellular uptake of SNWT conjugates 5 and 6 using L1210FR leukemia cells [42] in a manner similar to that for fluorescent Probe A described above. Figure 16A & B show CFM images of L1210FR cells after treatment with 10 μg/ml of conjugates 5 and 6, respectively, for 3 h at 37°C, followed by thorough washing with PBS and analysis [42]. It is apparent that L1210FR cells treated with conjugate 6 yielded far more intense fluorescence than those incubated with conjugate 5. This observation was quantified by FACS analysis, which clearly showed that the fluorescence intensity of the cells treated with conjugate 6 increased by one order of magnitude as compared with conjugate 5 (Figure 16C) [42]. Also, the RME mechanism was confirmed [42] by the temperature dependence, sodium azide inhibition test and saturation of the vitamin receptors with pre-incubation of excess biotin in the same manner as those performed for Probes A–C described above. The results indicate markedly enhanced internalization of conjugate 6 into the cancer cells through highly efficient RME that can be attributed to multivalent binding of the biotin modules to the vitamin receptors. At the same time, the result practically eliminates the ‘piece through mechanism’ known for unfunctionalized and lightly functionalized SWNTs [59].
Figure 16. Confocal fluorescence microscopy and fluorescence-activated cell sorting analysis of L1210FR cells treated with single-walled carbon nanotube-conjugates 5 and 6.

(A) Confocal fluorescence microscopy image after incubation with SWNTs–FITC (5) and (B) that with biotin–SWNTs–FITC (6) at the final concentration of 10 μg/ml at 37°C for 2 h. (C) Comparison of fluorescence intensities of L1210FR cells by fluorescence-activated cell sorting upon treatment with pristine SWNTs 0 (purple), conjugate 5 (blue) and 6 (red) at the final concentration of 10 μg/ml in each case. Background data for untreated cells are plotted in black.
FITC: Fluorescein isothiocyanate; SWINT: Single-walled carbon nanotube.
Adapted with permission from [42].
Next, we examined the efficacy of conjugate 7 for cellular uptake and drug release inside the leukemia cells [42] in a manner similar to that for fluorescent Probe C described above. To evaluate the efficacy of 7 for its internalization and drug release, L1210FR cells were incubated with 7 (50 μg/ml) for 3 h at 37°C, followed by thorough washing with PBS and analysis. The CFM image of the cells shows intense fluorescence, confirming the efficient internalization of 7 (Figure 17a) [42]. Then, the cells were treated with GSH-OEt for an additional 2 h at 37°C in order to ensure cleavage of the disulfide bond covalently linking the taxoid to the biotin–SWNT moiety. Figure 17b shows that the fluorescent taxoid binds to the target protein (microtubules) fluorescently activating large bundles of microtubules [42], which provides ultimate proof of the designed drug release in the same manner as that for Probe C described above.
Figure 17. Confocal fluorescence microscopy analysis of L1210FR cells treated with single-walled carbon nanotube conjugate 7.

Confocal fluorescence microscopy image of L1210FR cells incubated with conjugate 7 before (A) and after (B) the addition of glutathione (GSH)-OEt. Image (B) clearly highlights the presence of fluorescent microtubule networks in the living cells generated by the binding of the fluorescent taxoid 2 released by cleavage of the disulfide bond in the linker by either GSH or GSH-OEt.
Adapted with permission from [42].
We also evaluated the cell-specificity of 7 against L1210FR, L1210 and WI-38 cell lines [42]. As Table 4 shows, after 72 h of incubation, the IC50 value of 7 against the L1210FR cell line was 0.36 μg/ml, whereas those for L1210 and WI-38 cell lines were more than 50 μg/ml. As a control, the IC50 values of conjugates 5 and 7 against all three cell lines were determined, which were consistently >100 μg/ ml [42]. Therefore, the observed cytotoxicity is unambiguously attributed to fluorescent taxoid 2 released from 7 internalized. It should also be noted that taxoid–fluorescein molecules appear to be sufficiently released by endogenous GSH in L1210FR cells after 72 h of incubation in contrast to just 3 h exposure for the CFM study described above (see Figure 13).
