

Abstract
Adhesion-based cell capture on surfaces in microfluidic devices forms the basis of numerous biomedical diagnostics and in vitro assays. However, the performance of these platforms is partly limited by interfacial phenomena that occur at low Reynolds numbers. In contrast, cell homing to porous vasculature is highly effective in vivo during inflammation, stem cell trafficking, and cancer metastasis. Here, we show that a porous, fluid-permeable surface functionalized with cell-specific antibodies promotes efficient and selective cell capture in vitro. This architecture is advantageous due to enhanced transport as streamlines are diverted toward the surface. Moreover, specific cell-surface interactions are promoted due to reduced shear, allowing gentle cell rolling and arrest. Together, these synergistic effects enable highly effective cell capture at flow rates more than an order of magnitude larger than those provided by existing devices with solid surfaces.
Introduction
The identification, selection, and separation of a subpopulation of target cells from a larger heterogeneous population is essential for blood-based point-of-care diagnostics, personalized therapies, and cell biology (1–3). These cells of interest may be rare and present in extraordinarily low numbers relative to the general population, necessitating the processing of large sample volumes to accumulate a useful number. For instance, 1 mL of whole blood contains billions of red blood cells, millions of white blood cells, thousands of hematopoietic stem cells, hundreds of endothelial progenitor cells, and dozens of circulating tumor cells (4,5). Thus, even a perfectly efficient separation scheme requires at least 10 mL of whole blood to capture a usable sample of the rarest cell types, which must be rapidly processed to limit degradation and provide timely information to patients.
A number of approaches have been demonstrated to separate subpopulations of cells through their differential physical and biochemical phenotypes, which serve as handles for direct manipulation. For example, physical fields can partition a complex mixture of cells based on size, shape, deformability, density, electrical, magnetic, or optical properties (1,6). These approaches are advantageous because they can be label-free and relatively high-throughput, but are often confounded by the considerable variability found even within a specific cell type. Instead, one can achieve greater specificity using molecular recognition of unique cell surface markers. Cells in solution can be labeled and subsequently sorted with the use of fluorescent molecules (7) or magnetic beads (8). Alternatively, cells can be captured on solid surfaces functionalized with ligands that are complementary to a specific cell surface receptor (2,3). This approach has been used to isolate neutrophils (9,10), monocytes (10), lymphocytes (10–12), fibroblasts (13), endothelial progenitor cells (14), hematopoietic stem cells (15), mesenchymal stem cells (16), and circulating tumor cells (17–22). In these schemes, specific cell adhesion depends on the interactions between the cell and surface, and thus the operating conditions must be carefully controlled.
Microfluidic platforms have been widely explored for biomedical diagnostics because the samples can be precisely and reproducibly manipulated under well-defined physicochemical conditions. At these small length scales, the fluid dynamics are dominated by the high surface-to-volume ratio and interfacial phenomena (23,24). Although these effects have been cleverly exploited for various applications, they severely hinder sample throughput for analyte capture on solid surfaces (25,26). The first limitation in this regime arises because the transport of analytes to the surface may be too slow compared with the speed of transport through the microfluidic device. This is particularly problematic at high flow rates due to rapid advection of analytes through the device (analogous to a high Peclet number), as well as poor mixing of viscous flows (low Reynolds number). These issues can be partially overcome by increasing the effective surface area (17,20,21), as well as by using herringbone chaotic micromixers to disrupt fluidic streamlines through the microfluidic device (18,19,27).
The second limitation subsequently arises if the reaction of analytes with the surface does not have sufficient time to occur. This is particularly problematic for cells moving rapidly across the surface, because they require the formation of multiple adhesive bonds to be fully arrested (28). Indeed, any bonds that do form between cellular receptors and surface-immobilized ligands are more likely to dissociate at high shear rates (29). On the other hand, a certain threshold shear rate is necessary for adhesion-based capture to occur selectively (2), because weaker nonspecific molecular bonds are pulled apart more easily. This mechanism has been used to select for certain subpopulations with differential expression levels using a precisely controlled shear rate (11,12). Another danger is that cell sedimentation may dominate at low flow rates, which would further decrease selectivity. Overall, the effectiveness of adhesion-based capture is limited at high flow rates both by transport of cells to the surface and the subsequent reaction of the cells with the surface.
