

Abstract
Membrane pyrophosphatases (PPases), divided into K+-dependent and K+-independent subfamilies, were believed to pump H+ across cell membranes until a recent demonstration that some K+-dependent PPases function as Na+ pumps. Here, we have expressed seven evolutionarily important putative PPases in Escherichia coli and estimated their hydrolytic, Na+ transport, and H+ transport activities as well as their K+ and Na+ requirements in inner membrane vesicles. Four of these enzymes (from Anaerostipes caccae, Chlorobium limicola, Clostridium tetani, and Desulfuromonas acetoxidans) were identified as K+-dependent Na+ transporters. Phylogenetic analysis led to the identification of a monophyletic clade comprising characterized and predicted Na+-transporting PPases (Na+-PPases) within the K+-dependent subfamily. H+-transporting PPases (H+-PPases) are more heterogeneous and form at least three independent clades in both subfamilies. These results suggest that rather than being a curious rarity, Na+-PPases predominantly constitute the K+-dependent subfamily. Furthermore, Na+-PPases possibly preceded H+-PPases in evolution, and transition from Na+ to H+ transport may have occurred in several independent enzyme lineages. Site-directed mutagenesis studies facilitated the identification of a specific Glu residue that appears to be central in the transport mechanism. This residue is located in the cytoplasm-membrane interface of transmembrane helix 6 in Na+-PPases but shifted to within the membrane or helix 5 in H+-PPases. These results contribute to the prediction of the transport specificity and K+ dependence for a particular membrane PPase sequence based on its position in the phylogenetic tree, identity of residues in the K+ dependence signature, and position of the membrane-located Glu residue.
Keywords: Membrane Energetics, Membrane Enzymes, Proton Pumps, Proton Transport, Sodium Transport, H+ Pyrophosphatases, Na+ Pyrophosphatases, PPase, Primary Pump, Pyrophosphatases
Introduction
Membrane pyrophosphatases (PPases2; EC 3.6.1.1; Transporter Classification Database number 3.A.10) couple the hydrolysis of PPi to the active transport of cations across membranes and display no sequence homology to any known protein family. Membrane PPases consist of 70–81-kDa subunits that apparently form a homodimer (1) and are divided into K+-dependent and K+-independent subfamilies. The key determinant of K+ dependence is a single amino acid position located near the cytoplasm-membrane interface, which is occupied by Ala in the K+-dependent and Lys in the K+-independent enzymes (2). All PPases additionally require Mg2+ for function (3).
H+-transporting PPases (H+-PPases), discovered >40 years ago, are widespread among organisms in all three domains of life (4–6). Both K+-dependent and K+-independent H+-PPases have been characterized. In bacteria and archaea, these enzymes generate an ion-motive force for ATP synthesis and transport processes, particularly during stress and low energy conditions (7–9). In eukaryotes, H+-PPase acts in parallel with H+-ATPase and plays important roles in securing acidity and thus the functionality of cellular organelles, such as vacuoles in plants (1, 6, 10) and acidocalcisomes in protozoa (11). In plants, H+-PPase appears to be involved in auxin-dependent organ development (12, 13), and overexpression of the pump in agricultural plants confers resistance against water/nutrient deprivation, cold, and salinity (14–17). Furthermore, H+-PPase is a promising drug target in protozoan parasites (18).
In contrast, Na+-transporting PPases (Na+-PPases) were only recently identified in the mesophilic archaeon Methanosarcina mazei, the moderately thermophilic acetogenic bacterium Moorella thermoacetica, and the marine hyperthermophilic bacterium Thermotoga maritima (19). These enzymes are similar to the H+-PPases in many aspects but require both K+ and Na+ for activity (20).
The initial discovery of Na+-PPases in the contrasting ecological and evolutionary niches suggests that PPi-energized Na+ transport is a ubiquitous process. However, verification of this hypothesis has been severely hampered by our inability to predict transport specificity directly from protein sequence information. In this study, we overcame this limitation by experimentally screening transport specificity across different types of membrane PPases. These data, in conjugation with phylogenetic analysis, indicate that Na+ transport specificity is indeed a very common property and preceded H+ transport specificity in the evolutionary history of the protein family. We additionally employed site-directed mutagenesis to identify the structural elements defining membrane PPase transport specificity.
EXPERIMENTAL PROCEDURES
Sequence and Phylogenetic Analyses
Membrane PPase protein sequences were retrieved from the NCBI Protein Database with BLAST (21) using Rhodospirillum rubrum H+-PPase (YP_426905) as a query sequence. Protein sequences were aligned using default settings in MUSCLE Version 3.6 (22). The alignment was manually cured by eliminating incomplete and redundant sequences (leaving 121 taxa) and sequence regions, including indels and ambiguously aligned residues (leaving 333 amino acid columns). Input for Bayesian inference of protein phylogeny with MrBayes (23, 24) was either the alignment block or partitioned data set where the identity of the experimentally verified K+ dependence signature sequence was weighted by supplementing the sequence alignment with 398 columns of 0 or 1 character, depending on whether the full protein sequence harbored Ala (indicative of K+ dependence) or Lys (indicative of K+ independence), respectively, at position 460 (Carboxydothermus hydrogenoformans PPase (Ch-PPase) numbering). In the weighted tree, topology was estimated utilizing the information content of both partitions, whereas the amino acid alignment partition defined branch length. Four independent weighted and unconstrained phylogenetic analyses were initiated from random trees and run for 10 million generations (temperature option 0.15) on a computer cluster provided by the CSC-IT Center for Science Ltd. (Espoo, Finland). The average S.D. of split frequencies between independent MrBayes runs dropped below 0.01 during the first 2.5 million generations, indicating convergence of phylogenetic analysis. The consensus tree was summarized from the last 7.5 million generations. The likelihoods of constrained and unconstrained tree topologies were compared with the Shimodaira-Hasegawa test in Puzzle software using several different settings (25, 26). Ancestral protein sequences at internal nodes of the phylogenetic tree were inferred with the maximum likelihood method using PAML Version 4.4 (27). The likelihood of each possible amino acid state was calculated, given the sequence alignment, topology of the phylogenetic tree, JTT amino acid replacement model, and γ-distribution of among-site rate variations.