Table 4.
Cytotoxicity (IC50) of biotin–single-walled carbon nanotube–taxoid–fluorescein (7) against leukemia and normal human lung fibroblast cells.
| SWNT-conjugate | L1210 | L1210FR | WI38 |
|---|---|---|---|
| 7 | >50 μg/ml | 0.36 μg/ml | >50 μg/ml |
Based on further analysis, the IC50 value (0.36 μg/ml) of 7 corresponds to approximately 51 nM of taxoid 2 against the L1210FR cell line by assuming that all taxoid molecules attached to SWNTs are released. This means that the apparent cytotoxicity per taxoid is substantially increased by using the biotin–SWNT-based drug-delivery system (IC50 of taxoid 2 is 87.6 nM, see Table 3) [42]. The results clearly indicate that the mass drug delivery into the cytosol of the cancer cells using this unique drug-delivery system is superior to simple exposure of the drug itself to the same cancer cells. It is also worthy of note that the cell-specificity of SWNT–conjugate 7 is substantially higher than that of conjugate 1 (Table 3). When taxoids get into the cancer cells through the mass drug-delivery system via RME, the released taxoids can quickly and tightly bind to microtubules so that the effective intra cellular drug concentration is substantially higher than that achieved by extracellular exposure of the drug.
Future perspective
Numerous mAb-based anticancer drug immuno conjugates have been developed and are currently in human clinical trials, but still only a few drug immunoconjugates have become FDA-approved drugs. Nevertheless, it appears that the time has come for these tumor-targeting immunoconjugates to be clinically relevant drugs. Also, several folate–drug conjugates are currently undergoing clinical evaluations, and some of them might become FDA-approved drugs in a few years time. Since there are numerous cancer-specific receptors and bio-markers, tumor-targeting drug-delivery systems have a bright future for developments with a wise combination of TTMs, potent anticancer drugs and mechanism-based ‘smart’ linkers. Our future efforts will be concentrated on the design and synthesis of ‘tailor-made nano-medicines’, consisting of a well-defined vehicle, multiple targeting modules (including dual-targeting molecules), self-immolative smart linkers, highly potent cytotoxic agents (including a combination of different anticancer drugs) and imaging modules (for MRI, positron emission tomography and fluorescence imaging), which would enable us to perform diagnostics and therapy in the real time. We are certain that a variety of new and efficacious tumor-targeting drug conjugates will emerge in the coming years that specifically target different and challenging tumors. We predict that a number of such drug conjugates will go into clinical trials and eventually become powerful drugs in cancer chemotherapy with minimum adverse effects.
Key Terms.
Pluripotency: The potential of a cell to develop into more than one type of mature cell, depending on the environment.
Xenograft: The transplantation of living cells, tissues or organs from one species to another.
Omega-3 polyunsaturated fatty acids: Essential unsaturated fatty acids with a double bond (C=C) after the third carbon atom from the end of the carbon chain.
Key Term.
Human serum albumin: The most abundant protein in human blood plasma. It is produced in the liver. Albumin constitutes approximately half of the blood serum protein. It is soluble and monomeric, and transports hormones, fatty acids, and other compounds, buffers pH, and maintains osmotic pressure, among other functions.
Key Term.
Confocal fluorescence microscopy: An optical imaging technique used to increase optical resolution and contrast of microscopy by using point illumination and a spatial pinhole to eliminate out-of-focus light in specimens that are thicker than the focal plane. It enables the reconstruction of 3D structures from the images obtained.
Key Terms.
Fluorescence-activated cell sorting: A specialized type of flow cytometry. It provides a method for sorting a heterogeneous mixture of biological cells into two or more containers, one cell at a time, based upon the specific light scattering and fluorescent characteristics of each cell.
Flow cytometry: A technique for counting and examining microscopic particles, such as cells and chromosomes, by suspending them in a stream of fluid and passing them by an electronic detection apparatus. It allows simultaneous multiparametric analysis of the physical and/or chemical characteristics of up to thousands of particles per second.