Here, we show that microfluidic devices incorporating porous, fluid-permeable surfaces functionalized with cell-specific antibodies can be used to capture a rare subpopulation of target cells with excellent efficiency, selectivity, and throughput. The effectiveness of this platform arises both from enhanced mass transport to the porous surface (Fig. 1 A) and from enhanced cell-surface interactions that promote dynamic rolling adhesion with high specificity (Fig. 1 B). These cooperative mechanisms enable optimal performance at extremely fast flow rates. These flow rates are more than an order of magnitude faster than what can be achieved with conventional devices.
Figure 1.
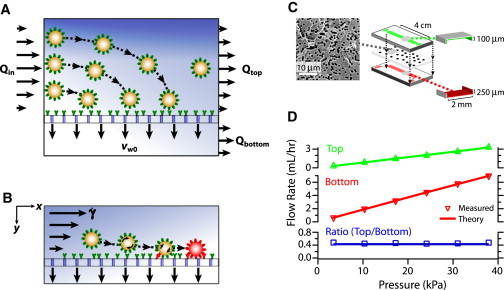
(A) Enhanced cell transport to a fluid-permeable capture surface is achieved by diverting streamlines. (B) Gentle cell rolling and arrest on the capture surface occur due to reduced shear and increased cell-surface interactions. (C) Scanning electron micrograph of polycarbonate surface with 200 nm pores and 10% porosity; schematic of microfluidic device assembly and dimensions. (D) Fluid flow rates through the top and bottom outlets vary linearly with increasing pressure; their ratio is constant and precisely controlled with the use of high-resistance outlets. Each marker is the average of five experiments per condition.
Materials and Methods
Device fabrication and surface modification
The microfluidic devices consisted of a polycarbonate membrane (200 nm pores, 10% porosity, 10 μm thick; GE Whatman, Piscatway, NJ) sandwiched between two polydimethylsiloxane layers (Fig. 1 C), as previously described (30). Each layer contained an independent inlet and outlet connected by a rectangular channel 100 μm or 250 μm high, 2 mm wide, and 4 cm long. The membranes were covalently functionalized with anti-EpCAM or anti-IgG (30 μg/mL) (R&D Systems, Minneapolis, MN) according to a previously described process (31). Anti-EpCAM is selective for EpCAM expression in cancer cells of epithelial origin but not blood cells (5), whereas anti-IgG is noncomplementary to both cancer cells and blood cells. See Supporting Material for additional details about the fabrication.
Sample preparation
Leukocytes (buffy coat) were isolated from whole blood via deterministic lateral displacement (32), fluorescently labeled (CellTrace Calcein Green; Invitrogen, Carlsbad, CA), and resuspended to a concentration of 500,000/mL. PC3 human prostate cancer cells were labeled with a different fluorescent dye (CellTracker Orange; Invitrogen) and spiked into the sample at a ratio of 1:250 (2000/mL). See Supporting Material for additional details.
Device operation
Samples were loaded into a 60 mL syringe, and a constant pressure syringe pump was used to apply a constant flow through the top inlet while the bottom inlet was closed. The top and bottom outlets were both open, and the ratio of transverse membrane flux and axial channel flux was regulated by means of the relative resistances of the outlet tubing (Supporting Material). After the sample had been processed, the bottom outlet was closed and phosphate-buffered saline was flowed through the top channel to remove nonspecifically bound cells.
Cell capture was visualized with an upright epifluorescence microscope (Nikon Eclipse 90i) using a 4X (Nikon Plan Fluor, NA = 0.13) or 10X objective (Nikon CFI Plan Apo, NA = 0.45) at 10 frames per second with a CCD camera (QImaging Retiga 2000R). At least 30 cells were tracked per condition with the use of commercial software (Nikon Elements AR3.1) and manually verified. We enumerated overall cell capture in the device (Nc) and waste collection (Nout) using three different emission spectra to identify cells (DAPI) and to distinguish spiked PC3 cells (TRITC) and background leukocytes (FITC). The capture efficiency was calculated as the captured cells divided by the total cells flowed through the device, i.e., Nc/(Nc+Nout) averaged over three experiments. We performed a mass balance on the number of PC3 cells that spiked into the cells by counting the cells that were injected into the device, the cells that were captured on the porous membrane, and the PC3 cells that exited the device into the collection well. There was a ∼4% difference between the intended number of cells injected into the device and the number of cells that were accounted for by mass balance.