Expression of Recombinant Membrane PPases
Genes encoding membrane PPases from Anaerostipes caccae (Ac-PPase), Chlorobium limicola (Cl-PPase), Clostridium thermocellum (Ct-PPase), Desulfuromonas acetoxidans (Da-PPase), Flavobacterium johnsoniae (Fj-PPase), Leptospira biflexa (Lb-PPase), Pyrobaculum aerophilum (Pa-PPase), and Streptomyces coelicolor were amplified from genomic DNA using the primers listed in supplemental Table S1. The codon usage-optimized gene for Clostridium tetani E88 membrane PPase (Ctet-PPase) was synthesized by Mr. Gene (Regensburg, Germany). Full-length PPase genes were inserted into the multiple cloning site of the pET36b(+) expression vector (Novagen) under control of the T7 promoter via the NdeI, XhoI, or HindIII restriction site (supplemental Table S1). The cloning of T. maritima PPase (Tm-PPase) and Ch-PPase has been described previously (2, 20). Amino acid substitutions were engineered with inverse PCR using Phusion DNA polymerase (Finnzymes), and mutated genes were transferred into the pET36b(+) vector. The PPase-encoding regions of the final expression constructs were sequenced.
Membrane PPase genes were expressed in Escherichia coli, and inner membrane vesicles (IMV) were isolated as described previously (20). IMV were suspended in storage buffer (10 mm MOPS-tetramethylammonium (TMA) hydroxide (pH 7.2), 1 mm MgCl2, 750–900 mm sucrose, 5 mm DTT, and 50 μm EGTA), frozen in liquid N2, and stored at −85 °C. The protein content of IMV was estimated with the Bradford assay (28).
Assay of Hydrolytic Activity
Because E. coli lacks endogenous membrane PPase and the IMV isolation protocol efficiently washes away E. coli soluble PPase contamination (with <1% remaining background PPase activity), the IMV prepared from cells carrying membrane PPase expression plasmids could be used directly for activity measurements. To assay membrane PPase hydrolytic activity, a thermostated reaction vessel was filled with 25 ml of reaction buffer typically containing 0.1 m MOPS-TMA hydroxide (pH 7.2), 5 mm MgCl2, 158 μm TMA4PPi, 40 μm EGTA, and variable concentrations of NaCl and KCl. In measurements of K+ or Na+ dependence of PPase activity at a constant concentration (50 mm) of the other ion, TMA hydroxide was replaced with NaOH and KOH, respectively. The reaction was triggered by the addition of IMV suspension (0.004–1.3 mg of protein), and liberation of inorganic phosphate was continuously recorded for 2–3 min with a flow-through phosphate analyzer (29). Reaction rates were calculated from the initial slopes of the Pi liberation traces. The results of duplicate measurements were usually consistent within 10%.
The dependences of PPi hydrolysis rates (v) on Na+ and K+ concentrations at saturating substrate and Mg2+ concentrations were analyzed in terms of Scheme 1 (A and B, respectively). ES represents the enzyme-substrate complex, M the alkali metal ligand (Na+ or K+), Ka and Ki the metal binding constants, and V1 and V2 the maximal velocities for the corresponding complexes. Scheme 1A assumes that binding of the first Na+ ion stimulates hydrolysis, whereas binding of the second Na+ ion inhibits the reaction. According to Scheme 1B, K+ binding modulates only hydrolytic activity (V1 ≠ V2). Assuming rapid equilibrium cation binding, rate equations for the mechanisms in Scheme 1 (A and B) are provided by Equations 1 and 2, respectively.
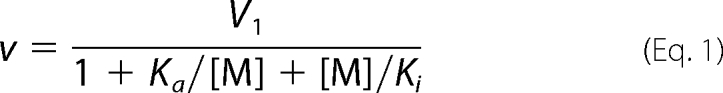 |
 |
Parameter values were determined by fitting these equations to rate data using Scientist software (Micromath), wherein [M] was treated as an independent variable; v as an experimentally measured variable; and V1, V2, Ka, and Ki as adjustable parameters.
SCHEME 1.
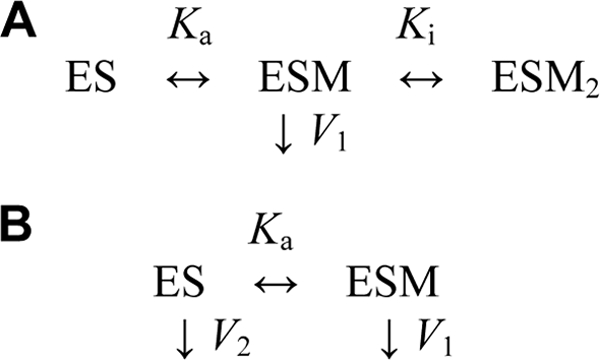
Alkali cation (M) binding to the enzyme-substrate complex (ES). A, Na+ binding; B, K+ binding.
Assay of Transport Activities
PPi-energized H+ transport into IMV was assayed by measuring the fluorescence quenching of the pH-sensitive probe 9-amino-6-chloro-2-methoxyacridine (Invitrogen) at 25 °C at excitation and emission wavelengths of 428 and 475 nm, respectively. IMV (0.2–1.2 mg) were incubated for 5 min in 2 ml of buffer (20 mm MOPS-TMA hydroxide (pH 7.2), 5 mm MgCl2, 8 μm EGTA, and 2 μm 9-amino-6-chloro-2-methoxyacridine) supplemented with 1.5–25 mm NaCl and 0–100 mm KCl before the reaction was initiated by the addition of 0.3 mm TMA4PPi. The reversibility of 9-amino-6-chloro-2-methoxyacridine fluorescence quenching was tested by disrupting the H+ gradient formed in IMV via the addition of 10 mm NH4Cl.
PPi-energized Na+ transport into IMV was assayed by determining the accumulation of 22Na+ within IMV at 22 °C. IMV (0.15–0.30 mg) were incubated for ∼1 h in 40 μl of buffer (100 mm MOPS-TMA hydroxide (pH 7.2), 5 mm MgCl2, 50 mm KCl, and 1 mm NaCl) containing 0.3–2.5 μCi of 22NaCl (PerkinElmer Life Sciences) before the transport reaction was initiated by the addition of 1 mm TMA4PPi. At various time points, IMV were separated from the reaction medium by passing 30-μl aliquots through a nitrocellulose filter (0.2-μm pore size, 13-mm diameter; Millipore). External 22Na+ was rinsed off by applying 1 ml of buffer (5 mm MOPS-TMA hydroxide (pH 7.2), 100 mm NaCl, 0.5 mm MgCl2) through the filter. The filter was transferred to a microcentrifuge tube, 1 ml of Ultima Gold mixture (PerkinElmer Life Sciences) was added, and the amount of 22Na+ trapped in IMV was determined via liquid scintillation counting.
