

Abstract
Aims
The highly expressed cell adhesion receptor CD29 (β1-integrin) is essential for cardiomyocyte growth and survival, and its loss of function causes severe heart disease. However, CD29-induced signalling in cardiomyocytes is ill defined and may involve reactive oxygen species (ROS). A decisive source of cardiac ROS is the abundant NADPH oxidase (NOX) isoform NOX2. Because understanding of NOX-derived ROS in the heart is still poor, we sought to test the role of ROS and NOX in CD29-induced survival signalling in cardiomyocytes.
Methods and results
In neonatal rat ventricular myocytes, CD29 activation induced intracellular ROS formation (oxidative burst) as assessed by flow cytometry using the redox-sensitive fluorescent dye dichlorodihydrofluorescein diacetate. This burst was inhibited by apocynin and diphenylene iodonium. Further, activation of CD29 enhanced NOX activity (lucigenin-enhanced chemiluminescence) and activated the MEK/ERK and PI3K/Akt survival pathways. CD29 also induced phosphorylation of the inhibitory Ser9 on the pro-apoptotic kinase glycogen synthase kinase-3β in a PI3K/Akt- and MEK-dependent manner, and improved cardiomyocyte viability under conditions of oxidative stress. The ROS scavenger MnTMPyP or adenoviral co-overexpression of the antioxidant enzymes superoxide dismutase and catalase inhibited CD29-induced pro-survival signalling. Further, CD29-induced protective pathways were lost in mouse cardiomyocytes deficient for NOX2 or functional p47phox, a regulatory subunit of NOX.
Conclusion
p47phox-dependent, NOX2-derived ROS are mandatory for CD29-induced pro-survival signalling in cardiomyocytes. These findings go in line with a growing body of evidence suggesting that ROS can be beneficial to the cell and support a crucial role for NOX2-derived ROS in cell survival in the heart.
Keywords: NOX, Reactive oxygen species, β1-Integrin, Cardiomyocytes, Glycogen synthase kinase-3β
1. Introduction
Connections to extracellular matrix (ECM) components are essential for the survival and the structural and functional integrity of cardiomyocytes.1,2 In particular, transmembrane cell surface adhesion receptors (e.g. integrins) bind ECM proteins such as laminin, collagen, and fibronectin.3,4 Integrins are heterodimeric transmembrane receptors composed of an α- and β-subunit. In total, 18 α- and 8 β-chain isoforms are known, which form 24 distinct receptors that are in a bent conformation in quiescent, and in an extended conformation in activated cells.5,6 The β1-integrin subunit (CD29) represents the most abundant β-isoform expressed in cardiomyocytes, where it can dimerize with different α-subunits.7–9 In vitro, CD29 is directly activated by the binding of ECM components or activating antibodies,10,11 which causes the rearrangement of the actin cytoskeleton and recruitment of signalling kinases (e.g. mitogen-activated protein kinases) to the cytosolic part of the integrin receptor4,12. Through activation of such signalling kinases, CD29 participates in the regulation of cardiomyocyte growth and survival. Specifically, CD29 facilitates α-adrenergic receptor (AR)-stimulated hypertrophy13 and inhibits β-AR-induced apoptosis in vitro.14,15 In vivo, deletion of CD29 is embryonically lethal,2,16 and cardiac-specific disruption or knock-out leads to disturbed cardiac function, impaired cardiac differentiation, fibrosis, and heart failure.16–18 Similarly, mouse hearts deficient in CD29 exhibit increased cardiac myocyte apoptosis in response to β-AR stimulation19 and after myocardial infarction20 when compared with wild-type (wt) hearts. Furthermore, integrin shedding, which is essential in cardiomyocyte growth, as well as the induction of apoptotic processes referred to as anoikis are typical features associated with the cellular detachment from ECM components.21 Similarly, cell adhesion of α5β1-integrin to fibronectin protects against apoptosis in modified Chinese hamster ovary cells expressing α5β1-integrin.22 These studies clearly point out the importance of CD29 in cardiomyocyte survival, but the understanding of the likely signalling pathways involved is poor.
Several studies affirmed the production of reactive oxygen species (ROS; oxidative burst) by various stimuli, including growth factors or G protein-coupled receptor agonists as well as by cell adhesion as seen in anchorage-dependent cell growth.23–26 These ROS act as essential mediators in redox-signalling activating signalling kinases implicated in cell growth, adhesion, and survival. They derive from various oxidase enzymes, including lipoxygenase, xanthine oxidase, or NADPH oxidase (NOX), the latter of which is one of the major sources of cardiac ROS.23,27–29 NOX is a multimeric enzyme that consists of the cytosolic subunits p40phox, p47phox, p67phox, and Rac1, all of which translocate to the membrane upon activation, as well as the transmembrane subunits gp91phox and p22phox. To date, seven gp91phox homologues, NOX1–5 and DUOX1–2, have been identified in mammals.30 NOX2 predominates in cardiomyocytes, although NOX1 and NOX4 are also expressed,31 NOX4 being constitutively active even in the absence of the cytosolic subunits. NOX catalyses a one-electron transfer to molecular oxygen to form the radical superoxide anion (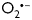 ).
).  is further reduced to hydrogen peroxide (H2O2), a reaction that occurs either spontaneously or is catalysed by superoxide dismutase (SOD), and finally degraded into water by catalase (cat).
is further reduced to hydrogen peroxide (H2O2), a reaction that occurs either spontaneously or is catalysed by superoxide dismutase (SOD), and finally degraded into water by catalase (cat).
