

Abstract
Autophagy contributes to the removal of prone-to-aggregate proteins, but in several instances these pathogenic proteins have been shown to interfere with autophagic activity. In the case of Huntington's disease (HD), a congenital neurodegenerative disorder resulting from mutation in the huntingtin protein, we have previously described that the mutant protein interferes with the ability of autophagic vacuoles to recognize cytosolic cargo. Growing evidence supports the existence of cross talk among autophagic pathways, suggesting the possibility of functional compensation when one of them is compromised. In this study, we have identified a compensatory upregulation of chaperone-mediated autophagy (CMA) in different cellular and mouse models of HD. Components of CMA, namely the lysosome-associated membrane protein type 2A (LAMP-2A) and lysosomal-hsc70, are markedly increased in HD models. The increase in LAMP-2A is achieved through both an increase in the stability of this protein at the lysosomal membrane and transcriptional upregulation of this splice variant of the lamp-2 gene. We propose that CMA activity increases in response to macroautophagic dysfunction in the early stages of HD, but that the efficiency of this compensatory mechanism may decrease with age and so contribute to cellular failure and the onset of pathological manifestations.
Introduction
Huntington's disease (HD) is a neurodegenerative disorder caused by a mutation that generates an expansion in the polyglutamine (polyQ) stretch normally present in the N-terminal end of the protein huntingtin (Htt) (Chen et al., 2002). The expanded polyQ region triggers the intranuclear and cytoplasmic accumulation of oligomeric and aggregated forms of the mutant protein, resulting in cellular toxicity that initially manifests as neuronal loss in the striatum and the cortex (Tobin and Signer, 2000) but also affects other organs because of the ubiquitous expression of Htt (Young, 2003; Landles and Bates, 2004).
Protein aggregates can be removed by macroautophagy, one of the lysosomal pathways that contribute to the degradation of cytosolic material in lysosomes (Webb et al., 2003; Fornai et al., 2008; Wang et al., 2009). Numerous studies support that macroautophagy contributes to the clearance of mutant Htt and, in fact, pharmacological upregulation of this autophagic pathway in animal models improves disease-related symptoms (Webb et al., 2003; Ravikumar et al., 2004; Sarkar et al., 2009). However, macroautophagy appears to be disrupted by mutant Htt. Recently, we reported that the abnormal interaction of mutant Htt with the vesicles that sequester cytosolic components in macroautophagy interferes with the recognition of cytosolic cargo, compromising the intracellular turnover of cytosolic organelles (Martinez-Vicente et al., 2010).
Many cell types respond to experimental blockage in macroautophagy by upregulating chaperone-mediated autophagy (CMA) (Kaushik et al., 2008), a type of autophagy conducive to degradation of cytosolic proteins in lysosomes through direct translocation across the membrane of these organelles (Dice, 2007; Cuervo, 2010). All CMA substrates, once recognized by the cytosolic chaperone hsc70 in the cytosol (Chiang et al., 1989), are targeted to lysosomes where they bind to the lysosome-associated membrane protein type 2A (LAMP-2A). This protein organizes into a multimeric complex that mediates translocation of the substrate protein across the membrane (Bandyopadhyay et al., 2008), assisted by a variant of hsc70 (lys-hsc70) resident in the lysosomal lumen (Agarraberes et al., 1997).
Although changes in CMA activity in HD have not been explored, overexpression of LAMP-2A in neuronal cells expressing post-translationally modified forms of mutant Htt reduces intracellular protein inclusions and improves cellular viability (Thompson et al., 2009). Furthermore, overexpression of a dimeric molecule bearing binding sites for both polyQ and hsc70 has proven effective in the targeting of mutant Htt to CMA and in reducing neuronal toxicity in HD mouse models (Bauer et al., 2010). This possibility of using CMA to eliminate pathogenic proteins makes it important to determine whether HD cells respond to the gradual decline in macroautophagic activity by enhancing CMA activity (Kaushik et al., 2008).
With that purpose, we have used a variety of cellular and mouse models of HD to investigate the status of CMA activity in HD cells. We have found that CMA is upregulated in HD cells and that increased levels of the CMA receptor at the lysosomal membrane result in the higher activity of this pathway. Furthermore, we have identified a negative effect of aging on this compensatory upregulation of CMA.
Materials and Methods
Animals and cells.
Male 18QHtt and 111QHtt knock-in mice and CD1 mice (6–8 weeks old) were from The Jackson Laboratory. Mouse embryonic fibroblasts (MEFs) from these animals were prepared as described previously (Xu, 2005). 7QHtt and 111QHtt striatal cell lines were from Coriell Cell Repositories (Camden, NJ). Human lymphoblasts from normal controls and HD patients were gifts from N. Wexler (Hereditary Disease Foundation, New York, NY), M. Andresen (Massachusetts Institute of Technology, Cambridge, MA) and J. Gusella (Massachusetts General Hospital, Boston, MA). Striatum neuronal cultures from HD94 mice were prepared as described previously (Petersén et al., 2001). Cells were maintained in DMEM (Sigma) in the presence of 10% newborn calf serum (NCS). Ventromedial neurons were obtained from days 0–2 postnatal mice. Cells were dissociated and cultured on monolayers of cortical astrocytes as described previously (Martinez-Vicente et al., 2010).
Chemicals.
Sources of chemicals and antibodies were as described previously (Kiffin et al., 2004; Cuervo and Dice, 1996; Martinez-Vicente et al., 2010). The antibody against the cytosolic tail of rat and mouse LAMP-2A was prepared in our laboratory (Cuervo and Dice, 1996), and the antibodies against LAMP-1 (clone 1D4B) and human LAMP-2 (clone H4B4) were from the Developmental Hybridoma Bank (University of Iowa, Iowa City, IA). The antibody against hexokinase was a generous gift from Dr. Erwin Knecht (CIPF, Valencia, Spain). The antibodies against hsc70, hsp90, and hsp40 were from Stressgen, against GAPDH and actin from Abcam, against flotillin-1 and α-synuclein from BD Transduction Laboratories, against IKB α (clone 21) from Santa Cruz Biotechnology, against the dopamine transporter and huntingtin from Millipore, against ribonuclease A (RNase A) and cathepsin A from Rockland Immunochemicals, against ubiquitin from Invitrogen, and against myc from Roche. The Amplex Red Cholesterol Assay Kit was from Invitrogen. Recombinant cholera toxin subunit B and the specific antibody against this subunit were from Invitrogen.
Lysosomal isolation and lysosomal subfractionation.
Lysosomes with high activity for CMA were isolated from mouse liver by centrifugation of a light mitochondrial–lysosomal fraction in a discontinuous metrizamide density gradient (Wattiaux et al., 1978) by the modified method described previously (Cuervo et al., 1997). A fraction enriched in lysosomes active for CMA was isolated from cultured cells after disruption of the plasma membrane by nitrogen cavitation and sequential centrifugation in Percoll/metrizamide discontinuous density gradients as described previously (Storrie and Madden, 1990). In all procedures, preparations with >10% broken lysosomes, measured by β-hexosaminidase latency (Storrie and Madden, 1990), were discarded. Lysosomal matrices and membranes were isolated after hypotonic shock of the lysosomal fraction followed by centrifugation as described previously (Ohsumi et al., 1983). Where indicated, membranes were subjected to washes of increasing stringency by incubation in the different buffers on ice for 30 min and subsequent centrifugation. Regions of different resistance to detergent solubilization were isolated from lysosomal membranes through flotation in a sucrose density gradient as described previously (Kaushik et al., 2006). Cytosolic and endoplasmic reticulum fractions were obtained by centrifugation for 1 h at 100,000 × g of the supernatant obtained after separating the mitochondria—lysosome-enriched fraction.
Intracellular protein turnover.
To measure degradation of long-lived proteins, confluent cells were labeled with [3H]leucine (2 μCi/ml) for 48 h at 37°C and then extensively washed and maintained in complete (10% NCS) or serum-deprived medium containing an excess of unlabeled leucine (2.8 mm) to prevent reutilization of radio-labeled leucine (Auteri et al., 1983). Aliquots of the medium taken at different times were precipitated with TCA, and proteolysis was measured as the percentage of the initial acid-insoluble radioactivity (protein) transformed into acid-soluble radioactivity (amino acids and small peptides) at the end of the incubation. Total radioactivity incorporated into cellular proteins was determined as the amount of acid-precipitable radioactivity in labeled cells immediately after washing. Where indicated serum was removed from the media during the chase, and 20 mm ammonium chloride and 100 μm leupeptin or 10 mm 3-methlyadenine were added (Kaushik and Cuervo, 2009). The kinetics of degradation of Htt in cultured cells was determined as described previously (Aniento et al., 1993; Cuervo et al., 1998). Briefly, cultured cells were radiolabeled with a [35S]methionine/cysteine mixture (0.2 μCi/ml) for 48 h and, after extensive washing, cells were maintained in the presence or absence of serum. At increasing times, cells were lysed in lysis buffer. Lysates were cleared by centrifugation, and supernatants were subjected to immunoprecipitation for Htt followed by SDS-PAGE. Gels were exposed to a PhosphorImager screen, and the half-life of the protein was calculated from the formula t1/2 = ln2/degradation rate.