RESULTS
Updated Phylogenetic Tree and Selection of PPases for Expression
The number of full-length putative membrane PPase sequences in the GenBankTM Data Bank has increased from ∼200 to 1000 since the previous phylogenetic analysis (19). Accordingly, a new tree has been constructed based on 121 non-redundant sequences. The known membrane PPases share 48% average and ≥33% pairwise amino acid identities. Sequence conservation is particularly strong in three cytoplasmically oriented loops forming the major portion of the PPi hydrolysis site (6). The polypeptides are very hydrophobic, typically containing 16 predicted transmembrane α-helices. In a few cases, more transmembrane α-helices have been identified in N- or C-terminal extensions to the core structure.
MrBayes analysis generated a tree largely consistent with the previously determined grouping of PPases into K+-dependent and K+-independent subfamilies (2). Notably, sequences from C. thermocellum and Thermotoga lettingae were grouped with K+-dependent enzymes near the boundary between the subfamilies, although they possessed sequence determinants (Lys478 and Thr481, Cl-PPase numbering) of K+ independence (Fig. 1). To assess the significance of anomalous grouping, we shifted the C. thermocellum and T. lettingae sequences to the K+-independent subfamily by heavily weighting the experimentally determined K+ dependence signature and comparing the resulting tree with an unconstrained tree using the Shimodaira-Hasegawa test implemented in Puzzle software (25). The constrained tree appeared to have a higher likelihood, whereas the unconstrained tree was significantly rejected (p < 0.05). Accordingly, the constrained tree was utilized for all further analyses.
FIGURE 1.

Phylogenetic tree, residue conservation. and functional classification of membrane PPases. The tree was arbitrarily rooted with the branch leading to the S. coelicolor clade of H+-PPase. The GenBankTM protein sequence number is indicated before the species name. The nodes are supplemented with clade credibility values and marked in selected cases with uppercase letters. The studied membrane PPases are boxed. The scale bar represents 0.2 substitutions per residue. For clarity, only 79 of 121 sequences used to construct the tree are shown. The residue columns 180, 242, and 246 (Cl-PPase numbering) to the right of the tree correspond to the positions of the pump specificity-linked Glu residue, whereas the residue columns 478 and 481 refer to the positions determining K+ requirements. The K+ dependence and Coupling ion columns present the inferred and experimentally verified K+ requirement and coupling ion specificity, respectively.
To comprehensively map the distribution of transport specificities among membrane PPases, we selected several key genes from the constructed phylogenetic tree for the expression and biochemical characterization of the corresponding expressed proteins. We focused on PPases located in hitherto unexplored clades within the phylogenetic tree. In total, seven PPase sequences were selected for analysis.
Expression of Functional PPases in E. coli
Novel membrane PPases were heterologically expressed in E. coli, and the recombinant proteins were isolated as E. coli IMV. We additionally expressed four previously described membrane PPases (from C. hydrogenoformans, P. aerophilum, S. coelicolor, and T. maritima). All PPases were expressed in the active form, as determined using two criteria. First, Western analysis of IMV proteins using rabbit antiserum raised against a conserved amino acid motif in the third cytoplasmic loop of the enzyme (19) revealed specific bands representing proteins of predicted sizes (supplemental Fig. S1). Second, the IMV exhibited increased PPi hydrolytic activity, which was suppressed by the membrane PPase inhibitor aminomethylene diphosphonate, but was insensitive to the E. coli soluble PPase inhibitor fluoride (Fig. 2) (30).
FIGURE 2.

Hydrolytic activities of recombinant membrane PPases in E. coli IMV. Hydrolytic activity was determined at 25 °C or 40 °C (insets). Unless indicated otherwise, the reaction medium contained 50 mm KCl and 10 mm NaCl (white bars). Gray and black bars depict PPase activity in the presence of 250 μm potassium fluoride and 20 μm aminomethylene diphosphonate, respectively. The K+ dose response of Ct-PPase activity was assayed in the absence of added Na+. Sc-PPase, S. coelicolor PPase.
The maximal PPi hydrolytic activities of Ch-PPase, Ct-PPase, Pa-PPase, and Tm-PPase were relatively low at 25 °C (5–50 nmol/min/mg) but increased at least 3-fold when the assay temperature was increased to 40 °C (Fig. 2), consistent with the fact that the genes encoding these membrane PPases were isolated from thermophilic host organisms. Importantly, Ct-PPase activity at 40 °C (and 25 °C) was independent of the K+ concentration (Fig. 2), leading to its classification as a member of the K+-independent subfamily. This finding further supports the validity of the constrained phylogenetic tree.
Ion Transport Activities of PPases
The H+ transport capacity of all PPases was assayed by monitoring the fluorescence of a pH indicator, 9-amino-6-chloro-2-methoxyacridine, at 25 °C. When PPi was added to the reaction mixture containing Lb-PPase-, Fj-PPase-, and Ch-PPase-harboring IMV, dye fluorescence progressively decreased to a steady-state level, indicating acidification of the IMV lumen (Fig. 3A). Upon disruption of the proton gradient with the addition of NH4Cl or consumption of all PPi, fluorescence returned to the initial level. Qualitatively similar fluorescence changes were previously observed for Ch-PPase IMV by employing acridine orange as the pH indicator (2). Given the lack of PPi-dependent IMV acidification in native E. coli vesicles devoid of membrane PPases, it is proposed that the observed H+ transport activity is specifically mediated by recombinant PPases.
FIGURE 3.

H+ transport by membrane PPases at 25 °C. The transport reaction was commenced by adding TMA4PPi to the otherwise complete reaction mixture. The fluorescence intensity at 475 nm returned to the initial level when the steady-state proton gradient was collapsed by the addition of 10 mm ammonium chloride. The incidence of PPi and ammonium chloride additions is indicated by arrows. A, wild-type PPases (measured at 10 mm Na+ and 50 mm K+). B, H+ transport by Lb-PPase as a function of Na+ concentration. The respective Lb-PPase hydrolytic activities were 320 (10 mm Na+ and 50 mm K+), 75 (25 mm Na+), 38 (10 mm Na+), 25 (5 mm Na+), and 13 (1.5 mm Na+) nmol/min/mg. C, Mutant PPases (10 mm Na+ and 100 mm K+). a.u., arbitrary units.