Due to the emerging role of ROS as regulators of cell survival, the crucial role of NOX as a potent source of ROS in the heart and the limited understanding of NOX-derived ROS in cardiomyocytes, we addressed the question if CD29-induced pro-survival signalling depends on NOX-derived ROS in neonatal cardiomyocytes.
2. Methods
Detailed information on materials and methods is given in the Supplementary material online.
2.1. Animal material
Wistar rats (2007–2010) were obtained from Harlan Laboratories and mice from the Jackson Laboratory. From January 2011, Sprague–Dawley rats originating from in-house breeding were used. All animal protocols were approved by the Veterinary Department of Basel (Switzerland) and conformed to the rules of the Swiss Federal Act on Animal Protection and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Neonatal rat ventricular myocytes (NRVMs) were isolated from the hearts of 1- to 3-day-old pups sacrificed by decapitation and isolation was performed as previously described.32,33 Neonatal mouse ventricular myocytes (NMVMs) were isolated from 1- to 2-day-old pup hearts of wt (C57BL/6), p47phox loss-of-function (LOF; homozygous Ncf1m1J) and NOX2 (homozygous Cybbtm1Din) knock-out strains as previously described.34,35 Genotyping of mouse tails was performed as recommended by the supplier (http://jaxmice.jax.org/pub-cgi/protocols/protocols.sh). For some experiments, H9c2 cardiomyoblasts (ATCC) were used.
2.2. Viral transfection
Viral constructs were purchased from the Gene Transfer Vector Core, University of Iowa, IA, USA. Serum-starved cells were transfected with the viral constructs Ad5CMVCatalase,36 Ad5CMVSOD137, or Ad5CMVcytoLacZ38 at concentrations of 50 or 100 multiplicity of infections (MOIs) for 36 h prior to α-CD29 (10 µg/mL) treatment for 15 min. Expression levels were 1.8 ± 0.3-fold for SOD and 9 ± 1.6-fold for cat at 50 MOI.
2.3. Western blotting
Sample protein concentrations were estimated using a Pierce protein BCA kit (Thermo Fisher Scientific). 10–35 µg protein was loaded on a 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto a polyvinylidene fluoride membrane (GE Healthcare). Immunoreactive bands were quantified with Image J version 1.37 (http://rsb.info.nih.gov/ij/).39
2.4. Flow cytometry
Cardiomyocytes were loaded with 12.5 µmol/L (5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester, CM-H2DCF-DA; Invitrogen) in phenol-red-free (prf) DMEM with Hepes (25 mmol/L) and l-glutamine (4 mmol/L) for 20 min at 37°C with 5% CO2 in the dark. After washing, cells were detached with accutase (PAA Laboratories) and kept in 500 µL prf-DMEM on ice. Green fluorescence was measured with FL-1 488 nm excitation and 530 nm emission on a DAKO flow cytometer and DAKO Summit version 4.3 software.
2.5. Lucigenin assay
NOX activity was measured using lucigenin-enhanced chemiluminescence. H9c2 cells were treated with α-CD29 antibody or IgG (10 μg/mL, 15 min), then washed three times with cold PBS and scraped into Jude Krebs buffer (see Supplementary material online) containing protease inhibitor cocktail (Roche). Lysates were sonicated (3 s, four times on ice) and centrifuged at 3000 rpm at 4°C for 4 min. Measurements of NOX activity were performed by a luminescence assay in a microplate luminometer using 40 μg of lysate with 5 μl DMSO, 10 μmol/L Lucigenin (Sigma), 100 μmol/L NAD(P)H (Sigma), and 50 mmol/L Tiron (Fluka) per well (96-well plate). Data were recorded as relative light units over time, and integrated and calculated as area under the curve using Image J software for statistical analysis.
2.6. RNA interference
NRVMs were transfected with 2 μg/106 cells ON-TARGETplus using the Amaxa Neonatal Rat Cardiomyocytes Nucleofector kit (VPE-1002), according to the manufacturer's instructions, and efficiency of knockdown was assessed using quantitative RT–PCR.
2.7. Cell viability
Cell viability was measured by a tetrazolium (MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-based colorimetric assay using Cell Proliferation Kit I (Roche) according to the manufacturer's instructions.
2.8. Statistical analyses
All statistical analyses were performed in Graphpad Prism version 4.03 and P-values <0.05 were considered to indicate significant differences. Significance was tested in repeated measures one-way ANOVA with Bonferroni's post-test (for sample sizes less than or equal to four) or Newman–Keuls (for more than four samples to compare) correction. For analysing flow cytometry data, the geometric mean was used and overlays were performed in FloJo software version 7.5.5. (Tri Star Inc.).
3. Results
3.1. CD29 activation induces a time-dependent oxidative burst, which is impaired by apocynin and diphenylene iodonium in cardiomyocytes
To assess whether CD29 activation generates ROS in NRVM, the redox-sensitive fluorescent dye 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (DCF) was used. Flow cytometry analyses demonstrated a significant increase in DCF fluorescence indicative of an oxidative burst after 30 min of incubation of NRVM with an activating α-CD29 antibody, which subsided after 2 h (Figure 1A and B). A qualitatively similar increase in DCF fluorescence was observed in response to exogenous ROS (H2O2, 100 µmol/L) and to 12-phorbol 13-myristate acetate (PMA, 1 µmol/L), which induces endogenous ROS in other cell types40,41 (Figure 1A).
Figure 1.