Degradation of substrate proteins by intact lysosomes in vitro.
Degradation of radiolabeled proteins by isolated intact lysosomes was measured as described previously (Kaushik and Cuervo, 2009). Briefly, lysosomes isolated from mice livers or the different cells in culture were incubated with radiolabeled proteins in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer (10 mm MOPS, pH 7.3, 0.3 m sucrose, 1 mm dithiothreitol, and 5.4 m cysteine) for 30 min at 37°C. Reactions were stopped with 10% TCA and filtered through a Millipore Multiscreen Assay System (Millipore) and detected in a WinSpectral 1414 liquid scintillation analyzer (PerkinElmer/Wallac). Proteolysis was measured as the percentage of the initial acid-insoluble radioactivity transformed into acid-soluble radioactivity.
Binding and uptake of CMA substrate proteins by isolated lysosomes.
Uptake of GAPDH, RNase A, and α-synuclein proteins by isolated lysosomes was analyzed as described previously (Kaushik and Cuervo, 2009). Briefly, freshly isolated lysosomes were incubated with the substrate protein in MOPS buffer at 37°C for 20 min. Where indicated, lysosomes were preincubated with a cocktail of protease inhibitors for 10 min on ice. Lysosomes were collected by centrifugation, washed with MOPS buffer, and subjected to SDS-PAGE and immunoblotted for the used substrate protein. Binding is calculated from densitometric analysis as the amount of substrate protein bound to the lysosomal membrane in the absence of protease inhibitors. Uptake was calculated by subtracting the amount of protein associated with lysosomes in the presence (protein bound to the lysosomal membrane and taken up by lysosomes) and absence (protein bound to the lysosomal membrane) of protease inhibitors.
Measurement of CMA activity in intact cells.
Striatal cell lines were infected with a lentivirus carrying the CMA reporter (KFERQ-photoswichable-CFP2) (Koga et al., 2011). Cells were photoactivated 48 h postinfection by light-emitting diode (Norlux) for 10 min with the intensity of 3.5 mA (current constant). After 16 h, cells were fixed with 4% paraformaldehyde and the number of fluorescent puncta by cell was quantified using ImageJ software (NIH) in individual frames after thresholding.
LAMP-2A stability.
Rates of degradation of LAMP-2A in the isolated membranes were determined by immunoblotting using a specific antibody against the cytosolic tail of LAMP-2A as described previously (Cuervo and Dice, 2000a). Briefly, isolated lysosomal membranes were incubated in MOPS buffer at 37°C, and at different times aliquots were removed and subjected to SDS-PAGE and immunoblotted for LAMP-2A.
Immunofluorescence and immunohistochemistry.
Cells grown on coverslips were fixed with a 3% formaldehyde solution, blocked, and then incubated with the primary and corresponding fluoresce-conjugated secondary antibodies (Kaushik et al., 2006). Mounting medium contained DAPI (4′,6-diamidino-2-phenylindole) to highlight the cellular nucleus. Images were acquired with an Axiovert 200 fluorescence microscope (Carl Zeiss), subjected to deconvolution with the manufacture's software, and prepared using Adobe Photoshop 6.0 software (Adobe Systems). Quantification was carried on using ImageJ (NIH) in three different Z-stack sections for each picture after thresholding. For immunofluorescence in tissue slides, flash frozen mouse brains were sectioned to include the striatal region. Sections were fixed in precooled acetone at −20°C and blocked with 3% fetal bovine serum in PBS containing 0.01% Triton X-100. Sections were treated with the appropriate primary and fluorophore-tagged secondary antibody and visualized as described above.
mRNA quantification.
Total RNA was extracted from rat livers using the RNeasy Protect Mini Kit (Qiagen) following the manufacturer's indications, and stored at −80°C until use. The first strand cDNA was synthesized from 0.5 μg of the total RNA with the SuperScript II RNase H Reverse Transcriptase (Invitrogen) and oligo(dT)12–18 primers. Actin and a region of the exon 8 of LAMP-A were amplified with specific primers (LAMP-2A, 5′-GCAGTGCAGATGAAGACAAC-3′, 5′-AGTATGATGGCGCTTGAGAC-3′;actin, 5′-AA GGACTCCTATAGTGGGTGACGA-3′, 5′-ATCTTCTCCATGTCGTCCCAGTTG-3′) using the SYBR Green PCR kit (PE Biosystems). Amplification of the LAMP-2A and actin DNA products (120 and 108 bp, respectively) was measured in real time in a SmartCycler (Cepheid). The expression levels of LAMP-2A in different samples were normalized with respect to those of actin in the same samples. Differences between samples were calculated based on the differences in the number of cycles required to reach a threshold fluorescence intensity level.
Statistical analysis.
All numerical results are reported as mean + SE, and represent data from a minimum of three independent experiments unless otherwise stated. We determined the statistical significance in instances of single comparisons by the two-tailed unpaired Student's t test of the means with the SigmaPlot software (Jandel Scientific). In instances of multiple means comparisons, we used one-way ANOVA followed by the Bonferroni post hoc test to determine statistical significance.
Results
Autophagic pathways are differently altered in HD cells
To determine possible changes in the rates of protein degradation through autophagic pathways other than macroautophagy in HD cells, we first used mouse embryonic fibroblasts from knock-in mice in which the entire exon 1 of huntingtin has been replaced by the human exon of the protein bearing 18 glutamine-encoding CAG tracks (control; 18QHtt) or 111 CAG tracks (mutant; 111QHtt). In agreement with our previous studies, we found that although both 18QHtt and 111QHtt MEFs were still able to increase total rates of degradation of long-lived proteins in response to serum removal, protein degradation under these conditions was consistently lower in 111QHtt MEFs (Fig. 1A). These differences between MEFs expressing control and mutant Htt were more pronounced when we compared the percentage of protein degradation occurring in lysosomes (sensitive to inhibition of lysosomal proteolysis by ammonium chloride and leupeptin) (Fig. 1B). Analysis of the fraction of the lysosomal proteolysis occurring through macroautophagy (sensitive to inhibition by the PI3-kinase inhibitor, 3-methyladenine) confirmed the previously described compromised ability to activate this autophagic pathway in response to starvation in 111QHtt MEFs (Fig. 1C).
Figure 1.
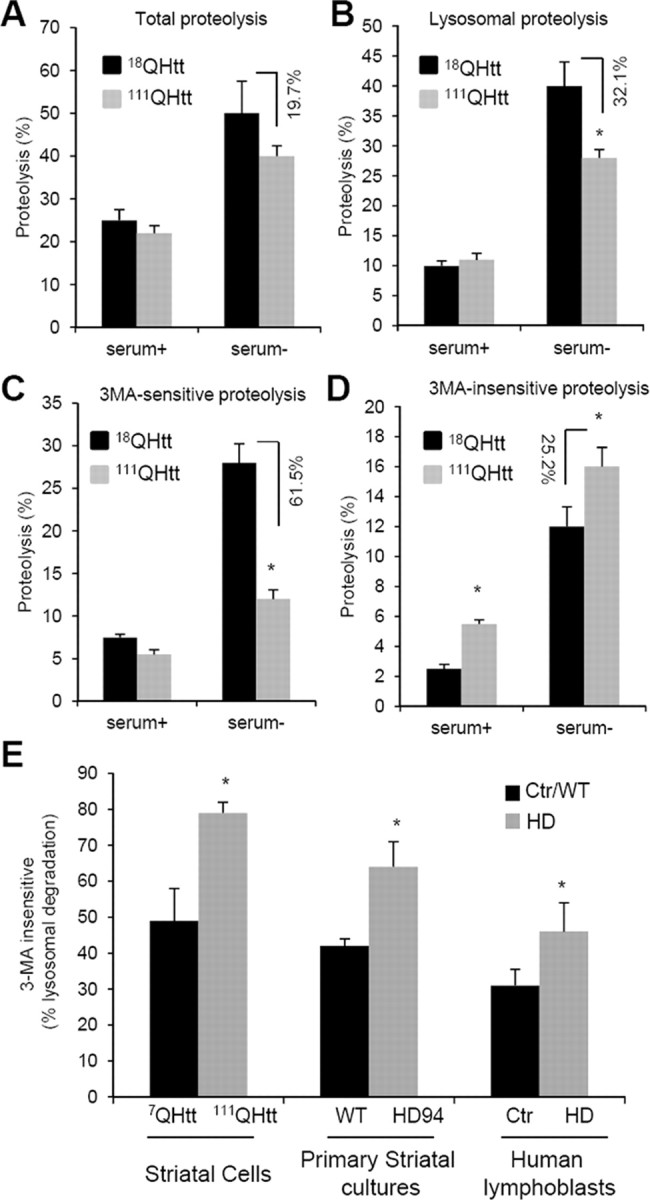
Non-macroautophagic lysosomal degradation is increased in HD cells. A–D, MEFs from 18QHtt and 111QHtt knock-in mice were subjected to metabolic labeling (as described in Materials and Methods), and the total rates of long-lived protein degradation (A), rates of lysosomal-dependent degradation (sensitive to inhibition by NH4Cl and leupeptin) (B), rates of macroautophagy-dependent degradation (lysosomal degradation sensitive to inhibition by 3-methyladenine) (C), and rates of non-macroautophagic lysosomal degradation (D) were calculated. E, The percentage of lysosomal degradation insensitive to 3-methyladenine (3MA) was calculated using similar procedures in striatal neuronal cell lines derived from 7QHtt and 111QHtt knock-in mice (left), primary striatal cultures from HD94 mice expressing mutant exon-1 Htt with 94 CAG repeats (middle), and lymphoblasts from healthy control (Ctr) and HD patients (right). All values are expressed as mean ± SE and are mean of at least three different experiments. WT, Wild type.