Key Term.
Carbon nanotubes: Allotropes of carbon with a cylindrical nanostructure. These cylindrical carbon molecules have unusual properties, which are valuable for nanotechnology, electronics, optics and other fields of materials science and technology. Most single-walled nanotubes have a diameter of close to 1 nm, with a tube length that can be many millions of times longer. The structure of a single-walled nanotubes can be conceptualized by wrapping a one-atom-thick layer of graphite called graphene into a seamless cylinder.
Executive summary.
Background
■ There is a critical need to develop highly efficacious tumor-targeting drug conjugates to overcome the shortcomings of conventional chemotherapy.
■ Polyunsaturated fatty acids, vitamins (e.g., biotin and folic acid) and monoclonal antibodies can be used as tumor-targeting modules (TTMs) and single-walled carbon nanotubes can be used as unique vehicles for tumor-targeting drug-delivery systems.
■ Various cancer cells overexpress vitamin receptors and tumor-specific antigens such as growth factor receptors, which can be exploited as targets for tumor-specific drug delivery.
New-generation taxoids as the drugs to be delivered specifically to tumors
■ A number of new-generation taxoids possess two to three orders of magnitude greater potency than paclitaxel and docetaxel, against multidrug-resistant cancer cell lines.
■ Treatment of colon cancer stem cells with a new-generation taxoid (SB-T-1214) led to effective suppression of cell growth and significant downregulation of the pluripotency gene expression in cancer stem cells.
Polyunsaturated fatty acid as a TTM of drug-delivery systems
■ Several polyunsaturated fatty acid–taxoid drug conjugates exhibited high efficacy in vivo. Among those conjugates, docosahexaenoic acid–SB-T-1214 demonstrated exceptional activity against highly drug-resistant DLD-1 colon cancer xenograft in severe-combined immune deficiency mice with minimal systemic toxicity. Investigational New Drug application will be filed for FDA approval shortly and this drug conjugate will go into clinical trials in the near future.
Monoclonal antibody as a TTM of drug-delivery systems
■ A taxoid–mAb immunoconjugate showed excellent antitumor activity in vivo against A-431 human epidermoid carcinoma xenografts in severe-combined immune-deficiency mice without noticeable toxicity to the animals, and there was no trace of cancer cells based on histopathological analysis at the end of the experiment.
Self-immolative disulfide linkers for tumor-targeting drug-delivery system
■ Highly efficient self-immolative disulfide linkers have been designed and successfully incorporated into various tumor-targeting drug conjugates.
Vitamins as TTMs for drug-delivery systems
■ A novel biotin–linker–taxoid conjugate exhibited excellent target-specific activity via receptor-mediated endocytosis (RME) for a leukemia cell line as compared with a normal human cell line. The mechanism of internalization (i.e., validation of RME), drug release and drug binding to the target protein (i.e., microtubules) was unambiguously confirmed using fluorescent probes and monitoring by confocal fluorescence microscopy and fluorescence-activated cell sorting.
Single-walled carbon nanotubes as a unique vehicle for tumor-targeting drug delivery
■ The use of multifunctionalized single-walled carbon nanotubes as a unique drug-delivery system bearing multiple cytotoxic payloads and multiple targeting modules has shown highly promising results, demonstrating the benefit of mass delivery of anticancer drug molecules to cancer cells with high specificity via enhanced RME by multivalent binding of TTM. The mechanism of internalization, drug release and drug binding to the target protein was verified using fluorescent probes and monitoring by confocal fluorescence microscopy and fluorescence-activated cell sorting.
Acknowledgments
Financial & competing interests disclosure
This research has been supported by grants from the NIH (National Cancer Institute and National Institute of General Medical Sciences) in the USA. Generous support from Indena, SpA is also gratefully acknowledged.
