

Abstract
Mild cognitive impairment (MCI) is rapidly becoming one of the most common clinical manifestations affecting the elderly. The pathologic and molecular substrate of people diagnosed with MCI is not well established. Since MCI is a human specific disorder and neither the clinical nor the neuropathological course appears to follows a direct linear path, it is imperative to characterize neuropathology changes in the brains of people who came to autopsy with a well-characterized clinical diagnosis of MCI. Herein, we discuss findings derived from clinical pathologic studies of autopsy cases with various subtypes of MCI antemortem. The heterogeneity of clinical MCI imparts significant challenges to any review of this subject. The pathologic substrate of MCI is equally complex and must take into account not only conventional plaque and tangle pathology but also a wide range of cellular biochemical and molecular deficits many of which relate to cognitive decline as well as compensatory responses to the progressive disease process. The multifaceted nature of the neuronal disconnection syndrome associated with MCI, suggests that there is no single event, which precipitates this prodromal stage of AD. In fact, it can be argued that neuronal degeneration initiated at different levels of the central nervous system drive cognitive decline as a final common pathway at this stage of the dementing disease process.
Keywords: Alzheimer’s disease, amyloid, cholinergic, dementia, MCI, neurofibrillary tangles, neuropathology, molecular, neurotrophins, synapses
Mild Cognitive Impairment
The development of the concept of mild cognitive impairment (MCI) was derived from memory clinics, which attracted milder cases of dementia than Alzheimer’s disease (AD) and epidemiological studies of elderly populations followed over time, in which subjects were evaluated annually for cognitive status. Memory clinic researchers noticed that many of the earlier, milder cases of cognitive loss in the elderly did not meet the “two cognitive domains impaired” criteria required for an NINDS/ADRDA diagnosis of AD established by McKhann and coworkers [98]. In the 1990’s such cases, were found most frequently but not always to be characterized by an amnestic disorder and the term “Mild Cognitive Impairment” or MCI, was popularized by Petersen [133] becoming the category in which these ‘single domain impaired’ individuals (who did NOT meet criteria for dementia/AD) were placed. The term MCI, however, was devoid of any pathological implications and was more descriptive than specific. Thus, while it emerged from the study of AD and the implication was that “all MCI was a precursor to AD”, increasing studies led to the identification of multiple types of MCI-some static (e.g., because of an injury, most commonly vascular), and some progressive, most commonly due to AD but possibly to vascular or other neurodegenerative disease processes, such as FrontoTemporal Dementia (FTD) [129].
While amnestic MCI was the most common form of MCI, especially in Memory Disorders Clinics, it was clear that MCI with other affected single cognitive domains were a small, but significant, component of this clinical presentation. While some presented with rare neurodegenerative disorders (e.g., dysexecutive or aphasic dysfunction as the sole cognitive problem in the MCI phase of FTD), some were vascular and many progress to AD. Moreover, the nature of MCI might differ by the nature of the research entity. Thus. Memory Disorders clinics report amnestic MCI (aMCI) as the most common form of MCI. In epidemiological populations, in which subjects have vascular disease, chronic medical diseases (e.g., congestive heart failure) and require medications for chronic illness, which may affect cognition, do not have aMCI as the predominant condition [90, 91]. However, people with a clinical diagnosis of MCI comprise a heterogeneous cohort of which, those who present solely with memory deficit are classified as single domain amnestic MCI (aMCI), while those who have a deficit in memory as well as other cognitive domains are categorized as multi-domain aMCI [76, 130]. Those with aMCI are at a higher risk of developing AD [76, 130]. Those with impairment in cognitive domains other than memory are designated as either single or multi-domain non-amnestic MCI (naMCI) [131, 188].
If the pathobiology of MCI is to be determined, autopsies of subjects with well-characterized MCI of different types are a necessity. Our goal, in this review, is to extend beyond the many excellent neuropathologic reviews of MCI [64, 137, 148]. Here, in addition to discussing the gross brain features of MCI, we will concentrate on pathomechanistic signature(s) of this clinical construct including biochemical and neuroanatomical alterations, synaptodegeneration, cell loss, neurotrophic failure, cellular genetics, neuronal selective vulnerability and other factors that occur in the MCI brain. Since, to date, there are no true animal models of MCI, we emphasize relevant research derived from clinical pathologic investigations of the human condition, the gold standard for the field. Understanding the complete neuropathological status of individuals with MCI will be essential for appropriate therapeutic interventions and realistic expectations for slowing or stopping the clinical decline.
Gross Morphologic features of MCI
Although there have been many advances in defining the clinical definition of MCI [5, 43, 168], distinguishing brains at autopsy from people who died with a clinical diagnosis of NCI and MCI is challenging. For example, the cortical gyral and sulcal patterns appear similar between NCI (Fig. 1a) and nonamnestic MCI (naMCI) (Fig. 1b) brains. However, we have observed a widening of sulci, such as the ventral ramus of the lateral fissure as well as a blunting of the anterior tip of the temporal pole in aMCI (Fig. 1c) and mild AD (Fig. 1d) compared to the NCI or naMCI brain. These gross morphological changes are magnified and extended to other cortical regions in late stage AD (Fig. 1e). Brains harvested from those who came to autopsy with a clinical diagnosis of MCI are providing the material needed to unravel the structural and cellular pathobiologic substrate of this prodromal stage of the dementia.
Fig. 1.
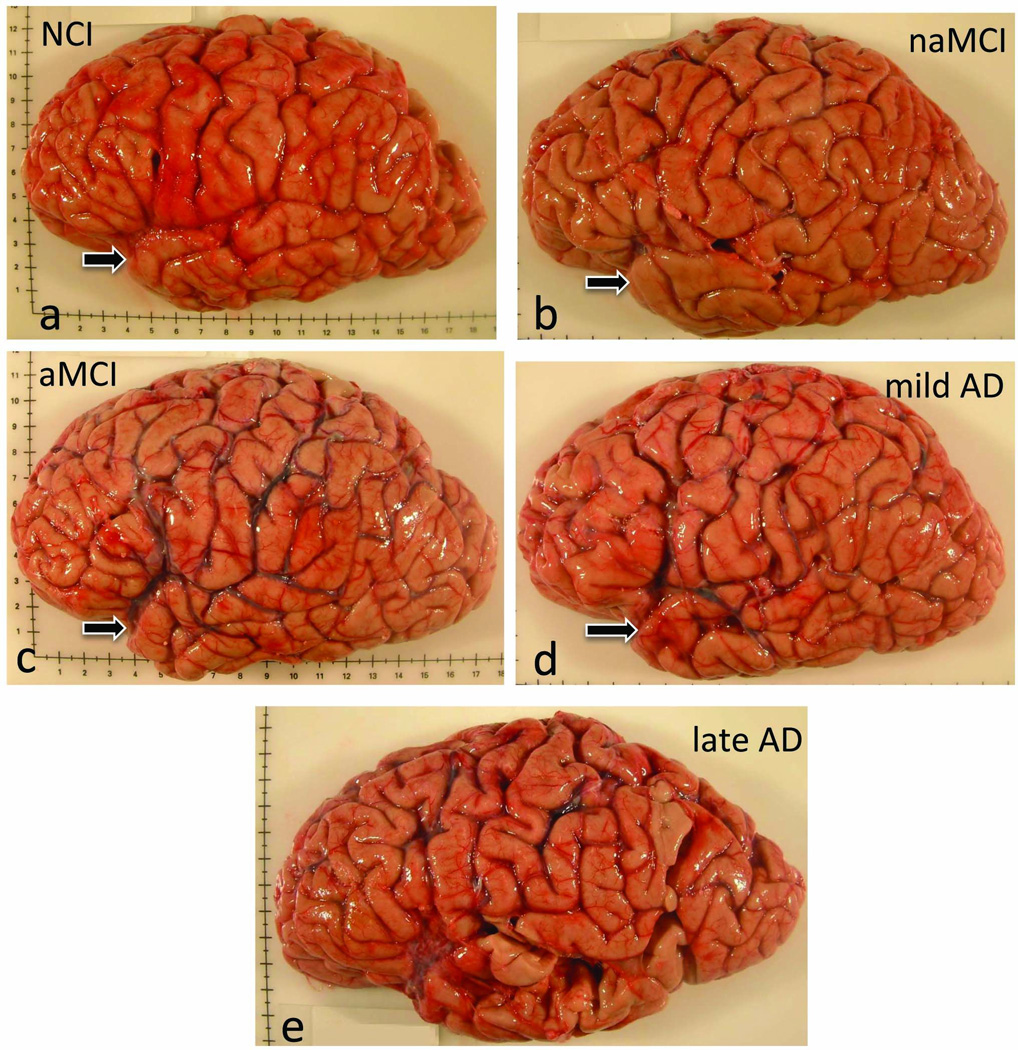
Gross anatomy showing similarities and differences between the brain of a person who dies with a clinical diagnosis of (a) no cognitive impairment (NCI), (b) non amnestic mild cognitive impairment (naMCI), (c) amnestic MCI (aMCI), (d) mild and (e) severe Alzheimer’s disease
Amyloid Pathology in MCI
Brain amyloid beta (Aβ) plaques, are a hallmark lesion of people with a clinical diagnosis of MCI. The distribution of Aβ deposits changes with time and reflects the spread of Aβ deposition in the diseased brain [181]. Senile plaques first appear as diffuse and “fleecy” [180] plaques throughout the neocortex and extend hierarchically into other brain regions. In a second stage, Aβ plaques occur in allocortical areas (e.g. entorhinal cortex and subiculum/CA1 region). In the third phase, plaques appear in the basal ganglia, thalamus, and hypothalamus. In the fourth phase, amyloid reaches the midbrain and medulla oblongata. Finally, in a fifth stage, senile plaques appear in the pons and cerebellum. Later stages feature neuritic (Fig. 2a) and amyloid cored (Fig. 2b) plaques. Whether cored plaques evolve from dying neurons and their role in the onset of dementia remain unclear. Furthermore, whether amyloid plaque pathology defines the substrate underlying MCI is still unanswered. The distribution of amyloid deposits in MCI appears to be intermediate between the changes seen in the NCI and AD brain [93]. Many aged control and MCI brains exhibit a similar degree of Aβ deposition as shown in postmortem brain tissue [137] as well as in clinical imaging studies [4], limiting the use of these lesions as a true pathologic marker for the distinction between normal aging and MCI. Early clinical pathological studies revealed that people with clinical dementia rating (CDR) scores of 0.5 (questionable dementia/MCI) displayed significant increases in the density of diffuse plaques (DPs) in the temporal cortex [105]. The density of plaques increased with increasing dementia and the proportion of plaques shifted from diffuse to neuritic. By contrast, a recent study using samples from the University of Kentucky Alzheimer’s Disease Research Center (UKADRC) revealed that the brains of people without cognitive impairment and with a Braak score of II showed no differences in the number of DPs in the neocortex or limbic medial temporal lobe (MTL) structures compared to MCI cases [94] as defined by the CDR criteria [105, 131]. Neuritic plaque (NP) counts were significantly greater in the medial frontal cortex and amygdala in MCI compared to normal controls, whereas no differences were found in the number of NPs within the hippocampal CA1 region, subiculum or entorhinal cortex (ERC). Numbers of DPs were unchanged in most cortical regions between controls and MCI, whereas NP number was significantly increased in the above limbic cortical regions when MCI and early AD were compared suggesting that NPs mark this transition. A clinical pathologic study using a mixed cohort of MCI (aMCI and mdMCI) cases from the Rush Religious Orders Study (RROS), a clinical pathologic longitudinal study of aging and dementia [15, 109] found that ERC Aβ-amyloid peptide load in MCI cases appeared intermediate between controls and early AD, but the difference was not statistically significant [109] lending support to the suggestion that β-amyloid deposition is not the major pathological factor defining the transition to MCI [93]. Moreover, age-adjusted group comparisons of cortical insoluble Aβ40 and Aβ42 levels correlated with neuropathologic AD status (CERAD and Braak staging) but were not predictive of a clinical diagnosis of MCI [46]. In the precuneus region of the medial parietal cortex [Brodmann area 7, (BA7)], soluble Aβ42 concentration and [3H] PiB binding to insoluble Aβ aggregates were elevated in MCI cases; both measures correlated with lower Mini-Mental State Exam (MMSE) scores [71]. Although MCI and early AD are distinguishable biochemically from aged controls in terms of the Aβ42/Aβ40 ratio (both soluble and insoluble) [183], removal of amyloid plaques following Aβ vaccination therapy failed to prevent further cognitive decline in people with a clinical diagnosis of early AD [67]. Interestingly, decreased levels of Aβ1–42 and increased phospho-tau levels detected in cerebral spinal fluid may provide a biomarker for those destined to develop dementia [107, 173], but this remains to be more fully validated. Therefore, solely targeting this protein for the treatment of dementia may not be entirely feasible, and anti-amyloid therapies may need to be initiated prior to dementia onset. In light of the recent finding that Aβ normally functions as an antimicrobial peptide in the innate immune system in response to clinically relevant pathogenic micro-organisms, its removal may result in increased vulnerability to infection [167] and continued brain dysfunction. Perhaps the field requires a major paradigm shift to go beyond the problems of Aβ protein cleavage, processing and aggregation [78, 142] in MCI.