Interestingly, the decrease in macroautophagy activity in HD cells in response to serum removal was greater than that observed for lysosomal proteolysis (61.5% reduction versus 32.1%; compare Fig. 1, B and C). This discrepancy can only be reconciled if proteolysis through alternate mechanisms also depending on lysosomes is upregulated. In fact, in 111QHtt MEFs, we found a significant increase in the percentage of lysosomal proteolysis insensitive to 3-methyladenine in response to serum removal when compared to 18QHtt MEFs (Fig. 1D). Furthermore, although under basal conditions we did not find marked differences in total rates of protein degradation (Fig. 1A) or in lysosomal degradation (Fig. 1B) between 18QHtt MEFs and 111QHtt MEFs, and only a discrete but consistent reduction in macroautophagy activity in 111QHtt MEFs under these conditions (Fig. 1C), the percentage of lysosomal degradation independent of macroautophagy was significantly higher in these cells even when maintained in the presence of serum (Fig. 1D). These findings support a switch in the contribution of non-macroautophagic pathways to total lysosomal degradation from 24.5% in 18QHtt MEFS to 50.1% in 111QHtt MEFs under basal conditions and from 30.0% in 18QHtt MEFs to 57.2% in 111QHtt MEFs upon serum removal (compare Fig. 1, B and D).
We confirmed similar increases in the percentage of lysosomal degradation not attributable to macroautophagy in striatal cells derived from 111QHtt mice when compared to the ones derived from 7QHtt mice. This was also the case in primary striatal cultures from HD94 mice expressing mutant human exon-1 Htt with 94 CAG repeats when compared to those from wild-type mice, and in lymphoblasts from HD patients when compared to lymphoblasts from age-matched healthy individuals (Fig. 1E).
CMA is upregulated in HD cells
Insensitivity to PI3-K inhibitors is a characteristic of CMA-dependent proteolysis, but we cannot discard the possibility that other autophagic pathways, such as microautophagy, could contribute to the observed increase in 3-methyladenine-insensitive degradation in HD cells. To directly compare CMA activity in control and HD cells, we used a novel photoswitchable reporter (KFERQ-PS-CFP2) that allows tracking the selective delivery to lysosomes of an artificial substrate bearing the KFERQ motif (Koga et al., 2011). A change in the intracellular fluorescent pattern from cytosolic distribution to fluorescent puncta is indicative of delivery of the artificial substrate to lysosomes via CMA. As shown in Figure 2, removal of serum for 12 h in control 7QHtt striatal cells transfected with the KFERQ-PS-CFP2 construct increased the number of fluorescent puncta, supporting CMA upregulation. Comparatively, the number of fluorescent puncta per cell was significantly higher in 111QHtt striatal cells, not only after serum removal but also in cells maintained in serum-supplemented media (Fig. 2A,B). The KFERQ-PS-CFP2-positive puncta did not colocalize with LC3, a marker for autophagic vacuoles, in any of the two groups of cells, discarding the idea that changes in macroautophagy in HD cells were responsible for the observed increase in puncta positive for the CMA reporter (Fig. 2C). In addition, the absence of colocalization of KFERQ-PS-CFP2 with ubiquitin-positive puncta discarded possible aggregation of the CMA reporter in these cells (Fig. 2D). These results support that, in contrast to the observed decrease in macroautophagy, both basal CMA and inducible CMA are upregulated in HD cells.
Figure 2.

Augmented CMA activity in HD cells monitored by a CMA fluorescence reporter. A, Changes in the fluorescence pattern of a photoswitchable fluorescent CMA reporter in striatal cells derived from 7QHtt and 111QHtt knock-in mice maintained in the presence (Serum+) or absence (Serum−) of serum for 24 h. Left, Representative micrographs (insets show higher magnification and arrows point to fluorescent puncta). Scale bar, 5 μm. B, Quantification of the total number of fluorescent puncta in cells maintained in the presence or absence of serum. Values are mean ± SE of three different experiments with >50 cells counted per experiment. Micrographs are shown as inverted grayscale mode to better appreciate the vesicular pattern. Differences with control are significant for *p < 0.05. C,D, Colocalization of photo-converted KFERQ-PS-CFP2 (green) with LC3 (C) and ubiquitin (Ub) (D) in striatal cells from 7QHtt and 111QHtt knock-in mice maintained in the presence or absence of serum. Scale bars, 5 μm.
The most direct way to monitor CMA activity, independent of changes in other autophagic and proteolytic pathways, is to use a well established in vitro system with isolated lysosomes (Kaushik and Cuervo, 2009). This assay analyzes the proteolytic breakdown of radiolabeled CMA substrates by intact lysosomes, which recapitulates their binding, uptake, and proteolysis in the lysosomal lumen. An important requirement for these types of assays is the preservation of the stability of the lysosomal membrane during the incubation. Analysis of the amount of hexosaminidase released from lysosomes into the incubation media at different times of incubation did not reveal significant differences between lysosomes isolated from livers of 18QHtt and 111QHtt knock-in mice (Fig. 3A). Incubation of these lysosomes with two 14C-labeled, well characterized CMA substrates, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and RNase A, revealed significantly higher rates of substrate uptake/degradation by 111QHtt lysosomes (Fig. 3B). The fact that these differences in proteolysis between 18QHtt and 111QHtt CMA active lysosomes were no longer evident if the lysosomal membrane was disrupted (Fig. 3C) supports the possibility that changes were mainly occurring at the level of binding and uptake rather than proteolysis per se. To directly assess differences in substrate binding and uptake separately from possible changes in proteolysis, we used a second in vitro assay in which association of substrate proteins to lysosomes is monitored by immunoblot (Kaushik and Cuervo, 2009). The amount of substrate recovered in lysosomes not treated with protease inhibitors represents substrate bound to the lysosomal membrane, since any internalized substrate is rapidly degraded by the luminal proteases. Substrate uptake is calculated as the difference in the amount of substrate recovered in lysosomes treated with protease inhibitors (bound to the membrane and present in the lumen) and that detected in untreated lysosomes (bound to the membrane). Using these criteria, we found that binding and uptake of both GAPDH (Fig. 3D) and α-synuclein (Fig. 3E) were higher in lysosomes isolated from liver of 111QHtt knock-in mice. These results confirm that the higher proteolysis rates of radiolabeled CMA substrates observed in lysosomes of 111QHtt knock-in mice were mainly due to faster lysosomal binding and uptake rather than enhanced proteolysis once inside the lysosomal lumen.
Figure 3.
