

Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder frequent at old age characterized by atrophy of the nigrostriatal projection. Overexpression and A53T-mutation of the presynaptic, vesicle-associated chaperone alpha-synuclein are known to cause early-onset autosomal dominant PD. We previously generated mice with transgenic overexpression of human A53T-alpha-synuclein (A53T-SNCA) in dopaminergic substantia nigra neurons as a model of early PD. To elucidate the early and late effects of A53T-alpha-synuclein on the proteome of dopaminergic nerve terminals in the striatum, we now investigated expression profiles of young and old mice using two-dimensional fluorescence difference in gel electrophoresis (2D-DIGE) and mass spectrometry. In total, 15 proteins were upregulated and 2 downregulated. Mice before the onset of motor anomalies showed an upregulation of the spot containing 14-3-3 proteins, in particular the epsilon isoform, as well as altered levels of chaperones, vesicle trafficking and bioenergetics proteins. In old mice, the persistent upregulation of 14-3-3 proteins was aggravated by an increase of glial fibrillary acidic protein (GFAP) suggesting astrogliosis due to initial neurodegeneration. Independent immunoblots corroborated GFAP upregulation and 14-3-3 upregulation for the epsilon isoform, and also detected significant eta and gamma changes. Only for 14-3-3 epsilon a corresponding mRNA increase was observed in midbrain, suggesting it is transcribed in dopaminergic perikarya and accumulates as protein in presynapses, together with A53T-SNCA. 14-3-3 proteins associate with alpha-synuclein in vitro and in pathognomonic Lewy bodies of PD brains. They act as chaperones in signaling, dopamine synthesis and stress response. Thus, their early dysregulation probably reflects a response to alpha-synuclein toxicity.
Electronic supplementary material
The online version of this article (doi:10.1007/s00702-011-0717-3) contains supplementary material, which is available to authorized users.
Keywords: 14-3-3 protein, A53T-alpha-synuclein overexpression, Striatal proteome, Transgenic mice
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder mostly of elderly people which displays characteristic motor symptoms including muscular rigidity, resting tremor, slowing of movements and postural instability. The main cause of these motor symptoms is the progressive loss of the pigmented dopaminergic neurons of the substantia nigra (SN) pars compacta (Fahn 2003) which project to the striatum, whereas non-motor symptoms, such as hyposmia, sleep disorder, autonomic dysfunctions, depression and dementia mirror the progressive neuronal loss which starts from the enteric, autonomic and olfactory nervous systems via the brainstem to the cortex (Braak et al. 2003).
While the molecular causes are currently unknown in most cases of old-age PD, the underlying genetic factors have been elucidated in some rare familial cases, usually with earlier disease onset (Gasser 2001; Huang et al. 2004). In the past 13 years, 16 genes or chromosomal loci (PARK1-16) have been linked to familial PD, commencing with the discovery of an A53T missense mutation in the alpha-synuclein gene (SNCA) (Polymeropoulos et al. 1997) and the later description of SNCA gene duplication and triplication events as the cause of autosomal dominant early-onset PD (Singleton et al. 2003). Also for the pathogenesis of late-onset PD, a central role of alpha-synuclein was established by the observation that the majority of brain autopsies show pathological aggregation of alpha-synuclein as the main component of Lewy bodies (LBs) and Lewy neurites (Spillantini et al. 1997) in the absence of any SNCA mutations. Abnormal protein-inclusion bodies containing alpha-synuclein have since been demonstrated in several other neurodegenerative diseases, and alpha-synuclein aggregates are now the defining molecular feature for one class of human neurodegenerative diseases, the synucleinopathies (reviewed in Maries et al. 2003).
Alpha-synuclein is mainly a neuronal protein of 140 amino acids and is primarily concentrated in the nerve terminals both in soluble form and in association with membranes, especially with synaptic vesicles (Clayton and George 1998). The reversible binding to lipid membranes is mediated by the alpha-helical N-terminal domain consisting of seven repeats of 11-amino acids (consensus sequence XKTKEGVXXXX) (Bussell and Eliezer 2003). Alpha-synuclein was claimed to act as a small molecular chaperone assisting the SNARE (soluble N-ethylmaleimide sensitive fusion protein attachment receptor) protein complex during rapid rounds of vesicle exocytosis to refold into the necessary conformation (Chandra et al. 2005). Interestingly, the currently known missense mutations of SNCA (A30P, A53T, E46K) are situated in the lipid-binding motifs and are thought to facilitate a conformational shift from an alpha-helical to a beta-sheet structure (Narhi et al. 1999) that strengthens its association to synaptic vesicles, favors fibrillation and may finally lead to protein deposition (Conway et al. 1998; Kessler et al. 2003). The beta-sheet containing alpha-synuclein oligomers or fibrils, rather than the insoluble inclusion bodies such as LBs, were proposed to mediate the neurotoxicity via formation of pore-like structures that permeabilize membranes (Volles and Lansbury 2003), a process which might lead to pathological neurotransmission as well as affect mitochondria. Alpha-synuclein toxicity appears aggravated by mitochondrial dysfunction, oxidative stress, and finally leads to neurodegeneration (Schapira and Tolosa 2010).
The aggregation of alpha-synuclein and the process of neurodegeneration are mitigated in transgenic mice, where the mutant human alpha-synuclein protein coexists with the wild-type (WT) murine alpha-synuclein (Cabin et al. 2005). Indeed, our alpha-synuclein transgenic mouse lines do not show the end-stage features of human PD such as loss of nigrostriatal dopaminergic neurons and alpha-synuclein aggregates visible by light microscopy in affected neurons (Kahle et al. 2001; Gispert et al. 2003). They were selected in view of their approximately 1.5-fold overexpression of A53T-SNCA driven by the neuronal PrP promoter in the substantia nigra dopaminergic projection neurons and their absence of transgene expression from striatal neurons, with the aim to dissect the functional anomalies of the dopaminergic nerve terminals and postsynaptic neurons in the striatum prior to the onset of structural pathology. These mice have an interesting phenotype, since they display progressive Parkinsonian features such as impairment of spontaneous motor activity from the age of 6 months onward (Gispert et al. 2003). This locomotor syndrome correlates with progressive changes in the striatum, where an abnormally high DA content is accompanied by postsynaptic and glial efforts to maximize the DA signal suggesting a neurotransmission deficit in the presynaptic compartment. The cellular compensatory efforts appear insufficient, since a progressive loss of corticostriatal synaptic plasticity is observed (Kurz et al. 2010). A global transcriptome profile of the striatum in these mice at old age corroborated the prominent signaling alteration, demonstrated a reduced DA response of postsynaptic neurons and detected early changes in cell adhesion and in protein aggregation pathways (Kurz et al. 2010).
Since a global description of the protein network changes underlying this synaptic dysfunction in response to alpha-synuclein toxicity is lacking, we aimed to document the proteome of the striatal target region of the dopaminergic nigrostriatal nerve terminals by application of two-dimensional difference gel electrophoresis (2D-DIGE). 2D-DIGE is a powerful method to differentially display dysregulated proteins in a complex biological context. We assessed the progression of proteome changes from the early adult age of 6 months to the advanced age of 22 months, and identified in total 17 dysregulated proteins by mass spectrometry.
Materials and methods
Mice
Transgenic mice (the PrPmtA line on the inbred FVB/N background) expressing the human A53T-SNCA cDNA under control of the murine neuron-specific PrP promoter (Gispert et al. 2003) were used for differential proteome studies. The mice were housed at the FELASA-certified Central Animal Facility (ZFE) of Frankfurt Goethe University Medical School according to the animal husbandry guidelines of the German Animal Welfare Act, the Council Directive of 24 November 1986 (86/609/EWG) with Annex II and the ETS123 (European Convention for the Protection of Vertebrate Animals). All the experiments were conducted in conformity with the European Communities Council Directive of November 1986 (86/609/ECC).
The mice were bred to homozygosity and housed in individually ventilated cages under special-pathogen-free conditions. Studies were performed in 6 and 22 month-old mice and the respective age- and sex-matched controls (male, wild-type FVB/N, n = 5 each). After cervical dislocation, animals were decapitated, brains removed, striatal tissue dissected, directly frozen in liquid nitrogen, stored at −80°C and shipped on dry ice.