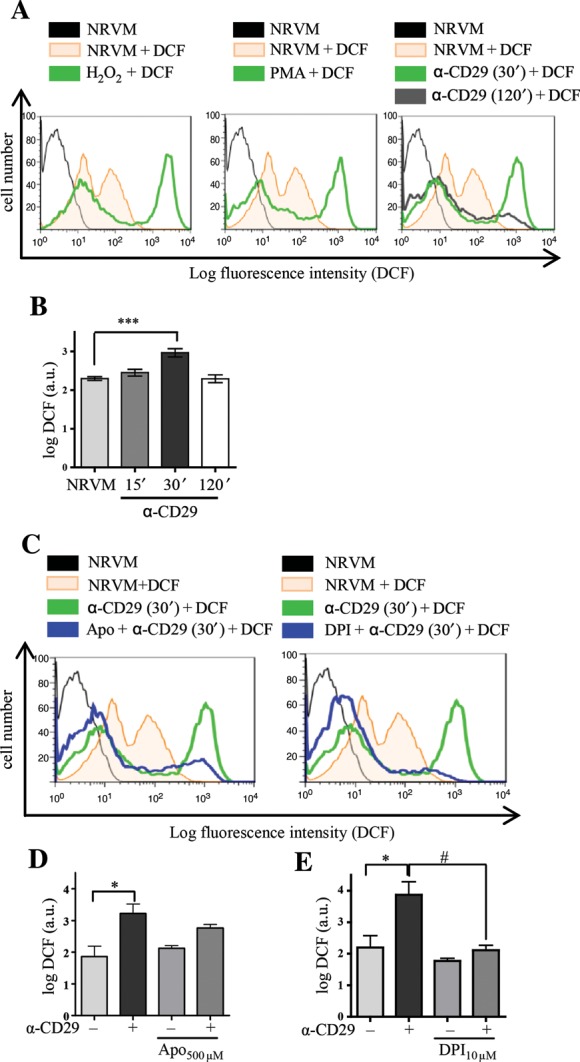
CD29 activation induces an oxidative burst in NRVM, which is inhibitable by apocynin and DPI. (A and B) (A) Representative flow cytometric histograms depict NRVM loaded with the ROS indicator DCF (12.5 µmol/L). Unstimulated cells (NRVM) without DCF in black. Fluorescence intensity is shown for unstimulated cells (NRVM + DCF, orange line) and cardiomyocytes stimulated with H2O2 (100 µmol/L), PMA (1 µmol/L), or α-CD29 (5 µg/mL) for 30 min (green line). The shift to the right indicates an increase in ROS. CD29 activation induced a time-dependent burst that peaked at 30 min (A and B, 30′) and declined after 2 h (120′, grey line). (B) Depicts α-CD29-induced fluorescence intensity at different time points. Data are mean ± SEM. Unless indicated otherwise, comparisons were not significant. ***P< 0.001 (n= 8). (C–E) Cells were pretreated (blue) with either apocynin (Apo; 500 µmol/L, 14–16 h), or DPI (10 µmol/L, 45 min) prior to α-CD29 administration (5 µg/mL, 30 min, C). (D and E) Pretreatment suppressed the α-CD29-induced oxidative burst partially (apocynin, n= 4) and completely (DPI, n= 5). Data are presented as mean ± SEM. Unless indicated otherwise, comparisons were not significant. * and #P< 0.05.
We further investigated the NOX-dependency of this burst using the inhibitors apocynin (apo, 500 µmol/L) and diphenylene iodonium (DPI, 10 µmol/L). Apocynin is, besides known to inhibit the regulatory NOX subunit p47phox in leukocytes, implicated as an antioxidant in endothelial and smooth muscle cells,42 and DPI inhibits flavoenzymes, including NOX.43 Both compounds markedly decreased DCF fluorescence intensity when administered prior to α-CD29 (Figure 1C–E).
3.2. CD29 activation induces NOX activity in cardiomyocytes
As both apocynin and DPI are non-specific, H9c2 cardiomyoblasts were treated with α-CD29 or non-immune IgG (10 μg/mL, 15 min) and NOX activity was measured by lucigenin-enhanced chemiluminescence. Cross-linkage of CD29 via activating antibody significantly enhanced NADPH-dependent lucigenin chemiluminescence in H9c2 cells (Figure 2A and B), thus confirming activation of NOX in response to CD29 stimulation. A similar observation was made in NRVM plated on dishes precoated with laminin, the physiological ligand for CD29. These cells exhibited higher NOX activity when compared with NRVM plated on non-coated dishes (data not shown).
Figure 2.

CD29 induces activation of NOX. H9c2 cells were treated with α-CD29 or non-immune IgG (10 μg/mL) for 15 min and NOX activity was measured by lucigenin-enhanced chemiluminescence. Kinetics of lucigenin-enhanced chemiluminescence is shown in (B). Basal fluorescence was measured five times before adding first NAD(P)H and later tiron, respectively (arrows). Data were recorded as relative light units over time and calculated as area under the curve for statistical analysis (A, n= 3). Data are given as mean ± SEM. **P< 0.01.
3.3. CD29 activation induces phosphorylation of ERK1/2 (Thr202/Tyr204), Akt (Ser473), and GSK-3β (Ser9)
CD29 activates the MEK/ERK and the PI3K/Akt pathways in various cell types.12 In NRVM, increased phosphorylation of ERK and Akt was observed upon CD29 stimulation, which peaked at 15 min and was no longer detectable at 45 min (Supplementary material online, Figure S1A and B). Phosphorylation of serine 9 of glycogen synthase kinase (GSK)-3β, which is inhibitory to its kinase activity, was used to measure CD29-induced pro-survival signalling, because (i) it is involved in the CD29-induced anti-apoptotic effect in adult cardiomyocytes44 and (ii) it is downstream of the PI3K/Akt pathway.45 Using a phosphorylation site-specific GSK-3β (Ser9) antibody, immunoreactive bands were observed at 15 min (Supplementary material online, Figure S1C). No changes in phosphorylation were detectable when cardiomyocytes were incubated with either an inhibitory α-CD29 antibody or with a non-specific IgG (Supplementary material online, Figure S1A–C).