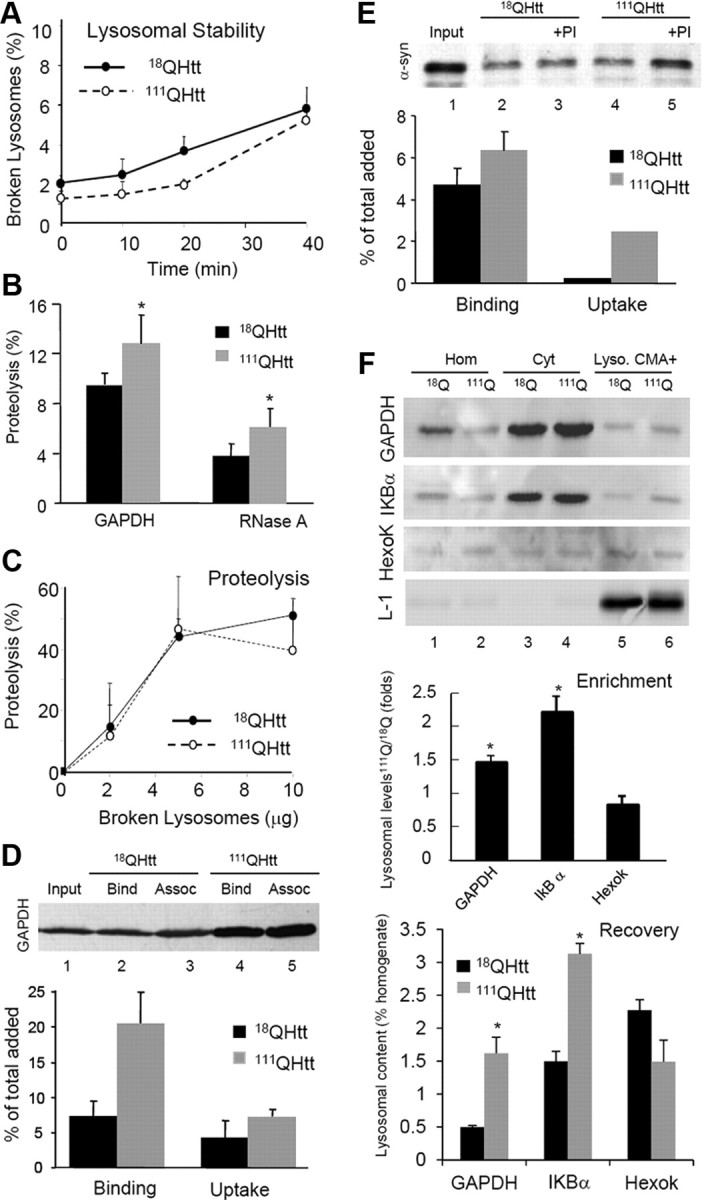
Uptake of CMA substrate proteins is increased in HD. A, Stability of the lysosomal membrane of intact lysosomes isolated from liver of 18QHtt and 111QHtt knock-in mice calculated by the hexosaminidase latency assay as described under Materials and Methods. Values are mean ± SE of three experiments with triplicate samples. B, Proteolysis of [14C] GAPDH and [14C]RNase A in the same samples. Values are expressed as percentage of the protein added and are mean ± SE of five different experiments with triplicates. C, Proteolysis of a pool of radiolabeled cytosolic proteins by increasing concentrations of disrupted lysosomes isolated from the same mice. Values are mean ± SE of three different mice. D, E, Association of GAPDH (D) or α-synuclein (E) with the same isolated lysosomes untreated or pretreated with protease inhibitors (PI). Input (lane 1), 1/10 of total protein added to lysosomes. Bottom graphs show the mean value of the binding and uptake of both proteins by isolated lysosomes calculated as described under Material and Methods. Values are expressed as percentage of the total protein added to the reaction and are mean ± SE of three different experiments. Assoc, Association. F, Immunoblots for the indicated proteins of homogenates (Hom), cytosol (Cyt), and lysosomes with high CMA activity (Lyso. CMA+) isolated from livers of the same animals. Hexok, Hexokinase. Graphs show quantification of the enrichment (top) and recovery (bottom) for each protein in the lysosomal fractions. Values are expressed as relative to the values in 18QHtt lysosomes and are mean ± SE of three different experiments. Differences with control are significant for *p < 0.05.
To further confirm that enhanced CMA also occurs in vivo, we compared the levels of different endogenous cytosolic proteins in lysosomes isolated from the liver of 18QHtt and 111QHtt knock-in mice. We can separate two populations of lysosomes with different activity for CMA depending on their content of hsc70, the chaperone required to complete substrate translocation into lysosomes via CMA (Cuervo et al., 1997). Immunoblot for GAPDH and IKBα, previously characterized as CMA substrates (Aniento et al., 1993; Cuervo et al., 1998), revealed higher content for both in CMA-active lysosomes isolated from 111QHtt knock-in mice (Fig. 3F). Differences between control and HD lysosomes were even more marked when the percentage of total cellular protein present in lysosomes (recovery) was calculated (Fig. 3D, bottom). In contrast, other cytosolic proteins that do not have a KFERQ-motif such as hexokinase were detected at comparable levels in lysosomes from both groups of mice and less enriched in lysosomes from 111QHtt knock-in mice (Fig. 3D, bottom).
Higher levels of CMA components in lysosomes from HD cells
To analyze the origin of the constitutive upregulation of CMA observed in HD cells, we examined levels of CMA components in different HD models. Higher levels of CMA activity are often mediated by an increase in the lysosomal levels of the CMA receptor LAMP-2A and the chaperone hsc70 (Agarraberes et al., 1997; Cuervo and Dice, 2000a). We found that intracellular levels of LAMP-2A, but not of other lysosomal proteins (LAMP-1 shown here), were markedly higher in 111QHtt MEFs than in 18QHtt MEFs both when maintained in the presence of serum or after 24 h serum removal (Fig. 4A). Likewise, comparative analysis of CMA markers in livers from 18QHtt and 111QHtt knock-in mice revealed higher cellular levels of LAMP-2A in hepatocytes from the latter group (Fig. 4B). Differences in the LAMP-2A content between control and HD cells were even more pronounced (30–40% increase) when isolated mouse liver lysosomes were compared, whereas levels of other lysosomal membrane proteins (LAMP-1 shown here) remain unchanged (Fig. 4B). Although total cellular levels of most chaperones involved in CMA were similar in control and HD cells, we found a consistent marked increase in the lysosomal content of hsc70 in lysosomes from 111QHtt mouse liver (Fig. 4B). Lysosomal levels of hsp90 and hsp40, both involved in nonlimiting CMA steps, were comparable in both groups of animals (Fig. 4B).
Figure 4.

Increased levels of CMA components in lysosomes from HD cells. A, Immunoblots for the indicated proteins of MEFs from 18QHtt and 111QHtt knock-in mice maintained in medium supplemented (Serum+) or not (Serum−) with serum. Right, Quantification of the changes in LAMP-2A and LAMP-1 relative to their values in 18QHtt serum+. Values are mean ± SE, n = 5. B, Immunoblots for the indicated proteins of homogenates (Hom), cytosol (Cyt), and lysosomes with high CMA activity (Lys) isolated from livers of 18QHtt and 111QHtt knock-in mice. Right, Quantification of the changes in the indicated proteins in the lysosomal fraction relative to their values in 18QHtt lysosomes. Values are mean ± SE n = 4. C, Immunoblots for LAMP-2A and hsc70 of total lysosomes (Lys total), lysosomal membranes (Lys mb), and lysosomal matrices (Lys mtx) isolated from the same animals. Right, Percentage of lysosomal LAMP-2A present at the lysosomal membrane (mb) and matrix (mtx) in each group of lysosomes. Differences with control are significant for *p < 0.05.
Separate analysis of lysosomal membranes and matrices, isolated by centrifugation after disrupting lysosomes with a hypotonic shock, confirmed that most of the additional molecules of LAMP-2A in lysosomes from 111QHtt cells were indeed present at the lysosomal membrane (Fig. 4C). In fact, analysis of the distribution of LAMP-2A between the lysosomal membrane and lumen revealed a preferential localization of this protein at the membrane of HD lysosomes. A similar redistribution of LAMP-2A from the lysosomal lumen toward the membrane has been described under conditions leading to maximal activation of CMA, such as prolonged starvation (beyond 48 h) (Cuervo and Dice, 2000a). Levels of hsc70 were higher in both membranes and lumen of HD lysosomes (Fig. 4C), likely reflecting the different functions of this chaperone in both compartments and the need to upregulate both functions to sustain the higher CMA activity observed in this group.
