

Abstract
Deposits of iron and hemosiderosis in the kidney have been observed in diseases with intravascular hemolysis, including paroxysmal nocturnal hemoglobinuria, and valvular heart diseases and prosthetic heart valve implants. However, the decrease in kidney function associated with hemolysis caused by cardiac valvular disease or prostheses is less well recognized. We present a case of intravascular hemolysis after repair and banding of the mitral valve that resulted in massive renal tubular deposition of hemosiderin with decreased kidney function. We discuss the pathophysiologic process of both acute and chronic tubular injury from heme and heme proteins, including injury to organelles resulting in autophagic vacuoles containing damaged organelles, such as mitochondria. We conclude that tubular injury resulting from heme proteins should be considered as a cause of decreased kidney function in all patients with a cardiac valvular disease or prosthesis.
INDEX WORDS: Hemolysis, renal hemosiderosis, acute tubular necrosis, mitral valve annuloplasty, renal failure
Renal deposits of iron, hemosiderosis, have been observed in a number of diseases featuring intravascular hemolysis.1 A classic example is paroxysmal nocturnal hemoglobinuria, in which acute kidney injury can occur during episodes of acute hemolysis, and chronic kidney disease (CKD) can evolve during months to years.2 Varying degrees of intravascular hemolysis also are associated commonly with severe valvular heart diseases and prosthetic heart valve implants.3-5 However, in patients with renal hemosiderosis caused by native or prosthetic valve–induced hemolysis, there is a common perception that such hemolysis does not lead to significant impairment in kidney function.
We present a case of intravascular hemolysis after repair and banding of a mitral valve with a Cosgrove-Edwards annuloplasty band prosthesis (Edwards Lifesciences, Irvine, CA) that resulted in massive renal tubular deposition of hemosiderin and a progressive decrease in kidney function.
CASE REPORT
Clinical History and Initial Laboratory Data
A 70-year-old man with a history of hypertension, hyperlipidemia, atherosclerotic vascular disease, and CKD stage 3b (baseline serum creatinine, 1.8 mg/dL [159.1 μmol/L]; estimated glomerular filtration rate [GFR], 40 mL/min/1.73 m2 [0.67 mL/s/1.73 m2] using the 4-variable Modification of Diet in Renal Disease [MDRD] Study equation) was admitted to the hospital because of dark urine, anemia, and decreasing GFR. Seven months ago, he underwent mitral valve repair with quadrangular resection of a segment of the posterior leaflet and implantation of a 32-mm Cosgrove-Edwards annuloplasty band for severe mitral valve regurgitation caused by myxoid degeneration. The immediate perioperative course was uneventful. Repeated transthoracic echocardiography showed mild residual mitral regurgitation. However, 10 days later, he developed anemia when hemoglobin concentration decreased from 11.6 g/dL (116 g/L) preoperatively to 7.6 g/dL (76 g/L) and thrombocytopenia when platelet count decreased to 77 × 103/μL (77 × 109/L) from a normal level of 168 × 103/μL (168 × 109/L). His serum creatinine concentration increased to 2.2 mg/dL (194.5 μmol/L; estimated GFR, 29 mL/min/1.73 m2 [0.48 mL/s/1.73 m2]), and he was nonoliguric. Further evaluation showed an increase in lactate dehydrogenase level to 1,700 IU/L and schistocytosis on peripheral-blood smear. A provisional diagnosis of microangiopathic hemolytic anemia possibly caused by thrombotic thrombocytopenic purpura was made. The patient was started on steroid therapy and discharged from the hospital. Despite steroid therapy, the anemia worsened during the subsequent months, necessitating multiple blood transfusions, and there was a further decrease in kidney function.