Dissection of striatum and midbrain
Immediately after the removal of the brain, an anterior block was isolated (including olfactory bulb, frontal cortex, striatum, thalamus, hypothalamus) by a coronal cut through the median eminence of the hypothalamus. The easily distinguishable structure of striatum and accumbens was separated from the surrounding cortex and basal ganglia using a curved forceps along axon bundles. In the corresponding posterior block, the remaining cortex and hippocampus was removed, a cut from the median eminence to the upper end of the superior colliculi was carried out, and the midbrain was isolated from brainstem and cerebellum with a second cut above the pons, according to the mouse brain atlas of Paxinos and Franklin (Paxinos and Franklin 2001).
Protein extraction and labeling
For 2D-DIGE, frozen tissue samples were transferred to a mortar cooled with liquid nitrogen, incubated with 1.2 volumes of lysis buffer (7 M urea, 2 M thiourea, 4% CHAPS, 30 mM Tris, pH 8.5) and ground to a fine powder. The suspension was transferred to a 1.5-ml reaction tube and mixed for 30 min at 4°C. The homogenate was centrifuged (30 min, 4°C, 226,000 g) (centrifuge: Beckman Optima, rotor: MLT130, 65,000 rpm) and the supernatant was carefully transferred to a 1.5-ml empty reaction tube. The pellet was poured back into the mortar, incubated with 1.2 volumes of lysis buffer and ground again. The process was repeated as described above and both supernatants were combined. The collected supernatants were extensively mixed and centrifuged again under the described conditions. The resulting supernatant was used for labeling after protein determination. The tagging of the proteins using the CyDye minimal dyes (GE Healthcare, Freiburg, Germany) was performed essentially according to Sitek et al. (2005). In brief, the dyes were added to the protein extract (8 pmol/g protein). An internal standard was prepared from all samples (equivalent amounts) included in this study and labeled with Cy2. After vortexing, centrifugation, and incubation (0.5 h in the dark), 10 mM l-lysine (1 l-lysine/400 pmol dye) (Sigma, St. Louis, USA) were admixed to stop the reaction (10 min in the dark). A potential dye bias as described by (Karp et al. 2007) was excluded by performing a preceding same–same experiment.
Two-dimensional fluorescence difference in gel electrophoresis
Proteins were separated by 2D-PAGE (polyacrylamide gel electrophoresis) essentially according to the method described by Stuhler et al. (2006). After rehydration of the IPG (immobilized pH gradient)-strips (24 cm, pH 3–11, NL) in rehydration buffer overnight (8 M urea, 2 M thiourea, 2% CHAPS, 0.8% Servalyte, 100 mM DTT) the fluorescently tagged proteins were loaded via cup loading (150 μg of protein, 50 μg of each labeled sample). Following focusing (at max. 20°C for 110 kVh as follows: 6 h at 100 V/step, 1 h at 300 V/gradient, 3 h at 300 V/step, 6 h at 1,000 V/gradient, 1.5 h at 6,000 V/gradient, 16 h at 6,000 V/step) and equilibration (15 min in equilibration buffer A: pH 8.8, 6 M urea, 50 mM Tris, 2% SDS, 30% glycerin, 65 mM DTT, 15 min in equilibration buffer B: pH 8.8, 15 min in equilibration buffer, pH 8.8, 6 M urea, 50 mM Tris, 2% SDS, 30% glycerin, 280 mM iodoacetamide), the IPG-strips were transferred to the top of a 12% SDS polyacrylamide gel (20 × 30 × 7 cm), sealed with agarose sealing solution (50 mM Tris–HCl, 0.1% SDS, 192 mM glycine, 0.3% (w/v) agarose, bromophenol blue) and run on a Desaphor VA 300 system (Klose and Kobalz 1995) at 75 mA per two gels at 20°C. The internal standard was a mixture of lysates from A53T-alpha-synuclein mice and control mice of one age stage. It implies there were in total two internal standards one for the 6 and one for the 22-month-old mice.
Scanning and image analysis
Gels were scanned using a Typhoon 9400 scanner (GE Healthcare). Excitation and emission wavelengths were chosen specifically for each dye according to the manufacturer’s recommendations. Images were preprocessed using the ImageQuant™ software. Intra-gel spot detection and inter-gel matching were performed using the Differential In-gel Analysis mode (Butor et al. 1993) and Biological Variation Analysis (BVA) mode of DeCyder™ software (all GE Healthcare), respectively. The spot intensities were normalized and differentially expressed protein spots were determined applying Student’s t test by pair-wise comparison of the transgenic with the respective control data. Every protein spot with p ≤ 0.05 and reproducible expression change in at least four of five independent experiments was considered as significantly differentially expressed and was subsequently analyzed by mass spectrometry (MS).
Protein identification by mass spectrometry
Protein spots were excised from the gel, washed, tryptically (Promega, Madison, WI, USA) digested overnight as described previously (Schaefer et al. 2003, 2006) and sequenced by MALDI-TOF/TOF–MS (matrix-assisted laser desorption/ionization—time-of-flight—mass spectrometry) and nanoLC–ESI–MS/MS (liquid chromatography directly coupled to electrospray ionization/tandem mass spectrometry).
MALDI-TOF/TOF–MS
Peptide extraction was done by sonication for 15 min in 15 μl 0.1% trifluoroacetic acid (TFA) followed by repeated extraction with 15 μl 0.1% TFA/100% ACN (acetonitrile) in case of MALDI-TOF/TOF analysis using the Ultraflex II (Bruker Daltonik, Bremen, Germany). The MALDI target was prepared as follows: ten microliters of α-cyano-4-hydroxycinnamic acid (HCCA) saturated matrix solution (in 97% acetone/3% TFA) was added to the anchor chip target and constantly drawn over 24 anchor positions. One microliter of the peptide extract was pipetted onto one prepared anchor. The dried sample was recrystallized using 0.7 μl recrystallisation solution (6:3:1, ethanol:acetone:washing solution; washing solution: 10 mM NH4H2PO4, 0.1% TFA) and subsequently washed for 1 min with a small droplet of washing solution. MALDI-TOF/TOF–MS analyses were performed using the following parameters: target-voltage: 25 kV, acceleration voltage: 21.8 kV, reflector voltage: 26.3 kV, detector voltage: 13.9 kV, lens voltage: 10.35 kV, laser-offset: 68 + 15%. MS- and fragmentation spectra were recorded manually. Data were collected using ProteinScape™ (Bruker Daltonik) (Chamrad et al. 2003; Stephan et al. 2006). Peptide mass fingerprinting (PMF) spectra interpretation was done using the Profound (Zhang and Chait 2000) and Mascot (Perkins et al. 1999) algorithms with the following parameters: 50 ppm mass tolerance, tryptic digest, two missed cleavage sites, carbamidomethylation at cysteine residues (+57 Da) (fixed), oxidation of methionine (variable). Proteins were considered as identified if the MetaScore (ProteinScape) (Chamrad et al. 2003) was higher than 3.0. Fragmentation spectra were analyzed with the SEQUEST™ (TurboSEQUEST v3.2) (Eng et al. 1994; Yates et al. 1995) and Mascot (version 2.0.0.4) search algorithms, respectively. The mass tolerance was set to 0.5 Da both for parent ions and fragment ions and the other parameters were set as described above. All searches were performed using the IPI.mouse.V3.05 protein sequence database (http://www.ebi.ac.uk/IPI/).
NanoLC–ESI–MS/MS
The extracted peptides were first pre-concentrated (0.3 mm i.d. × 1 mm, PepMap-column) and separated (75 μm i.d. × 150 mm, PepMap column) on the Ultimate HPLC System consisting of Famos™, Switchos™ and Ultimate™ (all Dionex LC Packings, Idstein, Germany) online coupled to a Finnigan LCQdeca XP ion trap mass spectrometer (ThermoElectron Finnigan, San Jose, USA). The parameters used for nanoLC–ESI–MS/MS were already described in detail (Schaefer et al. 2004). ESI-MS/MS-spectra that are not interpreted were correlated with the IPI.mouse.v3.71 protein sequence database (http://www.ebi.ac.uk/IPI/) applying both the Mascot (version 2.0.0.4) and the SEQUEST™ (TurboSEQUEST v3.2) algorithm with the same parameters as described above considering a mass tolerance of ±1.5 Da for both parent and fragment ions. Proteins were considered to be identified if at least two peptides were explained by the spectra and if the SEQUESTMetaScore (ProteinScape) was higher than 3.0.