In contrast, no H+ transport activity was detected with Ac-PPase, Cl-PPase, Ct-PPase, Ctet-PPase, and Da-PPase. To ascertain whether this finding was due to low PPase activity at 25 °C, we estimated the sensitivity of the transport assay. To this end, we recorded the H+ transport signal mediated by Lb-PPase under conditions in which it was tuned to low activity by decreasing K+ and Na+ concentrations (Fig. 3B). In this setup, Lb-PPase IMV produced the lowest detectable H+ transport signal in the presence of 1.5 mm Na+, corresponding to a PPi hydrolysis rate of 13 nmol/min/mg. The hydrolytic activities of most PPases under conditions of H+ transport measurements were well above this limit, and the lack of the H+ transport signal for a particular PPase was strongly indicative of its inability to perform this function. The only exception was Ct-PPase, whose activity (5 nmol/min/mg) was too low to support measurable H+ transport activity. Unfortunately, high temperatures are unattainable for H+ transport experiments with E. coli IMV because the passive permeation rate across the membrane rises steeply with temperature.
Na+ transport activity was determined with a filtration assay utilizing 22Na. The active transport reaction was initiated by the addition of PPi to IMV pre-equilibrated with 22Na+. Significant transport rates were observed with Ac-PPase, Cl-PPase, Ctet-PPase, Da-PPase, and Tm-PPase (Fig. 4). These findings led to the identification of four new Na+-PPases (marked with asterisks in Fig. 4 and all subsequent figures and tables) and confirmed our earlier characterization of Tm-PPase as a Na+ pump using a different transport assay (19). No Na+ transport was observed with the other membrane PPases examined in this study (Fig. 4).
FIGURE 4.

Na+ transport by membrane PPases at 22 °C. The membrane PPase-catalyzed transport reaction was commenced by adding TMA4PPi to otherwise complete the reaction mixture and was allowed to proceed for 1 min. The inset shows the time dependence of Na+ transport into PPase-free IMV and IMV containing Tm-PPase or Lb-PPase. The error bars display S.D. for at least three independent measurements. The enzymes classified as Na+-PPases are indicated with asterisks. Sc-PPase, S. coelicolor PPase.
Previous studies have reported that membrane PPases from S. coelicolor, M. mazei (PPase 2), R. rubrum, and Arabidopsis thaliana (AVP2) belonging to the K+-independent subfamily and the K+-dependent enzyme from C. hydrogenoformans mediate PPi-dependent H+ transport activity (2, 31–34), whereas R. rubrum PPase has no Na+ transport activity (19). Our results consistently demonstrate that Pa-PPase, S. coelicolor PPase, and Ch-PPase do not mediate Na+ transport into IMV (Fig. 4). The activities of K+-independent H+-PPases and Ch-PPase thus appear to be specifically related to H+ transport.
Notably, the Na+ transport assay was more sensitive than the H+ transport assay. Despite the relatively low PPi hydrolytic activity of hyperthermophilic Tm-PPase (14 nmol/min/mg) at the assay temperature (22 °C), the enzyme catalyzed significant time-dependent 22Na+ accumulation into IMV (Fig. 4, see also the inset). Moreover, the 22Na+ transport signal was at least 3-fold higher than the background, even under conditions (1.5 mm K+ and 1 mm Na+) in which Tm-PPase hydrolytic activity was as low as 5 nmol/min/mg (data not shown). Because the hydrolytic activities of all PPases producing no Na+ transport signal were equal to or exceeded this value (Fig. 2), we concluded that these enzymes possess no Na+ transport activity. This result is consistent with the above finding that PPases (except Ct-PPase) are active as H+ transporters. Ct-PPase did not exhibit any transport activity, but given the lower sensitivity of the H+ transport assay, it is likely that Ct-PPase operates as a H+ transporter in C. thermocellum at its growth temperature (60 °C). This may also apply to P. aerophilum enzyme (Pa-PPase), another thermophilic enzyme for which no H+ transport signal was observed in previous studies (35).
Na+ and K+ Dependences of PPase Activity
H+-PPases either are active in the absence of alkali cations or require K+ for activity (6), whereas Na+-PPases absolutely require Na+ and are further activated by K+ (19). To determine the alkali cation requirements of the newly expressed membrane PPases, we measured their hydrolytic activities over a wide range of Na+ and K+ concentrations. No activity exceeding the background E. coli soluble PPase-attributable value was observed in the absence of both Na+ and K+.
The Na+ dose-response curves measured in the absence of K+ were similar for all PPases, except that low level inhibition of Cl-PPase was observed at high [Na+] (Fig. 5). These curves appear to reflect the findings that Na+-PPases absolutely require Na+ for activity and that Na+ can partially meet the K+ requirement of K+-dependent H+-PPases (3, 20).
FIGURE 5.

Dependence of the hydrolytic activities of membrane PPases on Na+ concentration at 25 °C. The K+ concentration was either zero (upper panel) or 50 mm (lower panel). Lines were obtained with Equation 1 using the best fit parameter values found in Table 1.
Significant alterations in the dose-response curves were observed when PPi hydrolysis was assayed in the presence of 50 mm K+. First, the maximal activity values increased in all cases. Second, the curves for the Na+-PPases shifted to lower Na+ concentrations. Third, the H+-PPases lost their response to Na+ concentrations (Lb-PPase), or Na+ became inhibitory (Fj-PPase). The latter observation is consistent with a lower capacity of Na+ to activate K+-dependent H+-PPases compared with K+ (3). Additionally, inhibition by excess Na+ was more pronounced with Cl-PPase. The parameter values for Scheme 1A (one activating and one inhibiting Na+) obtained using Equation 1 are summarized in Table 1. In terms of maximal activity (V1) and the activating Na+ binding constant (Ka), K+ enhanced the maximal rate of PPi hydrolysis by 2–11-fold and Na+ binding affinity by 40–400-fold in different Na+-PPases (Table 1). The novel Na+-PPases thus appear to utilize the same bifunctional mechanism of K+ activation as those described previously (19, 20), i.e. K+ increases both the Na+ binding affinity and catalytic constant. In contrast to Na+-PPases, which require both Na+ and K+ for maximal activity, K+ alone is sufficient for full activation of H+-transporting Fj-PPase and Lb-PPase (Fig. 5). Accordingly, these PPases have been classified as K+-dependent enzymes.