To further elucidate the pathways of kinase activation upon CD29 stimulation, the MEK inhibitor U0126 (U), the PI3K inhibitors Wortmannin (W) and LY294002 (LY), and the Akt inhibitor Akti1/2 (Akti) were used to inhibit selected parts of the signalling pathway. Of the four inhibitors, U suppressed the α-CD29-induced phosphorylation of ERK (Supplementary material online, Figure S2A–D) and W, LY, and Akti (but not U) suppressed the α-CD29-induced phosphorylation of Akt (Supplementary material online, Figure S2E–H). No cross talk could be observed between the PI3K/Akt and the MEK/ERK signalling pathways. However, all inhibitors significantly reduced α-CD29-induced GSK-3β (Ser9) phosphorylation (Supplementary material online, Figure S2I–L). This observation places GSK-3β (Ser9) downstream of both the MEK/ERK and the PI3K/Akt pathways.
3.4. CD29-induced pro-survival signalling is ROS-dependent
Because activation of CD29 induced an oxidative burst in cardiomyocytes (Figure 1), the possibility exists that CD29-stimulated signalling is ROS-regulated. To test this hypothesis, MnTMPyP (MnT), a mimetic of the antioxidant enzymes SOD and cat, was used to reduce ROS levels. Pretreatment with MnT significantly inhibited the α-CD29-induced phosphorylation of ERK1/2 (Supplementary material online, Figure S3A), Akt (Supplementary material online, Figure S3B), and GSK-3β (Supplementary material online, Figure S3C). Similarly, co-overexpression of SOD and cat using adenoviral constructs significantly reduced MEK-, Akt-, and GSK-3β-phosphorylation (Figure 3A–C). To assess the respective contribution of O2•− and H2O2 to CD29-induced signalling, similar experiments were performed in NRVM expressing either SOD or cat. Although either enzyme inhibited phospho-MEK1/2 and phospho-GSK-3β, persistence of phospho-Akt was observed with overexpression of SOD, but not of cat, up to 100 MOI (Figure 3D and Supplementary material online, Figure S4).
Figure 3.

CD29-induced pro-survival signalling depends on ROS. (A–C) NRVM were infected with adenoviral constructs to express β-galactosidase (LacZ100) at 100 MOI or co-overexpress SOD (S50) and cat (C50) at 50 MOI 36 h prior to α-CD29 administration (10 µg/mL, 15 min). Equal protein amounts were subjected to western blotting using phosphorylation-specific antibodies for MEK1/2 (Ser217/221), Akt (Ser473), and GSK-3β (Ser9). Data are presented as mean ± SEM. *** and ###P< 0.001, ** and ##P< 0.01, and #P< 0.05 (n= 5 for A and B, n= 3 for C). Unless indicated otherwise, comparisons were not significant. (D) Western blot of CD29-induced signalling with single-enzyme overexpression at two different MOI, including the protein expressions of SOD and cat.
3.5. CD29-induced pro-survival signalling is inhibited by apocynin and DPI
Primarily, a likely role of NOX in CD29-induced survival signalling was investigated using apo and DPI. Both inhibitors suppressed CD29-induced ERK1/2- (Supplementary material online, Figure S5A and D) and Akt-phosphorylation (Supplementary material online, Figure S5B and E). Although both apo and DPI also inhibited the CD29-induced GSK-3β phosphorylation, the reliable assessment of this inhibition is hindered, because both substances affected basal kinase phosphorylation (Supplementary material online, Figure S5C and F). However, these findings suggest that NOX might serve as the source of CD29-induced ROS that trigger ROS-dependent pro-survival signalling in cardiomyocytes.
3.6. gp91phox- and p47phox-deficient cardiomyocytes show loss of α-CD29-induced pro-survival signals
In another set of experiments, NOX was corroborated as source of ROS by examining α-CD29-induced signalling in NMVMs isolated from gp91phox (NOX2) knock-out mice and from mice lacking a functional catalytic subunit p47phox. Upon CD29 stimulation, significantly lower levels of phospho-ERK1/2, phospho-Akt, and phospho-GSK-3β were observed in NOX2-knock-out mouse cardiomyocytes when compared with wt (Figure 4A–C). In p47phox LOF NMVM, CD29 activation failed to induce phosphorylation of Akt (Ser473; Figure 4E) and GSK-3β (Ser9; Figure 4F). In contrast, CD29-induced phosphorylation of ERK1/2 (Figure 4D) was unaffected by the loss of functional p47phox, suggesting the activation of a p47phox-independent and/or compensatory pathway.
Figure 4.

CD29-induced signal transduction requires NOX2-derived ROS. α-CD29 administration (10 µg/mL, 15 min) induced phosphorylation of ERK1/2 (p44/42), Akt and GSK-3β in wild-type (wt) NMVM (A–F). α-CD29-induced phosphorylation was absent in NOX2 knock-out (NOX2k.o.) cardiomyocytes (A–C); and partially diminished in p47phox loss of function (p47LOF) NMVM (D–F). Data are presented as mean ± SEM. Unless indicated otherwise, comparisons were not significant. * and #P< 0.05, ** and ##P< 0.01, *** and ###P< 0.001 (n= 5 for A, C, and D–F; n= 6 for B).