Immunoblotting—chemiluminescent detection of 14-3-3
Following tissue homogenization with radioimmunoprecipitation assay (RIPA), buffer (50 mM TRIS pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS) supplemented with complete protease inhibitor cocktail (Roche, Mannheim, Germany), samples were incubated at 4°C, rotating for 20 min. Homogenates were centrifuged at 4°C for 15 min at 4,500 g and supernatants were collected. Twenty micrograms of protein were separated in 10% tris–glycine polyacrylamide gels and transferred onto a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Munich, Germany). Membranes were blocked for 1 h at room temperature (RT) in a commercially available ready-to-use blocking buffer (Rockland Immunochemicals Inc., Gilbertsville, PA, USA) and incubated with the primary antibodies (dilution 1:1,000) for the specific 14-3-3 protein isoforms [anti-14-3-3 epsilon (#sc-1020), anti-14-3-3 beta/alpha (#9636), anti-14-3-3 gamma (#9637), anti-14-3-3 tau (#9638), anti-14-3-3 zeta/delta (#9639) and anti-14-3-3 eta (#9640); with the exception of the 14-3-3 epsilon antibody (Santa Cruz Biotechnology/Santa Cruz, CA, USA)], all 14-3-3 antibodies were obtained from Cell Signaling Technology®/Danvers, MA, USA) or anti-beta-actin (dilution 1:15,000; (#A5441), Sigma-Aldrich/Hamburg, Germany) in ready-to-use blocking buffer at 4°C overnight (ON), washed thrice with PBS/Tween (0.1%) and incubated with the secondary antibody (alkaline phosphatase conjugated anti-rabbit antibody, 1:15,000 dilution (#NA934V, GE Healthcare, Munich, Germany) for 1 h at RT. After washing, the secondary antibody was detected with a chemiluminescent substrate (Pierce SuperSignal West Pico Chemiluminescent; Thermo Fisher Scientific Inc., Rockford, IL, USA). Densitometric analyses utilized TotalLab software (v.2.0; Amersham Biosciences, Freiburg, Germany).
Immunoblotting—infrared detection of GFAP, beta-actin, alpha-synuclein
Five micrograms of protein lysate were separated using 10% Bis–Tris gels according to manufacturer’s recommendations (Invitrogen, Karlsruhe, Germany). Proteins were electro-transferred to nitrocellulose membranes using semidry-blotting with a discontinuous buffer system and incubated in StartingBlock™ (Pierce, Woburn, MA, USA) for 2 h. Primary antibodies were diluted in TBS-buffer, the nitrocellulose membranes probed with the primary antibodies for 1 h and washed in TBS-buffer 3× for 20 min. The membranes were incubated with the fluorescent secondary antibody solution (0.5× Starting-Block™) for 1 h and finally washed in TBS-buffer 3× for 20 min. The fluorescence read out was performed on an Odyssey™ (LI-COR Biosciences GmbH, Bad Homburg, Germany). Beta-Actin was used as internal standard. Nonlinear data regression was done using the Origin™ software package (LI-COR). To control genotyping by immunoblot analyses, we applied an anti-human monoclonal antibody to alpha-synuclein (clone LB409; 1:2,000; Zymed®, Invitrogen) as well as the anti-mouse and anti-human monoclonal antibody to alpha-synuclein (1:2,000; clone 42; BD Transduction Laboratories™, San Diego, CA, USA). The anti-glial fibrillary acidic protein (GFAP) polyclonal antibody (1:2,000; Z0334) was purchased from DakoCytomation (Dako Germany GmbH, Hamburg, Germany), and the anti-mouse monoclonal antibody to beta-actin (1:10,000; clone AC-15) was from Sigma-Aldrich (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). Secondary anti-mouse IRDye800CW or Alexa® 680 antibodies (1:15,000), as well as the anti-rabbit Alexa® 680 antibody (1:30,000) were obtained from LI-COR.
Immunohistochemistry
Detection of 14-3-3 epsilon was carried out after heat antigen retrieval either through treatment in a Bull’s Eye Decloaker (BioCare Medical) (Fig. 5) or through microwave cooking in citrate buffer, with similar results. Endogenous peroxidase was then blocked with 3% hydrogen peroxide in Tris–HCl pH 7.6 for 30 min. After washing with 0.1% Tris–HCl and blocking with 10% goat serum, 0.5% PBST for 90 min at 25°C, sections were incubated with the primary antibody in 0.1% Tris–HCl overnight at 4°C. After washing with 0.1% Tris–HCl, sections were incubated with biotinylated goat anti-mouse (for 14-3-3 epsilon) or anti-rabbit (for GFAP) IgG (1:200; Vector Laboratories) for 90 min at 25°C. After washing with 0.1% Tris–HCl, sections were then incubated with Vectastain ABC (Vector Laboratories) for 2 h and finally washed with 0.1% Tris–HCl. Peroxidase activity was then detected with Vector Nova Red SK4800 (Linaris). Pictures were taken with 20× or 65× (with oil immersion) objectives on an Axiovert 200 m microscope, using an Axiocam CCD camera and the Axiovision Rel. 4.8 software.
Fig. 5.
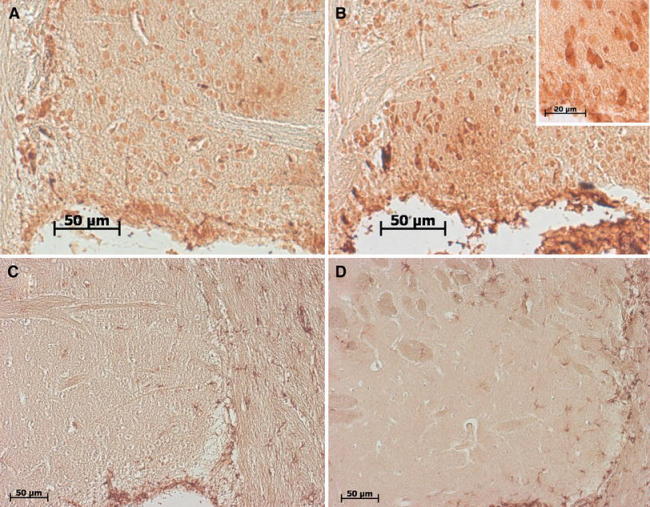
Immunohistochemical validation of upregulated striatal proteins. Striatal tissue from late age animals was analyzed. 14-3-3 epsilon staining showed neuronal immunoreactivity throughout the cytoplasm in wild-type (a) and A53T-SNCA overexpressing mice (b) (corpus callosum on the left side and the ventricle below). In the mutant tissue, the neuronal cytoplasmic staining was enhanced, in agreement with the proteome results, with stronger signal intensity at the cell border (see high-magnification inset). GFAP staining showed comparable astrogliosis throughout the corpus callosum of wild-type (c) and mutant (d) aged animals (corpus callosum on the right side and the ventricle below), while the astrogliosis in the striatum appeared enhanced in the A53T-SNCA overexpressing mice, again in concordance with the proteome data
Analyses of transcript levels via qPCR
Total RNA was extracted from midbrain derived from old mice with TRIZOL (Invitrogen, Karlsruhe, Germany) and digested with DNase (amplification grade I; Invitrogen) following the manufacturer’s instructions. In case of the striata derived from old animals, RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany) was used for RNA extraction. Two micrograms of DNase-treated RNA were reverse transcribed in a 36-μl reaction, using pd(N)6 and NotI-d(T)18 primers (First Strand cDNA Synthesis Kit; Amersham Biosciences, Munich, Germany).
For analyses of 14-3-3 protein expression on transcript level, all qPCR reactions were carried out in a total volume of 20 μl containing 5 μl cDNA template, 10 μl 2× TaqMan® Universal Master Mix (Applied Biosystems, Darmstadt, Germany) and 1 μl 20× TaqMan® gene expression assay (Applied Biosystems) on an ABI Prism 5700 sequence detection system (SDS5700; Applied Biosystems).
Expression levels of the transcript for TATA binding protein (Tbp) were used as endogenous control for normalization. Expression changes were analyzed with the 2−ΔΔCt method (Livak and Schmittgen 2001). The following commercially available TaqMan® assays were employed: Mm01247811_m1 (Ywhab = 14-3-3 beta transcript), Mm00777369_m1 and Mm03047428_m1 (Ywhag = 14-3-3 gamma transcript), Mm00494246_m1 (Ywhae = 14-3-3 epsilon transcript), Mm00834297_m1 and Mm01714763_gH (Ywhah = 14-3-3 eta transcript), Mm01231061_g1 (Ywhaq = 14-3-3 theta transcript), Mm01158417_g1 (Ywhaz = 14-3-3 zeta transcript) and Mm00446973_m1 (Tbp).
Statistical analysis
Statistical analysis of 14-3-3 validation via Immunoblotting and qPCR was performed using unpaired Student’s t tests via the Prism 3 software (GraphPad, La Jolla, CA, USA). Data are presented as mean ± SEM. Significant differences were highlighted with asterisks (*p < 0.05).