On examination, the patient appeared pale, blood pressure was 144/92 mm Hg, pulse rate was 90 beats/min, respiratory rate was 18 breaths/min, he was afebrile, lung fields were clear, and there was 1+ pitting edema in the lower extremities. Laboratory studies showed the following values: hemoglobin, 7.2 g/dL (72 g/L); platelet count, 95 × 103/μL (95 × 109/L); white blood cell count, 6.1 × 103/μL (6.1 × 109/L) with a normal differential count; bilirubin (total/indirect), 3.1/2.8 mg/dL (53/47.9 μmol/L); sodium, 129 mEq/L (129 mmol/L); potassium, 5.9 mEq/L (5.9 mmol/L); blood urea nitrogen, 65 mg/dL (23.2 mmol/L); creatinine, 4.5 mg/dL (397.8 μmol/L; estimated GFR, 13 mL/min/1.73 m2 [0.25 mL/s/1.73 m2]); lactate dehydrogenase, 4,002 IU/L; fractional excretion of sodium, 0.3%, and haptoglobin, 4 mg/dL (0.04 g/L; reference, 45-32 mg/dL [0.45-0.32 g/L]). Urine appeared dark brown, osmolality was 357 mOsm/kg (357 mmol/kg), and there was large heme, 1-3 red blood cells/highpower field, occasional granular casts, and 1-3 renal epithelial cells/high-power field. Urine protein-creatinine ratio was 0.13. Direct Coombs test was negative, and cold agglutinin titer was within the reference range. Blood smear showed schistocytosis. To further ascertain the cause of anemia and kidney failure, a percutaneous diagnostic kidney biopsy was performed.
Kidney Biopsy
Three cores containing both renal cortex and medulla were received for light microscopy. Fourteen glomeruli were present; 3 glomeruli were globally sclerotic. Glomeruli with segmental sclerosis were not present. Glomeruli did not show significant changes, and there was no evidence of crescents, fibrinoid necrosis, thrombosis, or endocapillary proliferation. The mesangium was unremarkable. The interstitium showed tubular injury with distention of tubules and debris in the lumen. The most prominent finding was the presence of numerous brown pigment deposits on hematoxylin and eosin and periodic acid–Schiff stains, forming small reabsorption-like granules in tubular epithelial cells, particularly in the proximal tubular epithelial cells (Fig 1A and B). Iron using the Perls ferrocyanide staining method showed extensive hemosiderin deposits in tubular cells (Fig 1C). There was no significant interstitial inflammation. Approximately 20% of the interstitium showed tubular atrophy and interstitial fibrosis. Arteries and arterioles showed mild sclerosis of the intima. There was no evidence of thrombosis. Immunofluorescence studies were negative for immune deposits in glomeruli.
Figure 1.
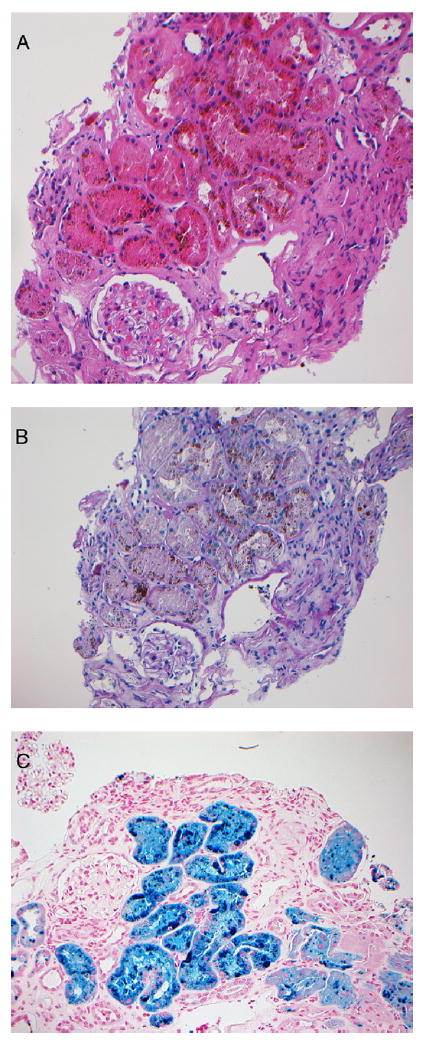
(A) Hematoxylin and eosin and (B) periodic acid–Schiff sections show brown pigment deposits in the proximal tubular epithelial cells accompanied by tubular degeneration and necrosis. (C) Prussian blue iron stain shows hemosiderin deposits in the tubular epithelial cells (A-C: original magnification, ×20).