3.7. The CD29-induced oxidative burst is impaired in p47phox-LOF, but not in NOX2-knock-out cardiomyocytes
Because NOX2-derived ROS play an important role in CD29-induced pro-survival signalling in NMVM, we tested whether the CD29-induced oxidative burst was impaired in NMVM deficient in NOX2 or p47phox. Upon activation of CD29, a shift in DCF fluorescence was observed in wt NMVM that was missing in p47phox-LOF NMVM (Figure 5A and C). In contrast, in NOX2-knock-out cells, CD29 activation enhanced DCF fluorescence (Figure 5B and D). Therefore, another p47phox-dependent NOX (e.g. NOX1)46 might be involved in CD29-induced ROS formation in NMVM.
Figure 5.
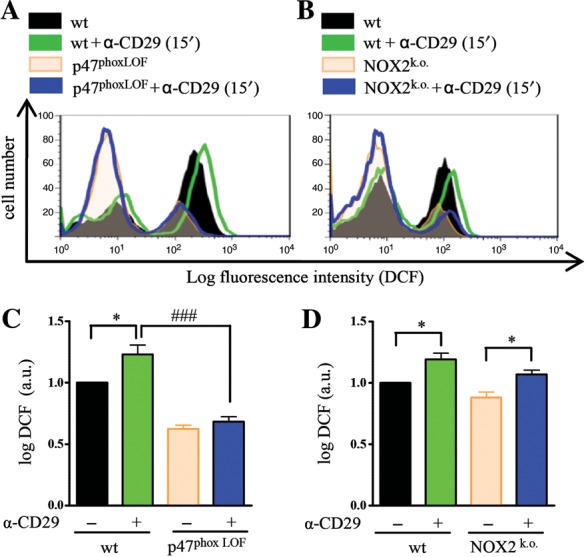
Lack of CD29-induced oxidative burst in p47phox-LOF cardiomyocytes. Serum-starved NMVM were incubated with DCF (12.5 µmol/L, 20 min) before detaching with accutase. Wt cardiomyocytes and cells deficient in functional NOX subunits were treated with α-CD29 antibody (15 µg/mL, 15 min) and DCF intensity was measured by flow cytometry. Representative histograms of p47phoxLOF (A and C) and NOX2 knock-out (B and D). NMVM depict untreated wt (black), α-CD29 treated wt (green), untreated (orange), and treated (blue) (A) p47phoxLOF or (B) NOX2k.o. cardiomyocytes. Quantified data (C and D) are presented as mean ± SEM. Unless indicated otherwise, comparisons were not significant. *P< 0.05; ###P< 0.001 (n= 4 each).
4. Discussion
This work demonstrates that NOX-derived ROS regulate cardiomyocyte survival pathways. Similar to what has been described for growth factors and cytokines,29 transient endogenous ROS were formed in cardiomyocytes in response to CD29 activation (Figure 6) and this ROS formation was impaired by NOX inhibitors. These findings are in agreement with DPI-based data implicating NOX in CD29-induced ROS production in the colic adenocarcinoma cell line (caco-2),47 but not with findings from mouse embryonic fibroblasts (NIH-3T3), where synergistically growth factor- and integrin-driven ROS were mostly lipoxygenase-dependent.48 In cardiac myocytes, however, CD29 activation induced NOX activity and an additional line of evidence for NOX-involvement was obtained using gene knock-outs (see below).
Figure 6.

Proposed model for the NOX2- and ROS-dependency of CD29-induced pro-survival signalling in neonatal cardiomyocytes. For details, see text.
ROS at moderate levels may serve as signalling molecules participating in the regulation of cell survival,24,49,50 because of their ability to activate pro-survival related cascades, including the PI3K/Akt and the MEK/ERK pathway.51 A prominent target of Akt is GSK3,52 of which two isoforms, GSK-3α and GSK-3β, are expressed in cardiomyocytes. Inhibition of GSK-3 via the PI3K pathway promotes cell survival in Drosophila or neurones.53 Additionally, CD29-dependent, PI3K-mediated phosphorylation of the inhibitory GSK-3β Ser9 protects adult rat cardiomyocytes from β-AR-induced apoptosis.44 Although GSK-3β is reportedly directly phosphorylated by Akt, several other kinases, including the ERK1/2 target p90 ribosomal S6 kinase have also been implicated in GSK-3β inhibition in the heart.45 Our data show activation of both, the PI3K/Akt and the MEK/ERK pathway upon CD29 engagement in NRVM and uncover GSK-3β as a target of both these pathways.
Clearance of ROS by antioxidants elicited apoptotic pathways in leukaemia cells, inhibiting the phosphorylation of Akt or ERK.54 In accordance with these findings, co-overexpression of both SOD and catalase (Figure 3) significantly diminished CD29-induced phosphorylation of MEK1/2, Akt, and GSK-3β. It is of note that at higher expression levels single-enzyme overexpression was equally effective with regard to induced phospho-MEK/ERK and -GSK-3β inhibition, implicating both 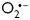 and H2O2 as second messengers in CD29-induced pro-survival signal transduction in NRVM. However, SOD appeared less efficient to suppress Akt-phosphorylation than catalase. Although this difference could be attributed to the lower expression levels of SOD, differential regulation of the PI3K/Akt pathway through
and H2O2 as second messengers in CD29-induced pro-survival signal transduction in NRVM. However, SOD appeared less efficient to suppress Akt-phosphorylation than catalase. Although this difference could be attributed to the lower expression levels of SOD, differential regulation of the PI3K/Akt pathway through 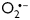 and H2O2 cannot be ruled out.
and H2O2 cannot be ruled out.