Higher content of CMA-active lysosomes (identified as those positive for LAMP-2A and hsc70) in HD cells was also confirmed by immunofluorescence staining for both proteins in neuronal cells (111QHtt striatal cells compared to 7QHtt cells) (Fig. 5A). These differences were manifested both upon serum removal, when CMA is maximally activated, and also even under basal conditions (Fig. 5A). A more detailed analysis of the immunofluorescence pattern of LAMP-2A-positive vesicles in these cells also revealed changes in the intracellular distribution of CMA-active lysosomes in HD cells compatible with upregulation of this pathway (Fig. 5B). We have previously reported that the relocation of CMA-active lysosome toward the perinuclear region is a hallmark of CMA activation that can be observed in most cell types upon prolonged serum removal (Agarraberes et al., 1997; Cuervo and Dice, 2000c) or exposure to oxidative stress (Kiffin et al., 2004), conditions known to activate CMA. Quantification of the average distance from the nucleus of LAMP-2A-positive vesicles in striatal cells revealed that a significantly higher percentage of LAMP-2A-positive lysosomes were located 0.5 μm or less from the nucleus in 111QHtt striatal cells, and once again the differences with 7QHtt cells were observed both during serum deprivation and also under basal conditions (Fig. 5B). These changes in LAMP-2A-enriched vesicles seem specific for these vesicles, because a comparative analysis of the acidic vesicular compartments (highlighted with LysoTracker) in striatal cells from 7QHtt and 111QHtt knock-in mice did not reveal marked differences between both groups (data not shown).
Figure 5.
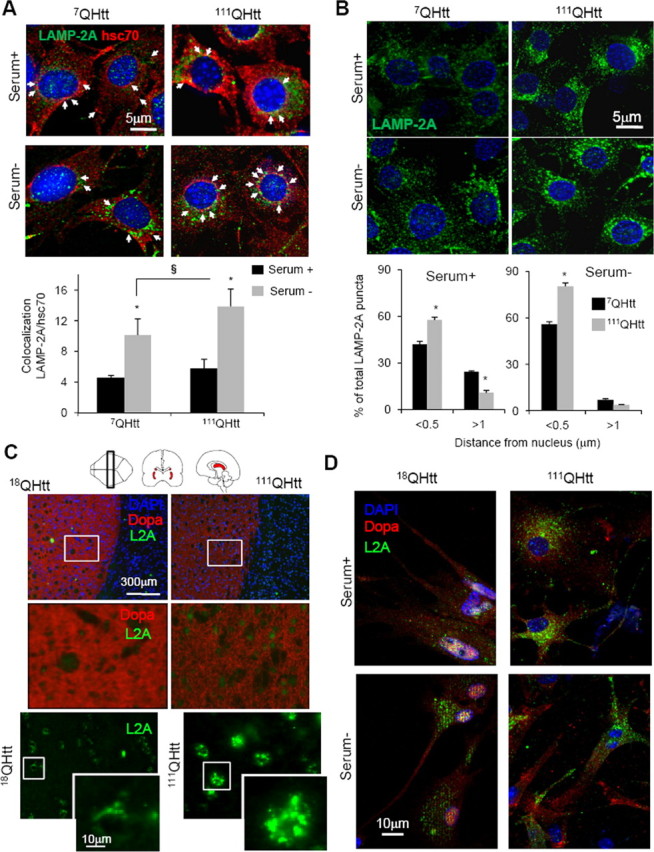
Changes in the group of lysosomes with high activity for CMA in neuronal HD cells. A, Striatal cells from 7QHtt and 111QHtt knock-in mice maintained in the presence (Serum +) or absence (Serum −) of serum were costained for LAMP-2A and hsc70. Top, Representative images. Nuclei are stained in blue with DAPI. Bottom, Quantification of the percentage of colocalization of both proteins. Values are mean ± SE of three different experiments and colocalization was calculated in >50 cells in each condition. Values are different respect serum+ (*) or 7QHtt (§) for p < 0.05. Arrows indicate colocalization of both fluorophores. B, Relocation of LAMP-2A-positive vesicles (CMA-active lysosomes) toward the perinuclear region in the same cells maintained in the presence or absence of serum. Top, Nuclei are stained in blue with DAPI. Bottom, Quantification of the distribution of lysosomes (LAMP-2A–positive puncta) with respect to the nucleus. The percentage of total vesicles at the indicated distances is shown. Values are mean of the quantification of 8–10 cells per condition in two independent experiments. Scale bars, 5 μm. C, Immunohistochemistry for LAMP-2A in brain sections from 18Qhtt and 111Qhtt knock-in mice. Nuclei are highlighted in blue with DAPI, green shows LAMP-2A staining, and red indicates dopaminergic-positive neurons (striatal region). Different magnification images are shown. D, Immunofluorescence for LAMP-2A of neuronal cultures from the same mice maintained in the presence or absence of serum. Dopaminergic neurons are stained in red and nuclei are highlighted in blue.
Finally, to examine whether an increase in CMA-active lysosomes in HD also occurs in vivo, we immunostained brain sections from 18QHtt and 111QHtt mice for LAMP-2A. Consistent with our findings in cultured cells and in liver fractions, we found a pronounced increase of LAMP-2A levels in the striatum (dopamine transporter-positive regions) of brain section from 111QHtt mice compared with those from 18QHtt mice (Fig. 5C). Staining for LAMP-2A in primary neuronal cultures from the same animals also confirmed that part of the observed increase in LAMP-2A levels in HD was in dopaminergic neurons and that the higher content of the CMA marker was evident both under basal conditions and upon removal of serum (Fig. 5D). These findings confirm that upregulation of CMA is likely a universal phenomenon in both neuronal and non-neuronal cells in HD.
Mechanism for CMA upregulation in HD
A variety of mechanisms have been shown to increase levels of LAMP-2A in lysosomes and consequently to upregulate CMA under different conditions. To determine the contribution of these mechanisms to the increased CMA activity observed in HD cells, we first measured levels of LAMP-2A mRNA by quantitative real-time PCR in different HD cells and their corresponding control cells. As shown in Figure 6A, LAMP-2A mRNA levels were higher in all types of HD cells, although the extent of the transcriptional upregulation of LAMP-2A appeared to be cell type dependent. Whereas in 111QHtt MEFs LAMP-2A mRNA levels were almost tenfold higher than those in 18QHtt MEFs, the increase in levels of LAMP-2A mRNA in HD striatal cells or liver was rather modest (Fig. 6A). Although it is possible that a small but sustained increase in de novo synthesis of LAMP-2A could contribute to the observed increase in the levels of this protein in HD cells, it is likely that mechanisms other than transcriptional upregulation contribute to the higher levels of LAMP-2A observed in these cells.
Figure 6.

Mechanisms of LAMP-2A upregulation in HD. A, Changes in the mRNA levels of LAMP-2A in striatal cell lines, MEFs, and liver from control and HD knock-in mice. Values are expressed relative to the value in their corresponding controls and are mean ± SE from 3–4 different experiments. B, Degradation of LAMP-2A in the membranes of lysosomes isolated from livers of 18QHtt/111QHtt knock-in mice at the indicated times of incubation in MOPS buffer at 37°C. Top, Representative immunoblot for LAMP-2A. Bottom, Kinetics of degradation of LAMP-2A calculated by densitometric analysis of immunoblots such as the one shown here. Values are expressed as a percentage of the total LAMP-2A at time 0 and are mean ± SE of three different experiments. C, Distribution of LAMP-2A and flotillin-1 in different fractions of the lysosomal membrane. Lysosomes from livers of 18QHtt/111QHtt knock-in mice were extracted with 1% Triton X-114 and then subjected to sucrose gradient centrifugation. Aliquots collected from top to bottom were grouped as the detergent-resistant (DR), intermediate (Int), and detergent-soluble (DS) fractions and subjected to SDS-PAGE and immunoblotted for LAMP-2A. The graph on the left shows densitometric quantification of the immunoblot. Values are expressed as percentage of the total LAMP-2A in lysosomes in each group. D, Cholesterol (cholest) levels in CMA active lysosomes (Lys) isolated from livers of 18QHtt/111QHtt knock-in mice. Values are mean ± SE of three different experiments. E, Dot blot analysis for the ganglioside GM1 in the same fractions used in C immobilized on a nitrocellulose membrane, incubated with 1 μg/ml recombinant toxin subunit B, and then with the antibody against this toxin. The graph shows densitometric quantification of the dot blots. Inset, Representative GM1 dot blots. Differences with control are significant for *p < 0.05.
Changes in the stability of LAMP-2A have been described to also contribute to modulate levels of this protein at the lysosomal membrane. LAMP-2A undergoes tightly regulated degradation in this compartment through the combined action of a pair of lysosomal proteases, cathepsin A and a yet unidentified metalloprotease (Cuervo and Dice, 2000a; Cuervo et al., 2003). Degradation of LAMP-2A occurs preferentially in discrete lipid microdomains of the lysosomal membrane enriched in both proteases (Kaushik et al., 2006). Active exclusion of LAMP-2A from these microdomains during CMA activation contributes to a rapid increase in levels of the CMA receptor in lysosomes by slowing down its degradation in this compartment (Kaushik et al., 2006). When we compared the kinetics of degradation of LAMP-2A in lysosomes isolated from livers of 18QHtt and 111QHtt knock-in mice we found that the stability of this protein was higher in the latter group (Fig. 6B).