Results
Cell transport to surface is enhanced by diverting streamlines
The effectiveness of conventional adhesion-based assays is strongly diminished at high flow rates because most cells do not reach the surface. To overcome this limitation, we developed a two-chamber microfluidic device with a porous capture surface sandwiched in between the chambers (Fig. 1 C). These surfaces consisted of commercially available polycarbonate membranes with an overall porosity of 10% and average pore diameter of ∼200 nm. Pores of this size allow fluid permeation but are small enough to prevent ∼10 μm cells from entering or becoming trapped (Fig. 1 C, left). This device geometry allows a controlled fraction of the incoming fluid flow to be diverted into the porous membrane (Fig. 1 D, middle), whereas the remainder continues to the outlet (Fig. 1 D, top). These fluid flow conditions were calibrated and found to be in good agreement with the expected values based on the applied pressure difference (Fig. 1 D, bottom). These devices were operated with samples consisting of fluorescently labeled PC3 cancer cells spiked at a ratio of 1:250 in a background population of white blood cells.
The limiting case of no fluid flux through the membrane is essentially equivalent to flow past a solid surface, because all of the fluid flux is exiting through the top outlet. The cell trajectories in this scenario are well described by pressure-driven Poiseuille flow in the axial direction, as well as a constant sedimentation velocity due to the density difference of the cells (33). The nondimensionalized axial and transverse fluid velocity field components Ux, Uy as a function of (nondimensionalized) coordinate system (X = x/h, Y = y/h), are given by:
| (1) |
The constant cell sedimentation perturbs the cell trajectories slightly from the fluid streamlines due to the density difference Δρ = 0.03 g/cm3 (Supporting Material). A representative case is shown in Fig. 2 A for a cell sedimentation velocity of 2 μm/s and an average flow velocity of <Ux> = 10,000 μm/s, corresponding to a flow rate Qin = 6 mL/h and cell radius Rc = 5 μm. At these high flow rates, cells are advected through the device so rapidly that they have very little time to sediment. Based on the calculated cell trajectories for these conditions, only those cells that are initially near the bottom of the channel (y < 10 μm) can reach the capture surface. Assuming that the cells are uniformly distributed at the 100 μm high entrance, this corresponds to a case in which ∼10% of the cells reach the surface, whereas the remaining ∼90% have no opportunity to interact with the surface and get captured. These calculations are consistent with experimental measurements of the cell trajectories under these flow conditions obtained by fluorescence microscopy, which show cells moving rapidly with constant axial velocity throughout the 3000 μm field of view (Fig. 2 C).
Figure 2.
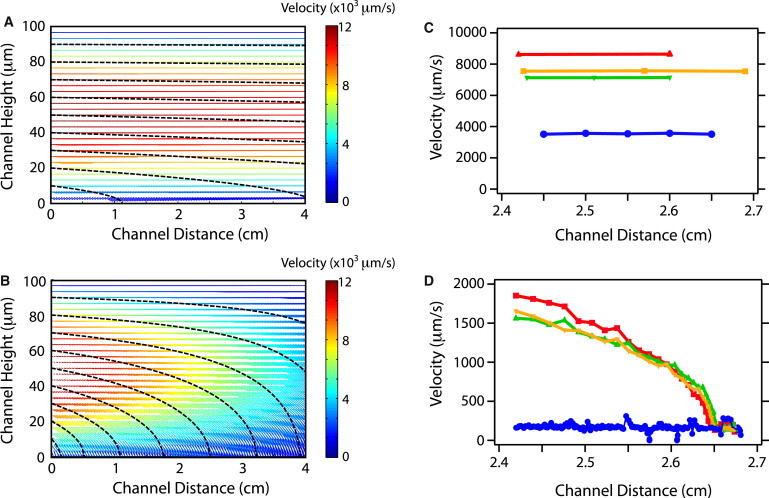
Theoretical particle trajectories (dashed black lines) and fluid velocity field vectors (color) in channels with (A) solid surface (A = 0%) and (B) fluid-permeable surface (A = 70%). The color bar corresponds to the magnitude of fluid velocity vectors. Experimentally measured particle velocities tracked in channels with (C) solid surface (A = 0%) and (D) fluid-permeable surface (A = 70%).