DPI and apocynin inhibited the CD29-induced phosphorylation of ERK1/2, Akt, and GSK-3β, implying NOX-derived ROS as signalling mediators. Mice carrying either a knock-out of the transmembrane NADPH oxidase subunit NOX2, or a LOF from the regulatory subunit p47phox were used to assess whether NOX2 is required for CD29-induced survival signalling in NMVM. Indeed, NOX2 knock-out NMVM showed reduced levels of CD29-induced ERK1/2 phosphorylation and the CD29-inducible phosphorylation of Akt and GSK-3β were nearly abolished in both NOX2 knock-out and p47phox-LOF cardiomyocytes (Figure 4A–F), highlighting the central role of NOX2- and p47phox-derived ROS in CD29-induced survival signalling. In contrast to NOX2 knock-out cardiomyocytes, however, where CD29-induced ERK-phosphorylation was mostly suppressed, induced phospho-ERK was still solid in p47phox-LOF cardiomyocytes. Although no oxidative burst was detectable in p47phox-LOF cardiomyocytes at 15 min, i.e. at the time of peak signalling, a compensatory pathway involving weaker, NOXO1-supported ROS release may account for ERK activation in the absence of functional p47phox. Alternatively, lack of ROS may allow for a ROS-independent pathway to activate ERK. In fact, co-existence of ROS-dependent and -independent pathways namely regarding ERK activation has previously been observed in other cell types.55,56
Interestingly, whereas the CD29-induced oxidative burst was absent in p47phox-LOF, it was maintained in NOX2 knock-out cardiomyocytes (Figure 5). Because both NOX1 and NOX2 may use p47phox to generate ROS in a stimulus-dependent manner,46 preservation of the oxidative burst in the absence of NOX2 implies another functional NOX homologue (for example NOX1) as an additional and/or compensatory ROS source. However, these additional ROS are unlikely to be involved in CD29-induced survival signalling, because this signalling is lacking in NOX2 knock-out mice. It is noteworthy that reciprocal compensation of NOX2 and NOX4 has been observed in human lung endothelial cells.57 Furthermore, there is evidence for spatiotemporal-defined localization of NOX isoforms at the plasma membrane (NOX2) or in subcellular compartments (e.g. endosomes, NOX1 and NOX2)58,59 and different NOX isoforms may elicit distinct signalling.60 Importantly, NOX2-derived ROS have been implicated as mediators of cardiomyocyte protection against ischaemia/reperfusion injury conferred by ischaemic preconditioning.61 These findings point out the importance of NOX-derived ROS and are in accordance with our findings that NOX2-derived ROS act as mediators of pro-survival signalling in cardiomyocytes. In fact, CD29 activation improved survival of NOX2-competent cardiomyocytes exposed to oxidative stress (H2O2), but appeared not to do so in NOX2-deficient cells (Supplementary material online, Figure S6). A proposed model for the role of NOX2-derived ROS in CD29-induced pro-survival signalling in cardiomyocytes is depicted in Figure 6.
In conclusion, CD29-induced pro-survival signalling in cardiomyocytes is ROS-dependent and NOX2 acts as source of these ROS. Moreover, CD29-induced inhibition of the pro-apoptotic kinase GSK-3β depends upon NOX2-derived ROS in rat and mouse cardiomyocytes. These findings support a beneficial role for NOX2-derived ROS in cell survival in the heart.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study was supported by a SCORE career development grant from the Swiss National Science Foundation (32323B-111352 and 32003B-111353 to G.M.K.), and project grants from the Swiss Heart Foundation and the SwissLife Jubiläumsstiftung (all to G.M.K.), as well as in part by the Mach-Gaensslen Foundation Switzerland (to O.P.) and the Stiftung für kardiovaskuläre Forschung Basel, Switzerland. The authors would like to acknowledge the University of Iowa Gene Transfer Vector Core, supported in part by the National Institute of Health and the Roy J. Carver Foundation, for viral vector preparations.
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We thank Dr Maria Filippova for providing non-specific IgG; and Prof Lucia Mori De Libero for generously providing Cybb animals. We are grateful to Drs Philipp Schlüter and Stephen Schauer for critical comments on the manuscript and to Dr Tracy Glass for English language editing.