To determine whether the observed changes in LAMP-2A degradation were consequence of differences in its mobilization of the lipid microdomains where this protein normally undergoes degradation, we subjected lysosomes from livers of 18QHtt and 111QHtt mice to extraction with Triton X-114 and recovered the detergent-resistant regions by floatation in discontinuous sucrose gradients. Consistent with the slower degradation of LAMP-2A in 111QHtt mice lysosomes, we found a consistently lower amount of LAMP-2A present in the membrane lipid microdomains enriched in flotillin-1 (Flot-1), a marker for detergent-resistant fractions in the cellular membranes, where it normally undergoes degradation when compared to those from 18QHtt knock-in mice (Fig. 6C).
Although the mechanisms that mediate mobilization of LAMP-2A to the discrete lysosomal membrane microdomains for degradation are for the most part unknown, it has been demonstrated that changes in total cholesterol levels at the lysosomal membrane have a direct impact on the amount of LAMP-2A detected in these regions (Kaushik et al., 2006). Analysis of the cholesterol content of the lysosomal membranes from 18QHtt and 111QHtt knock-in mice revealed a discrete but significant reduction in cholesterol in the HD lysosomes (Fig. 6D). Furthermore, we performed dot-blot analysis with cholera toxin B subunit to compare lysosomal membrane levels of ganglioside 1 (GM1), previously reported to be also in abundance in the LAMP-2A-positive microdomains (Kaushik et al., 2006). As shown in Figure 6E, the distribution of this lipid among the different subregions of the lysosomal membrane of 111QHtt mice was different when compared to 18QHtt mice. While GM1 was enriched in detergent-resistant fractions in 18QHtt mice, it dispersed toward the intermediate regions in 111QHtt mice.
In summary, the observed changes in cholesterol and GM1 levels in the lysosomal membrane of HD mice could explain, at least in part, the lower percentage of membrane LAMP-2A present in microdomains and its reduced degradation in these lysosomes. Future studies are needed to identify the molecular components that modulate the dynamic association of LAMP-2A with membrane microdomains and whether any of those components are primarily altered in HD cells.
Overall, our results support that although all HD cells are able to upregulate CMA by increasing their lysosomal levels of LAMP-2A, the mechanisms that contribute to the higher content of lysosomal LAMP-2A in these cells are cell type dependent. Thus, while in some cell types elevated de novo synthesis of LAMP-2A accounts for most of the increase in this protein in lysosomes, in other cells types the stabilization of LAMP-2A at the lysosomal membrane also contributes to this process.
CMA and intracellular HD turnover
Experimental upregulation of CMA by overexpressing LAMP-2A in cultured neuronal cells has been previously shown to be efficient in reducing levels of aggregate exon1-Htt and to improve neuronal survival (Thompson et al., 2009). In addition, artificial targeting of Htt for CMA degradation has also been shown to be protective against neuronal toxicity in mouse models (Bauer et al., 2010). However, the contribution of CMA to degradation of endogenous full-length Htt and possible changes on this degradation in the case of mutant Htt have not been explored yet. To gain insights on the possible degradation of Htt in lysosomes, we first determined the half-life of endogenous Htt in cultured fibroblasts and analyzed the effect that serum removal, the most potent physiological activator of CMA, had on the half-life of Htt. As shown in Figure 7A, although full-length Htt had a relatively long half-life of 63 h characteristic of many CMA substrates, upregulation of CMA through serum removal, failed to significantly shorten the half-life of this protein.
Figure 7.

Degradation of full-length and exon-1 Htt by CMA. A, Kinetics of degradation of Htt in fibroblasts maintained in the presence or absence of serum. Half-lives are indicated. B, Immunoblot for Htt in the indicated subcellular fractions isolated from human fibroblasts in culture, rat liver and kidney, and mouse liver. Hom, homogenate; Cyt, cytosol; ER, endoplasmic reticulum; Mit, mitochondria; Lys, lysosomes. C, Immunoblot for Htt and LAMP-1 (L-1) of homogenates and lysosomes from fibroblasts maintained in the presence or absence of serum. D, Immunoblot for the indicated proteins of lysosomal membranes (Memb.) and matrices isolated from mouse liver. Where indicated, mice were injected with leupeptin 2 h before lysosomal isolation. Cath A, Cathepsin A. E, Immunoblot for the indicated proteins of mouse lysosomes subjected to washes with MOPS, 1 m NaCl, 0.1 m NaCO2, or 1% Triton X-100. L-2, LAMP-2. F, Immunoblot for Htt and GAPDH of lysosomes incubated for the indicated times in isotonic media. G, Immunoblot for Htt of the indicated fractions isolated from liver and kidney of 18QHtt and 111QHtt knock-in mice. H, Immunoblots for the indicated proteins in cells expressing full-length or exon1 23QHtt and 145QHtt control (Ctr) or transfected with a vector expressing human LAMP-2A (hL-2A).
To directly determine the possible contribution of lysosomes and CMA to the degradation of full-length Htt, we analyzed the presence of Htt in this compartment through subcellular fractionation and immunoblot. A fraction of cellular Htt was consistently detected in lysosomes isolated from different cell types and from different species (Fig. 7B), but the association of Htt with lysosomes did not increase upon serum removal, contrary to what has been described for CMA substrates (Fig. 7C). In fact, analysis of the topology of Htt in lysosomes revealed that most of the protein is present at the lysosomal membrane rather than in the lumen (Fig. 7D). Failure to detect Htt inside lysosomes was not due to its rapid degradation in this compartment, because blockage of the proteolytic activity of lysosomes with leupeptin failed to reveal Htt in the lysosomal lumen (Fig. 7D). In fact, lysosome-associated Htt could not be washed out with high stringency washes, supporting its tight binding to the lysosomal membrane, uncharacteristic of CMA substrates (Fig. 7E). Furthermore, Htt remained stable in this compartment when lysosomes were incubated at 37°C, contrary to a well characterized CMA substrate protein that was internalized and degraded by lysosomes under these conditions (Fig. 7F). Overall, these results argue against an important contribution of CMA to the degradation of full-length Htt under normal conditions.
Analysis of the mutant form of the protein in different tissues of 111QHtt knock-in mice revealed that full-length mutant Htt also associated to lysosomes with similar properties to those described for the wild-type form (Fig. 7G). In further support of a rather limited contribution of CMA to the degradation of either wild-type or mutant Htt, we analyzed changes in the cellular levels of these proteins after overexpressing LAMP-2A in cultured cells, a manipulation previously shown to effectively upregulate CMA activity (Cuervo and Dice, 1996). As shown in Figure 7H, upregulation of CMA did not have a significant impact on the cellular levels of either wild-type or mutant full-length Htt. However, using similar manipulations in cells expressing exon-1 Htt, we found that upregulation of CMA markedly reduced the steady-state levels of both wild-type and mutant Htt (Fig. 7H).
Overall, these results support that although the increase in CMA that we observed in HD models may not directly contribute to the degradation of full-length mutant Htt in a large extent, it may be very efficient in the degradation of the truncated forms of this protein, previously shown to be cytotoxic.
Changes in CMA activity with age in a HD animal model
As with other neurodegenerative disorders, HD symptoms worsen with age. We have reported that CMA activity also decreases with age, due for the most part to reduced levels of LAMP-2A in lysosomes in older organisms (Cuervo and Dice, 2000b). To determine whether the compensatory upregulation of CMA that we observed in the 6-month-old HD knock-in mice is preserved at later ages, we analyzed CMA activity in a group of older 18QHtt and 111QHtt knock-in mice.
In the mouse strain of the HD knock-in model used in this study, CMA activity is preserved up to 12 months of age, when a gradual decline in LAMP-2A levels begins reaching a reduction of ∼ 40% by 20 months of age. As survival of the 111QHtt knock-in mice in our colony drops dramatically after the first year of age, we chose 12 months as the oldest age in our analysis.