Fig. 2.
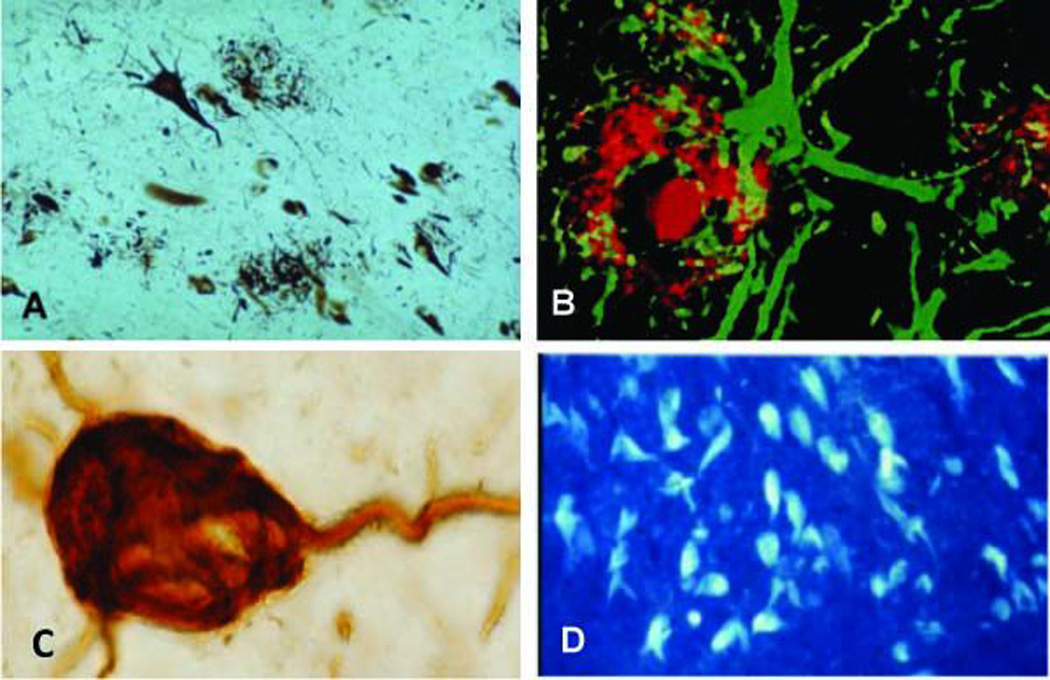
(a) Neuritic plaques (arrows) and neurofibrillary tangle (star) visualized by the Bielschowsky silver method in AD. (b) Fluorescent image of a cored amyloid beta immunostained plaque (red) surrounded by a GFAP positive glia cell (green) in the AD frontal cortex. (c) NFT containing the tau epitope Alz-50 in the frontal cortical in the AD frontal cortex. (d) Thioflavin-S positive staining of NFTS revealing the beta fibular configuration within entorhinal cortex layer 2 stellate neurons in AD
Recent studies suggest that Aβ monomers, dimers and higher order oligomeric forms may be an early and toxic form of amyloid [83]. In this regard, Aβ oligomers were found to accumulate in the frontal cortex of people with a clinical diagnosis of MCI (CDR= 0.5) and mild to moderate AD (CDR=1 or 2) compared to age matched controls (CDR=0) from the Alzheimer’s Disease Research Center at the University of California, San Diego [134]. Increased Aβ oligomer levels correlated with the severity of cognitive impairment (MMSE and Blessed Information Memory Concentration score), AD neurodegeneration (Braak staging), and lower levels of the presynaptic vesicle protein synaptobrevin and the postsynaptic density protein PSD-95 [134]. Perhaps Aβ monomers and oligomers underlie amyloid toxicity, causing a disruption of synaptic function in MCI, which initiates cognitive decline.
During the past several years, genomic analysis has revealed genetic risk factors not only for the development of dementia but which affect amyloid deposition during the early phase of the disease. The seminal findings of Roses and colleagues revealed that apolipoprotein E (APOE) e4 carriers are at increased risk for AD [32]. Imaging studies of presymptomatic e4 carriers have shown reduced cerebral glucose metabolism [162], increased cerebral amyloid deposition [139] and enhanced cortical atrophy [31] as well as accelerated memory decline [26]. Although these changes are milder, they qualitatively resemble those seen in AD suggesting that they may represent a very early stage that precedes the symptomatic expression of MCI. In fact, the total amyloid load in AD cases is higher in APOE e4 carriers than non-carriers [42]. Two studies, one examined a large Finnish cohort [85] and another a population from the Sun Health Brain Donation program [27], found that APOE e4 carriers without premortem diagnosis of dementia, including those with an MCI diagnosis at death displayed higher amyloid (but not NFT) burdens than non-carriers. PiB imaging of amyloid deposition seems to be predictive of decline in MCI cases, but not that predictive in pre-MCI cases [45, 166].
PiB-PET imaging data derived from four cortical regions known to have high amyloid deposition were used as a quantitative phenotype to identify genes related to fibrillar amyloid burden in 103 Alzheimer’s Disease Neuroimaging Initiative subjects [174]. Of the various genes examined only the intronic single nucleotide polymorphisms (SNP) of the DHCR24 gene was significantly associated with a lower average PiB uptake. The DHCR24 gene appears to play a neuroprotective role and has been shown to confer resistance to Aβ and oxidative stress induced apoptosis by inhibiting caspase-3 activation [55]. Another SNP located in the amyloid-associated LR11 gene (SORL1) is also associated with an increased risk for developing AD [16, 86], although this association is controversial [102]. LR11 (also termed sorLA) is a multi-functional neuronal receptor that binds apolipoprotein E (ApoE), plays a crucial regulatory role in the processing of the amyloid precursor protein (APP) and may maintain low levels of the pathological form of the Aβ peptide [6, 7, 41, 123]. A quantitative immunohistochemical report found that neuronal LR11 levels in MCI were intermediate between NCI and AD in superior frontal cortex (SFC) tissue obtained from the RROS [147]. MCI subjects with low LR11 levels displayed significant cognitive impairment compared to high LR11 level cases, suggesting that the loss of LR11 is an early alteration in the cascade of events underlying the development of MCI by regulating APP processing.
Neurofibrillary Tangle Pathology in MCI
NFTs are composed of aggregates of hyperphosphorylated forms of the protein tau [17] (Fig. 2c), display a beta pleated sheet conformation (Fig. 2d) and are argyrophilic (Fig. 2a). Recent biochemical reports suggest that NFT development progresses according to a linear sequence of molecular and conformational changes in the tau molecule before and during NFT formation and maturation [18, 47]. The Braaks [21] described the pathologic topographic spread of NFTs from the MTL to the neocortex, which composed six stages according to the location of the tangle-bearing neurons and the severity of changes (transentorhinal stages I–II: clinically silent cases; limbic stages III–IV: incipient AD; neocortical stages V–VI: fully developed AD). Clinical subtypes of MCI as well as many NCI cases have variable Braak staging scores ranging from a complete absence of NFTs (Stage 0) to stages V/VI representing AD [109, 112, 114, 132, 137]. Although these observations indicate that there is no clear demarcation between some control, MCI and AD brains using the Braak staging criteria, a study suggests that Braak scores are better at identifying aMCI from naMCI [138].
A broader evaluation of the distribution of NFTs indicate that they are significantly increased in the amygdala, entorhinal cortex (ERC), subiculum and the inferior parietal cortex (IPC) in MCI compared to controls suggesting that NFTs are critical for the transition to MCI [21, 58, 94, 118]. Alterations in the phosphorylation or conformation state of tau are related to MCI [64]. Clinical pathologic investigations using tissue from the RROS have shown a significant increase in tau positive NFTs and neuropil treads in the ERC and perirhinal cortex between MCI and NCI cases [103] coincident with a significant reduction in the number and atrophy of ERC layer 2 stellate neurons [87], which display prominent NFTs [69]. An examination of brains from two large autopsy populations, the Nun Study [140, 164, 165] and the National Alzheimer’s Coordinating Center (NACC) registry [13] failed to document an example of a truly end-stage NFT pathology coexisting with intact cognition [2]. These findings suggest a continuum of tau induced NFT pathology underlying the transition between normal aging, MCI and AD. A parsimonious corollary to the formation of NFTs within neurons, suggest that areas containing these lesions should exhibit significant neuronal loss in the MCI brain.
Neuronal loss in MCI
Numerous studies have examined changes in neuron numbers in MCI, focusing on structures in the MTL. Within MTL, the ERC, a major paralimbic cortical multimodal sensory relay region to the hippocampus undergoes cell loss in MCI [21, 40, 54, 69, 87]. Cases from the Washington University cohort showed no significant age related decrease in numbers of Nissl-stained ERC neurons or volume in healthy non-demented individuals [136]. Few or no differences were observed between healthy controls and what was termed “preclinical AD” or cases with normal cognition (CDR 0) despite plaques and NFTs at autopsy [136]. However, neuronal numbers were significantly decreased in the ERC and hippocampal CA1 subfield in very mild AD (CDR 0/0.5 or CDR 0.5) suggesting that neuronal atrophy and death has already occurred at a time when patients begin to manifest clinical symptoms [54]. Unbiased quantitative stereology revealed a significant loss of NeuN-immunoreactive ERC lamina II neurons and volume in MCI cases harvested from the RROS [87] in agreement with previous seminal findings [54, 136]. Furthermore, ERC atrophy correlated with impairment on MMSE and clinical tests of declarative memory [87, 106], indicating that ERC neurodegeneration impacts the progression of MCI. A likely consequence to neuronal loss is that it should lead to a decrease in the number of synapses.