TABLE 1.
Kinetic parameters for Na+ activation in the presence and absence of 50 mm K+
| Enzyme | Parameter value |
||||
|---|---|---|---|---|---|
| No K+a |
50 mm K+ |
||||
| V1 | Ka | V1 | Ka | Ki | |
| μmol/min/mg | mm | μmol/min/mg | mm | mm | |
| Ac-PPase* | 0.04 ± 0.01 | 23 ± 5 | 0.17 ± 0.01 | 0.43 ± 0.04 | NAb |
| Ctet-PPase* | 0.24 ± 0.01 | 27 ± 2 | 0.80 ± 0.08 | 0.45 ± 0.08 | NA |
| Da-PPase* | 0.48 ± 0.08 | 80 ± 2 | 0.78 ± 0.09 | 1.9 ± 0.4 | NA |
| Cl-PPase* | 0.26 ± 0.04 | 14 ± 3 | 1.1 ± 0.1 | 0.036 ± 0.002 | 100 ± 20 |
| Cl-PPase(E242D)* | 0.38 ± 0.08 | 190 ± 50 | 0.81 ± 0.06 | 16 ± 2 | 400 ± 110 |
| Fj-PPase | 0.09 ± 0.01 | 9 ± 2 | 0.99 ± 0.04 | NA | 130 ± 20 |
| Lb-PPase | 0.14 ± 0.02 | 9 ± 2 | 0.46 ± 0.05 | NA | NA |
a The Ki value was above 1000 mm in the absence of K+.
b NA, not attendant.
It is possible that in Fj-PPase and Cl-PPase, the Na+ binding constants (Ka) measured in the absence of K+ and Ki measured in the presence of K+ refer to the same binding site and that the difference between these constants reflects competition with K+ in the latter case. The K+ binding constant for Fj-PPase can thus be estimated from the data as 3.7 mm, compared with 9 mm for Na+.
At a fixed concentration of 50 mm Na+ and increasing K+ concentrations, the hydrolytic activities of all PPases increased from a non-zero basal level to a maximal level (Fig. 6). The activation profile is modeled in Scheme 1B assuming a single activating K+-binding site on the enzyme. The K+ binding constant (Ka) was in the range of 14–38 mm (Table 2) and did not change systematically between Na+- and H+-PPases.
FIGURE 6.

Dependence of the hydrolytic activities of membrane PPases on K+ concentration at 25 °C. The Na+ concentration was 50 mm. Lines were obtained with Equation 2 using the best fit parameter values found in Table 2.
TABLE 2.
Kinetic parameters for K+ activation in the presence of 50 mm Na+
| Enzyme | Parameter value |
||
|---|---|---|---|
| V1 | V2 | Ka | |
| μmol/min/mg | μmol/min/mg | mm | |
| Ac-PPase* | 0.25 ± 0.01 | 0.03 ± 0.01 | 14 ± 2 |
| Ctet-PPase* | 1.9 ± 0.2 | 0.44 ± 0.04 | 30 ± 9 |
| Da-PPase* | 1.31 ± 0.04 | 0.15 ± 0.01 | 38 ± 2 |
| Cl-PPase* | 1.2 ± 0.1 | 0.25 ± 0.02 | 32 ± 8 |
| Cl-PPase (E242D)* | 0.89 ± 0.03 | 0.06 ± 0.01 | 19 ± 2 |
| Fj-PPase | 1.45 ± 0.08 | 0.11 ± 0.01 | 20 ± 2 |
| Lb-PPase | 0.74 ± 0.06 | 0.14 ± 0.01 | 18 ± 5 |
Role of Membrane-located Glu in Ion Transport Specificity
An extensive manual or computational (Sequence Harmony) (36) search for sequence determinants correlating with enzyme transport specificities failed to identify a single pattern of residue conservation matching the observed transport specificities across the entire phylogenetic tree. The evolutionary history of membrane PPases therefore appears to include more than one change in transport specificity. Comparison of Na+-PPases and plant-type H+-PPases revealed a single conserved Glu residue (Glu242 in Cl-PPase) in all characterized Na+-PPases near the cytoplasmic end of transmembrane helix (TMH) 6, which serves as a membrane anchor for the highly conserved and functionally important cytoplasmic loop (residue 242 in Fig. 1) (31). In plant-type H+-PPases (including bacterial Lb-PPase), Glu is shifted by four residues toward the C-terminal end of the polypeptide and becomes membrane-buried. Interestingly, TMH6 is completely devoid of the relevant Glu residue in Fj-PPase-type H+ pumps (sequences from F. johnsoniae to Amoebophilus asiaticus in Fig. 1). In these enzymes, a conserved Glu residue is found instead in the preceding TMH5 in the position aligned with Cl-PPase residue 180. As conserved charged residues are not present in TMH5 in other types of membrane PPases, it is proposed that the Glu residue is specifically relocated from TMH6 to TMH5.
Because two independent Glu relocations are correlated with changes in ion transport specificity, we employed site-directed mutagenesis to study the functional consequences of engineered Glu substitutions and relocation. Initially, we relocated the Glu residues in TMH5 of Fj-PPase and TMH6 of Lb-PPase to the conventional Na+-PPase position at the cytoplasm-membrane interface in TMH6. We additionally replaced Gly246 of C. limicola Na+-PPase with Ala, as in F. johnsoniae H+-PPase, and transferred the Glu residue from TMH6 to TMH5 independently or in combination with the Gly-to-Ala substitution. The resulting recombinant variants (Fj-PPase E185S/G247E, Lb-PPase G249E/E253G, Cl-PPase S180E/E242G, Cl-PPase S180E/E242G/G246A, and Cl-PPase G246A) retained expression in E. coli (supplemental Fig. S1) but were inactive. Thus, membrane PPase poorly tolerates substitutions that increase amino acid sizes, possibly due to tight packing of the transmembrane helices.