We first compared the ability of intact lysosomes isolated from livers of 6- and 12-month-old 18QHtt and 111QHtt knock-in mice to degrade a radiolabeled pool of cytosolic proteins. As in the young animals, we did not find significant differences in the stability of the both groups (data not shown). As shown in Figure 8A, similar to our previous results with a single radiolabeled protein (Fig. 3A), the pool of cytosolic protein was also taken up and degraded faster by lysosomes of 6-month-old 111QHtt mice when compared to 18QHtt mice; however, differences between 18QHtt and 111QHtt were smaller in the 12-month-old group (notice that by 12 months the decrease in CMA activity is not evident yet in the control animals). Separate analysis of substrate binding and uptake using the in vitro studies described in previous sections revealed that membrane binding of substrates (GAPDH shown here) is modestly lower in lysosomes from 12-month-old 111QHtt mice when compared to the younger group, but net binding is still higher than in 18QHtt littermates (Fig. 8B, top). However, despite higher binding of substrates to lysosomes being still preserved in the older group of HD animals, the uptake capacity of these lysosomes is markedly compromised, reaching levels below those in lysosomes from 18QHtt littermates (Fig. 8B, bottom).
Figure 8.

Changes in CMA activity with age in an HD animal model. A, Proteolysis of a pool of radiolabeled cytosolic proteins by lysosomes isolated from 6- and 12-month-old 18QHtt and 111QHtt knock-in mice. Values are expressed as percentage of the amount of protein at time 0 and are mean ± SE of three different experiments. B, Binding and uptake of GAPDH by the same lysosomes. GAPDH (2 μg) was incubated with isolated lysosomes untreated or pretreated with protease inhibitors (PI). Top, Immunoblot for GAPDH in two different experiments. Bottom, Quantification of the binding or uptake of GAPDH by each group. Values are expressed relative to the values in 6-month-old 18QHtt and are mean ± SE of three different experiments. C, Immunoblots for LAMP-2A and LAMP-1 in membranes (same amount of protein loaded) from lysosomes isolated from livers of 6- or 12-month-old 18QHtt and 111QHtt knock-in mice. Bottom, Quantification of the changes in LAMP-2A and LAMP-1 content by densitometric analysis of the proteins in the lysosomal membrane and matrices. Values are expressed relative to the values in 6-month-old 18QHtt and are mean ± SE of 4–6 different experiments. D, Quantification of mRNA levels for LAMP-2A by semiquantitative real-time PCR in livers of old 18QHtt knock-in mice and are mean ± SE of 3–4 different experiments. E, Immunoblot for hsc70 and LAMP-1 of lysosomal membranes (Mb) and matrices (Mtx) from the same mice. Bottom, Quantification of the changes in hsc70. Values are expressed relative to the values in 6-month-old 18QHtt and are mean ± SE of 3–4 different experiments. F, Immunoblot for the indicated proteins in fibroblasts control (Ctr) or knocked-down for LAMP-2A transiently transfected with an empty vector (none) or vectors coding for 23Q or 145Q exon-1 Htt.
To determine whether changes in LAMP-2A levels were behind the pronounced decrease in CMA observed in the older 111QHtt mice, we analyzed levels of this protein in lysosomes isolated from the four different groups of mice. As described previously for other mice strains (Cuervo and Dice, 2000b), levels of LAMP-2A at 6 and 12 months were comparable in lysosomes isolated from 18QHtt mice (Fig. 8C). In contrast, levels of LAMP-2A in lysosomal membranes from 12 month 111QHtt mice were no longer higher than those in 18QHtt mice, and in fact they were consistently lower (∼35% decrease) than those observed in 6 month 111QHtt mice (Fig. 8C, middle). Real-time quantitative PCR in the liver of these animals confirmed that indeed, in contrast to the increased levels of LAMP-2A mRNA observed in young 111QHtt mice, levels of LAMP-2A mRNA in 12-month-old 111QHtt mice were lower than those in 12 month 18QHtt mice (Fig. 8D).
We also noticed that the higher levels of hsc70 observed in lysosomal membranes from young 111QHtt mice were no longer noticeable in the older group (Fig. 8E). We have previously described a progressive increase in the number of lysosomes recruited to perform CMA with age (Cuervo and Dice, 2000a), also observable in this study in the 12-month-old 18QHtt mice as an increase in levels of luminal hsc70 (Fig. 8E). In contrast, immunoblot analysis of the matrices of 111QHtt mice lysosomes revealed a failure of this compensatory mechanism in this group, because levels of hsc70 remained unchanged in this fraction (Fig. 8E).
In summary, our results suggest that the early upregulation of CMA observed in HD cells, likely to compensate at least partially for the reduced levels of macroautophagy in these cells, is not maintained in the later stages of pathology. Indeed, the age-dependent decrease in CMA we identified previously in normal cells is accelerated in HD cells. To determine the impact on cellular homeostasis of this accelerated decline in CMA with age when in the context of the pathogenic protein, we experimentally reduced the levels of LAMP-2A by knock-down in control cells and cells expressing wild-type (23QHtt) and pathogenic exon1 Htt (145QHtt). Analysis of the levels of protein aggregation in these cells, measured as the amount of polyubiquitinated proteins retained in the stacking gel when subjected to electrophoresis, revealed negligible levels of protein aggregates in control cells and cells expressing 23Q exon1 Htt (Fig. 8F, lanes 1 and 3). However, reduction of LAMP-2A levels and consequently lower CMA activity resulted in a marked increase in the amount of protein aggregates in cells expressing 145Q exon1 Htt that was not evident when CMA activity was preserved (Fig. 8F, lane 5). We thus propose that the more rapid decline in CMA activity with age in HD could be an aggravating factor in the course of the disease.
Discussion
Alterations in the cellular systems responsible for quality control—chaperones and proteolytic systems—have been recently described to underlie the basis of different neurodegenerative disorders, or at least contribute to aggravate the course of the disease (Wong and Cuervo, 2010). Although macroautophagy can remove protein aggregates of mutant Htt (Iwata et al., 2005; Wong et al., 2008), primary defects in the autophagic machinery and its regulation in HD cells may limit the efficiency of this pathway as a cellular clearance mechanism (Martinez-Vicente et al., 2010). Cells often respond to compromise in one quality control pathway by upregulation of another (Massey et al., 2006; Kaushik et al., 2008; Kirkin et al., 2009; Korolchuk et al., 2009). In this work we have identified constitutive upregulation of CMA in multiple HD cell types. Interestingly, although we found higher rates of CMA activity in 2- to 7-month-old HD mouse models, this upregulation is no longer observed at older ages.
Although CMA cannot compensate for macroautophagy, as it is unable to degrade organelles or aggregated proteins, the unique selectivity of CMA for the degradation of cytosolic soluble proteins could be advantageous for cells as macroautophagy decreases. In fact, despite CMA contributing to about 30% of protein degradation, experimental blockage of this pathway in cultured cells affects specific transcriptional programs and metabolic pathways such as glycolysis and modifies cell growth and cell survival (Massey et al., 2006; Kaushik et al., 2008; Kirkin et al., 2009; Korolchuk et al., 2009). A maintained decrease in CMA activity, even in the order of 20–30%, is enough to compromise cellular homeostasis and the ability of cells to respond to stress (Massey et al., 2006; Kaushik et al., 2008; Kirkin et al., 2009; Korolchuk et al., 2009). In contrast, preventing the age-dependent decline in CMA has proven to be enough to preserve normal organ function and slow down aging (Zhang and Cuervo, 2008).
The relevance of our findings is twofold. First, our findings support the value of the recently proposed use of CMA for the removal of pathogenic Htt, since we demonstrate here that this pathway is preserved and is indeed upregulated in HD cells. We have found that despite the presence of 3 KFERQ-like motifs in the sequence of Htt, the full-length wild-type or mutant protein is not normally degraded via CMA. However, both wild-type and mutant exon 1 Htt are susceptible to degradation by CMA. Although further studies are needed to determine the basis for the different CMA susceptibility of full-length and exon-1 Htt, it is possible that changes in the conformation around the regions containing the CMA-targeting motives and/or posttranslational modifications in these nearby regions contribute to facilitate the interaction with the targeting chaperone and the subsequent delivery to lysosomes. Also of interest is the identification in this study of a lysosome-associated form of Htt resident on the cytosolic side of the lysosomal membrane. In light of the recently identified role of Htt in intracellular vesicular trafficking, it is possible that this protein may contribute to lysosomal mobilization. Whether or not the association of mutant Htt to lysosomal membrane contributes to alter lysosomal function requires future investigation.