Synaptodegeneration in the hippocampus in MCI
The hippocampus, an integral component of a trisynaptic memory circuit, displayed a significant decline in the plasticity related post-synaptic density protein 95 (PSD 95) in a small aMCI cohort compared to subjects without dementia [172]. Quantitative immunoblotting found that synaptophysin, a key player in membrane trafficking events which precede exocytosis, as well as synaptotagmin, a Ca2+ microsensor that modulates activity-dependent exocytosis [172], were relatively preserved in aMCI cases from the RROS [33]. On the other hand, drebrin, an f-actin postsynaptic binding protein [66] involved in synaptic plasticity [66], was decreased in the hippocampus of MCI cases and correlated with cognitive decline [65]. Taken together, these findings suggest that the hippocampus displays decreased synaptic plasticity in MCI. Whether synaptic protein changes in hippocampus are reflected in changes to synapse number has great relevance to the level of cognitive decline seen in MCI.
Studies aimed at defining synapse loss in the hippocampus have been performed using tissue harvested at the UKADRC [158] or RROS [15] and processed for unbiased stereological sampling coupled with transmission electron microscopy. These investigations revealed no significant differences in synapse number in the outer molecular layer of the dentate gyrus between cognitively intact subjects and aMCI, whereas a milder AD group showed a significant loss of synapses compared to both NCI and aMCI [154]. A subsequent investigation using the same cases showed synaptic changes in the stratum radiatum region of the hippocampal CA1 subfield [153]. Unlike the outer molecular layer of the dentate gyrus, estimation of total CA1 synaptic numbers was more pronounced in aMCI. Importantly, synaptic counts strongly correlated with antemortem cognitive scores. It would be anticipated that synaptic defects are reflected in a change in the gross structure of these limbic regions.
Structural changes in the MTL in MCI
Support for the involvement of the ERC and hippocampus early in the disease process also comes from structural MRI investigations, which demonstrate atrophy of these regions in aMCI compared to cognitively healthy elderly controls [81, 170, 176]. Recent, voxel based morphometry studies demonstrated a significant decrease in the volume of the parahippocampal white matter a region that includes the perforant pathway projections to the hippocampus in aMCI compared to healthy control subjects [171]. Furthermore, the rate of MTL atrophy seems to be related to the extent of declarative memory dysfunction [171]. These in vivo results are in line with postmortem tissue investigations suggesting that AD-related pathology affects the entorhinal region before the hippocampus [169, 176] and that white matter volume change reflects not only loss of afferent and efferent fibers in the region of the parahippocampal gyrus, but may also be due to partial demyelination in remaining fibers in MCI [77, 143].
Synaptodegeneration in the cortex in MCI
Although neocortical synapse loss is a prominent feature of AD [97, 151], very few studies have reported changes in the MCI neocortex. Evaluation of cortical tissue from individuals with CDR scores of 0.5–1.0 containing probable MCI and early AD cases compared to aged non-demented controls revealed a 25% loss in synaptophysin immunostaining in the frontal cortex but no change in synaptotagmin and GAP-43 [95]. Quantitative protein immunoblotting using tissue from clinically diagnosed MCI and either mild/moderate or severe AD from the RROS revealed decreased levels of the presynaptic vesicle marker, synaptophysin selectively in the superior temporal cortex (STC) and inferior parietal cortex (IPC) in AD compared to NCI supporting findings in mild or severe AD [68, 96, 160, 179]. Synaptophysin protein levels in the STC and IPC correlated with MMSE scores, suggesting that the loss of this presynaptic protein within select neocortical areas mark a clinical progression of the disease. However, levels of the predominantly presynaptic protein alpha-synuclein (α-syn), which is involved in neuronal plasticity, was significantly reduced in the SFC only in AD compared to MCI and NCI [186]. In contrast, cortical presynaptic synaptotagmin levels remained stable from NCI to MCI to AD independent of region with no association with cognition. Taken together, these findings suggest that select differential alterations of presynaptic proteins contribute to early perturbations in synaptic homeostasis in MCI cortex.
With respect to postsynaptic cortical proteins, studies using tissue from the RROS revealed that levels of drebrin were significantly reduced in the STC in MCI compared to controls. On the other hand, SFC drebrin levels were increased in MCI compared to NCI and AD. Drebrin is localized to postsynaptic dendritic spines at excitatory synapses [8, 66, 161], where it may regulate spine morphogenesis and postsynaptic densities [66, 175]. Since drebrin levels in the anterior cingulate, STC and visual cortex correlated with cognitive impairment, the progression of dementia may, in part, be related to a loss of neocortical excitatory postsynaptic contacts in the cortex similar to the hippocampus.
A recent ultrastructural study demonstrated a significant loss of synaptic contacts from NCI to aMCI in the inferior temporal cortex (ITC, BA 20) in samples from UKADRC and RROS (Fig. 3) [155]. There was a strong association between the number of synapses in the ITC and the individual performance on both the MMSE and tests of verbal fluency [155]. Immediately adjacent tissue showed a significant loss of different pre and post-synaptic proteins including synaptophysin and PSD-95 in aMCI, which correlated with MMSE score, supporting a role for the ITC in the early stages of disease progression [155]. Despite MTL synaptodegeneration, many other cellular processes are likely to play important roles in MCI-associated neuronal dysfucntion.
Fig. 3.
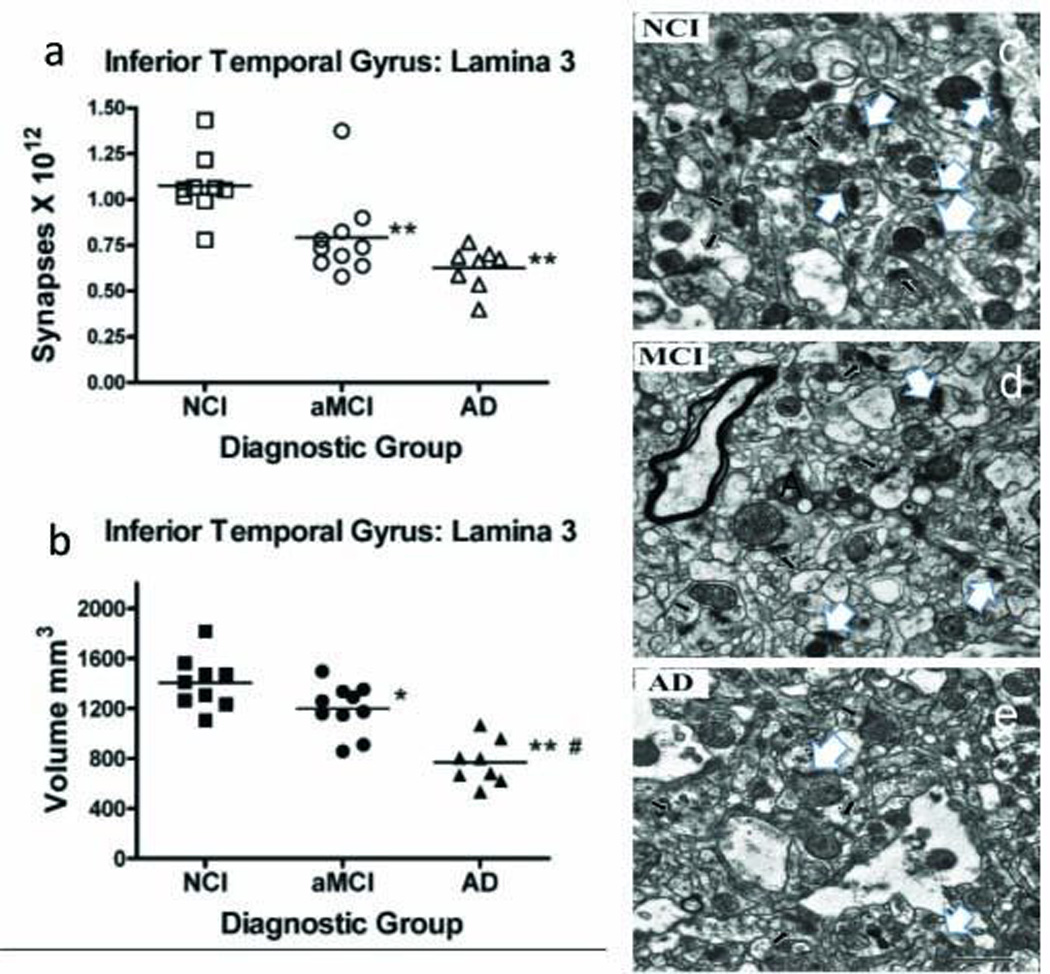
Graphs showing the estimate of total synapse number in layer 3 of the inferior temporal gyrus (ITG) across clinical diagnostic group using unbiased stereology coupled with electron microscopy. (a) Total volume of lamina 3 of the inferior temporal gyrus was estimated with the Cavalieri method from tissue sections immediately adjacent to regions used for synaptic counts (b). Single points represent individual subjects. Horizontal lines indicates group median. *p < 0.05; **p < 0.005 compared to NCI; #p < 0.005 compared to MCI. Electron micrographs of ITG layer 3 showing synaptic complexes (white arrows) in tissue from (c) no cognitive impairment (NCI), (d) amnestic mild cognitive impairment (aMCI) and (e) AD cases. In all tissue, the synaptic complexes appeared normal with synaptic vesicles observed in the presynaptic component and the synaptic density observed in the postsynaptic component. Scale bar = 1.0 mm Adapted with permission from [152]
Cholinotrophic basal forebrain systems in MCI
The MTL receives input from various neurotransmitter systems including cholinergic innervation from the basal forebrain (Fig. 4a, b) [100]. Biochemical [37] and histologic [187] investigations revealed a reduction in cortical cholinergic activity and loss of cholinergic basal forebrain (CBF) neurons (Fig. 4c), which led to the cholinergic hypothesis of AD [11]. Sustaining the function of the impaired central cholinergic system is the primary mechanism of action for currently effective FDA-approved drugs for AD [110]. This emphasizes the importance of defining the status of the cholinergic system in subjects with MCI as well as cognitively normal elderly people who are at risk for developing AD. However, whether or not cholinergic system degeneration is an early or late feature of AD is still an open question. In MCI, CBF neurons also contain early and late tau conformational markers of NFTs [99, 185]. Furthermore, cell expression profiling studies indicated that the ratios of 3-repeat tau (3Rtau)/4-repeat tau (4Rtau) are significantly decreased due to a reduction in expression of 3Rtau rather than an increase in 4Rtau tau in CBF neurons in both MCI and AD [52]. Such a shift in ratio may lead to an increased propensity of tau to form toxic aggregates; this is suggested by in vitro studies indicating that 4Rtau is more prone to aggregate than is 3Rtau [82]. Hence, the CBF degenerative process(es) appear to be initiated early in the disease. However, quantitative stereological studies revealed that the number of CBF nucleus basalis (NB) perikarya expressing either choline acetyltransferase (ChAT, the synthetic enzyme for acetylcholine) or the vesicular acetylcholine transporter was stable in MCI in cases from the RROS (Fig. 4d) suggesting that frank NB cell loss is a later stage event despite the early appearance of NFTs [50]. Furthermore, George and colleagues [48] found that the volume of the substantia innominata (SI), which contains the cholinergic neurons of the NB, was significantly reduced in participants from the Rush Alzheimer’s Disease Center clinic and RROS diagnosed with AD, as reported previously [61–63, 150]; however, the NCI and aMCI groups did not differ from each other. The lack of change in SI volume in individuals with MCI is consistent with postmortem tissue investigations showing the preservation of SI cholinergic neuron number early in the disease [50]. However, others found significant MRI-derived atrophy of the SI in patients with MCI [116]. These apparent differences may be due to the region of the SI examined or methodologies used by different investigators. Whether deficits in forebrain cholinergic projection neurons are mimicked in their cortical and hippocampal projection sites is an active area of investigation.
Fig. 4.