To avoid potential steric clashes, we replaced Glu with Asp in the second round of mutagenesis. The variant enzymes Fj-PPase E185D, Lb-PPase E253D, and Cl-PPase E242D retained 24, 32, and 37% of the wild-type PPi hydrolytic activity, respectively. The transport data recorded for the variant enzymes indicated decreased transport rates, in keeping with decreased enzymatic activity, but no changes in transport specificity (Figs. 3 and 4). Most importantly, the Na+ dose-response curve of the Cl-PPase E242D variant was dramatically shifted to higher Na+ concentrations relative to the wild-type enzyme (Fig. 5). Accordingly, the Na+ binding constant (Ka) for the activating Na+ ion increased by up to 450-fold (Table 1). In contrast, K+ binding affinity was not significantly affected (Fig. 6 and Table 2).
In the third round of mutagenesis, the carboxyl group of the Glu residue under consideration was eliminated by substitution with Ser. The E242S substitution appeared to structurally destabilize Cl-PPase because no variant polypeptides were detected in the IMV fraction by Western analysis. The Fj-PPase E185S and Lb-PPase E253S variants retained 20–30% of the wild-type PPase activity, respectively, but exhibited no detectable PPi-dependent H+ or Na+ transport activity (Figs. 3 and 4). The hydrolytic activities of both variant PPases were sufficiently high (>85 nmol/min/mg) to generate a recordable H+ or Na+ transport signal (see above). Furthermore, IMV leakage was not significantly affected by membrane PPase substitutions because, in all tested cases, the IMV supported comparable levels of the ATP-dependent H+ gradient generated by E. coli housekeeping enzymes. These results suggest that Glu242 (Cl-PPase numbering) is a critical but not the sole determinant of transport specificity in membrane PPases.
DISCUSSION
The membrane PPase family consists of two independently evolving subfamilies, one (K+-dependent) requiring millimolar concentrations of K+ to attain catalytic activity and the other being K+-independent (2). Both subfamilies were originally believed to operate as H+ pumps until the recent discovery of Na+-PPases within the K+-dependent subfamily (19). The four novel Na+-PPases uncovered in this study have further increased the total number of identified Na+-PPases to seven. Moreover, Biegel and Müller (37) recently reported PPi-dependent Na+ accumulation in IMV prepared from Acetobacterium woodii, suggesting that this bacterium also contains a Na+-PPase. Notably, Ctet-PPase was previously reported to operate as a H+ pump (38). We did not observe a PPi-induced H+ transport signal with Ctet-PPase IMV but detected a strong Na+ transport signal. Ctet-PPase has thus been classified as a Na+ pump, consistent with the findings that C. tetani employs Na+ as the major coupling ion for membrane transport processes, lacks primary H+ pumps, and has a V-type Na+-coupled ATPase instead of H+-ATPase (39).
K+-dependent PPases can be phylogenetically classified into four major clades rooting at nodes A, B, C, and D (Fig. 1). Clade A branches at the interface of K+-dependent and K+-independent subfamilies, whereas clades B, C, and D group monophyletically at node N further within the K+-dependent subfamily. Clade A contains H+-PPases, clades B and C are populated by Na+-PPases, and clade D possesses both H+- and Na+-PPases. Na+-PPases thus dominate the K+-dependent subfamily, whereas Na+ pumping and dependence are traced to node N and thus are considered as an ancestor of the N cluster (groups of clades B, C, and D). At the functional level, the monophyletic origin of all existing Na+-PPases is supported by the finding that the activities of the enzymes characterized are regulated similarly by a complex Na+- and K+-dependent mechanism affecting both maximal enzymatic activity and ligand binding affinities. Two scenarios are envisaged to explain the presence of H+-PPases in clade D, specifically placement of H+-PPase in clade D is an artifact of phylogenetic reconstitution, or the H+ specificity of some PPases in clade D is not homologous to that in clade A and K+-independent H+-PPases but a result of homoplasy. Our analysis of the Glu residues in TMH5 and TMH6 favors the latter possibility.
The three subtypes of clade D membrane PPases are represented by Na+-transporting Cl-PPase, H+-transporting Fj-PPase, and plant-type H+-PPases. Cl-PPase is similar to enzymes from evolutionarily diverse bacteria. Fj-PPase has close homologs only in Bacteroidetes, whereas the plant category of H+-PPases additionally includes enzymes from unicellular protozoa and the spirochaete bacterium Leptospira. H+-transporting Fj-PPase and plant enzymes group polyphyletically and differ from all Na+-PPases and the clade D ancestor enzyme in that Glu242 (Cl-PPase numbering) is relocated from its cytoplasm-membrane interface position in TMH6 to a membrane-embedded position in TMH5 or TMH6, respectively. Several lines of evidence support the hypothesis that Glu242 relocations are key, albeit not the only structural modifications during the evolutionary pathways responsible for the H+ transport specificity of Fj-PPase-type and Leptospira/protozoan/plant-type enzymes. First, site-directed mutagenesis data indicate an essential role for TMH5 and TMH6 carboxyl groups in maintaining PPi hydrolysis coupling to H+ transport across the membrane. This conclusion is further supported by the recently published results of an alanine scanning mutagenesis study of transmembrane domain 6 of mung bean H+-PPase (40). Second, N,N′-dicyclohexylcarbodiimide, which preferentially reacts with protonated carboxyl groups, targets the relevant Glu residue in A. thaliana H+-PPase (AVP1) (41). Finally, the E242D substitution leads to a dramatic increase in the Na+ concentration required for the maximal activity of Na+-PPase in the presence of K+, indicating changes in the affinity of the Na+-specific binding site that presumably functions as the cytoplasmic acceptor for the transported Na+ (42).
K+-independent enzymes from C. thermocellum and T. lettingae are phylogenetically the most closely related to K+-dependent PPases. Ct-PPase and Pa-PPase do not transport Na+ and may thus perform H+ transport activity similar to other characterized K+-independent membrane PPases, but this is yet to be directly demonstrated. Because five characterized K+-independent H+-PPases represent distinct divergent clades within the subfamily and no K+-independent Na+-PPases have been encountered so far, it is highly likely that all members of the K+-independent subfamily operate as H+ pumps.