Footnotes
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J. Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011. CA Cancer J. Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 3.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005;13(17):5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 4.Ducry L, Stump B. Antibody–drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 5.Xia W, Low PS. Folate-targeted therapies for cancer. J. Med. Chem. 2010;53:6811–6824. doi: 10.1021/jm100509v. [DOI] [PubMed] [Google Scholar]
- 6.Farokhzad C, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl Acad. Sci. USA. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ojima I. Use of fluorine in the medicinal chemistry and chemical biology of bioactive compounds: a case study on fluorinated taxane anticancer agents. Chem. Biochem. 2004;5:628–635. doi: 10.1002/cbic.200300844. [DOI] [PubMed] [Google Scholar]
- 8■.Ojima I. Guided molecular missiles for tumor-targeting chemotherapy: case studies using the second-generation taxoid as warheads. Acc. Chem. Res. 2008;41(1):108–119. doi: 10.1021/ar700093f. [Concise review of the discovery and development of efficient mechanism-based tumor-targeting drug-delivery systems, termed ‘guided molecular missiles.’.] [DOI] [PubMed] [Google Scholar]
- 9■■.Chen S, Zhao X, Chen J, et al. Mechanism-based tumor-targeting drug delivery system. Validation of efficient vitamin receptor-mediated endocytosis and drug release. Bioconjugate Chem. 2010;21:979–987. doi: 10.1021/bc9005656. [Describes a highly efficient tumor-targeting drug-delivery system for new-generation taxoids. Mechanism of internalization and drug release was verified by using fluorescent probes.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vredenburg MR, Ojima I, Veith J, et al. Effects of orally active taxanes on P-glycoprotein modulation and colon and breast carcinoma drug resistance. J. Natl Cancer Inst. 2001;93:1234–1245. doi: 10.1093/jnci/93.16.1234. [DOI] [PubMed] [Google Scholar]
- 11.Ojima I, Slater JC, Michaud E, et al. Syntheses and structure–activity relationships of the second-generation antitumor taxoids: exceptional activity against drug-resistant cancer cells. J. Med. Chem. 1996;39:3889–3896. doi: 10.1021/jm9604080. [DOI] [PubMed] [Google Scholar]
- 12.Ojima I, Slater JC, Kuduk SD, et al. Syntheses and structure–activity relationships of taxoids derived from 14-β-hydroxy-10-deacetylbaccatin III. J. Med. Chem. 1997;40(3):267–278. doi: 10.1021/jm960563e. [DOI] [PubMed] [Google Scholar]
- 13.Ojima I, Wang T, Miller ML, et al. Syntheses and structure-activity relationships of new second-generation taxoids. Bioorg. Med. Chem. Lett. 1999;9:3423–3428. doi: 10.1016/s0960-894x(99)00629-0. [DOI] [PubMed] [Google Scholar]
- 14.Ojima I, Lin S, Wang T. The recent advances in the medicinal chemistry of taxoids with novel β-amino acid side chains. Curr. Med. Chem. 1999;6:927–954. [PubMed] [Google Scholar]
- 15.Ferlini C, Distefano M, Pignatelli F, et al. Antitumor activity of novel taxanes that act as cytotoxic agents and P-glycoprotein inhibitors at the same time. Brit. J. Cancer. 2000;83:1762–1768. doi: 10.1054/bjoc.2000.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geney R, Chen J, Ojima I. Recent advance in the new generation taxane anticancer agents. Med. Chem. 2005;1:125–139. doi: 10.2174/1573406053175292. [DOI] [PubMed] [Google Scholar]
- 17.Ehrlichova M, Vaclavikova R, Ojima I, et al. Transport and cytotoxicity of paclitaxel, docetaxel and novel taxanes in human breast cancer cells. Arch. Pharmacol. 2005;372:95–105. doi: 10.1007/s00210-005-1080-4. [DOI] [PubMed] [Google Scholar]