(a) Coronal brain slab showing the location of cholinergic neurons within the anterior nucleus basalis subfield (arrow). (b) Artist’s drawing showing the cholinergic innervation from the medial septal/vertical limb of the diagonal band (MS/VDB; Ch1-2) to the hippocampus (red) and the nucleus basalis of Meynert (NBM) to the cortex and amygdala (blue). Above the schematic are images showing nucleus basils neurons containing choline acetyltransferase (ChAT), nerve growth factor (NGF), trkA and p75NTR. (c) Low power image showing the extensive loss of cholinergic neurons in the nucleus basalis in AD, higher magnification of the cholinergic cell loss (d) and NFT containing cholinergic neurons revealed by thioflavin-S histochemistry (yellow) (e). (f) Composite histogram showing phenotypic differences in the number of ChAT, vesicular acetylcholine transporter (VAChT), trkA and p75NTR-immunopositive neurons in MCI and AD individuals. Note a significant reduction in NGF receptor and not ChAT containing neurons within the NB of people with MCI and AD. This difference is not acerbated during the transition from MCI to early AD. (g) All NB neurons were dual immunostained for ChAT (pink) and p75NTR (blue) in aged non-demented controls, which appeared as purple. (h) In contrast, in MCI (i, pink neurons) and AD (J; open arrows) many more ChAT-only immune reactive neurons compared to dual stained neurons (thin arrow in j)
Biochemical analyses of ChAT [39] and acetyltransferase (AChE) [38] activities in neocortical regions from cases from the Jewish Home and Hospital in New York or the RROS, found no changes in MCI while significant deficits were detected in severely demented AD cases. By contrast, ChAT activity is increased in the MCI superior frontal [39] but reduced in the primary visual cortex, an area relatively spared in late stage [49] and mild-moderate AD cases [72]. Interestingly, the visual cortex displays significant loss of cholinergic fibers even in the absence of frank synaptic pathology [12]. The precuneus is another cortical area found to display a significant cholinergic deficit in AD cases from the RROS [71]. Collectively these studies demonstrate that early stages of AD dementia are associated with cholinergic deficits, however, these changes are detectable only in selected brain regions.
Of great interest are findings showing elevated ChAT activity in the hippocampus (Fig. 6a) and superior frontal cortex (SFC) in MCI subjects from the RROS [39, 73]. It appears that select hippocampal and cortical cholinergic projection systems are capable of compensatory responses during prodromal dementia. Increased hippocampal and SFC ChAT activity in MCI may be important in promoting biochemical stability, or compensating for neurodegenerative defects, which may delay the transition of these subjects to AD. Interestingly, hippocampal ChAT activity was increased selectively in MCI cases scored as a Braak III/IV stage, suggesting that upregulation of ChAT occurs, at least in part, due to the disconnection of glutamatergic ERC input to the hippocampus early in the disease process (Fig. 6b). The source of these compensatory responses may be the cholinergic neurons, which preserve their cholinergic phenotype in MCI [50] (Fig. 4d) resulting in a biochemical upregulation of ChAT protein or enzyme activity, which accommodates for reduced regional cholinergic fibers and axon varicosities [71]. The biochemical cortical cholinergic upregulation in MCI [39] is not paralleled by a structural plasticity of cortical cholinergic fibers [70], suggesting increased SFC ChAT activity precedes the loss of cholinergic axonal input to this brain region during the clinical progression of AD. Further evidence of cortical plasticity is reflected by a paradoxical increase in the number of glutamatergic synapses in the MCI midfrontal gyrus versus NCI in cases from the RROS [14].
Fig. 6.

(a) ChAT activity increased in the hippocampus in MCI and returned to control levels in mild AD. (b) Schematic drawing illustration the loss of innervation to the hippocampus arising from the glutamatergic layer II entorhinal cortex neurons (red) triggering a cholinergic plasticity response (blue) which likely originates from the septal cholinergic projection neurons into the denervated glutamatergic sites in the hippocampus in MCI
The relationship between changes in cholinergic activity and regions of the brain, which display high levels of amyloid burden defined by PiB-PET binding in MCI has not been examined extensively. Although both frontal and precuneus cortex develop amyloid pathology early in the disease process [84, 182], ChAT activity is only upregulated in the former [39] and stable in the latter cortical region in MCI [71]. This disparity may reflect the differential ability of these two regions to launch a compensatory response. While the progression of amyloid pathology does not appear to affect regional cholinergic enzyme activity in the frontal cortex [39], there is a strong association between reduced ChAT activity levels and increased Aβ load in the precuneus as well as an association with impaired cognitive performance on the MMSE [71]. An analysis of frontal and parietal cortex demonstrated a reduction in ChAT activity and the coupling of M1 muscarinic acetylcholine receptor (mAChR) to G-proteins in non-demented elderly subjects with plaques compared to those without plaques using tissue from the Banner Institute Brain Bank, Phoenix, AZ, suggesting that cortical cholinergic dysregulation is initiated at the preclinical stage in parallel with increased amyloid plaque deposition [135]. Since anticholinesterase drugs are widely prescribed for early AD, it is imperative to determine the extent to which acetycholinesterase (AChE), the cholinergic degrading enzyme, is affected in MCI. PET imaging using [11C] MP4A or [11C]PMP, has been used to measure alterations in acetycholinesterase (AChE) activity in vivo. These studies revealed a reduction in the hippocampus and cortex with the most pronounced reduction in temporal polar region in MCI (CDR 0.5) [59] and early AD [141] (Fig. 5). A parsimonious event that most likely occurs in the cholinergic system would be impairments to cholinergic receptors in MCI.
Fig. 5.
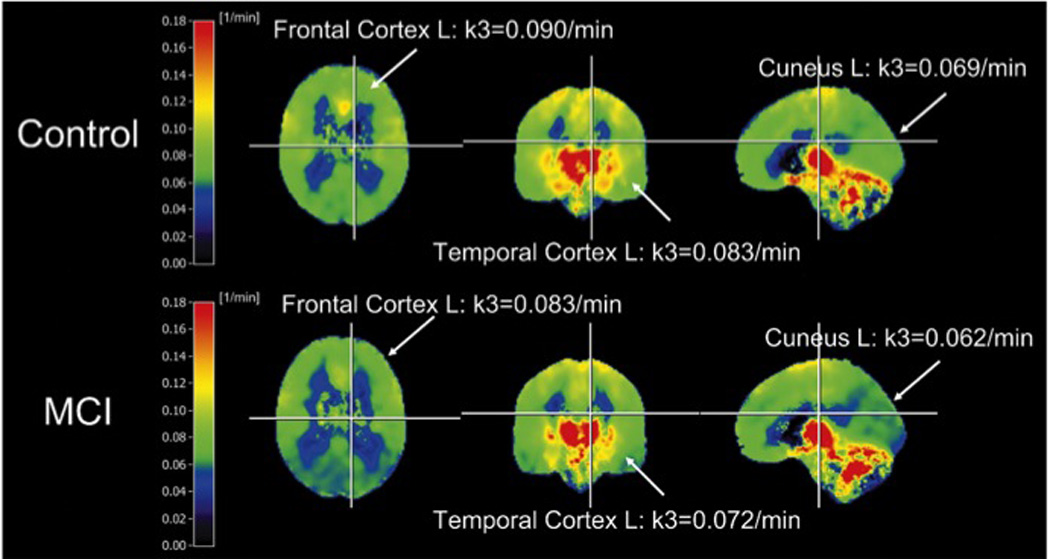
Mean parametric images of [11C] MP4A hydrolysis rates [1/minute] used to measure acetycholinesterase activity in controls and mild cognitive impairment (MCI) patients (orthogonal slices, L= left hemisphere, R = right hemisphere). Reproduced with permission from [59]
Acetylcholine receptors (nAChR) in MCI
Currently there is no clear consensus on whether presynaptic or post-synaptic (or both) receptor components of cholinergic neurotransmission are defective in MCI. The nicotinic acetylcholine receptor (nAChR) system is altered in AD [122], however, these receptors have not been studied extensively in MCI. Using an in vitro binding assay with [H-3] methyllycaconitine (MLA, a potent ligand for α-bungarotoxin sensitive nAChR), α7 receptor binding was examined in the superior frontal cortex of RROS cases and found to be unchanged in MCI and mild/moderate AD compared to NCI subjects [74]. Interestingly, this study indicated an association between increased α7 nAChR binding and cortical neuritic plaques. This finding is in agreement with a previous observation that α-bungarotoxin binding correlates positively with Aβ plaque density in AD brains [128], and may reflect up regulation of α7 nAChR mRNA in cortical-projecting cholinergic neurons in early AD [34]. These results support a role for α7 nAChR in the development of cortical Aβ pathology [117, 177] and potential functional interaction between α7nAChR and Aβ [89]. The role of α4β2 nAChRs in MCI and AD is somewhat more controversial. A postmortem study of nAChRs, using samples from the Sun Health Research Institute Brain Donation Program, found that [3H](+/−) epibatidine binding was stable in the midfrontal cortex suggesting the preservation of cortical α4β2 nAChRs [146]. Likewise, a PET study using 2-[(18)F]fluoro-3-(2(S)-azetidinylmethoxy) pyridine (2-[(18)F]FA-85380) on 15 mild AD (CDR<2) and healthy control subjects found no evidence of α4β2 reduction in early AD [44]. In contrast, another (18)FA-85380 PET study reported that MCI (CDR=0.5) and mild-moderate AD patients had significant reductions in cortical and hippocampal α4β2 nAChR binding; interestingly, only those MCI who converted later to AD had deficient binding [80]. In this regard, α4β2 nAChRs were also examined using 123I-5-IA-85380 SPECT confirming reductions of these receptors in temporal lobe [178] or their preservation in the MCI brain [104]. Therefore, it appears that reductions in the predominant brain nAChR subtypes are altered early during disease progression and likely impact cholinergic function.
It is generally accepted that M1 mAChR densities are unchanged in AD; however, details about their role in disease progression are limited [184]. [(35)S]GTPgammaS binding/immunocapture assay revealed that M1 mAChR function in MCI frontal (BA 10) cortex samples from the RROS were unchanged, but increased significantly in AD relative to controls [124]. Another study measured postsynaptic M1 mAChR coupling, using displacement of 3H-pirenzepine binding by oxotremorine-M in the presence and absence of GppNHp, and ChAT activity in frontal and parietal cortices from cognitively normal (preclinical aged subjects with either, the presence, or absence of amyloid plaques at postmortem examination) and clinical AD cases [135]. Both cholinergic markers were reduced in preclinical cases with amyloid burden, and this reduction was accelerated further in clinical AD. Therefore during AD pathogenesis, cholinergic deficits may emerge early, together with the development of amyloid plaques and prior to the onset of clinical symptoms. The mechanisms driving degeneration of the cholinotrophic system in MCI have recently received a great deal of attention.
Neurotrophic abnormalities in MCI
A central concept underlying the survival of CBF neurons is their dependence upon the neurotrophic substance nerve growth factor (NGF), which is synthesized from its precursor (proNGF) molecule and interacts with the cognate NGF receptor (TrkA) principally for pro-survival and the pan-neurotrophin receptor (p75NTR) principally for pro-cell death functions [110, 111]. Examination of cases from the RROS revealed that the number of NB perikarya expressing either the TrkA or the pan-neurotrophin receptor p75NTR was reduced ~50% in MCI and mild AD compared to NCI (Fig. 4d) and this deficit correlated with impaired performance on the MMSE and tests of working memory and attention [113, 114]. Many cholinergic NB neurons appear to undergo a phenotypic silencing of NGF receptor expression (Fig. 4e–g) in the absence of frank neuronal loss [149] during the early stages of cognitive decline. Moreover, TrkA gene expression, but not p75NTR, was reduced in NB neurons in MCI and AD (Fig. 7a,c) [53]. These alterations may signify an early deficit in neurotrophic support during the progression to MCI. In MCI, NB neurons also contain early and late tau markers of NFTs [99, 185].