The increase in CMA in HD cells described in this study justifies future attempts to pharmacologically mimic these posttranslational modifications in the pathogenic protein as well as other artificial procedures that enhance targeting of Htt to CMA. Also of value would be future studies aimed at identifying the molecular components that regulate the compensatory upregulation of CMA, since they could become potential therapeutic targets. Growing evidence supports the existence of cross talk among different autophagic pathways and of autophagy with other proteolytic systems, such as the ubiquitin/proteasome system (Massey et al., 2006; Kaushik et al., 2008; Kirkin et al., 2009; Korolchuk et al., 2009). Although the mechanisms that modulate this cross talk are not completely understood, reduced macroautophagic activity has been shown to have a negative impact on the ubiquitin/proteasome system (Korolchuk et al., 2009), which could explain in part the decrease in the activity of this system observed in HD cells. We have recently reported that blockage of the proteasome catalytic activity in cultured cells also elicits upregulation of CMA (Koga et al., 2011). It is thus possible that compromised activity of both macroautophagy and the ubiquitin/proteasome system described in HD is behind the increase in CMA activity observed in the affected cells.
The other important finding from our study is the possibility that there is a relatively narrow window of opportunity to apply these procedures, since at more advanced stages of the disease, upregulation of CMA is no longer sustained in HD cells. It is possible that if efficient removal of the pathogenic protein via CMA is attained in the early stages of this disease, the accelerated decline in the activity of this pathway may be prevented, because in the absence of the pathogenic protein, other proteolytic systems and cellular homeostasis in general may be preserved.
The reasons for the accelerated decline in CMA activity observed in the group of older HD mice are unclear. It is possible that sustained upregulation of this autophagic pathway, even under basal conditions, for long periods of time leads to exhaustion of effectors and regulators of CMA. For example, rapid functional decline after maintained upregulation has been described for macroautophagy in diabetic cells or after HIV-1 infection in dendritic cells (Zhou and Spector, 2008; Zhou et al., 2010). Alternatively, although we have not found a direct toxic effect of mutant Htt on CMA active lysosomes, it is possible that abnormal interactions of the pathogenic protein with regulators of this pathway may with time inhibit its activity. Finally, because we have identified one of the main factors that contributes to the decline in CMA with age is an altered stability of the receptor associated with changes in the lipid composition of the lysosomal membrane (Kiffin et al., 2007), it may be that alterations in lipid metabolism reported in HD may contribute to a faster decline of CMA. In support of this possibility, a marked decrease in the lysosomal levels of LAMP-2A was the most pronounced difference in the lysosomes from 12-month-old HD mice compared to control mice. In this respect, we have recently shown that macroautophagy contributes to intracellular lipid metabolism and that this function is altered in different HD cells (Martinez-Vicente et al., 2010). It is possible that the inability to properly handle cellular lipids due to the compromise of macroautophagy in HD cells could, in time, affect the lipid composition of intracellular membranes, including those of the CMA active lysosomes. In fact, cholesterol levels and the distribution GM1 are different in lysosomes from 111QHtt mice when compared to 18QHtt mice.
An additional finding of this study is that although the upregulation of CMA in HD cells was detected in both neuronal cells (striatal cell lines and primary striatal neuronal cultures) and non-neuronal cells (mouse embryonic fibroblasts and hepatocytes) and in both mouse and human samples, the mechanisms behind this upregulation may be more cell type dependent. We have previously reported different mechanisms for CMA upregulation depending on the type of stimuli—transcriptional upregulation of LAMP-2A occurs in response to oxidative stress (Kiffin et al., 2004)—whereas increased stability of LAMP-2A at the lysosomal membrane is a preferred mechanism for activation of CMA during nutrient starvation (Cuervo and Dice, 2000a). Although some level of transcriptional upregulation for LAMP-2A was observed in all HD cells, the extent of this upregulation was very variable, depending on the cell type. Indeed, in hepatocytes and striatal cells the modest increase in LAMP-2A de novo synthesis seems to be complemented by the stabilization of LAMP-2A in lysosomes. The adoption of these different mechanisms of activation requires further investigation.
In light of our findings, we propose that approaches that use targeting of pathogenic proteins to CMA for clearance in HD cells or those aimed at preventing the accelerated decline in the activity of this pathway with age in HD cells may provide a means to prevent the protein toxicity associated with this devastating neurodegenerative disorder.
Footnotes
This work was supported by National Institutes of Health/National Institute on Aging Grants AG021904 and AG031782 (to A.M.C.) and a National Institute of Neurological Disorders and Stroke Udall Center of Excellence Grant (to D.S.), a Huntington's Disease Society of America Grant (to D.S. and A.M.C.), and a Hirschl/Weill-Caulier Career Scientist Award (to A.M.C.). E.A. is supported by Micinn/Fulbright Fellowship 2008-0128.
The authors declare no competing financial interests.
References
- Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997;137:825–834. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento F, Roche E, Cuervo AM, Knecht E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J Biol Chem. 1993;268:10463–10470. [PubMed] [Google Scholar]
- Auteri JS, Okada A, Bochaki V, Dice JF. Regulation of intracellular protein degradation in IMR-90 human diploid fibroblasts. J Cell Physiol. 1983;115:167–174. doi: 10.1002/jcp.1041150210. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–5763. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, Miyazaki H, Matsumoto G, Kino Y, Nagai Y, Nukina N. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010;28:256–263. doi: 10.1038/nbt.1608. [DOI] [PubMed] [Google Scholar]
- Chen S, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci U S A. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2010;21:142–150. doi: 10.1016/j.tem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000a;1:570–583. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000b;275:31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000c;113:4441–4450. doi: 10.1242/jcs.113.24.4441. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997;272:5606–5615. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Hu W, Lim B, Dice JF. IkB is a substrate for a selective pathway of lysosomal proteolysis. Mol Biol Cell. 1998;9:1995–2010. doi: 10.1091/mbc.9.8.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Mann L, Bonten EJ, d'Azzo A, Dice JF. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003 doi: 10.1093/emboj/cdg002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3:295–299. doi: 10.4161/auto.4144. [DOI] [PubMed] [Google Scholar]
- Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, Modugno N, Siciliano G, Isidoro C, Murri L, Ruggieri S, Paparelli A. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105:2052–2057. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009;452:297–324. doi: 10.1016/S0076-6879(08)03619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Massey AC, Cuervo AM. Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. EMBO J. 2006;25:3921–3933. doi: 10.1038/sj.emboj.7601283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19:2179–2192. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey AC, Martinez-Vicente M, Cuervo AM. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci. 2007;120:782–791. doi: 10.1242/jcs.001073. [DOI] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Koga H, Martinez-Vicente M, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Comm. 2011;2:386. doi: 10.1038/ncomms1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5:958–963. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006;103:5805–5810. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y, Ishikawa T, Kato K. A rapid and simplified method for the preparation of lysosomal membranes from rat liver. J Biochem. 1983;93:547–556. [PubMed] [Google Scholar]
- Petersén A, Hansson O, Puschban Z, Sapp E, Romero N, Castilho RF, Sulzer D, Rice M, DiFiglia M, Przedborski S, Brundin P. Mice transgenic for exon 1 of the Huntington's disease gene display reduced striatal sensitivity to neurotoxicity induced by dopamine and 6-hydroxydopamine. Eur J Neurosci. 2001;14:1425–1435. doi: 10.1046/j.0953-816x.2001.01765.x. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington's disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- Storrie B, Madden EA. Isolation of subcellular organelles. Meth Enzymol. 1990;182:203–225. doi: 10.1016/0076-6879(90)82018-w. [DOI] [PubMed] [Google Scholar]
- Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, O'Rourke JG, Khashwji H, Lukacsovich T, Zhu YZ, Lau AL, Massey A, Hayden MR, Zeitlin SO, Finkbeiner S, Green KN, LaFerla FM, Bates G, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AJ, Signer ER. Huntington's disease: the challenge for cell biologists. Trends Cell Biol. 2000;10:531–536. doi: 10.1016/s0962-8924(00)01853-5. [DOI] [PubMed] [Google Scholar]
- Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattiaux R, Wattiaux-De Coninck S, Ronveaux-Dupal MF, Dubois F. Isolation of rat liver lysosomes by isopycnic centrifugation in a metrizamide gradient. J Cell Biol. 1978;78:349–368. doi: 10.1083/jcb.78.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ES, Tan JM, Soong WE, Hussein K, Nukina N, Dawson VL, Dawson TM, Cuervo AM, Lim KL. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet. 2008;17:2570–2582. doi: 10.1093/hmg/ddn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol. 2005;28:28–1. doi: 10.1002/0471142727.mb2801s70. [DOI] [PubMed] [Google Scholar]
- Young AB. Huntingtin in health and disease. J Clin Invest. 2003;111:299–302. doi: 10.1172/JCI17742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Spector SA. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS. 2008;22:695–699. doi: 10.1097/QAD.0b013e3282f4a836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Wu S, Li X, Xue Z, Tong J. Rapamycin induces autophagy and exacerbates metabolism associated complications in a mouse model of type 1 diabetes. Indian J Exp Biol. 2010;48:31–38. [PubMed] [Google Scholar]