- 18■■.Ojima I, Chen J, Sun L, et al. Design, synthesis and biological evaluation of new-generation taxoids. J Med. Chem. 2008;51:3203–3221. doi: 10.1021/jm800086e. [Reports the development of an efficient mechanism-based tumor-targeting drug delivery system, based on tumor-specific vitamin receptor-mediated endocytosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovar J, Ehrlichova M, Smejkalova B, Zanardi I, Ojima I, Gut I. Comparison of cell death-inducing effect of novel taxane SB-T-1216 and paclitaxel in breast cancer cells. Anticancer Res. 2009;29:2951–2960. [PMC free article] [PubMed] [Google Scholar]
- 20.Beer M, Lenaz L, Amadori D. Phase II study of ortataxel in taxane-resistant breast cancer. J. Clin. Oncol. (Suppl.) 2008;26:1066. [Google Scholar]
- 21.Kamb A, Wee S, Lengauer C. Why is cancer drug discovery so difficult? Nat. Rev. Drug Discov. 2007;6:115–120. doi: 10.1038/nrd2155. [DOI] [PubMed] [Google Scholar]
- 22.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu. Rev. Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 23■.Botchkina GI, Zuniga ES, Das M, et al. New-generation taxoid SB-T-1214 inhibits stem cell-related gene expression in 3D cancer spheriods induced by purified colon tumor-initiating cells. Mol. Cancer. 2010;9:192–204. doi: 10.1186/1476-4598-9-192. [Study revealed that a second-generation taxoid, SB-T-1214, effectively inhibited cancer stem cell growth and suppressed the expression of stem cell-related genes.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauer LA, Dauchy RT, Blask DE. Mechanism for the antitumor and anticachectic effects of n-3 fatty acids. Cancer Res. 2000;60:5289–5295. [PubMed] [Google Scholar]
- 25.Wigmore SJ, Ross JA, Falconer JS, et al. The effect of polyunsaturated fatty acids on the progress of cachexia in patients with pancreatic cancer. Nutrition. 1996;12:S27–S30. doi: 10.1016/0899-9007(96)90014-3. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins RA, Sangster K, Arends MJ. Apoptotic death of pancreatic cancer cells induced by polyunsaturated fatty acids varies with double bond number an involves an oxidative mechanism. J. Pathol. 1998;185:61–70. doi: 10.1002/(SICI)1096-9896(199805)185:1<61::AID-PATH49>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 27.Grammatikos SI, Subbaiah PV, Victor TA, Miller WM. n-3 and n-6 fatty acid processing and growth effects in neoplastic and noncancerous human mammary epithelial cell lines. Br. J. Cancer. 1994;70:219–227. doi: 10.1038/bjc.1994.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diomede L, Colotta F, Piovani B, Re F, Modest EJ, Salmona M. Induction of apoptosis in human leukemic cells by the ether lipid 1-octadecyl-2-methyl-racglycero-3-phosphocholine. A possible basis for its selective action. Int. J. Cancer Res. 1993;53:124–130. doi: 10.1002/ijc.2910530123. [DOI] [PubMed] [Google Scholar]
- 29■.Bradley MO, Webb NL, Anthony FH, et al. Tumor targeting by covalent conjugation of a natural fatty acid to taxol. Clin. Cancer Res. 2001;7:3229–3238. [Presents the full preclinical evaluation of docosahexaenoic acid–paclitaxel.] [PubMed] [Google Scholar]
- 30.Sparreboom A, Wolff AC, Verweij J, et al. Dispostition of docosahexaenoic acid: paclitaxel, a novel taxane, in blood. Clin. Cancer Res. 2003;9:151–159. [PubMed] [Google Scholar]
- 31.Ibrahim NK, Desai N, Legha S, et al. Phase I and pharmacokinetic study ABI-007, a cremophor-free, protein-stabilized nanoparticle formulation of paclitaxel. Clin. Cancer Res. 2002;8:1038–1044. [PubMed] [Google Scholar]