Fig. 7.

(a) Pre and post microaspirated cholinergic nucleus basalis (NB) and (b) neurofilament (NF)-immunoreactive hippocampal CA1 neurons employed for microarray analysis following terminal continuation (TC) RNA amplification. Scale bar: 50 µm. (c) Color-coded heatmap illustrating no significant differences in ChAT, p75NTR and GAPDH gene expression compared to a significant down regulation (asterisk) of trkA, trkB, and trkC in MCI and AD. Expressed sequence-tagged cDNAs (ESTs) identifying extracellular domains (ECD) and tyrosine kinase (TK) domains display down regulation in MCI and AD. The decrement of trk gene expression in MCI is intermediate relative to AD. (d) Color-coded heatmap illustrating a significant up regulation (asterisk) for endosomal and trafficking compartments rab GTPases rab4, rab5, rab7, and rab24 in MCI and AD compared to a down regulation (double asterisk) for the synaptic marker rab3 and the BDNF receptor TrkB within NF-immunoreactive CA1 neurons
Although mature NGF (mNGF) levels are preserved in neocortex CBF neuron projections sites in MCI [112], proNGF is elevated relative to non-demented controls in MCI [127] and TrkA levels increase in mild AD, whereas p75NTR levels are stable in tissue harvested from the RROS [35]. Recent studies suggest that the putative pro-apoptotic effect(s) of p75NTR-mediated proNGF signaling is dependent on interactions between p75NTR and the neurotensin receptor sortilin, a Vps10p domain trafficking protein that acts as a cell surface co-receptor with p75NTR to mediate proNGF-induced cell death [92, 144]. Evaluation of cortical sortilin protein levels revealed no changes between NCI and MCI cases from the RROS [115]. These alterations in cholinotrophic activity may favor a shift in the pro-apoptotic proNGF:p75NTR:sortilin trimeric ratio during the earliest stages of AD (Fig. 8). Increased cortical proNGF levels in MCI also suggest potentially pathogenic alterations in metabolic pathways mediating the maturation and degradation of mNGF [23]. In MCI cases from the RROS, there was up-regulation of MMP-9 protein levels and activity, that correlated inversely with cognitive status [24] suggesting yet another defect in the NGF biosynthetic pathway in MCI (Fig. 8), which may contribute to the selective vulnerability of CBF neurons in MCI. However, neurotrophic dysfunction is not the only and not the earliest biochemical defect associated with CBF cellular dysfunction in MCI adding to the complexity of the disease process underlying neuronal selective vulnerability.
Fig. 8.
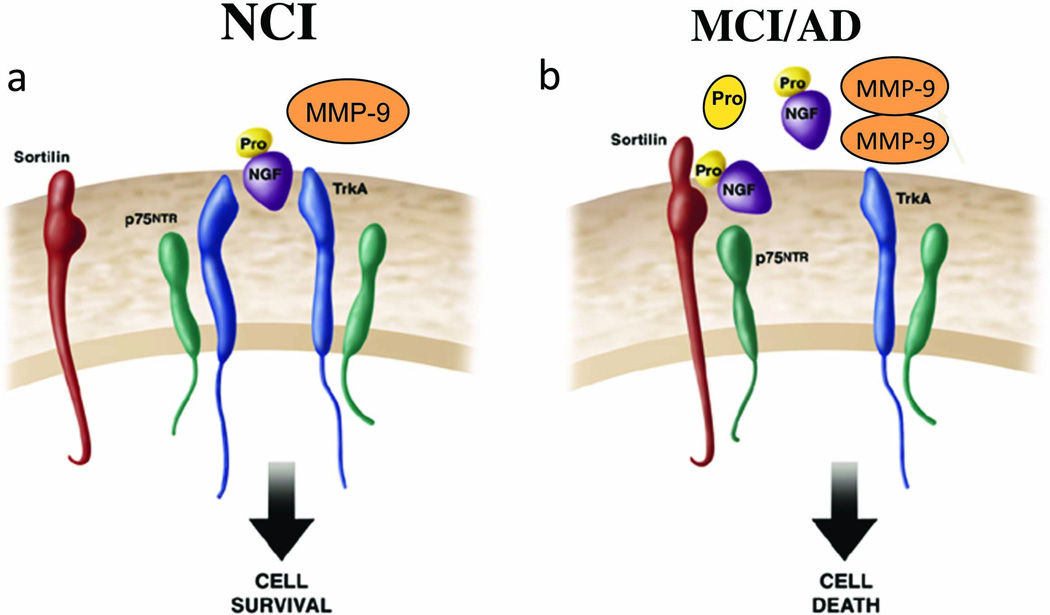
Illustration of changes related to the nerve growth factor system during the progression from non-demented (a) to MCI (b) within the cortex. During the progression of AD, elevated cortical proNGF in the face of reduced TrkA may enhance binding of proNGF to p75NTR/sortilin complex. Since sortlin acts as molecular switch governing a p75NTR mediated pro-apoptotic signal, increased proNGF may trigger cell death in the face of decreased TrkA. In addition, increased MMP-9 may also facilitate the degradation of mature NGF further impairing cholinergic trophic support
Endosomal and oxidative stress in MCI
In this regard, several studies indicate that endosomal and oxidative stress dysregulation are among the earliest pathological changes observed in cortical, hippocampal CA1 pyramidal and NB CBF neurons [28–30, 119–121, 163], precede clinical symptoms and appear prior to substantial deposition of cerebral amyloid, vascular amyloid and tau pathology in AD [30, 120]. With regard to endosomal pathways, laser capture microdissection (LCM) coupled with microarray analysis of CA1 pyramidal neurons and NB cholinergic neurons revealed an upregulation of markers of endosomal activation, including select rab GTPases in MCI and mild/moderate AD tissue from the RROS (Fig. 7b,c) [34, 51]. In NB cholinergic neurons [51, 53], select rab GTPase expression levels were selectively increased as antemortem measures of cognition declined and occurred in parallel with a down regulation of TrkB and TrkC, the cognate receptors for brain-derived neurotrophic factor (BDNF) and NT3, respectively, both members of the NGF neurotrophin family [9]. However, unlike the NGF/proNGF complex, proBDNF and mautre BDNF are both reduced in the MCI cortex [126]. Hence, increased endocytic pathway activity, driven by elevated rab GTPase expression, may result in long-term deficits in hippocampal and basal forebrain neurotrophic signaling and represent a key pathogenic mechanism underlying the onset of MCI.
With regards to oxidative stress, heme oxygenase (HO-1, an indirect marker of oxidative stress), together with biliverdin reductase-A (BVR-A, a pleiotropic enzyme that plays a pivotal role in antioxidant defense against free radicals) are elevated in MCI hippocampus and temporal neocortex samples obtained from the RROS [156, 157] and UKADC [10]. Astroglial HO-1 immunoreactivity in the temporal neocortex, but not hippocampus, correlated with the burden of neurofibrillary pathology [157]. In addition, there was a significant reduction in protein carbonyl-derivatives of BVR-A and an up regulation of inducible nitric oxide synthase in the MCI and AD hippocampus [156, 157]. MCI individuals also show increased levels of lipid peroxidation and nucleic acid oxidation in hippocampus [79]. Redox proteomics revealed excessive oxidative modification of the oxidative proteins A-enolase, glutamine synthetase, pyruvate kinase M2 and peptidyl-prolyl cis/trans isomerase 1 in MCI hippocampus compared to control samples accrued from the UKADRC [25]. These findings indicate the functional interaction of these oxidative proteins in energy metabolism, synaptic plasticity and mitogenesis/proliferation pathways and strengthen the suggestion that oxidative stress is an early cellular event, perhaps inducing cells to compensate for the increase of intracellular oxidative events during the prodromal stages of dementia [19]. Interestingly, Smith and coworkers [88] proposed that tau phosphorylation represents a compensatory neuronal response against oxidative stress, which acts as a neuroprotective event during the early phases of cellular insult. This mechanism may explain reports indicating that tangle bearing neurons survive for at least two decades [108], suggesting that NFT are not initiators of cell death [88] but may play a role in altering nuclear transcription factors.
An intriguing report using western blot analysis of isolated cytosolic and nuclear fractions prepared from postmortem human hippocampi harvested from the UKADRC with a premortem clinical diagnosis of NCI and MCI [131], demonstrated a shift of nuclear factor of activated T-cells (NFAT) 1 and 2 to nuclear compartments at different stages of AD neuropathology and cognitive decline, whereas NFAT2 remained unchanged [1]. Changes in NFAT3 were directly correlated to soluble amyloid-β (Aβ1–42), and oligomeric Aβ levels in hippocampus. These findings add to the growing number of factors that are dysregulated in MCI and suggest that NFAT signaling may play an important role in driving Aβ-mediated neurodegeneration early in the development of dementia.
Concomitant Pathogenic Factors in MCI
Several clinical pathological studies have reported that some patients with aMCI had other concomitant neuropathologic features, which may contribute to the clinical presentation of the subjects. For example, aMCI cases also display argyrophilic grain disease [20, 132], hippocampal sclerosis and vascular disease [132, 159]. Macroscopic cerebral infarcts without a pathologic diagnosis of AD, were found to be more common in naMCI (18.6%) compared to aMCI (13.3%) [159]. Certain vascular dementia subtypes, particularly those related to subcortical microvascular disease, may be preceded by MCI, which exhibit similar domains of cognitive impairment and an overlapping progressive course that may mimic AD [101]. A recent report demonstrated a small number of MCI brains that also displayed neocortical Lewy bodies; however, no person displayed a combination of AD pathology, macroscopic infarcts and neocortical Lewy bodies [101, 159]. Therefore, as with other human disorders, for example, cardiovascular disease, there may be many contributing pathologies that create a vulnerability state for MCI and neurodegeneration as well as in other neurologic disorders.
Neuropathology in Parkinson’s disease with MCI
A clinical study revealed that patients with Parkinson’s disease (PD) and MCI, defined by the Peterson criteria, had a higher risk of developing dementia than cognitively intact PD subjects, suggesting that MCI in PD is an early manifestation of dementia [75]. However, the pathology of MCI in PD is only now receiving attention. In a preliminary report, the majority of PD-MCI cases were found to be Braak AD stages III–IV (two amnestic MCI cases being stage IV) [3]. However, the major pathologic features were limbic and/or neocortical Lewy body and AD histopathology and possibly cerebrovascular pathology. This is an area that requires extensive clinical pathologic investigation using well-characterized patient populations.
Monoaminergic and Serotonergic Pathology in MCI
Although most reviews of MCI have concentrated on the pathology of cortical and basal limbic forebrain regions, there is mounting evidence suggesting that the brainstem harbors the earliest cellular degenerative events, even before those seen in neo and limbic cortex. Clinical pathological investigations of the norepinephrine (NE) containing locus coeruleus (LC) demonstrate NFTs occur during aging and in the earliest Braak pathological stages [22]. In MCI, LC neurons display sequential early and late tau conformational epitopes linked to NFT formation [22, 33, 57]; abnormal tau aggregates occur within proximal axons of LC projection neurons in the absence of either NFTs or neuropil threads in the transentorhinal cortex [22]. Since LC cytopathology correlates with overall cognition, noradrenergic dysfunction should be considered among the earliest cytopathologic lesions mediating the onset of cognitive decline in the aging-MCI continuum [57]. Most likely this occurs by disrupting ascending LC noradrenergic input to the thalamus, hippocampus and cortex. [22]. Similar to the LC, clinical pathologic investigations reveal the involvement of the brain stem serotonergic raphe cortical projection neurons in AD [36, 60, 125]. In the early Braak stages 0, II, and III, phospho-tau cytoskeletal changes occur in the supratrochlear subnucleus of the dorsal raphe nucleus (ST-DR) [56, 145], suggesting that the ST-DR plays a key role in the induction and spread of AD-related cytoskeletal pathology. Currently, information on the state of raphe neurons in MCI is lacking. Once these regions of the brain are fully investigated in MCI, it may be that the currently accepted stages of the cytopathology of MCI may require reclassification based on the emerging concept of a neuron-to-neuron transynaptic propagation, which maybe initiated in the brainstem and over time spreads to the telencephalon [22].