References
- 1.Meredith J, Fazeli B, Schwartz M. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hynes R. Targeted mutations in cell adhesion genes: what have we learned from them? Dev Biol. 1996;180:402–412. doi: 10.1006/dbio.1996.0314. [DOI] [PubMed] [Google Scholar]
- 3.Ross R, Borg T. Integrins and the myocardium. Circ Res. 2001;88:1112–1119. doi: 10.1161/hh1101.091862. [DOI] [PubMed] [Google Scholar]
- 4.Hynes R. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 5.Hynes R. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 6.Askari JA, Tynan CJ, Webb SED, Martin-Fernandez ML, Ballestrem C, Humphries MJ. Focal adhesions are sites of integrin extension. J Cell Biol. 2010;188:891–903. doi: 10.1083/jcb.200907174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carver W, Price R, Raso D, Terracio L, Borg T. Disruption of b-1 integrin in the developing heart. J Histochem Cytochem. 1994;42:167–175. doi: 10.1177/42.2.8288862. [DOI] [PubMed] [Google Scholar]
- 8.Hemler M. VLA proteins in the integrin family: structures, functions, and their role in leukocytes. Annu Rev Immunol. 1990;8:365–400. doi: 10.1146/annurev.iy.08.040190.002053. [DOI] [PubMed] [Google Scholar]
- 9.Ross R. The extracellular connections: the role of integrins in myocardial remodeling. J Card Fail. 2002;8:S326–S331. doi: 10.1054/jcaf.2002.129263. [DOI] [PubMed] [Google Scholar]
- 10.Diamond M, Springer T. The dynamic regulation of integrin adhesiveness. Curr Biol. 1994;4:506–517. doi: 10.1016/s0960-9822(00)00111-1. [DOI] [PubMed] [Google Scholar]
- 11.Byron A, Humphries JD, Askari JA, Craig SE, Mould AP, Humphries MJ. Anti-integrin monoclonal antibodies. J Cell Sci. 2009;122:4009–4011. doi: 10.1242/jcs.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark E, Brugge J. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 13.Ross R, Pham C, Shai S-Y, Goldhaber J, Fenzik C, Glembotski C, et al. b1 integrins participate in the hypertrophic response of rat ventricular myocytes. Circ Res. 1998;82:1160–1172. doi: 10.1161/01.res.82.11.1160. [DOI] [PubMed] [Google Scholar]
- 14.Communal C, Singh M, Menon B, Xie Z, Colucci W, Singh K. b1 integrins expression in adult rat ventricular myocytes and its role in the regulation of b-adrenergic receptor stimulated apoptosis. J Cell Biochem. 2003;89:381–388. doi: 10.1002/jcb.10520. [DOI] [PubMed] [Google Scholar]
- 15.Menon B, Singh M, Ross R, Johnson J, Singh K. b-adrenergic receptor-stimulated apoptosis in adult cardiac myocytes involves MMP2-mediated disruption of b1 integrin signaling and mitochondrial pathway. Am J Physiol Cell Physiol. 2005;290:254–261. doi: 10.1152/ajpcell.00235.2005. [DOI] [PubMed] [Google Scholar]
- 16.Fässler R, Rohwedel J, Maltsev V, Bloch W, Lentini S, Guan K, et al. Differentiation and integrity of cardiac muscle cells are impaired in the absence of b1 integrin. J Cell Sci. 1996;109:2989–2999. doi: 10.1242/jcs.109.13.2989. [DOI] [PubMed] [Google Scholar]
- 17.Keller R, Shai S-Y, Babbitt C, Pham C, Solaro R, Valencik M, et al. Disruption of integrin function in the murine myocardium leads to perinatal lethality, fibrosis, and abnormal cardiac performance. Am J Pathol. 2001;158:1079–1090. doi: 10.1016/S0002-9440(10)64055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shai S-Y, Harpf A, Babbitt C, Jordan M, Fishbein M, Chen J, et al. Cardiac myocyte-specific excission of the b1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 19.Krishnamurthy P, Subramanian V, Singh M, Singh K. b1 integrin modulate b-adrenergic receptor stimulated cardiac myocyte apoptosis and myocardial remodeling. Hypertension. 2007;49:865–872. doi: 10.1161/01.HYP.0000258703.36986.13. [DOI] [PubMed] [Google Scholar]
- 20.Krishnamurthy P, Subramanian V, Singh M, Singh K. Deficiency of b1integrins results in increased myocardial dysfunction after myocardial infarction. Heart. 2006;92:1309–1315. doi: 10.1136/hrt.2005.071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldsmith EC, Carver W, McFadden A, Goldsmith JG, Price RL, Sussman M, et al. Integrin shedding as a mechanism of cellular adaption during cardiac growth. Am J Physiol Heart Circ Physiol. 2003;284:2227–2234. doi: 10.1152/ajpheart.00920.2002. [DOI] [PubMed] [Google Scholar]
- 22.Matter ML, Ruoslahti E. A signaling pathway from the a5b1 and avb3 integrins that elevate bcl-2 transcription. J Biol Chem. 2001;276:27757–27763. doi: 10.1074/jbc.M102014200. [DOI] [PubMed] [Google Scholar]
- 23.Chiarugi P, Fiaschi T. Redox signalling in anchorage-dependent cell growth. Cell Signal. 2007;19:672–682. doi: 10.1016/j.cellsig.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- 25.Xiao L, Pimentel D, Wang J, Singh K, Colucci W, Sawyer D. Role of reactive oxygen species and NAD(P)H oxidase in a1-adrenoreceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol. 2002;282:C926–C934. doi: 10.1152/ajpcell.00254.2001. [DOI] [PubMed] [Google Scholar]
- 26.Finkel T. Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol. 1999;65:337–340. doi: 10.1002/jlb.65.3.337. [DOI] [PubMed] [Google Scholar]
- 27.Sawyer D, Siwik D, Xiao L, Pimentel D, Singh K, Colucci W. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol. 2002;34:379–388. doi: 10.1006/jmcc.2002.1526. [DOI] [PubMed] [Google Scholar]
- 28.Bendall J, Cave A, Heymes C, Gall N, Shah A. Pivotal role of gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 29.Rhee S. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 30.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 31.Hingtgen S, Tian X, Yang J, Dunlay S, Peek A, Wu Y, et al. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics. 2006;26:180–191. doi: 10.1152/physiolgenomics.00029.2005. [DOI] [PubMed] [Google Scholar]
- 32.Thaik C, Calderone A, Takahashi N, Colucci W. Interleukin-1b modulates the growth and phenotype of neonatal rat cardiac myocytes. J Clin Invest. 1995;96:1093–1099. doi: 10.1172/JCI118095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim C, Zuppinger C, Guo X, Kuster G, Helmes M, Eppenberger M, et al. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem. 2004;279:8290–8299. doi: 10.1074/jbc.M308033200. [DOI] [PubMed] [Google Scholar]