Electron microscopy of a glomerulus was unremarkable and showed well-preserved foot processes of the epithelial cells. Glomerular basement membranes appeared to be of normal thickness. Electron-dense deposits were not present along the capillary walls or in the mesangium. Tubules showed dark irregularly shaped granular deposits grouped in a ring-like structure likely made up of heme proteins (Fig 2; white arrows) and with a pale center. In addition, tubules showed lysosomal granules containing remnants of mitochrondria as evidenced by cristae within the vacuoles (autophagic vacuoles, black arrows; Fig 2).
Figure 2.
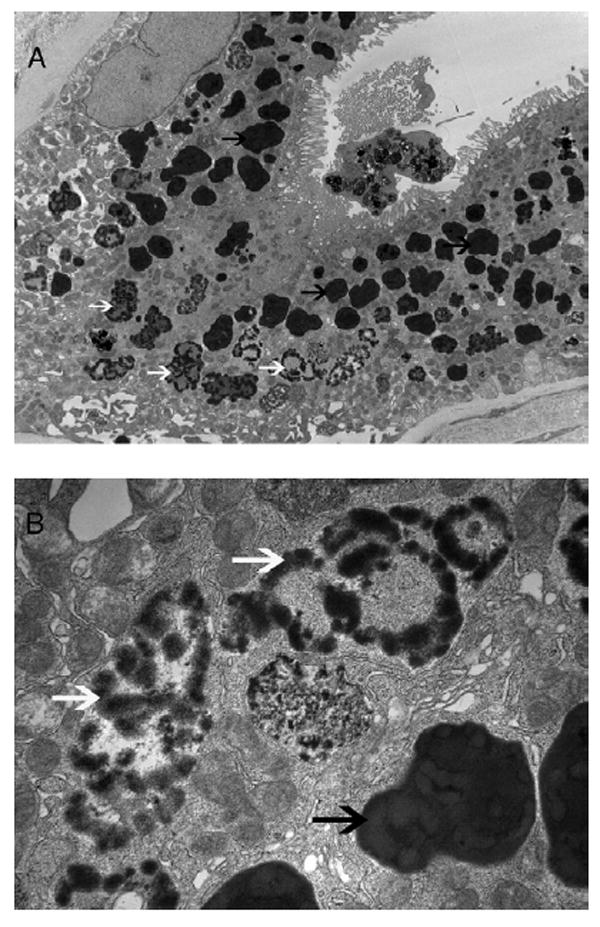
Electron microscopy of tubular epithelial cells shows groups of dark granules likely made up of heme proteins (white arrows) and autophagolytic lysosomal vacuoles (black arrows) containing mitochondria (original magnification, A: ×3,400; B: ×17,500). An apoptotic cell shedding into the tubular lumen also can be seen.
Diagnosis
Renal hemosiderosis with acute and chronic tubulointerstitial injury.
Clinical Follow-up
The patient has been followed up for 13 months. He is active and able to carry out daily life activities. Hemoglobin level has been 8-9 g/dL (80-90 g/L) and serum creatinine level is 3 mg/dL (265.2μmol/L; estimated GFR, 20 mL/min/1.73 m2 [0.33 mL/s/1.73 m2]). He is not on dialysis or other renal replacement therapy. He has received 8 units of packed red blood cells since discharge from the hospital after surgery.
DISCUSSION
Intravascular hemolysis occurs often in patients with severe cardiac valvular abnormalities or after replacement of heart valves with mechanical prostheses.3 Although acute kidney injury and CKD are recognized complications in diseases typified by recurrent hemolysis (hemoglobinuric nephropathy) such as paroxysmal nocturnal hemoglobinuria,2,6 a GFR decrease associated with hemolysis caused by cardiac valvular disease or prostheses is less well recognized. We present a patient with underlying CKD who developed a decrease in kidney function attributable to hemolysis associated with mitral valve repair and implantation of a Cosgrove-Edwards annuloplasty band.