As the fluid permeation flux through the membrane is increased, more and more of the streamlines are diverted from the top outlet to the porous membrane (Fig. 2 B). As a result, cells traveling along the streamlines are now rapidly advected directly to the capture surface, in contrast to the slow and ineffective sedimentation in the previous scenario (Fig. 2 A). For the fractional permeation flux through the membrane A, the nondimensionalized velocity field components are given by:
| (2) |
The calculated cell trajectories for this scenario (Supporting Material) are illustrated for a representative case of 70% permeation flux through the membrane, but with the same flow rate as in the previous example (Qin = 6 mL/h). In this case, the calculated cell trajectories do not deviate significantly from the streamlines (<1%) because advection (∼10,000 μm/s) dominates over sedimentation (∼2 μm/s) and hydrodynamic effects. These calculations are corroborated by experimental measurements of the cell trajectories (Fig. 2 D), which show that the axial velocity Ux rapidly decreases from an initial value of 2000 μm/s at x = 2.4 cm to ∼200 um/s at x = 2.7 cm as the cell is transported to the surface and the transverse velocity Uy becomes more significant. Because of the device geometry and the rapid axial velocity of the cell, it is difficult to directly measure the height of the cell during its trajectory. However, a qualitative comparison of the calculated cell trajectories (Fig. 2 B) and the experimentally measured cell axial velocities (Fig. 2 D) suggests that these cells entered the device at a height roughly halfway between the bottom and top. According to the calculated streamlines, all cells that enter the channel within 70 μm of the bottom surface (∼70%) should be captured, and the remaining 30% of cells should exit the device at the top outlet. Experimentally, the total fraction of cells that are transported to the porous capture surface scales linearly with the percentage of the fluid flux into the membrane (Fig. S3).
An important implication of the streamline calculation is that when permeation occurs, the axial fluid velocity Ux decreases linearly along the length of the device. As a result, the shear rate near the surface is also expected to decrease with increasing distance x, reaching a minimum at the end of the porous surface. To verify this trend, we experimentally measured the cell surface velocity uc(x) at three locations along the length of the device for various values of A at a constant flow rate (Qin = 6 mL/h) and fit the data using linear regression (Fig. 3). We then compared these measurements with the hydrodynamic model proposed by Goldman et al. (34) for a particle moving near a solid surface due to a shear field, using the following scaling for cell surface velocity:
| (3) |
where uc,0 is the cell surface velocity at the entrance and L is the channel length. The measured velocities and Goldman model show good agreement for A = 50% (Fig. S4). However, at larger permeation fluxes, the experimental cell surface velocities are consistently slower than the expected values by several hundred microns per second. This suggests that the cell surface velocity is not solely dictated by the local shear field, but may have additional inhibitory interactions with a porous surface that do not occur on a solid surface. These cell-surface interactions are examined in more detail in the following section.
Figure 3.
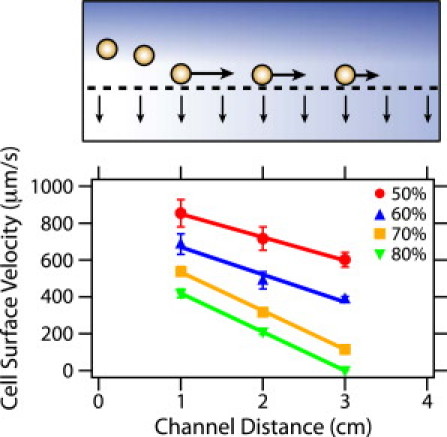
Experimental measurements of cell surface velocity (markers) as a function of percentage permeation flux A and channel distance. Porous surface was not functionalized. Solid lines are best-fit linear regressions. Each marker and error bar is the average and standard deviation of 30 cells per condition.