- 34.Springhorn J, Claycomb W. Preproenkephalin mRNA expression in developing rat heart and in cultured ventricular cardiac muscle cells. Biochem J. 1989;258:73–78. doi: 10.1042/bj2580073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiang F, Sakata Y, Cui L, Youngblood J, Nakagami H, Liao J, et al. Transcription factor CHF1/Hey2 suppresses cardiac hypertrophy through an inhibitory interaction with GATA4. Am J Physiol Heart Circ Physiol. 2006;290:1997–2006. doi: 10.1152/ajpheart.01106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam E, Zwacka R, Seftor E, Nieva D, Davidson B, Engelhardt J, et al. Effects of antioxidant enzyme overexpression on the invasive phenotype of hamster cheek pouch carcinoma cells. Free Radiac Biol Med. 1999;27:572–579. doi: 10.1016/s0891-5849(99)00109-4. [DOI] [PubMed] [Google Scholar]
- 37.Zwacka RM, Dudus L, Epperly MW, Greenberger JS, Engelhardt JF. Redox gene therapy protects human IB-3 lung epithelial cells against ionizing radiation-induced apoptosis. Hum Gene Ther. 1998;9:1381–1386. doi: 10.1089/hum.1998.9.9-1381. [DOI] [PubMed] [Google Scholar]
- 38.Davidson B, Allen E, Kozarsky K, Wilson J, Roessler B. A model system for in vivo gene transfer into the central nervous system using an adenoviral vector. Nat Genet. 1993;3:219–223. doi: 10.1038/ng0393-219. [DOI] [PubMed] [Google Scholar]
- 39.Burger W, Burge M. Digital imaging processing: an algorithmic introduction using Java. New York: Springer-Verlag; 2007. [Google Scholar]
- 40.Babior B. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 41.Li J-M, Mullen A, Yun S, Wientjes F, Brouns G, Thrasher A, et al. Essential role of the NADPH oxidase subunit p47phox in endothelial cell superoxide production in response to phorbol esters and tumor necrosis factor-a. Circ Res. 2002;90:143–150. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- 42.Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HHHW, Busse R, Schröder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 43.Lambeth JD, Krause K-H, Clark RA. NOX enzymes as novel targets for drug development. Semin Immunopathol. 2008;30:339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 44.Menon B, Johnson J, Ross RS, Singh M, Singh K. Glycogen synthase kinase-3b plays a pro-apoptotic role in b-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes: role of b1-integrin. J Mol Cell Cardiol. 2007;42:653–661. doi: 10.1016/j.yjmcc.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugden S, Fuller S, Weiss S, Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br J Pharmacol. 2008;153:S137–S153. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bánfi B, Clark RA, Steger K, Krause K-H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 47.Honoré S, Kovacic H, Pichard V, Briand C, Rognoni J-B. a2b1-integrin signaling by itself control G1/S transition in a human adenocarcinoma cell line (Caco-2) implication of NADPH oxidase-dependent production of ROS. Exp Cell Res. 2003;285:59–71. doi: 10.1016/s0014-4827(02)00038-1. [DOI] [PubMed] [Google Scholar]
- 48.Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161:933–944. doi: 10.1083/jcb.200211118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiarugi P. Survival or death: the redox parodox. Antioxid Redox Signal. 2009;11:2651–2654. doi: 10.1089/ars.2009.2770. [DOI] [PubMed] [Google Scholar]
- 50.Groeger G, Quiney C, Cotter T. Hydrogen peroxide as a cell survival signaling molecule. Antioxid Redox Signal. 2009;11:2655–2671. doi: 10.1089/ars.2009.2728. [DOI] [PubMed] [Google Scholar]
- 51.Sugden P, Clerk A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal. 2006;8:2111–2124. doi: 10.1089/ars.2006.8.2111. [DOI] [PubMed] [Google Scholar]
- 52.Cross D, Alessi D, Cohen P, Andjelkovich M, Hemmings B. Inhibition of glycogen synthase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 53.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maraldi T, Prata C, Fiorentini D, Zambonin L, Landi L, Hakim G. Induction of apoptosis in human leukemic cell line via reactive oxygen species modulation by antioxidants. Free Radiac Biol Med. 2009;46:244–252. doi: 10.1016/j.freeradbiomed.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 55.Lee RL, Westendorf J, Gold MR. Differential role of reactive oxygen species in the activation of mitogen-activated protein kinases and Akt by key receptors on B-lymphocytes: CD40, the B cell antigen receptor, and CXCR4. J Cell Commun Signal. 2007;1:33–43. doi: 10.1007/s12079-007-0006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frank GD, Eguchi S, Yamakawa T, Tanaka S, Inagami T, Motley ED. Involvement of reactive oxygen species in the activation of tyrosine kinase and extracellular signal-regulated kinase by angiotensin II. Endocrinology. 2000;141:3120–3126. doi: 10.1210/endo.141.9.7630. [DOI] [PubMed] [Google Scholar]
- 57.Pendayala S, Gorshkova I, Usatyuk P, He D, Pannathur A, Lambeth JD, et al. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal. 2009;11:747–764. doi: 10.1089/ars.2008.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ushio-Fukai M. Localizing NADPH oxidase-derived ROS. Sci STKE. 2006;re8 doi: 10.1126/stke.3492006re8. [DOI] [PubMed] [Google Scholar]
- 59.Oakley F, Abbott D, Li Q, Engelhardt J. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11:1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anilkumar N, Weber R, Zhang M, Brewer A, Shah A. Nox4 and Nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol. 2008;28:1347–1354. doi: 10.1161/ATVBAHA.108.164277. [DOI] [PubMed] [Google Scholar]
- 61.Bell RM, Cave AC, Johar S, Hearse DJ, Shah AM, Shattock MJ. Pivotal role of NOX2-containing NADPH oxidase in early ischemic preconditioning. FASEB J. 2005;19:2037–2039. doi: 10.1096/fj.04-2774fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.