Results
Our approach to use adult, young and old A53T-alpha-synuclein overexpressing mouse striatal tissue to study the global proteome expression changes identified 17 dysregulated proteins which represent mainly signaling pathways as well as vesicle dynamics and bioenergetics.
We applied 2D-DIGE minimal labeling to striatal protein lysate from mice at age 6 and 22 months, with n = 5 each (control and A53T-alpha-synuclein mice) in order to exclude effects in differential protein expression due to general biological variances. Altogether we could identify 17 differentially expressed proteins with consistency and high statistical relevance (p ≤ 0.05). Fifteen of those proteins were upregulated and two were downregulated (Fig. 1).
Fig. 1.
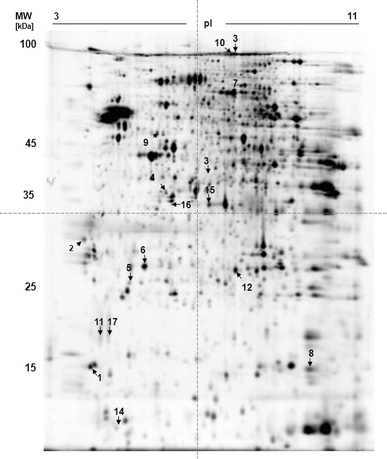
2D-DIGE gel of striatal cell lysate 2D-DIGE of cytosolic whole-cell striatal lysate (150 μg) of A53T-alpha-synuclein-overexpressing mice (PrPmtA line on the inbred FVB/N background) versus controls after CyDye minimal fluorescent labeling revealed in total 17 protein spots (indicated by arrows) with a significant expression change. All protein spots could be identified. The respective identification data are summarized in Table 1
Differential protein spots were picked from the gels, washed and proteins were tryptically digested. All 17 proteins could be identified using LC-ESI–MS (Table 1). Identified proteins are grouped with respect to a change in expression at either early (6 months) or late age (22 months).
Table 1.
Proteins identified in striata of cytosolic lysates of A53T transgenic mice by nanoLC–ESI–MS/MS analysis
| # | Protein name | SWISS-PROT accession number | MW (kDa) | Fold change TG/WT in 6-month-old mice | Fold change TG/WT in 22-month-old mice | Student’s t test p values of 6-month-old mice | Student’s t test p values of 22-month-old mice | [Peptides]2+ sequenced; sequence coverage [%] | Location | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Cytosol | ||||||||||
| 1 | Alpha-synuclein (human) | P37840 | 14.5 | 4.65 | 5.15 | 0.0045 | 0.00079 | 5; 60.7 | Cy, PM |
Highly expressed in presynaptic nerve terminals. May be involved in the regulation of dopamine release and transport Molecular chaperone. Anti-apoptotic, in high concentration accumulated to fibrils with pro-apoptotic effect. Interacts with UCHL1 |
| 2 | 14-3-3 epsilon | P62259 | 29.2 | 4.68 | 11.71 | 0.43 | 0.00015 | 17; 54.9 | Cy |
Adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways Modulated in neuronal differentiation |
| 3 | Glial fibrillary acidic protein | P03995 | 49.9 | – | 3.27 | – | 0.008 | 20; 41.9 | Cy | Class-III intermediate filament, is a cell-specific marker that, during the development of the central nervous system, distinguishes astrocytes from other glial cells |
| 4 | Serine (or cysteine) proteinase inhibitor, clade B, member 1a | Q9D0S8 | 42.6 | – | 1.17 | – | 0.02 | 2; 7.7 | Cy | Regulates the activity of the neutrophil proteases and thus, forms complexes with chymotrypsin, elastase, cathepsin G and proteinase-3 |
| 5 | Mu-crystallin homolog | O54983 | 33.5 | – | −1.5 | – | 0.017 | 8; 22.4 | Cy | Accumulates in the brains of patients with Alexander’s disease; HSP 20 family |
| Cytoskeleton | ||||||||||
| 6 | Isoform 4 of Dynamin-1 | P39053-4 | 97.8 | 1.31 | – | 0.0066 | – | 8; 19.0 | Cy, E, V | Microtubule-associated force-producing protein. Participates in various endocytotic events and vesicular traffic |
| 7 | Cofilin 1 (non-muscle) | P18760 | 18.6 | – | 1.79 | – | 0.0336 | 5; 19.2 | Cy, Nu | Major component of intranuclear and cytoplasmic actin rods. Controls reversibly actin polymerization and depolymerization in a pH-sensitive manner |
| Mitochondria | ||||||||||
| 8 | Ubiquinol-cytochrome c reductase complex 11 kDa protein, mitochondrial precursor | P99028 | 10.4 | 1.20 | – | 0.00048 | – | 5; 23.6 | Mt |
Part of the mitochondrial respiratory chain. May mediate formation of the complex between cytochromes c and c1 |
| Plasma membrane | ||||||||||
| 9 | Ezrin | P26040 | 69.4 | – | 1.29 | – | 0.036 | 6; 15.0 | PM | Probably involved in connections of major cytoskeletal structures to the plasma membrane. Expressed in cerebrum and cerebellum, mostly in emryonal stage E5, E8, E11 and E12 |
| Protein degradation | ||||||||||
| 10 | Proteasome subunit alpha type 3 | O70435 | 28.4 | – | 1.32 | – | 0.026 | 10; 29.4 | Cy, Nu |
Proteasome component C8. Changes in the ubiquitin–proteasome system are linked to neurodegeneration |
| 11 | Ubiquitin carboxyl-terminal hydrolase isoenzyme L1 | Q9R0P9 | 24.8 | – | 1.19 | – | 0.07 | 6; 26.9 | Cy |
Ubiquitin-protein hydrolase involved both in the processing of ubiquitin precursors and of ubiquitinated proteins. Mutation in UCHL1 causes PARK5 mutation |
| 12 | Charged multivesicular body protein 4b | Q9D8B3 | 24.9 | 1.20 | – | 0.033 | – | 3; 17.9 | Cy |
Component of the ESCRT-III complex, which is required for multivesicular bodies (MVBs) formation and sorting of endosomal cargo proteins into MVBs The MVB pathway mediates delivery of transmembrane proteins into the lumen of the lysosome for degradation |
| Energy and nucleotide metabolism | ||||||||||
| 13 | Glycogen phosphorylase, brain form | Q8CI94 | 96.7 | 1.31 | – | 0.0066 | – | 6; 8.9 | Cy | Involved in carbohydrate metabolism |
| 14 | Triosephosphate isomerase | P17751 | 26.7 | 1.27 | – | 0.025 | – | 11; 58.8 | Cy | From carbohydrate biosynthesis and gluconeogenesis. Belongs to the endogenously nitrated proteins in mouse brain which are considered to be linked to neurodegenerative diseases |
| 15 | Inosine triphosphate pyrophosphatase | Q9D892 | 21.9 | – | 1.26 | – | 0.0032 | 3; 10.6 | Cy | Involved in nucleotide biosynthesis |
| 16 | Malate dehydrogenase, mitochondrial | P08249 | 35.6 | – | 1.20 | – | 0.0034 | Mt |
Has oxidoreductase activity. From the tricarboxylic acid cycle |
|
| 17 | l-lactate dehydrogenase B chain | P16125 | 36.4 | – | −1.34 | – | 0.0018 | 14; 36.6 | Cy | Involved in the final step of the anaerobic glycolysis |
Cy cytosol, E endosome, ECM extra cellular matrix, ER endoplasmic reticulum, Go golgi apparatus, Nu nucleus, Mt mitochondrion, MW molecular weight, PM plasma membrane, Ri ribosome, V vesicles
Proteome changes at young age
At the age of 6 months, seven non-redundant proteins were slightly or moderately upregulated in the transgenic compared to control mice, representing different functional pathways or subcellular localizations:
Changes of 14-3-3 chaperones
The main change concerned the transgenic human A53T-alpha-synuclein, which was (as expected) absent in controls and strongly expressed in transgenic tissue, while no significant changes were detected for murine endogenous alpha-synuclein [as observed previously, see Kurz et al. 2010] protein, consistent with unchanged brain mRNA levels for murine endogenous alpha-synuclein (data not shown). The 14-3-3 protein spot was found upregulated to a comparable degree. An even higher upregulation of the 14-3-3 protein spot was observed at the age of 22 months (Fig. 2a). In the present and previous proteomic studies of mouse models for neurodegenerative diseases, the effects of the transgene on the overall brain proteome (even when analyzing histologically homogeneous regions) produced relatively low effects [e.g., (Palacino et al. 2004) and own unpublished results]. Thus, the high regulation factor for 14-3-3 protein suggests an important and selective effect. To elucidate the identity of 14-3-3 protein isoforms responding to A53T-alpha-synuclein overexpression, mass spectrometric analysis was performed and indicated 14-3-3 epsilon to be the differentially expressed isoform (Fig. 2a/b).