Significantly, the Na+-PPase and K+-independent H+-PPase genes are adjacent in the M. mazei genome, suggesting that the gene cluster is a footprint of an ancient gene duplication event resulting in the formation of the two deeply rooted subfamilies. To date, the classification of membrane PPases has been based on their requirement for K+. However, we speculate that the initial specialization of the duplicates leading to emergence of two subfamilies was, in fact, driven by changes in transported cation specificity and followed by secondary loss of K+ dependence in H+-PPases. Indeed, the absence of K+-independent Na+-PPases and our failure to generate such enzymes via mutagenesis3 suggest that K+ independence is incompatible with Na+ pumping. The change in transport specificity adds to the capability of the organism to utilize PPi for the generation of both Na+ and H+ gradients, whereas changes in K+ dependence do not provide an immediate fitness benefit. We further propose that the ancestor enzyme is possibly a Na+ pump. Thus, the evolution of a membrane PPase coupling ion specificity from Na+ to H+ parallels the scenario suggested for Na+- and H+-coupled F- and V-type ATP synthases/ATPases and adds support to the concept of global evolutionary primacy of Na+-coupled bioenergetics (43–45, 53).
Further research is required to establish the physiological role of Na+-PPase. A literature search has revealed two interesting trends. First, Na+-PPase is more frequently found in anaerobic organisms than H+-PPase. Thus, 23 of the 30 characterized and predicted Na+-PPases in the phylogenetic tree (Fig. 1) are derived from anaerobic hosts, whereas only 14 of 49 H+-PPases belong to anaerobes. Thus, a change from an anaerobic to aerobic environment may have provided the driving force for evolution from PPi-energized Na+ pumping to H+ pumping. This is most evident in clade D, which contains four of seven aerobic Na+-PPases (compared with only 3 of 25 Na+-PPases from aerobic hosts in clades B and C). Second, at least half of the anaerobic and all except one of the aerobic Na+-PPase hosts are halophilic or halotolerant compared with only one-quarter of the H+-PPases. Accordingly, it is proposed that Na+-PPase function is related to creating salt tolerance, especially in aerobic host organisms. This theory may be extended to speculate that because Na+-PPase is naturally preferred in halophiles, this enzyme may prove better than H+-PPase (6) in engineering salt tolerance in plants/microbes.
In summary, our results confirm that a wide range of microorganisms employ PPi-energized Na+ extrusion systems. We provide a comprehensive reconstruction of membrane PPase evolution and predict the transport specificity of membrane PPases as follows: (i) K+-independent enzymes are proton pumps, (ii) K+-dependent enzymes belonging to clade N are sodium pumps if Glu is present at the TMH6 interface and proton pumps if the Glu residue is relocated, and (iii) K+-dependent enzymes belonging to clade A are proton pumps. Notably, the sequence determinants governing the differences in transport specificity between clades A and N are not fully clarified at present, and therefore, the position of the sequence in the phylogenetic tree is an essential part of the prediction algorithm.
Supplementary Material
Acknowledgments
We are sincerely grateful to Dr. Maria Muntyan for helpful advice on the Na+ transport assay and to Dr. Mathieu Picardeau for providing genomic DNA of L. biflexa.
The work was supported by Academy of Finland Grants 114706, 130581, and 139031; a grant from the Ministry of Education of Finland (for the National Graduate School in Informational and Structural Biology); and Russian Foundation for Basic Research Grant 09-04-00869.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1 and Table S1.
G. A. Belogurov and H. H. Luoto, unpublished data.
- PPase
- pyrophosphatase
- Ch-PPase
- C. hydrogenoformans PPase
- Ac-PPase
- A. caccae PPase
- Cl-PPase
- C. limicola PPase
- Ct-PPase
- C. thermocellum PPase
- Da-PPase
- D. acetoxidans PPase
- Fj-PPase
- F. johnsoniae PPase
- Lb-PPase
- L. biflexa PPase
- Pa-PPase
- P. aerophilum PPase
- Ctet-PPase
- C. tetani PPase
- Tm-PPase
- T. maritima PPase
- IMV
- inner membrane vesicle(s)
- TMA
- tetramethylammonium
- TMH
- transmembrane helix.
REFERENCES
- 1. Maeshima M. (2000) Biochim. Biophys. Acta 1465, 37–51 [DOI] [PubMed] [Google Scholar]
- 2. Belogurov G. A., Lahti R. (2002) J. Biol. Chem. 277, 49651–49654 [DOI] [PubMed] [Google Scholar]
- 3. Baykov A. A., Bakuleva N. P., Rea P. A. (1993) Eur. J. Biochem. 217, 755–762 [DOI] [PubMed] [Google Scholar]
- 4. Baltscheffsky M. (1967) Nature 216, 241–243 [DOI] [PubMed] [Google Scholar]
- 5. Baltscheffsky H., Von Stedingk L. V., Heldt H. W., Klingenberg M. (1966) Science 153, 1120–1122 [DOI] [PubMed] [Google Scholar]
- 6. Serrano A., Pérez-Castiñeira J. R., Baltscheffsky M., Baltscheffsky H. (2007) IUBMB Life 59, 76–83 [DOI] [PubMed] [Google Scholar]
- 7. Baltscheffsky M., Schultz A., Baltscheffsky H. (1999) FEBS Lett. 457, 527–533 [DOI] [PubMed] [Google Scholar]
- 8. López-Marqués R. L., Pérez-Castiñeira J. R., Losada M., Serrano A. (2004) J. Bacteriol. 186, 5418–5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. García-Contreras R., Celis H., Romero I. (2004) J. Bacteriol. 186, 6651–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drozdowicz Y. M., Rea P. A. (2001) Trends Plant Sci. 6, 206–211 [DOI] [PubMed] [Google Scholar]
- 11. Docampo R., de Souza W., Miranda K., Rohloff P., Moreno S. N. (2005) Nat. Rev. Microbiol. 3, 251–261 [DOI] [PubMed] [Google Scholar]