Cell-surface interactions are promoted by a fluid permeable surface
The effectiveness of cell capture assays also depends on the arrest of cell motion on the surface, which occurs through the competition of specific biomolecular bond formation and local shearing forces. Cells near a surface have been observed to roll at constant velocity due to hydrodynamic interactions, which is essential for selective capture both in vitro and in vivo. We examined representative individual cell trajectories near a porous surface functionalized with noncomplementary IgG antibodies (Fig. 4 A). Up until time t = 1 s, the measured axial velocity Ux rapidly decreases as the cell approaches the surface, after which it maintains a reduced, constant velocity (Fig. 4 B). Because the IgG antibodies cannot form strong, specific biomolecular bonds with the cellular receptors, motion is never completely arrested on the surface. However, the velocity shows surprising fluctuations where the cell appears to temporarily pause its motion on the surface (Fig. 4 A, inset). The magnitude of this fluctuation (Δuc ∼ 350 μm/s) is considerably larger than the standard deviation (SD) of the velocity before and after this event (σu ∼ 40 μm/s). These fluctuations may occur because of local differences in surface porosity, which apply slightly stronger suction forces to the cell to slow down its motion. This mechanism is clearly insufficient to capture cells permanently, but it is likely to influence the binding kinetics in a way that does not occur on a solid surface.
Figure 4.
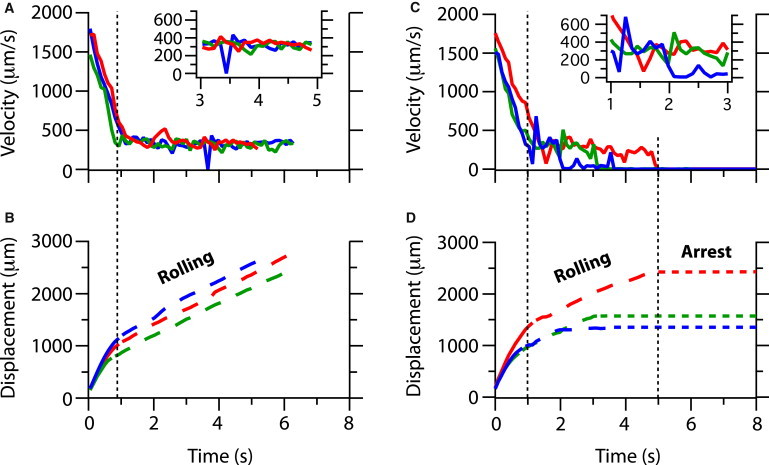
Instantaneous velocity and displacement trajectories for PC3 cancer cells transported to (A and B) noncomplementary anti-IgG, exhibiting rolling motion at constant speed, and (C and D) anti-EpCAM fluid-permeable surfaces at x = 3 cm with A = 70%, exhibiting rolling before complete arrest.
These velocity fluctuations increase in frequency and duration on porous surfaces that have been functionalized with anti-EpCAM antibodies (σu ∼ 130 μm/s; Fig. 4 C), which are complementary to cancer cells of epithelial origin but not leukocytes (5). In these representative trajectories, cells again reach the porous surface around time t = 1 s and show a similar decrease in axial velocity, indicating they are being advected to the surface. However, rather than continuing at a constant diminished velocity (Fig. 4 B), these cells continue to decelerate and are completely arrested within a few seconds (Fig. 4 D). The permeation flux through a porous surface thus appears to promote the kinetics of strong, specific biomolecular bond formation by slowing down cell rolling across the surface. As evidenced by the previous example with noncomplementary anti-IgG, this mechanism is not strong enough to irreversibly stabilize cell motion against the local shear field. However, in the presence of complementary anti-EpCAM, this enhanced cell-surface interaction allows specific cell capture to continue to occur even at fast total flow rates. Essentially, these conditions of highly reduced shear and an additional braking suction mechanism near a permeable surface lead to conditions comparable to those found on a solid surface at dramatically lower flow rates and shear.
Cell capture on fluid-permeable surfaces exceeds solid surfaces at elevated flow rates
The capture efficiency of PC3s on both porous and solid surfaces functionalized with either noncomplementary anti-IgG or complementary anti-EpCAM are plotted as a function of total flow rate in Fig. 5 A. With a mixture of PC3s and leukocytes at a ratio of 1:250, nonspecific capture of PC3s is minimal (∼10%) on a solid anti-IgG surface due to the lack of complementary bonds and decreases rapidly with increasing flow rates. For a porous anti-IgG surface (A = 70%), the cell capture rate is slightly higher (∼20%) and decreases more gradually with increasing flow rate, reflecting nonspecific adsorption due to the enhanced transport to the surface as well as suction effects. For the solid anti-EpCAM surface, the capture efficiency achieves a maximum value of ∼60% only at low flow rates (Qin < 0.3 mL/h). The capture efficiency drops off rapidly with increasing flow rate, becoming negligible by Qin = 1.5 mL/h. In comparison, the porous anti-EpCAM surface (A = 70%) achieves the optimal capture efficiency of 70 ± 3% up to relatively high flow rates of Qin = 6 mL/h (Fig. 5 A). This is consistent with the theoretical maximum of 70% based on the fraction of streamlines that are expected to reach the surface. In comparison, at this flow rate, no cells are captured on the flat surface for both anti-EpCAM and anti-IgG, and only a few cells are nonspecifically adsorbed on the anti-IgG porous surface (Fig. 5, C–F). As the flow rate is further increased with the anti-EpCAM porous surface, the cell capture efficiency decreases to 15% at Qin = 30 mL/h. Nevertheless, the maximum effective flow rate of the porous anti-EpCAM surface is 20-fold higher than that on the solid anti-EpCAM surface, enabling a potentially transformative enhancement in processing throughput.