Fig. 2.
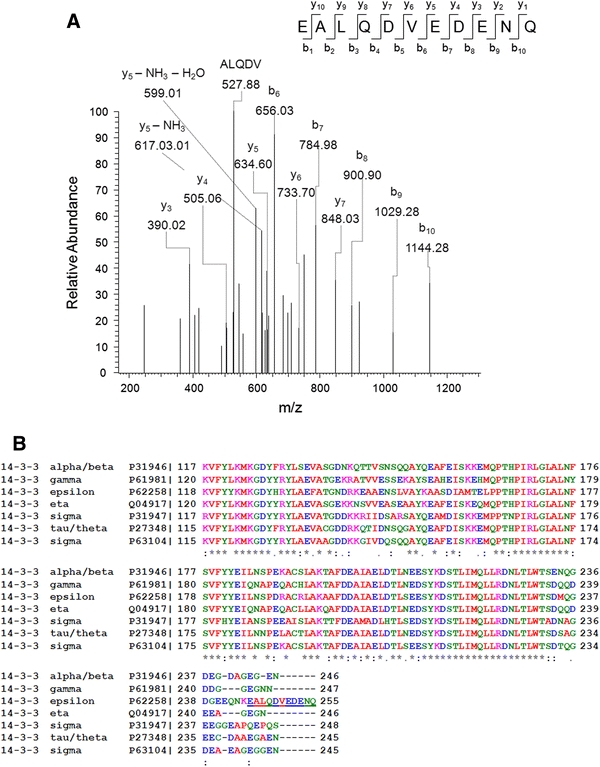
Detailed description of the expression changes of 14-3-3 proteins. a Mass spectrum of a peptide identifying 14-3-3 epsilon. Mass spectrometric analysis of the protein spot 2 (see Fig. 1) unequivocally identified the epsilon isoform of 14-3-3 protein family based on the C-terminal peptide sequence EALQDVEDENQ. b Alignments of the C-termini of several 14-3-3 protein isoforms demonstrate that the peptide identified by MS/MS (a) corresponds to the epsilon isoform of the 14-3-3 protein family
Changes of proteins involved in synaptic vesicle trafficking
For all other proteins, much smaller expression change ratios were observed in comparison, but always a consistent upregulation was found (Table 1). The elevated levels of vesicle fission protein dynamin-1 isoform 4 that mediates endocytosis (Ferguson et al. 2007) can be expected to affect signaling, similar to 14-3-3. An upregulation was also found for Chmp4b (charged multivesicular body protein 4b) which is implicated in vesicle dynamics.
Changes of proteins related to bioenergetics
Particularly noteworthy were the triosephosphate isomerase (TPI), a protein engaged in glycolysis and the mitochondrial tricarboxylic acid cycle, and the glycogen phosphorylase, which is involved in energy mobilization from astrocytic glycogen reserves (Wiesinger et al. 1997), as they both suggest an alteration of glycolytic flux in the mutant striatal tissue. The upregulation of mitochondrial ubiquinol-cytochrome C reductase complex 11 kDa protein also suggests altered bioenergetic turnover.
Proteome changes at late age
At the age of 22 months, 12 non-redundant proteins were slightly or moderately upregulated and 2 were downregulated compared to controls.
Changes of 14-3-3 chaperones
An even higher upregulation of the 14-3-3 protein spot was observed at this advanced age (Table 1). To validate this result, antibodies for all 14-3-3 protein isoforms were employed in immunoblots in order to assess the expression levels in old striatum from independent animals (Fig. 3a and Supplementary Figure 1). A significant upregulation was confirmed for 14-3-3 epsilon. For further validation, transcript levels of the different 14-3-3 isoforms were quantified in additional animals. Both the striatum and the midbrain were analyzed, since the axons and the presynaptic compartments of dopaminergic A53T-alpha-synuclein-overexpressing neurons projecting to the striatum are almost devoid of mRNA, while their cell bodies located in the midbrain are the site of most mRNA translation. Indeed, a significant upregulation was again observed for the 14-3-3 epsilon isoform with commercial quantitative real-time reverse-transcriptase-PCR (qPCR) assays in the midbrain tissue, but not in the striatum (Fig. 3b). Immunohistochemical analyses of striatal tissue at late age were compatible with a neuronal localization of 14-3-3 epsilon and showed increased 14-3-3 epsilon signals at the neuronal cytoplasmic rim in transgenic mice (Fig. 5). Thus, different technical approaches in independent animal groups all support the selective dysregulation of the 14-3-3 epsilon isoform. The protein levels of 14-3-3 alpha/beta, gamma, delta/zeta, theta/tau and eta isoforms were also analyzed, revealing a significant upregulation also for 14-3-3 eta, as well as a significant downregulation for 14-3-3 gamma (Fig. 3c). In the midbrain of our A53T-SNCA mice, the mRNA levels of the gamma, theta and eta 14-3-3 isoforms were not significantly altered (data not shown), in contrast to a previous report on epsilon, gamma and theta mRNA downregulation in the cortex of transgenic alpha-synuclein mice (Yacoubian et al. 2011).
Fig. 3.
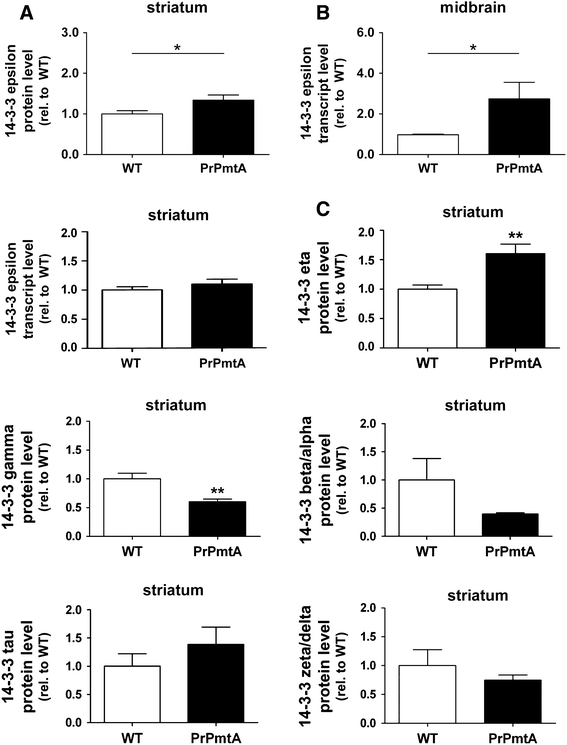
Validation of elevated 14-3-3 epsilon levels. The overexpression of 14-3-3 epsilon detected in the 2D-DIGE study was validated with independent techniques in the nigrostriatal projection via immunoblotting and qPCR analyses. a Analysis of 14-3-3 epsilon protein levels in the striatum via immunoblotting confirmed increased 14-3-3 epsilon expression. Beta-actin was used as endogenous control for normalization. b Analysis of 14-3-3 epsilon transcript levels in the midbrain via qPCR showed significantly increased 14-3-3 epsilon expression, while the levels in the striatum were not increased. TATA-binding protein (Tbp) was used as endogenous control. c Analysis of protein levels of the different 14-3-3 isoforms in aged striatum via immunoblotting demonstrated significantly increased 14-3-3 eta levels and significantly decreased 14-3-3 gamma levels. Data sets were normalized to the corresponding WT mean values, Student’s t test was applied for statistical analyses and significant alterations were highlighted with asterisks (*p < 0.05). N = 3–8 animals/genotype, a shows the combined data derived from two independent experiments
Changes of aggregation and cytoskeleton proteins
The consistently increased expressions of UCH-L1 (ubiquitin carboxy-terminal hydrolase isoenzyme L1), Psma3 (proteasome subunit alpha type 3), Serpin B1a (serine/cysteine proteinase inhibitor clade B member 1a, being a member of a gene family implicated in several conformational diseases as well as PD, Crowther 2002) and Mu-crystallin homolog at this advanced age represent selective changes of protein degradation/aggregation molecules, and might reflect the dysfunction of the ubiquitin/proteasome system in later stage PD (Olanow and McNaught 2006). Besides 14-3-3, the upregulated astocytic cytoskeletal protein glial fibrillary acidic protein (GFAP) in old transgenic animals compared to control animals represents the second strong expression change and is of high interest, since it is a well-established biomarker of astrocyte proliferation that responds in a sensitive manner to neural degeneration processes during aging and in brain degeneration disorders. This GFAP increase was detected on 2D gels (Table 1) and validated by immunoblotting (Fig. 4) as well as immunohistochemistry (Fig. 5). A slight upregulation was also observed for the actin-cytoskeleton interactor ezrin.