The mechanism of intravascular hemolysis in patients with cardiac valvular disease or prosthetic device implantation is mechanical, resulting from turbulent blood flow and shear forces through the diseased valve or prostheses, in which velocity and pressure are rapidly changing. In most cases, as in the present patient, valvular regurgitation is a prominent feature, and such regurgitant flow greatly increases the frequency with which cells are traumatized.7 In our patient, mechanical destruction of erythrocytes likely was exacerbated by tightening of the mitral valve area with the banding operation, causing a more rapid change in blood velocity that increased shear force across the valve area and led to erythrocyte fragmentation.
Mechanical trauma to erythrocytes liberates hemoglobin into plasma, where hemoglobin is bound by haptoglobin, thereby forming a haptoglobin-hemoglobin protein complex.6,8 This complex, too large to be filtered by the glomerulus, is taken up by the reticuloendothelial cells of the liver, spleen, and bone marrow and degraded. When plasma haptoglobin is fully saturated in hemoglobin complexes, free plasma hemoglobin dissociates from its usual tetrameric globin structure to dimeric hemoglobin. Dimeric hemoglobin is filtered more easily by the glomerulus than tetrameric hemoglobin, and after filtration, hemoglobin is incorporated into proximal tubules through the megalin-cubulin receptor system present on the apical surface of these cells.6,8 Intracellular hemoglobin then dissociates into heme and globin. The increased intracellular levels of heme are potentially cytotoxic. Heme is degraded by heme oxygenase (HO), and the inducible isoform, HO-1, is upregulated rapidly in the kidney exposed to heme proteins. Such induction of HO-1 in the kidney has been described previously in case reports of patients with paroxysmal nocturnal hemoglobinuria2 and autoimmune hemolytic anemia.9 Induction of HO-1 is accompanied by increased ferritin synthesis, and this dual response allows the degradation of heme by HO and binding of iron by ferritin, thereby minimizing cellular exposure to heme and catalytically active “free” iron.
The acute and chronic nephrotoxicity of heme proteins and heme is well recognized.6,8 Heme proteins can cause acute kidney injury through 3 main mechanisms: decreased renal perfusion, direct cytotoxicity, and intratubular casts formed from the interaction of heme proteins with Tamm-Horsfall protein. Cytotoxic effects of large amounts of heme result from its lipophilic, oxidant, proinflammatory, and apoptotic effects. Studies in an experimental model of heme protein–induced kidney injury show that mitochondria are particularly vulnerable to heme-mediated damage.10 In these studies, mitochondria show markedly increased amounts of heme and impaired oxygen consumption; the exposure of normal mitochondria to such concentrations of heme impairs mitochondrial respiration, thereby incriminating heme as a mediator of such mitochondrial dysfunction in heme protein–induced kidney injury. 10 In this model, mitochondria also show evidence of ultrastructural injury, and some injured mitochondria are engulfed within autophagic vacuoles. Interestingly, electron microscopic studies of the kidney in this case report show dark intracellular vacuoles, some of which may represent autophagic vacuoles containing damaged organelles, such as mitochondria.10
Episodes of acute kidney injury, especially those that fail to completely resolve, predispose to CKD. Interestingly, after exposure to heme proteins, the kidney shows markedly increased expression of MCP-1 (monocyte chemoattractant protein 1) and transforming growth factor β1 isoform 1 (TGF-β1), cytokines that potently recruit monocytes/macrophages and provoke fibrosis, respectively.11 Moreover, induction of MCP-1 and TGF-β1 can occur in vitro when cells are exposed to heme or iron-catalyzed oxidative stress.11 Persistent or intermittent upregulation of MCP-1 and TGF-β1, incurred by exposure to heme proteins, thereby can contribute to tubulointerstitial disease.
Hemolysis associated with hemoglobinuria and hemosiderinuria in the context of cardiac valvular diseases or valvular prostheses has often been considered a well-tolerated benign condition. However, our patient clearly experienced a decrease in kidney function from hemosiderosis temporally linked to persistent hemolysis after mitral valve repair and banding. We postulate that our patient was more susceptible to tubular injury inflicted by heme proteins and heme because of existing tubulointerstitial disease for several reasons. CKD may impair the elicitation of adaptive mechanisms that protect the kidney against heme proteins. In addition, pre-existing upregulation of chemotactic and fibrogenic cytokines (such as MCP-1 and TGF-β1) in CKD may render the diseased kidney more vulnerable to the injurious effects of heme proteins and heme.