- 32.Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 2006;12:1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- 33.Seitz JD, Ojima I. Drug conjugates with polyunsaturated fatty acids. In: Kratz F, Senter P, Steinhagen H, editors. Drug Delivery in Oncology: From Basic Research to Cancer Therapy. Wiley-VCH; Weinheim, Germany: 2011. pp. 1323–1360. [Google Scholar]
- 34.Rose DP, Connolly JM. Omega-3 fatty acids as cancer chemopreventive agents. Pharmacol. Ther. 1999;83:217–244. doi: 10.1016/s0163-7258(99)00026-1. [DOI] [PubMed] [Google Scholar]
- 35.Whelan J. Targeted taxane therapy for cancer. Drug Discov. Today. 2002;7:90–92. doi: 10.1016/s1359-6446(01)02149-3. [DOI] [PubMed] [Google Scholar]
- 36■■.Kuznetsova L, Chen J, Sun X, et al. Syntheses and evaluation of novel fatty acid: 2nd-generation taxoid conjugates as promising anticancer agents. Bioorg. Med. Chem. Lett. 2006;16:974–977. doi: 10.1016/j.bmcl.2005.10.089. [Reports the in vivo evaluation of polyunsaturated fatty acid–taxoid drug conjugates, which exhibited strong activity against highly multidrug-resistant colon cancer xenografts in mice.] [DOI] [PubMed] [Google Scholar]
- 37■.Chari RVJ. Targeted delivery of chemotherapeuitcs: tumor-activated prodrug therapy. Adv. Drug Deliv. Rev. 1998;31:89–104. doi: 10.1016/s0169-409x(97)00095-1. [Discusses potent and tumor-specific monoclonal antibody–drug conjugates, which act as tumor-activated prodrugs for chemotherapy.] [DOI] [PubMed] [Google Scholar]
- 38.Doronina SO, Mendelsohn BA, Bovee TD, et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjugate Chem. 2006;17:114–124. doi: 10.1021/bc0502917. [DOI] [PubMed] [Google Scholar]
- 39.Beck A, Haeuw JF, Wurch T, Goetsch L, Bailly C, Corvaia N. The next generation of antibody–drug conjugates comes of age. Discov. Med. 2010;10:329–339. [PubMed] [Google Scholar]
- 40.Kigawa J, Minagawa Y, Kanamori Y, et al. Glutathione concentration may be a useful predictor of response to second-line chemotherapy in patients with ovarian cancer. Cancer. 1998;82:697–702. doi: 10.1002/(sici)1097-0142(19980215)82:4<697::aid-cncr12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 41■.Ojima I, Geng X, Wu X, et al. Tumor-specific novel taxoid–monoclonal antibody conjugates. J Med. Chem. 2002;45:5620–5623. doi: 10.1021/jm025540g. [Reports the synthesis and biological evaluation of first-generation mAb–taxoid drug conjugates, which exhibited remarkable tumor-specific anti-tumor activity in vivo.] [DOI] [PubMed] [Google Scholar]
- 42■■.Chen J, Chen S, Zhao X, Kuznetsova L, Wong SS, Ojima I. Functionalized single-walled carbon nanotubes as rationally designed vehicles for tumor-targeted drug delivery. J. Am. Chem. Soc. 2008;130:16778–16785. doi: 10.1021/ja805570f. [Reports the successful use of covalently multifunctionalized single-walled carbon nanotubes as unique vehicle for tumor-targeting drug-delivery systems. Excellent cancer cell-specificity and potency of a vitamin–single-walled carbon nanotube–taxoid conjugate was demonstrated.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weinstein SJ, Hartman TJ, Stolzenberg-Solomon R, et al. Null association between prostate cancer and serum folate, vitamin B6, vitamin B12 and homocysteine. Cancer Epidemiol. Biomarkers Prev. 2003;12:1271–1272. [PubMed] [Google Scholar]
- 44■.Reddy JA, Leamon CP. Folate receptor targeted cancer chemotherapy. In: Jackman AL, Leamon CP, editors. Targeted Drug Strategies for Cancer and Inflammation. Springer Science+Business Media, LLC; NY, USA: 2011. pp. 135–150. [Provides up-to-date summary of the promising preclinical evaluations of folate receptor-targeted drug conjugates.] [Google Scholar]