Fig. 4.
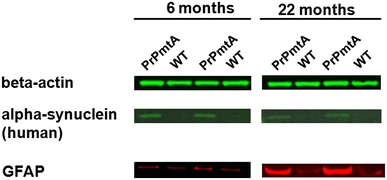
Immunoblot analyses of GFAP protein levels in the striatum of A53T-alpha-synuclein-overexpressing mice. The overexpression of glial fibrillary acidic protein (GFAP) detected in the 2D-DIGE study was validated by immunoblotting using Odyssey near-IR detection. Immunoblot analysis showed a several-fold increase of total GFAP in the 22-month-aged PrPmtA mice. Beta-actin was used as internal control, and human alpha-synuclein discriminated transgenic from WT mice
Discussion
Studying the striatal brain proteome of A53T-alpha-synuclein overexpressing mice at young and old age with 2D-DIGE and mass spectrometry, we found strong evidence for an early and progressive upregulation of the molecular chaperone 14-3-3 epsilon suggesting presynaptic signaling pathology, and a modest upregulation of the astrocytic cytoskeleton protein GFAP at old age suggesting some neurodegenerative changes to take place in the striatum of this PD mouse model. Other expression changes were observed in early adult life for proteins representing chaperones, vesicle trafficking and bioenergetics pathways, while changes in protein aggregation pathways became apparent in later life. Although these changes were too slight for independent validation by immunoblotting, they showed consistency in independent mice.
Changes of 14-3-3 chaperones as an early consequence of alpha-synuclein toxicity
The most relevant result of our study is the upregulation of 14-3-3 epsilon mRNA in the region containing the dopaminergic nigral neurons and of the corresponding protein in the region containing the dopaminergic terminals. 14-3-3 epsilon is known to be associated with synaptic membranes (Martin et al. 1994), similar to alpha-synuclein (Clayton and George 1998). Our data also indicate 14-3-3 epsilon to be transported to the presynapse, possibly in association with the pathogenic alpha-synuclein, and to follow its expression change. This seems plausible, since a co-localization of alpha-synuclein and 14-3-3 protein was previously observed in homozygous A53T-alpha-synuclein transgenic mice with severe movement disorder and neuronal synucleinopathy (Shirakashi et al. 2006). Furthermore, in vitro models showed that elevated alpha-synuclein leads to an increase of 14-3-3 protein and demonstrated the association of both proteins within 54–83 kDa complexes which mediate DA neurotoxicity in susceptible neurons (Xu et al. 2002). Our data are consistent with these findings and extend them, indicating that upregulation of the signaling modulator 14-3-3 protein in response to A53T-alpha-synuclein overexpression is a prominent effect in vivo already at an age where only subtle behavior impairment is noticeable, well before the advent of detectable neurodegeneration in the brain. In addition, our data clearly identify the principal 14-3-3 isoform, while both studies cited above failed to define the disease stage and the specific 14-3-3 protein variants. The 14-3-3 protein family includes six brain-expressed proteins forming homo- and heterodimers (beta/alpha, gamma, epsilon, delta/zeta, theta/tau, eta), which are encoded by different genes (YWHAB, YWHAG, YWHAE, YWHAZ, YWHAQ, YWHAH). Interestingly, 14-3-3 epsilon was previously found dysregulated also in mice with deficiency of alpha-synuclein (Chandra et al. 2004). This suggests that through their protein interaction, 14-3-3 epsilon senses the physiological function of alpha-synuclein and responds to any dysfunction by a corresponding expression change. Furthermore, our data show that 14-3-3 epsilon increases its expression with advancing pathology, and a previous report found it by immunohistochemistry in the LB halo of PD patient brains (Berg et al. 2003) suggesting it to be accumulating as a biomarker of Parkinson progression. The relevance of 14-3-3 proteins for the pathogenesis of PD seems to extend beyond alpha-synuclein, since they were recently identified as interactors of LRRK2, another disease gene responsible of autosomal dominant PD variants which has been implicated in phospho-signaling (Dzamko et al. 2010; Nichols et al. 2010; Rudenko and Cookson 2010; Titz et al. 2010). Thus, all available data point to an important role of this particular isoform for alpha-synuclein physiological function and for Parkinson neurotoxicity.
What is the functional impact of this 14-3-3 protein dysregulation? 14-3-3 proteins are signal transducers and were initially discovered as biosynthesis activators of neurotransmitters such as DA, leading to their synonymous name tyrosine-3-monooxygenase/tryptophan-5-monooxygenase activation proteins. It is important to note that 14-3-3 proteins share physical and functional properties with alpha-synuclein (Ostrerova et al. 1999), constituting possibly the basis for related chaperone functions. They were detected in protein association with alpha-synuclein (Xu et al. 2002), and in interaction with tyrosine hydroxylase (TH), the rate-limiting enzyme of DA biosynthesis (Perez et al. 2002). While 14-3-3 proteins activate TH, wild-type alpha-synuclein was observed to dose-dependently inhibit TH and DA synthesis (Perez et al. 2002) and may thus act as an antagonist. The notion of 14-3-3 upregulation to act as a cellular compensation of alpha-synuclein toxicity is supported by in vitro data which show the pharmacological inhibition of 14-3-3 proteins to enhance neurotoxic vulnerability, whereas 14-3-3 overexpression in C. elegans has a protective effect (Yacoubian et al. 2011). These properties of 14-3-3 proteins may exert a relevant effect in our A53T-alpha-synuclein-overexpressing mice, since significantly elevated striatal DA levels were previously observed in these animals from the age of 6 months onward (Kurz et al. 2010), a finding that could not be explained by the overexpression of A53T-alpha-synuclein, but is easily understood as a correlate of 14-3-3 protein upregulation. Thus, our documentation of a consistent upregulation of 14-3-3 epsilon in 2D-DIGE analyses, mass spectrometry, immunoblots and qPCRs explains the previously reported early increase of striatal DA in these mice. Regarding the contrasting changes of 14-3-3 gamma, which showed decreased protein levels in the striatum in spite of unchanged mRNA levels in the substantia nigra, it is possible that this isoform is sequestered into insolubility through the aggregation process of alpha-synuclein detectable in the striatum of our mice (Kurz et al. 2010).
Changes of proteins involved in synaptic vesicle trafficking
In the context of synaptic dysfunction, the small expression alterations of two proteins involved in synaptic vesicle trafficking, the dynamin-1 isoform 1 and the charged multivesicular body protein 4b, might be a specific early consequences of A53T-SNCA overexpression. Alpha-synuclein has been claimed to act as a chaperone for the SNARE complex and thus modulate neurotransmitter vesicle dynamics in the presynaptic compartment (Chandra et al. 2005). Data from our mouse model and from other alpha-synuclein mice suggest a scenario of progressive synaptic failure, where SNARE proteins redistribute pathologically and neurotransmitter is no longer released appropriately (Gillardon et al. 2008; Garcia-Reitbock et al. 2010), with concomitant pre- and postsynaptic changes in the neurotransmitter biosynthesis, degradation and receptor levels (Kurz et al. 2010). Thus, the two proteins identified might represent this vesicle exocytosis/endocytosis/trafficking pathology.
Changes of proteins related to bioenergetics
In contrast, the apparent bioenergetic alteration is likely an unspecific reflection of cell stress. Only the very early increase of the triosephosphate isomerase might be specific, since loss-of-function mutations of this protein lead to dystonia and neurodegenerative disease (Poll-The et al. 1985; Ahmed et al. 2003).