Summary and conclusions
The data presented in this review indicate that the neuropathologic substrate(s) of MCI is complex and must take into account not only senile plaque and NFT pathology but also cellular dysfunction and the initiation of neuroplastic responses. The wide range of cellular dysregulation that occurs prior to and during the prodromal MCI stage of the disease process suggests that there is no “silver bullet” at this time, which best fits the diverse pathologic, molecular and cellular constellation of events that occur in the MCI brain. Just as there are subtypes of clinical MCI, there may be multiple pathological entities that drive the onset of MCI. It will clearly require much more investigation to tease the pathology underlying these states apart. Emerging data suggest that MCI pathology is initiated via a trans-synaptic neuron-to-neuron disconnection syndrome affecting multiple levels within the central nervous system. In any case the pathologic mechanism underlying MCI most likely begins years before the onset of cognitive decline. The data presented herein suggest that it is too simplistic to attribute a single event to the precipitation of MCI. Instead, the clinical pathologic data suggest that the brain undergoes multisystem dysfunction and that in some instances the disease process triggers cellular and biochemical repair mechanisms in an attempt to slow the disease process. Understanding the molecular pathogenesis of these compensatory processes will provide novel clues about how the brain naturally responds to the signals that propagate the onset of MCI pathology, and give insight on how to potentially treat this complex, prodromal disease state with novel therapeutic approaches and cognitive remediation.
Acknowledgements
Supported by NIA grants PO1 AG14999, PO1 AG09466, AG025204, AG10688 and AG025204. We thank all our collaborators and the participants in each Alzheimer’s Disease Center, institute and organization without whom the information reviewed would not have been possible.
References
- 1.Abdul HM, Sama MA, Furman JL, et al. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abner EL, Kryscio RJ, Schmitt FA, et al. "End-stage" neurofibrillary tangle pathology in preclinical Alzheimer's disease: fact or fiction? Journal of Alzheimer's disease : JAD. 2011;25:445–453. doi: 10.3233/JAD-2011-101980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adler CH, Caviness JN, Sabbagh MN, et al. Heterogeneous neuropathological findings in Parkinson's disease with mild cognitive impairment. Acta neuropathologica. 2010;120:827–828. doi: 10.1007/s00401-010-0744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of neurology. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen OM, Reiche J, Schmidt V, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen OM, Schmidt V, Spoelgen R, et al. Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry. 2006;45:2618–2628. doi: 10.1021/bi052120v. [DOI] [PubMed] [Google Scholar]
- 8.Aoki C, Sekino Y, Hanamura K, et al. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. The Journal of comparative neurology. 2005;483:383–402. doi: 10.1002/cne.20449. [DOI] [PubMed] [Google Scholar]
- 9.Barbacid M. Neurotrophic factors and their receptors. Current opinion in cell biology. 1995;7:148–155. doi: 10.1016/0955-0674(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 10.Barone E, Di Domenico F, Cenini G, et al. Oxidative and nitrosative modifications of biliverdin reductase-a in the brain of subjects with Alzheimer's disease and amnestic mild cognitive impairment. Journal of Alzheimer's disease : JAD. 2011;25:623–633. doi: 10.3233/JAD-2011-110092. [DOI] [PubMed] [Google Scholar]
- 11.Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 12.Beach TG, McGeer EG. Cholinergic fiber loss occurs in the absence of synaptophysin depletion in Alzheimer's disease primary visual cortex. Neuroscience letters. 1992;142:253–256. doi: 10.1016/0304-3940(92)90385-k. [DOI] [PubMed] [Google Scholar]
- 13.Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer disease and associated disorders. 2004;18:270–277. [PubMed] [Google Scholar]
- 14.Bell KF, Ducatenzeiler A, Ribeiro-da-Silva A, et al. The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol Aging. 2006;27:1644–1657. doi: 10.1016/j.neurobiolaging.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 15.Bennett DA, Wilson RS, Schneider JA, et al. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- 16.Bettens K, Brouwers N, Engelborghs S, et al. SORL1 is genetically associated with increased risk for late-onset Alzheimer disease in the Belgian population. Human mutation. 2008;29:769–770. doi: 10.1002/humu.20725. [DOI] [PubMed] [Google Scholar]
- 17.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Bonda DJ, Wang X, Perry G, et al. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59:290–294. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Braak H, Braak E. Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathology and applied neurobiology. 1989;15:13–26. doi: 10.1111/j.1365-2990.1989.tb01146.x. [DOI] [PubMed] [Google Scholar]
- 21.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Del Tredici K. Alzheimer's pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol. 2011;121:589–595. doi: 10.1007/s00401-011-0825-z. [DOI] [PubMed] [Google Scholar]
- 23.Bruno MA, Cuello AC. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc Natl Acad Sci U S A. 2006;103:6735–6740. doi: 10.1073/pnas.0510645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruno MA, Mufson EJ, Wuu J, Cuello AC. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J Neuropathol Exp Neurol. 2009;68:1309–1318. doi: 10.1097/NEN.0b013e3181c22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butterfield DA, Poon HF, St Clair D, et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiology of disease. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Caselli RJ, Dueck AC, Osborne D, et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. The New England journal of medicine. 2009;361:255–263. doi: 10.1056/NEJMoa0809437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caselli RJ, Walker D, Sue L, Sabbagh M, Beach T. Amyloid load in nondemented brains correlates with APOE e4. Neuroscience letters. 2010;473:168–171. doi: 10.1016/j.neulet.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cataldo AM, Paskevich PA, Kominami E, Nixon RA. Lysosomal hydrolases of different classes are abnormally distributed in brains of patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1991;88:10998–11002. doi: 10.1073/pnas.88.24.10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cataldo AM, Peterhoff CM, Troncoso JC, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K, Reiman EM, Alexander GE, et al. Correlations between apolipoprotein E epsilon4 gene dose and whole brain atrophy rates. The American journal of psychiatry. 2007;164:916–921. doi: 10.1176/ajp.2007.164.6.916. [DOI] [PubMed] [Google Scholar]
- 32.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 33.Counts SE, B H, Nadeem M, Wuu J, Mufson EJ. Hippocampal drebrin loss in mild cognitive impairment. Neurodeg Dis. doi: 10.1159/000333122. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Counts SE, He B, Che S, et al. Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Archives of neurology. 2007;64:1771–1776. doi: 10.1001/archneur.64.12.1771. [DOI] [PubMed] [Google Scholar]
- 35.Counts SE, Nadeem M, Wuu J, et al. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer's disease. Annals of neurology. 2004;56:520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- 36.D'Amato RJ, Zweig RM, Whitehouse PJ, et al. Aminergic systems in Alzheimer's disease and Parkinson's disease. Annals of neurology. 1987;22:229–236. doi: 10.1002/ana.410220207. [DOI] [PubMed] [Google Scholar]
- 37.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 38.Davis KL, Mohs RC, Marin D, et al. Cholinergic markers in elderly patients with early signs of Alzheimer disease. Jama. 1999;281:1401–1406. doi: 10.1001/jama.281.15.1401. [DOI] [PubMed] [Google Scholar]
- 39.DeKosky ST, Ikonomovic MD, Styren SD, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Annals of neurology. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- 40.Delacourte A, David JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease. Neurology. 1999;52:1158–1165. doi: 10.1212/wnl.52.6.1158. [DOI] [PubMed] [Google Scholar]
- 41.Dodson SE, Gearing M, Lippa CF, et al. LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:866–872. doi: 10.1097/01.jnen.0000228205.19915.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drzezga A, Grimmer T, Henriksen G, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72:1487–1494. doi: 10.1212/WNL.0b013e3181a2e8d0. [DOI] [PubMed] [Google Scholar]
- 43.Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer's disease: a new lexicon. Lancet neurology. 2010;9:1118–1127. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- 44.Ellis JR, Villemagne VL, Nathan PJ, et al. Relationship between nicotinic receptors and cognitive function in early Alzheimer's disease: a 2-[18F]fluoro-A-85380 PET study. Neurobiology of learning and memory. 2008;90:404–412. doi: 10.1016/j.nlm.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 45.Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain : a journal of neurology. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 46.Forman MS, Mufson EJ, Leurgans S, et al. Cortical biochemistry in MCI and Alzheimer disease: lack of correlation with clinical diagnosis. Neurology. 2007;68:757–763. doi: 10.1212/01.wnl.0000256373.39415.b1. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. Journal of Alzheimer's disease : JAD. 2003;5:65–77. doi: 10.3233/jad-2003-5201. [DOI] [PubMed] [Google Scholar]
- 48.George S, Mufson EJ, Leurgans S, et al. MRI-based volumetric measurement of the substantia innominata in amnestic MCI and mild AD. Neurobiol Aging. 2011;32:1756–1764. doi: 10.1016/j.neurobiolaging.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geula C, Mesulam MM. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer's disease. Cerebral cortex. 1996;6:165–177. doi: 10.1093/cercor/6.2.165. [DOI] [PubMed] [Google Scholar]
- 50.Gilmor ML, Erickson JD, Varoqui H, et al. Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol. 1999;411:693–704. [PubMed] [Google Scholar]
- 51.Ginsberg SD, Alldred MJ, Counts SE, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry. 2010;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ginsberg SD, Che S, Counts SE, Mufson EJ. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Neurochem. 2006;96:1401–1408. doi: 10.1111/j.1471-4159.2005.03641.x. [DOI] [PubMed] [Google Scholar]
- 53.Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- 54.Gomez-Isla T, Price JL, McKeel DW, Jr, et al. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greeve I, Hermans-Borgmeyer I, Brellinger C, et al. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer's disease-associated neurodegeneration and oxidative stress. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:7345–7352. doi: 10.1523/JNEUROSCI.20-19-07345.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grinberg LT, Rub U, Ferretti RE, et al. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer's disease. A precocious onset? Neuropathology and applied neurobiology. 2009;35:406–416. doi: 10.1111/j.1365-2990.2009.00997.x. [DOI] [PubMed] [Google Scholar]
- 57.Grudzien A, Shaw P, Weintraub S, et al. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28:327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 58.Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Archives of neurology. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 59.Haense C, Kalbe E, Herholz K, et al. Cholinergic system function and cognition in mild cognitive impairment. Neurobiology of aging. 2010 doi: 10.1016/j.neurobiolaging.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 60.Halliday GM, McCann HL, Pamphlett R, et al. Brain stem serotonin-synthesizing neurons in Alzheimer's disease: a clinicopathological correlation. Acta neuropathologica. 1992;84:638–650. doi: 10.1007/BF00227741. [DOI] [PubMed] [Google Scholar]
- 61.Hanyu H, Asano T, Sakurai H, et al. MR analysis of the substantia innominata in normal aging, Alzheimer disease, and other types of dementia. AJNR Am J Neuroradiol. 2002;23:27–32. [PMC free article] [PubMed] [Google Scholar]