- 45.Leamon CP, Reddy JA. Folate-targeted chemotherapy. Adv. Drug Deliv. Rev. 2004;56:1127–1141. doi: 10.1016/j.addr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 46.Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Deliv. Rev. 2002;54:675–693. doi: 10.1016/s0169-409x(02)00042-x. [DOI] [PubMed] [Google Scholar]
- 47.Lu Y, Sega E, Leamon CP, Low PS. Folate receptor-targeted immunotherapy of cancer: mechanism and therapeutic potential. Adv. Drug Deliv. Rev. 2004;56:1161–1176. doi: 10.1016/j.addr.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Leamon CP, Reddy JA, Vlahov IR, et al. Synthesis and biological evaluation of EC72: a new folate-targeted chemotherapeutic. Bioconjugate Chem. 2005;16(16):803–811. doi: 10.1021/bc049709b. [DOI] [PubMed] [Google Scholar]
- 49.Reddy JA, Westrick E, Vlahov I, Howard SJ, Santhapuram HK, Leamon CP. Folate receptor specific anti-tumor activity of folate–mitomycin conjugates. Cancer Chemother. Pharmacol. 2006;58:229–236. doi: 10.1007/s00280-005-0151-z. [DOI] [PubMed] [Google Scholar]
- 50.Zempleni J, Wijeratne SS, Hassan YI. Biotin. Biofactors. 2009;35(1):36–46. doi: 10.1002/biof.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51■■.Russell-Jones G, McTavish K, McEwan J, Rice J, Nowotnik D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J. Inorg. Biochem. 2004;98(10):1625–1633. doi: 10.1016/j.jinorgbio.2004.07.009. [Discusses the overexpression of vitamin receptors (folate, vitamin B12 and biotin) on cancer cell surfaces of a variety of cancer cell lines. Discovery of the biotin receptors as viable biomarker for tumor-targeting drug delivery.] [DOI] [PubMed] [Google Scholar]
- 52.Mukherjee S, Ghosh RN, Masfield FR. Endocytosis. Physiol. Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 53.Zuniga E, Ojima I. Design, synthesis and biological evaluation of tumor-targeting drug conjugates bearing dual warheads.. Presented at: The American Chemical Society National Meeting; Denver, CO, USA. 28 August–1 September 2011. [Google Scholar]
- 54■.Prato M, Kostarelos K, Bianco A. Functionalized carbon nanotubes in drug design and discovery. Acc. Chem. Res. 2008;41(1):60–68. doi: 10.1021/ar700089b. [Critical and concise review of the development of functionalized carbon nanotubes and their applications to biomedical purposes.] [DOI] [PubMed] [Google Scholar]
- 55.Bianco A, Kostarelos K, Prato M. Applications of carbon nanotubes in drug delivery. Curr. Opin Chem. Biol. 2005;9(6):674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 56.Kam NWS, Jessop TC, Wender PA, Dai H. Nanotube molecular transporters internalization of carbon-nanotube-protein conjugates into mammalian cells. J. Am. Chem. Soc. 2004;126:6850–6851. doi: 10.1021/ja0486059. [DOI] [PubMed] [Google Scholar]
- 57.Duncan R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 58.Bottini M, Cerignoli F, Dawson MI, Magrini A, Rosato N, Mustelin T. Full-length single-walled carbon nanotubes decorated with streptavidin-conjugated quantum dots as multivalent intracellular fluorescent nanoprobes. Biomacromolecules. 2006;7:2259–2263. doi: 10.1021/bm0602031. [DOI] [PubMed] [Google Scholar]
- 59.Kostarelos K, Lacerda L, Pastorin G, et al. Cellular uptake of functionalized carbon nanotubes is independent of functional group and cell type. Nat. Nanotechnol. 2007;2(2):108–113. doi: 10.1038/nnano.2006.209. [DOI] [PubMed] [Google Scholar]
- 101.ClinicalTrialsFeeds www.clinicaltrialsfeeds.org/clinical-trials/results/term=Drug+Taxoprexin.