Regulation of protein aggregation markers and the neurodegeneration marker GFAP
It is interesting that elevated expression of UCH-L1 was found in the striatum of the old mice with manifest locomotor phenotype, since mutations affecting the activity of UCH-L1 have previously been shown to constitute a risk factor for the development of PD, leading to the recognition of the UCH-L1 gene as PARK5 (Leroy et al. 1998; Setsuie and Wada 2007). The altered levels of three additional aggregation proteins is consistent with our previous report about dysregulation of several gene transcripts involved in alpha-synuclein aggregation at this age in the striatum of these mice, such as Fkbp1a, Sept4, Tmeff2 and the alpha-synuclein interactor Sncaip (Kurz et al. 2010), and our demonstration of increased insolubility of alpha-synuclein in old nigrostriatal tissue of these mice (Kurz et al. 2010). This suggests that alpha-synuclein aggregation is a feature of disease progression in these mice, becoming apparent together with subtle signs of neurodegeneration such as the GFAP upregulation observed. This upregulation marks the hypertrophy of astrocytes in an effort to fill the gaps left behind by the insidious atrophy of neuronal processes such as the dopaminergic presynapses in the striatum.
Synopsis of alpha-synuclein overexpression triggered proteome changes
Proteome studies have the potential to identify novel key molecules in a pathological process such as PD (Licker et al. 2009), but invariably represent only a small part of the huge proteome complexity and are known to suffer from low reproducibility. This is well illustrated by the previous studies into proteomic consequences of alpha-synuclein overexpression. In the Drosophila melanogaster fly model, a concordant expression change at the protein and mRNA level was observed only for the GTP cyclohydrolase gene (Xun et al. 2007b), which is involved in dopamine biosynthesis together with the 14-3-3 proteins, but this dysregulation at postnatal day 1 was no longer observed at later ages. The protein showing the most reproducibly altered levels at days 10, 30, 50 and 60 in the same fly model, elongation factor 1a48D, was reported upregulated at two ages and downregulated at two ages (Xun et al. 2007a). However, none of these changes reappeared when the same team studied the A53T mutant instead of the A30P mutant (Xun et al. 2008). A concordant upregulation of the levels of several ribosomal proteins in this latter study contrasts with a reported downregulation of several ribosomal proteins by human alpha-synuclein in Caenorhabditis elegans (Ichibangase et al. 2008). This lack of reproducibility is worsened by differing mutation effects, transgenic overexpression patterns and levels, as well as a technical variance up to 33% just by inconsistent sample preparation/fractionation and detection methods (Baumann et al. 2005). Proteome studies of the mouse brain have therefore focused on the carbonylated proteins only (Poon et al. 2005) or on the synaptosomal proteins only (Gillardon et al. 2008) and emphasized the importance of mitochondrial protein pathology, in agreement with a proteome study in mitochondrial fractions of a cell culture model (Pennington et al. 2010).
In contrast, our study used non-fractionated striatal tissue from the mouse brain, accepting the disadvantage that synaptic/neural effects of A53T-alpha-synuclein overexpression would be diluted by the tissue heterogeneity and in particular the glial proteome, increasing the threat of false-negative data. In exchange, we sought to obtain the overview and to elucidate, which effects of A53T-alpha-synuclein overexpression dominate throughout the tissue at early and late stages, thus enabling us to strengthen the proteome data through independent validation by Western blots and histology from brain tissue. The predominant effect of A53T-alpha-synuclein overexpression on 14-3-3 levels in the striatum already at early age in our transgenic mouse model is reflected by two independent proteome studies of cells exposed to elevated alpha-synuclein. Rat mesencephalic/neuroblastoma hybrid cells with alpha-synuclein aggregation after oxidative stress were found to have decreased 14-3-3 levels in Nonidet P-40 soluble fractions, among many other effects such as altered chaperone levels (Zhou et al. 2004). In a separate investigation, brain microglia cells cultured with recombinant nitrated alpha-synuclein added to the medium displayed a reduction of 14-3-3 protein levels, again besides effects on chaperones and bioenergetics (Reynolds et al. 2009). Both cell culture studies are compatible with the notion of a direct interaction and sequestration of these proteins at short term. According to our mouse study, in the long-term a transcriptional regulation of 14-3-3 levels seems a main consequence of alpha-synuclein gain-of-function. Thus, in spite of inherent difficulties of proteome studies in animal or cell models of disease, the altered levels of 14-3-3 in response to alpha-synuclein toxicity seem to be among the rare reproducible and validated findings.
Taken together, our data indicate that A53T-alpha-synuclein overexpression in the mouse striatum early on leads to a strong upregulation of 14-3-3 epsilon signaling together with slight alteration of synaptic vesicle turnover and glycolysis, while later the progression of toxicity is apparent by the modest astrocyte proliferation and by subtle elevation of protein aggregation molecules.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors were supported by the Federal Ministry of Education and Research (BMBF) within the framework of the National Genome Research Network (NGFN2 and NGFNplus) (Förderkennzeichen 01GR0440 and 01GS08138) and by the DFG (GI342/1-1 and 1-2). We are grateful to Birgitt Meseck–Selchow for technical support.
Conflict of interest
The authors declare no financial/commercial conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
A. Kurz, C. May, G. Auburger and K. Marcus contributed equally to this work.
Contributor Information
Georg Auburger, Phone: +49-69-63017428, FAX: +49-69-63017142, Email: auburger@em.uni-frankfurt.de.
Katrin Marcus, Phone: +49-234-3228444, FAX: +49-234-3214554, Email: katrin.marcus@rub.de.
References
- Ahmed N, Battah S, Karachalias N, Babaei-Jadidi R, Horanyi M, Baroti K, Hollan S, Thornalley PJ. Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochim Biophys Acta. 2003;1639:121–132. doi: 10.1016/j.bbadis.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Baumann S, Ceglarek U, Fiedler GM, Lembcke J, Leichtle A, Thiery J. Standardized approach to proteome profiling of human serum based on magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chem. 2005;51:973–980. doi: 10.1373/clinchem.2004.047308. [DOI] [PubMed] [Google Scholar]
- Berg D, Holzmann C, Riess O. 14–3-3 proteins in the nervous system. Nat Rev Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329:763–778. doi: 10.1016/S0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- Butor C, Diaz S, Varki A. High level O-acetylation of sialic acids on N-linked oligosaccharides of rat liver membranes. Differential subcellular distribution of 7- and 9-O-acetyl groups and of enzymes involved in their regulation. J Biol Chem. 1993;268:10197–10206. [PubMed] [Google Scholar]
- Cabin DE, Gispert-Sanchez S, Murphy D, Auburger G, Myers RR, Nussbaum RL. Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiol Aging. 2005;26:25–35. doi: 10.1016/j.neurobiolaging.2004.02.026. [DOI] [PubMed] [Google Scholar]
- Chamrad DC, Koerting G, Gobom J, Thiele H, Klose J, Meyer HE, Blueggel M. Interpretation of mass spectrometry data for high-throughput proteomics. Anal Bioanal Chem. 2003;376:1014–1022. doi: 10.1007/s00216-003-1995-x. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Sudhof TC. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci USA. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/S0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Crowther DC. Familial conformational diseases and dementias. Hum Mutat. 2002;20:1–14. doi: 10.1002/humu.10100. [DOI] [PubMed] [Google Scholar]
- Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, Nichols RJ. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14–3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JM, McCornack AL, Yates JR., 3rd An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wolfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, O’Toole E, Flavell R, Cremona O, Miesenbock G, Ryan TA, De Camilli P. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Garcia-Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della Corte L, Spano P, Tofaris GK, Goedert M, Spillantini MG. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain. 2010;133:2032–2044. doi: 10.1093/brain/awq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser T. Genetics of Parkinson’s disease. J Neurol. 2001;248:833–840. doi: 10.1007/s004150170066. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Mack M, Rist W, Schnack C, Lenter M, Hildebrandt T, Hengerer B. MicroRNA and proteome expression profiling in early-symptomatic alpha-synuclein (A30P)-transgenic mice. Proteomics. 2008;2:697–705. doi: 10.1002/prca.200780025. [DOI] [PubMed] [Google Scholar]
- Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, Nussbaum RL. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–429. doi: 10.1016/S1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- Huang Y, Cheung L, Rowe D, Halliday G. Genetic contributions to Parkinson’s disease. Brain Res Brain Res Rev. 2004;46:44–70. doi: 10.1016/j.brainresrev.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Ichibangase T, Saimaru H, Takamura N, Kuwahara T, Koyama A, Iwatsubo T, Imai K. Proteomics of Caenorhabditis elegans over-expressing human alpha-synuclein analyzed by fluorogenic derivatization-liquid chromatography/tandem mass spectrometry: identification of actin and several ribosomal proteins as negative markers at early Parkinson‘s disease stages. Biomed Chromatogr. 2008;22:232–234. doi: 10.1002/bmc.931. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar HA, Haass C. Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol. 2001;159:2215–2225. doi: 10.1016/S0002-9440(10)63072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp NA, McCormick PS, Russell MR, Lilley KS. Experimental and statistical considerations to avoid false conclusions in proteomics studies using differential in-gel electrophoresis. Mol Cell Proteom. 2007;6:1354–1364. doi: 10.1074/mcp.M600274-MCP200. [DOI] [PubMed] [Google Scholar]
- Kessler JC, Rochet JC, Lansbury PT., Jr The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry. 2003;42:672–678. doi: 10.1021/bi020429y. [DOI] [PubMed] [Google Scholar]
- Klose J, Kobalz U. Two-dimensional electrophoresis of proteins: an updated protocol and implications for a functional analysis of the genome. Electrophoresis. 1995;16:1034–1059. doi: 10.1002/elps.11501601175. [DOI] [PubMed] [Google Scholar]
- Kurz A, Double KL, Lastres-Becker I, Tozzi A, Tantucci M, Bockhart V, Bonin M, García-Arencibia M, Nuber S, Schlaudraff F, Liss B, Fernández-Ruiz J, Gerlach M, Wüllner U, Lüddens H, Calabresi P, Auburger G, Gispert S. A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS ONE. 2010;5:e11464. doi: 10.1371/journal.pone.0011464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Licker V, Kovari E, Hochstrasser DF, Burkhard PR. Proteomics in human Parkinson’s disease research. J Proteom. 2009;73:10–29. doi: 10.1016/j.jprot.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. The role of alpha-synuclein in Parkinson’s disease: insights from animal models. Nat Rev Neurosci. 2003;4:727–738. doi: 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- Martin H, Rostas J, Patel Y, Aitken A. Subcellular localisation of 14-3-3 isoforms in rat brain using specific antibodies. J Neurochem. 1994;63:2259–2265. doi: 10.1046/j.1471-4159.1994.63062259.x. [DOI] [PubMed] [Google Scholar]
- Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR, Alessi DR. 14–3-3 binding to LRRK2 is disrupted by multiple Parkinson‘s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430:393–404. doi: 10.1042/BJ20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, McNaught KS. Ubiquitin-proteasome system and Parkinson’s disease. Mov Disord. 2006;21:1806–1823. doi: 10.1002/mds.21013. [DOI] [PubMed] [Google Scholar]
- Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, Wolozin B. alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci. 1999;19:5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The Mouse Brain in Stereotaxic Coordinates., Second edn. Academic Press, San Diego
- Pennington K, Peng J, Hung CC, Banks RE, Robinson PA. Differential effects of wild-type and A53T mutant isoform of alpha-synuclein on the mitochondrial proteome of differentiated SH-SY5Y cells. J Proteom Res. 2010;9:2390–2401. doi: 10.1021/pr901102d. [DOI] [PubMed] [Google Scholar]
- Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Poll-The BT, Aicardi J, Girot R, Rosa R. Neurological findings in triosephosphate isomerase deficiency. Ann Neurol. 1985;17:439–443. doi: 10.1002/ana.410170504. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Poon HF, Frasier M, Shreve N, Calabrese V, Wolozin B, Butterfield DA. Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice–a model of familial Parkinson’s disease. Neurobiol Dis. 2005;18:492–498. doi: 10.1016/j.nbd.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Proteomic studies of nitrated alpha-synuclein microglia regulation by CD4+ CD25+ T cells. J Proteome Res. 2009;8:3497–3511. doi: 10.1021/pr9001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko IN, Cookson MR. 14–3-3 proteins are promising LRRK2 interactors. Biochem J. 2010;430:e5–e6. doi: 10.1042/BJ20101200. [DOI] [PubMed] [Google Scholar]
- Schaefer H, Marcus K, Sickmann A, Herrmann M, Klose J, Meyer HE. Identification of phosphorylation and acetylation sites in alphaA-crystallin of the eye lens (mus musculus) after two-dimensional gel electrophoresis. Anal Bioanal Chem. 2003;376:966–972. doi: 10.1007/s00216-003-1983-1. [DOI] [PubMed] [Google Scholar]
- Schaefer H, Chervet JP, Bunse C, Joppich C, Meyer HE, Marcus K. A peptide preconcentration approach for nano-high-performance liquid chromatography to diminish memory effects. Proteomics. 2004;4:2541–2544. doi: 10.1002/pmic.200300801. [DOI] [PubMed] [Google Scholar]
- Schaefer H, Chamrad DC, Herrmann M, Stuwe J, Becker G, Klose J, Blueggel M, Meyer HE, Marcus K. Study of posttranslational modifications in lenticular alphaA-Crystallin of mice using proteomic analysis techniques. Biochim Biophys Acta. 2006;1764:1948–1962. doi: 10.1016/j.bbapap.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Tolosa E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat Rev Neurol. 2010;6:309–317. doi: 10.1038/nrneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- Setsuie R, Wada K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int. 2007;51:105–111. doi: 10.1016/j.neuint.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Shirakashi Y, Kawamoto Y, Tomimoto H, Takahashi R, Ihara M. alpha-Synuclein is colocalized with 14-3-3 and synphilin-1 in A53T transgenic mice. Acta Neuropathol. 2006;112:681–689. doi: 10.1007/s00401-006-0132-2. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Sitek B, Apostolov O, Stuhler K, Pfeiffer K, Meyer HE, Eggert A, Schramm A. Identification of dynamic proteome changes upon ligand activation of Trk-receptors using two-dimensional fluorescence difference gel electrophoresis and mass spectrometry. Mol Cell Proteom. 2005;4:291–299. doi: 10.1074/mcp.M400188-MCP200. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stephan C, Reidegeld KA, Hamacher M, van Hall A, Marcus K, Taylor C, Jones P, Muller M, Apweiler R, Martens L, Korting G, Chamrad DC, Thiele H, Bluggel M, Parkinson D, Binz PA, Lyall A, Meyer HE. Automated reprocessing pipeline for searching heterogeneous mass spectrometric data of the HUPO Brain Proteome Project pilot phase. Proteomics. 2006;6:5015–5029. doi: 10.1002/pmic.200600294. [DOI] [PubMed] [Google Scholar]
- Stuhler K, Pfeiffer K, Joppich C, Stephan C, Jung K, Muller M, Schmidt O, van Hall A, Hamacher M, Urfer W, Meyer HE, Marcus K. Pilot study of the Human Proteome Organisation Brain Proteome Project: applying different 2-DE techniques to monitor proteomic changes during murine brain development. Proteomics. 2006;6:4899–4913. doi: 10.1002/pmic.200600089. [DOI] [PubMed] [Google Scholar]
- Titz B, Low T, Komisopoulou E, Chen SS, Rubbi L, Graeber TG. The proximal signaling network of the BCR-ABL1 oncogene shows a modular organization. Oncogene. 2010;29:5895–5910. doi: 10.1038/onc.2010.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- Wiesinger H, Hamprecht B, Dringen R. Metabolic pathways for glucose in astrocytes. Glia. 1997;21:22–34. doi: 10.1002/(SICI)1098-1136(199709)21:1<22::AID-GLIA3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- Xun Z, Sowell RA, Kaufman TC, Clemmer DE. Lifetime proteomic profiling of an A30P alpha-synuclein Drosophila model of Parkinson’s disease. J Proteome Res. 2007;6:3729–3738. doi: 10.1021/pr0700504. [DOI] [PubMed] [Google Scholar]
- Xun Z, Sowell RA, Kaufman TC, Clemmer DE. Protein expression in a Drosophila model of Parkinson’s disease. J Proteome Res. 2007;6:348–357. doi: 10.1021/pr060488o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xun Z, Sowell RA, Kaufman TC, Clemmer DE. Quantitative proteomics of a presymptomatic A53T alpha-synuclein Drosophila model of Parkinson disease. Mol Cell Proteom. 2008;7:1191–1203. doi: 10.1074/mcp.M700467-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian TA, Slone SR, Harrington AJ, Hamamichi S, Schieltz JM, Caldwell KA, Caldwell GA, Standaert DG (2011) Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson’s disease. Cell Death Dis 1 [DOI] [PMC free article] [PubMed]
- Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- Zhang W, Chait BT. ProFound: an expert system for protein identification using mass spectrometric peptide mapping information. Anal Chem. 2000;72:2482–2489. doi: 10.1021/ac991363o. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gu G, Goodlett DR, Zhang T, Pan C, Montine TJ, Montine KS, Aebersold RH, Zhang J. Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J Biol Chem. 2004;279:39155–39164. doi: 10.1074/jbc.M405456200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.