- 62.Hanyu H, Shimizu S, Tanaka Y, et al. MR features of the substantia innominata and therapeutic implications in dementias. Neurobiol Aging. 2007;28:548–554. doi: 10.1016/j.neurobiolaging.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 63.Hanyu H, Tanaka Y, Sakurai H, Takasaki M, Abe K. Atrophy of the substantia innominata on magnetic resonance imaging and response to donepezil treatment in Alzheimer's disease. Neurosci Lett. 2002;319:33–36. doi: 10.1016/s0304-3940(01)02507-1. [DOI] [PubMed] [Google Scholar]
- 64.Haroutunian V, Hoffman LB, Beeri MS. Is there a neuropathology difference between mild cognitive impairment and dementia? Dialogues in clinical neuroscience. 2009;11:171–179. doi: 10.31887/DCNS.2009.11.2/vharoutunian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatanpaa K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. Journal of neuropathology and experimental neurology. 1999;58:637–643. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 66.Hayashi K, Ishikawa R, Ye LH, et al. Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:7161–7170. doi: 10.1523/JNEUROSCI.16-22-07161.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 68.Honer WG, Dickson DW, Gleeson J, Davies P. Regional synaptic pathology in Alzheimer's disease. Neurobiology of aging. 1992;13:375–382. doi: 10.1016/0197-4580(92)90111-a. [DOI] [PubMed] [Google Scholar]
- 69.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer's disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 70.Ikonomovic MD, Abrahamson EE, Isanski BA, et al. Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Archives of neurology. 2007;64:1312–1317. doi: 10.1001/archneur.64.9.1312. [DOI] [PubMed] [Google Scholar]
- 71.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Precuneus amyloid burden is associated with reduced cholinergic activity in Alzheimer disease. Neurology. 2011;77:39–47. doi: 10.1212/WNL.0b013e3182231419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ikonomovic MD, Mufson EJ, Wuu J, Bennett DA, DeKosky ST. Reduction of choline acetyltransferase activity in primary visual cortex in mild to moderate Alzheimer's disease. Archives of neurology. 2005;62:425–430. doi: 10.1001/archneur.62.3.425. [DOI] [PubMed] [Google Scholar]
- 73.Ikonomovic MD, Mufson EJ, Wuu J, et al. Cholinergic plasticity in hippocampus of individuals with mild cognitive impairment: correlation with Alzheimer's neuropathology. Journal of Alzheimer's disease : JAD. 2003;5:39–48. doi: 10.3233/jad-2003-5106. [DOI] [PubMed] [Google Scholar]
- 74.Ikonomovic MD, Wecker L, Abrahamson EE, et al. Cortical alpha7 nicotinic acetylcholine receptor and beta-amyloid levels in early Alzheimer disease. Archives of neurology. 2009;66:646–651. doi: 10.1001/archneurol.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Janvin CC, Larsen JP, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson's disease: progression to dementia. Movement disorders : official journal of the Movement Disorder Society. 2006;21:1343–1349. doi: 10.1002/mds.20974. [DOI] [PubMed] [Google Scholar]
- 76.Johnson JK, Pa J, Boxer AL, et al. Baseline predictors of clinical progression among patients with dysexecutive mild cognitive impairment. Dementia and geriatric cognitive disorders. 2010;30:344–351. doi: 10.1159/000318836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kalus P, Slotboom J, Gallinat J, et al. Examining the gateway to the limbic system with diffusion tensor imaging: the perforant pathway in dementia. NeuroImage. 2006;30:713–720. doi: 10.1016/j.neuroimage.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 78.Kataturian Z. Revised criteria for diagnosis of Alzheimer’s disease: National Institute of Aging Alzheimer’s Association diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7:253–256. doi: 10.1016/j.jalz.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 79.Keller JN, Schmitt FA, Scheff SW, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 80.Kendziorra K, Wolf H, Meyer PM, et al. Decreased cerebral alpha4beta2* nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer's disease assessed with positron emission tomography. European journal of nuclear medicine and molecular imaging. 2011;38:515–525. doi: 10.1007/s00259-010-1644-5. [DOI] [PubMed] [Google Scholar]
- 81.Killiany RJ, Hyman BT, Gomez-Isla T, et al. MRI measures of entorhinal cortex vs hippocampus in preclinical AD. Neurology. 2002;58:1188–1196. doi: 10.1212/wnl.58.8.1188. [DOI] [PubMed] [Google Scholar]
- 82.King ME, Gamblin TC, Kuret J, Binder LI. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem. 2000;74:1749–1757. doi: 10.1046/j.1471-4159.2000.0741749.x. [DOI] [PubMed] [Google Scholar]
- 83.Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiology of aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 84.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Annals of neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 85.Kok E, Haikonen S, Luoto T, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Annals of neurology. 2009;65:650–657. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- 86.Kolsch H, Jessen F, Wiltfang J, et al. Association of SORL1 gene variants with Alzheimer's disease. Brain research. 2009;1264:1–6. doi: 10.1016/j.brainres.2009.01.044. [DOI] [PubMed] [Google Scholar]
- 87.Kordower JH, Chu Y, Stebbins GT, et al. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Annals of neurology. 2001;49:202–213. [PubMed] [Google Scholar]
- 88.Lee HG, Perry G, Moreira PI, et al. Tau phosphorylation in Alzheimer's disease: pathogen or protector? Trends in molecular medicine. 2005;11:164–169. doi: 10.1016/j.molmed.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 89.Lilja AM, Porras O, Storelli E, Nordberg A, Marutle A. Functional interactions of fibrillar and oligomeric amyloid-beta with alpha7 nicotinic receptors in Alzheimer's disease. Journal of Alzheimer's disease : JAD. 2011;23:335–347. doi: 10.3233/JAD-2010-101242. [DOI] [PubMed] [Google Scholar]
- 90.Lopez OL, Jagust WJ, DeKosky ST, et al. Prevalence and classification of mild cognitive impairment in the Cardiovascular Health Study Cognition Study: part 1. Archives of neurology. 2003;60:1385–1389. doi: 10.1001/archneur.60.10.1385. [DOI] [PubMed] [Google Scholar]
- 91.Lopez OL, Jagust WJ, Dulberg C, et al. Risk factors for mild cognitive impairment in the Cardiovascular Health Study Cognition Study: part 2. Archives of neurology. 2003;60:1394–1399. doi: 10.1001/archneur.60.10.1394. [DOI] [PubMed] [Google Scholar]
- 92.Mamidipudi V, Wooten MW. Dual role for p75(NTR) signaling in survival and cell death: can intracellular mediators provide an explanation? Journal of neuroscience research. 2002;68:373–384. doi: 10.1002/jnr.10244. [DOI] [PubMed] [Google Scholar]
- 93.Markesbery WR. Neuropathologic alterations in mild cognitive impairment: a review. Journal of Alzheimer's disease : JAD. 2010;19:221–228. doi: 10.3233/JAD-2010-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Markesbery WR, Schmitt FA, Kryscio RJ, et al. Neuropathologic substrate of mild cognitive impairment. Archives of neurology. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 95.Masliah E, Mallory M, Alford M, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 96.Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neuroscience letters. 1989;103:234–239. doi: 10.1016/0304-3940(89)90582-x. [DOI] [PubMed] [Google Scholar]
- 97.Masliah E, Terry RD, Mallory M, Alford M, Hansen LA. Diffuse plaques do not accentuate synapse loss in Alzheimer's disease. Am J Pathol. 1990;137:1293–1297. [PMC free article] [PubMed] [Google Scholar]
- 98.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 99.Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Annals of neurology. 2004;55:815–828. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- 100.Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- 101.Meyer JS, Xu G, Thornby J, Chowdhury MH, Quach M. Is mild cognitive impairment prodromal for vascular dementia like Alzheimer's disease? Stroke; a journal of cerebral circulation. 2002;33:1981–1985. doi: 10.1161/01.str.0000024432.34557.10. [DOI] [PubMed] [Google Scholar]
- 102.Minster RL, DeKosky ST, Kamboh MI. No association of dynamin binding protein (DNMBP) gene SNPs and Alzheimer's disease. Neurobiology of aging. 2008;29:1602–1604. doi: 10.1016/j.neurobiolaging.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mitchell TW, Mufson EJ, Schneider JA, et al. Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer's disease. Annals of neurology. 2002;51:182–189. doi: 10.1002/ana.10086. [DOI] [PubMed] [Google Scholar]
- 104.Mitsis EM, Reech KM, Bois F, et al. 123I-5-IA-85380 SPECT imaging of nicotinic receptors in Alzheimer disease and mild cognitive impairment. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2009;50:1455–1463. doi: 10.2967/jnumed.109.064030. [DOI] [PubMed] [Google Scholar]
- 105.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 106.Morris JC, Price AL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer's disease. J Mol Neurosci. 2001;17:101–118. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- 107.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of neurology. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morsch G, Maywald F, Wanner C. In vitro and in vivo studies with different precipitate filter cartridges for H.E.L.P.-LDL-apheresis. Optimization of precipitate filter cartridges. Bioseparation. 1995;5:11–18. [PubMed] [Google Scholar]
- 109.Mufson EJ, Chen EY, Cochran EJ, et al. Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol. 1999;158:469–490. doi: 10.1006/exnr.1999.7086. [DOI] [PubMed] [Google Scholar]
- 110.Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer's disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST. Human cholinergic basal forebrain: chemoanatomy and neurologic dysfunction. J Chem Neuroanat. 2003;26:233–242. doi: 10.1016/s0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 112.Mufson EJ, Ikonomovic MD, Styren SD, et al. Preservation of brain nerve growth factor in mild cognitive impairment and Alzheimer disease. Archives of neurology. 2003;60:1143–1148. doi: 10.1001/archneur.60.8.1143. [DOI] [PubMed] [Google Scholar]
- 113.Mufson EJ, Ma SY, Cochran EJ, et al. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol. 2000;427:19–30. doi: 10.1002/1096-9861(20001106)427:1<19::aid-cne2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 114.Mufson EJ, Ma SY, Dills J, et al. Loss of basal forebrain P75(NTR) immunoreactivity in subjects with mild cognitive impairment and Alzheimer's disease. J Comp Neurol. 2002;443:136–153. doi: 10.1002/cne.10122. [DOI] [PubMed] [Google Scholar]
- 115.Mufson EJ, Wuu J, Counts SE, Nykjaer A. Preservation of cortical sortilin protein levels in MCI and Alzheimer's disease. Neurosci Lett. 2010;471:129–133. doi: 10.1016/j.neulet.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Muth K, Schonmeyer R, Matura S, et al. Mild cognitive impairment in the elderly is associated with volume loss of the cholinergic basal forebrain region. Biol Psychiatry. 67:588–591. doi: 10.1016/j.biopsych.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 117.Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 118.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nixon RA. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 120.Nixon RA, Cataldo AM, Mathews PM. The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res. 2000;25:1161–1172. doi: 10.1023/a:1007675508413. [DOI] [PubMed] [Google Scholar]
- 121.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 122.Nordberg A. Nicotinic receptor abnormalities of Alzheimer's disease: therapeutic implications. Biological psychiatry. 2001;49:200–210. doi: 10.1016/s0006-3223(00)01125-2. [DOI] [PubMed] [Google Scholar]
- 123.Offe K, Dodson SE, Shoemaker JT, et al. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:1596–1603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Overk CR, Felder CC, Tu Y, et al. Cortical M1 receptor concentration increases without a concomitant change in function in Alzheimer's disease. Journal of chemical neuroanatomy. 2010;40:63–70. doi: 10.1016/j.jchemneu.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Parvizi J, Van Hoesen GW, Damasio A. The selective vulnerability of brainstem nuclei to Alzheimer's disease. Annals of neurology. 2001;49:53–66. doi: 10.1002/1531-8249(200101)49:1<53::aid-ana30>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 126.Peng S, Garzon DJ, Marchese M, et al. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer's disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol. 2004;63:641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- 128.Perry A, Cai DX, Scheithauer BW, et al. Merlin, DAL-1, and progesterone receptor expression in clinicopathologic subsets of meningioma: a correlative immunohistochemical study of 175 cases. Journal of neuropathology and experimental neurology. 2000;59:872–879. doi: 10.1093/jnen/59.10.872. [DOI] [PubMed] [Google Scholar]