Patients with CKD typically show decreased renal blood flow, increased renal vascular resistive index, and microvascular drop out, which likely result in varying degrees of parenchymal hypoxia. Our patient had a long-standing history of hypertension, atherosclerotic vascular disease, and arterial nephrosclerosis with CKD. These background comorbid conditions likely rendered him susceptible to additional kidney insults. During the course of his progressive GFR loss, intravascular hemolysis associated with the mitral valve annuloplasty and banding was the only identifiable cause of renal tubular injury.
In conclusion, intravascular hemolysis caused by cardiac valvular disease or prosthesis in patients with pre-existing kidney dysfunction can lead to a further decrease in kidney function. Heightened awareness of its occurrence amidst a global CKD epidemic is clinically relevant. The risk of its occurrence should be both anticipated by the providers before the surgery and explained to patients in advance.
Acknowledgments
We acknowledge Mr Michael Moore for expertise and help with electron microscopy.
Support: None.
Footnotes
Financial Disclosure: The authors declare that they have no relevant financial interests.
References
- 1.Leonardi P, Ruol A. Renal hemosiderosis in the hemolytic anemias: diagnosis by means of needle biopsy. Blood. 1960;16:1029–1038. [PubMed] [Google Scholar]
- 2.Nath KA, Vercellotti GM, Grande JP, et al. Heme proteininduced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int. 2001;59(1):106–117. doi: 10.1046/j.1523-1755.2001.00471.x. [DOI] [PubMed] [Google Scholar]
- 3.Roberts WC, Morrow AG. Renal hemosiderosis in patients with prosthetic aortic valves. Circulation. 1966;33(3):390–398. doi: 10.1161/01.cir.33.3.390. [DOI] [PubMed] [Google Scholar]
- 4.Ackermann D, Vogt B, Gugger M, Marti HP. Renal haemosiderosis: an unusual presentation of acute renal failure in a patient following heart valve prosthesis. Nephrol Dial Transplant. 2004;19(10):2682–2683. doi: 10.1093/ndt/gfh429. [DOI] [PubMed] [Google Scholar]
- 5.Concepcion B, Korbet SM, Schwartz MM. Intravascular hemolysis and acute renal failure after mitral and aortic valve repair. Am J Kidney Dis. 2008;52(5):1010–1015. doi: 10.1053/j.ajkd.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 6.Nath KA, Murali NS. Myoglobinuric and hemoglobinuric acute kidney injury. In: Greenberg A, editor. Primer on Kidney Diseases. 5. Philadelphia, PA: Saunders Elsevier; 2009. pp. 298–304. [Google Scholar]
- 7.Rubinson RM, Morrow AG, Gebel P. Mechanical destruction of erythrocytes by incompetent aortic valvular prosthesis; clinical, hemodynamic, and hematologic findings. Am Heart J. 1966;71(2):179–186. doi: 10.1016/0002-8703(66)90180-3. [DOI] [PubMed] [Google Scholar]
- 8.Tracz MJ, Alam J, Nath KA. Physiology and patho-physiology of heme: implications for kidney disease. J Am Soc Nephrol. 2007;18(2):414–420. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- 9.Fervenza FC, Croatt AJ, Bittar CM, et al. Induction of heme oxygenase-1 and ferritin in the kidney in warm anti-body hemolytic anemia. Am J Kidney Dis. 2008;52(5):972–977. doi: 10.1053/j.ajkd.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nath KA, Grande JP, Croatt AJ, Likely S, Hebbel RP, Enright H. Intracellular targets in heme protein-induced renal injury. Kidney Int. 1998;53(1):100–111. doi: 10.1046/j.1523-1755.1998.00731.x. [DOI] [PubMed] [Google Scholar]
- 11.Nath KA, Croatt AJ, Haggard JJ, Grande JP. Renal response to repetitive exposure to heme proteins: chronic injury induced by an acute insult. Kidney Int. 2000;57(6):2423–2433. doi: 10.1046/j.1523-1755.2000.00101.x. [DOI] [PubMed] [Google Scholar]