- 12. Fukuda A., Tanaka Y. (2006) Plant Physiol. Biochem. 44, 351–358 [DOI] [PubMed] [Google Scholar]
- 13. Li J., Yang H., Peer W. A., Richter G., Blakeslee J., Bandyopadhyay A., Titapiwantakun B., Undurraga S., Khodakovskaya M., Richards E. L., Krizek B., Murphy A. S., Gilroy S., Gaxiola R. (2005) Science 310, 121–125 [DOI] [PubMed] [Google Scholar]
- 14. Zhang J., Li J., Wang X., Chen J. (2011) Plant Physiol. Biochem. 49, 33–38 [DOI] [PubMed] [Google Scholar]
- 15. Lv S., Zhang K., Gao Q., Lian L., Song Y., Zhang J. (2008) Plant Cell Physiol. 49, 1150–1164 [DOI] [PubMed] [Google Scholar]
- 16. Li B., Wei A., Song C., Li N., Zhang J. (2008) Plant Biotechnol. J. 6, 146–159 [DOI] [PubMed] [Google Scholar]
- 17. Park S., Li J., Pittman J. K., Berkowitz G. A., Yang H., Undurraga S., Morris J., Hirschi K. D., Gaxiola R. A. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 18830–18835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Docampo R., Moreno S. N. (2008) Curr. Pharm. Des. 14, 882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malinen A. M., Belogurov G. A., Baykov A. A., Lahti R. (2007) Biochemistry 46, 8872–8878 [DOI] [PubMed] [Google Scholar]
- 20. Belogurov G. A., Malinen A. M., Turkina M. V., Jalonen U., Rytkönen K., Baykov A. A., Lahti R. (2005) Biochemistry 44, 2088–2096 [DOI] [PubMed] [Google Scholar]
- 21. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 22. Edgar R. C. (2004) Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ronquist F., Huelsenbeck J. P. (2003) Bioinformatics 19, 1572–1574 [DOI] [PubMed] [Google Scholar]
- 24. Huelsenbeck J. P., Ronquist F. (2001) Bioinformatics 17, 754–755 [DOI] [PubMed] [Google Scholar]
- 25. Shimodaira H., Hasegawa M. (1999) Mol. Biol. Evol. 16, 1114–1116 [Google Scholar]
- 26. Schmidt H. A., Strimmer K., Vingron M., von Haeseler A. (2002) Bioinformatics 18, 502–504 [DOI] [PubMed] [Google Scholar]
- 27. Yang Z. (2007) Mol. Biol. Evol. 24, 1586–1591 [DOI] [PubMed] [Google Scholar]
- 28. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 29. Baykov A. A., Avaeva S. M. (1981) Anal. Biochem. 116, 1–4 [DOI] [PubMed] [Google Scholar]
- 30. Baykov A. A., Dubnova E. B., Bakuleva N. P., Evtushenko O. A., Zhen R. G., Rea P. A. (1993) FEBS Lett. 327, 199–202 [DOI] [PubMed] [Google Scholar]
- 31. Mimura H., Nakanishi Y., Hirono M., Maeshima M. (2004) J. Biol. Chem. 279, 35106–35112 [DOI] [PubMed] [Google Scholar]
- 32. Bäumer S., Lentes S., Gottschalk G., Deppenmeier U. (2002) Archaea 1, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moyle J., Mitchell R., Mitchell P. (1972) FEBS Lett. 23, 233–236 [DOI] [PubMed] [Google Scholar]
- 34. Drozdowicz Y. M., Kissinger J. C., Rea P. A. (2000) Plant Physiol. 123, 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Drozdowicz Y. M., Lu Y. P., Patel V., Fitz-Gibbon S., Miller J. H., Rea P. A. (1999) FEBS Lett. 460, 505–512 [DOI] [PubMed] [Google Scholar]
- 36. Pirovano W., Feenstra K. A., Heringa J. (2006) Nucleic Acids Res. 34, 6540–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Biegel E., Müller V. (2011) J. Biol. Chem. 286, 6080–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang Y. T., Liu T. H., Chen Y. W., Lee C. H., Chen H. H., Huang T. W., Hsu S. H., Lin S. M., Pan Y. J., Lee C. H., Hsu I. C., Tseng F. G., Fu C. C., Pan R. L. (2010) J. Biol. Chem. 285, 23655–23664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bruggemann H., Baumer S., Fricke W. F., Wiezer A., Liesegang H., Decker I., Herzberg C., Martinez-Arias R., Merkl R., Henne A., Gottschalk G. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 1316–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pan Y. J., Lee C. H., Hsu S. H., Huang Y. T., Lee C. H., Liu T. H., Chen Y. W., Lin S. M., Pan R. L. (2011) Biochim. Biophys. Acta 1807, 59–67 [DOI] [PubMed] [Google Scholar]
- 41. Zhen R. G., Kim E. J., Rea P. A. (1997) J. Biol. Chem. 272, 22340–22348 [DOI] [PubMed] [Google Scholar]
- 42. Malinen A. M., Baykov A. A., Lahti R. (2008) Biochemistry 47, 13447–13454 [DOI] [PubMed] [Google Scholar]
- 43. Mulkidjanian A. Y., Galperin M. Y., Makarova K. S., Wolf Y. I., Koonin E. V. (2008) Biol. Direct 3, 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mulkidjanian A. Y., Dibrov P., Galperin M. Y. (2008) Biochim. Biophys. Acta 1777, 985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mulkidjanian A. Y., Galperin M. Y., Koonin E. V. (2009) Trends Biochem. Sci. 34, 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Belogurov G. A., Turkina M. V., Penttinen A., Huopalahti S., Baykov A. A., Lahti R. (2002) J. Biol. Chem. 277, 22209–22214 [DOI] [PubMed] [Google Scholar]
- 47. Drozdowicz Y. M., Shaw M., Nishi M., Striepen B., Liwinski H. A., Roos D. S., Rea P. A. (2003) J. Biol. Chem. 278, 1075–1085 [DOI] [PubMed] [Google Scholar]
- 48. Luo S., Marchesini N., Moreno S. N., Docampo R. (1999) FEBS Lett. 460, 217–220 [DOI] [PubMed] [Google Scholar]
- 49. Hill J. E., Scott D. A., Luo S., Docampo R. (2000) Biochem. J. 351, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruiz F. A., Marchesini N., Seufferheld M., Govindjee, Docampo R. (2001) J. Biol. Chem. 276, 46196–46203 [DOI] [PubMed] [Google Scholar]
- 51. Britten C. J., Zhen R. G., Kim E. J., Rea P. A. (1992) J. Biol. Chem. 267, 21850–21855 [PubMed] [Google Scholar]
- 52. Sarafian V., Kim Y., Poole R. J., Rea P. A. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 1775–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Holm N. G., Baltscheffsky H. (April 2, 2011) Orig. Life Evol. Biosph. 10.1007/s11084-011-9235-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.