- 129.Petersen R. Conceptual Overview. In: Petersen R, editor. Mild cognitive impairment Aging to Alzheimer's disease. New York: Oxford University Press; 2003. [Google Scholar]
- 130.Petersen RC. Mild cognitive impairment clinical trials. Nat Rev Drug Discov. 2003;2:646–653. doi: 10.1038/nrd1155. [DOI] [PubMed] [Google Scholar]
- 131.Petersen RC. Mild cognitive impairment as a diagnostic entity. Journal of internal medicine. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 132.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Archives of neurology. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 133.Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Archives of neurology. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 134.Pham E, Crews L, Ubhi K, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. Febs J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Potter PE, Rauschkolb PK, Pandya Y, et al. Pre- and post-synaptic cortical cholinergic deficits are proportional to amyloid plaque presence and density at preclinical stages of Alzheimer's disease. Acta neuropathologica. 2011;122:49–60. doi: 10.1007/s00401-011-0831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Price JL, Ko AI, Wade MJ, et al. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Archives of neurology. 2001;58:1395–1402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 137.Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Price JL, Morris JC. Tangles and plaques in nondemented aging and "preclinical" Alzheimer's disease. Annals of neurology. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 139.Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Riley KP, Snowdon DA, Markesbery WR. Alzheimer's neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Annals of neurology. 2002;51:567–577. doi: 10.1002/ana.10161. [DOI] [PubMed] [Google Scholar]
- 141.Rinne JO, Kaasinen V, Jarvenpaa T, et al. Brain acetylcholinesterase activity in mild cognitive impairment and early Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:113–115. doi: 10.1136/jnnp.74.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Robakis NK. Mechanisms of AD neurodegeneration may be independent of Abeta and its derivatives. Neurobiology of aging. 2011;32:372–379. doi: 10.1016/j.neurobiolaging.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rogalski EJ, Murphy CM, deToledo-Morrell L, et al. Changes in parahippocampal white matter integrity in amnestic mild cognitive impairment: a diffusion tensor imaging study. Behavioural neurology. 2009;21:51–61. doi: 10.3233/BEN-2009-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Roux PP, Barker PA. Neurotrophin signaling through the p75 neurotrophin receptor. Progress in neurobiology. 2002;67:203–233. doi: 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 145.Rub U, Del Tredici K, Schultz C, et al. The evolution of Alzheimer's disease-related cytoskeletal pathology in the human raphe nuclei. Neuropathology and applied neurobiology. 2000;26:553–567. doi: 10.1046/j.0305-1846.2000.00291.x. [DOI] [PubMed] [Google Scholar]
- 146.Sabbagh MN, Shah F, Reid RT, et al. Pathologic and nicotinic receptor binding differences between mild cognitive impairment, Alzheimer disease, and normal aging. Archives of neurology. 2006;63:1771–1776. doi: 10.1001/archneur.63.12.1771. [DOI] [PubMed] [Google Scholar]
- 147.Sager KL, Wuu J, Leurgans SE, et al. Neuronal LR11/sorLA expression is reduced in mild cognitive impairment. Annals of neurology. 2007;62:640–647. doi: 10.1002/ana.21190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Saito Y, Murayama S. Neuropathology of mild cognitive impairment. Neuropathology : official journal of the Japanese Society of Neuropathology. 2007;27:578–584. doi: 10.1111/j.1440-1789.2007.00806.x. [DOI] [PubMed] [Google Scholar]
- 149.Salehi A, Verhaagen J, Dijkhuizen PA, Swaab DF. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of Meynert neurons and their differential reduction in Alzheimer's disease. Neuroscience. 1996;75:373–387. doi: 10.1016/0306-4522(96)00273-4. [DOI] [PubMed] [Google Scholar]
- 150.Sasaki M, Ehara S, Tamakawa Y, et al. MR anatomy of the substantia innominata and findings in Alzheimer disease: a preliminary report. AJNR Am J Neuroradiol. 1995;16:2001–2007. [PMC free article] [PubMed] [Google Scholar]
- 151.Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiology of aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 152.Scheff SW, Ginsberg S, Counts SE, Mufson EJ. Synaptic integrity in mild cognitive impairment and Alzheimer’s disease. In: Sun M, editor. Research Progress in Alzheimer’s Disease and Dementia. New York: NOVA Scientific Publisher; (in press) [Google Scholar]
- 153.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 154.Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 155.Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ. Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer's disease. Journal of Alzheimer's disease : JAD. 2011;24:547–557. doi: 10.3233/JAD-2011-101782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schipper HM. Heme oxygenase expression in human central nervous system disorders. Free Radic Biol Med. 2004;37:1995–2011. doi: 10.1016/j.freeradbiomed.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 157.Schipper HM, Bennett DA, Liberman A, et al. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging. 2006;27:252–261. doi: 10.1016/j.neurobiolaging.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 158.Schmidt ML, DiDario AG, Otvos L, Jr, et al. Plaque-associated neuronal proteins: a recurrent motif in neuritic amyloid deposits throughout diverse cortical areas of the Alzheimer's disease brain. Exp Neurol. 1994;130:311–322. doi: 10.1006/exnr.1994.1209. [DOI] [PubMed] [Google Scholar]
- 159.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Annals of neurology. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shimohama S, Kamiya S, Taniguchi T, Akagawa K, Kimura J. Differential involvement of synaptic vesicle and presynaptic plasma membrane proteins in Alzheimer's disease. Biochem Biophys Res Commun. 1997;236:239–242. doi: 10.1006/bbrc.1997.6940. [DOI] [PubMed] [Google Scholar]
- 161.Shirao T, Inoue HK, Kano Y, Obata K. Localization of a developmentally regulated neuron-specific protein S54 in dendrites as revealed by immunoelectron microscopy. Brain research. 1987;413:374–378. doi: 10.1016/0006-8993(87)91032-8. [DOI] [PubMed] [Google Scholar]
- 162.Small GW, Mazziotta JC, Collins MT, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA : the journal of the American Medical Association. 1995;273:942–947. [PubMed] [Google Scholar]
- 163.Smith MA, Nunomura A, Lee HG, et al. Chronological primacy of oxidative stress in Alzheimer disease. Neurobiology of aging. 2005;26:579–580. doi: 10.1016/j.neurobiolaging.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 164.Snowdon DA, Gross MD, Butler SM. Antioxidants and reduced functional capacity in the elderly: findings from the Nun Study. The journals of gerontology Series A, Biological sciences and medical sciences. 1996;51:M10–M16. doi: 10.1093/gerona/51a.1.m10. [DOI] [PubMed] [Google Scholar]
- 165.Snowdon DA, Kemper SJ, Mortimer JA, et al. Linguistic ability in early life and cognitive function and Alzheimer's disease in late life. Findings from the Nun Study. JAMA : the journal of the American Medical Association. 1996;275:528–532. [PubMed] [Google Scholar]
- 166.Sojkova J, Zhou Y, An Y, et al. Longitudinal patterns of beta-amyloid deposition in nondemented older adults. Archives of neurology. 2011;68:644–649. doi: 10.1001/archneurol.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PloS one. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Stoub TR, Bulgakova M, Leurgans S, et al. MRI predictors of risk of incident Alzheimer disease: a longitudinal study. Neurology. 2005;64:1520–1524. doi: 10.1212/01.WNL.0000160089.43264.1A. [DOI] [PubMed] [Google Scholar]
- 170.Stoub TR, deToledo-Morrell L, Stebbins GT, et al. Hippocampal disconnection contributes to memory dysfunction in individuals at risk for Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10041–10045. doi: 10.1073/pnas.0603414103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stoub TR, Rogalski EJ, Leurgans S, Bennett DA, deToledo-Morrell L. Rate of entorhinal and hippocampal atrophy in incipient and mild AD: relation to memory function. Neurobiol Aging. 2010;31:1089–1098. doi: 10.1016/j.neurobiolaging.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease. Journal of neuroscience research. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sunderland T, Linker G, Mirza N, et al. Decreased beta-amyloid1-42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA : the journal of the American Medical Association. 2003;289:2094–2103. doi: 10.1001/jama.289.16.2094. [DOI] [PubMed] [Google Scholar]
- 174.Swaminathan S, Shen L, Risacher SL, et al. Amyloid pathway-based candidate gene analysis of [(11)C]PiB-PET in the Alzheimer's Disease Neuroimaging Initiative (ADNI) cohort. Brain imaging and behavior. 2011 doi: 10.1007/s11682-011-9136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Takahashi H, Sekino Y, Tanaka S, et al. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tapiola T, Pennanen C, Tapiola M, et al. MRI of hippocampus and entorhinal cortex in mild cognitive impairment: a follow-up study. Neurobiology of aging. 2008;29:31–38. doi: 10.1016/j.neurobiolaging.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 177.Teaktong T, Graham A, Court J, et al. Alzheimer's disease is associated with a selective increase in alpha7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia. 2003;41:207–211. doi: 10.1002/glia.10132. [DOI] [PubMed] [Google Scholar]
- 178.Terriere E, Dempsey MF, Herrmann LL, et al. 5-(123)I-A-85380 binding to the alpha4beta2-nicotinic receptor in mild cognitive impairment. Neurobiology of aging. 2010;31:1885–1893. doi: 10.1016/j.neurobiolaging.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 179.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Annals of neurology. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 180.Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Science of aging knowledge environment : SAGE KE. 2006 doi: 10.1126/sageke.2006.6.re1. 2006:re1. [DOI] [PubMed] [Google Scholar]
- 181.Thal DR, Holzer M, Rub U, et al. Alzheimer-related tau-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp Neurol. 2000;163:98–110. doi: 10.1006/exnr.2000.7380. [DOI] [PubMed] [Google Scholar]
- 182.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 183.Tremblay C, Pilote M, Phivilay A, et al. Biochemical characterization of Abeta and tau pathologies in mild cognitive impairment and Alzheimer's disease. Journal of Alzheimer's disease : JAD. 2007;12:377–390. doi: 10.3233/jad-2007-12411. [DOI] [PubMed] [Google Scholar]
- 184.Tsang SW, Lai MK, Kirvell S, et al. Impaired coupling of muscarinic M1 receptors to G-proteins in the neocortex is associated with severity of dementia in Alzheimer's disease. Neurobiology of aging. 2006;27:1216–1223. doi: 10.1016/j.neurobiolaging.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 185.Vana L, Kanaan NM, Ugwu IC, et al. Progression of Tau Pathology in Cholinergic Basal Forebrain Neurons in MCI and AD. American J Path. 179:2533–2550. doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang DS, Bennett DA, Mufson E, Cochran E, Dickson DW. Decreases in soluble alpha-synuclein in frontal cortex correlate with cognitive decline in the elderly. Neurosci Lett. 2004;359:104–108. doi: 10.1016/j.neulet.2004.01.044. [DOI] [PubMed] [Google Scholar]
- 187.Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Annals of neurology. 1981;10:122–126. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 188.Winblad B, Palmer K, Kivipelto M, et al. Mild cognitive impairment--beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. Journal of internal medicine. 2004;256:240–246. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]