Figure 5.
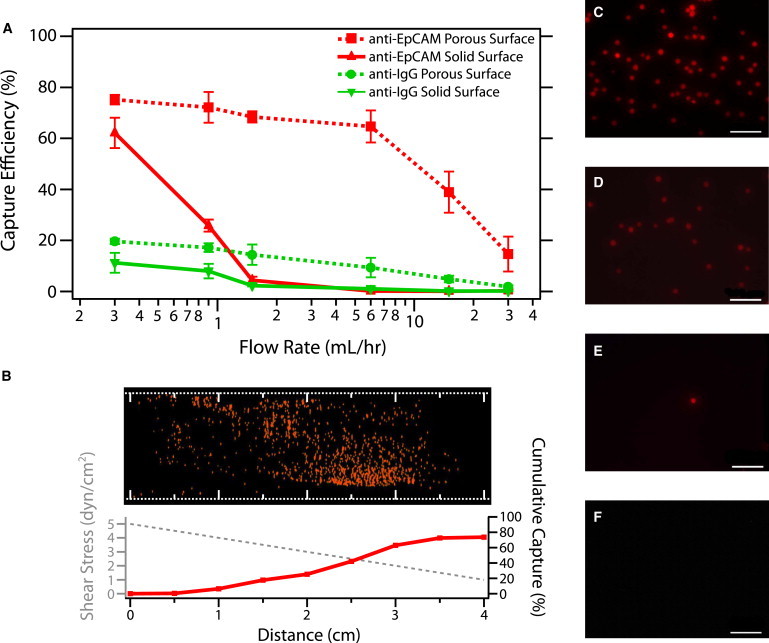
(A) Capture efficiency of PC3 cancer cells at increasing flow rates on complementary anti-EpCAM porous surfaces (red squares), anti-EpCAM solid surfaces (red triangles), noncomplementary anti-IgG porous surfaces (green circles), and anti-IgG solid surfaces (green triangles). Each marker and error bar is the average and SD of three experiments. (B) Capture profile varies along the channel length on an anti-EpCAM porous capture surface at Qin= 6 mL/h and A = 70%. The transverse wall velocity vw0 = 141 μm/s. (C–F) Representative fluorescence micrograph of captured PC3 cells at x = 3 cm for (C) anti-EpCAM porous surface, (D) anti-IgG porous surface, (E) anti-EpCAM solid surface, and (F) anti-IgG solid surface. Scale bar is 100 μm.
The shear-dependent capture of cells is illustrated by the concentration profile along the length of the channel (Fig. 5 B). A stitched image of the entire length of the device at representative flow conditions (A = 70%, Qin = 6 mL/h) shows that cell capture increases cumulatively with distance, reaching ∼70% at x = 4 cm as the shear field linearly decreases.
Optimizing experimental conditions to maximize capture efficiency and selectivity
One consideration unique to this device architecture is that cells may be advected downward so rapidly that they overwhelm the capture surface. This would suppress the effectiveness of the device because layers of accumulated cells would block access to the capture surface (“caking”) and ultimately impede transverse fluid flow through the surface. To avoid this scenario, the flux of cells that are being advected to the surface must not exceed the flux of cells that are translating across the surface. One can achieve this condition at a given bulk cell concentration by making the permeation flux as large as possible while retaining sufficient axial flow to drive cell rolling due to shear stress. Romero and Davis (35) previously considered the scenario of hard spheres accumulating at a porous surface, and derived an expression for a critical distance xcr where particles become close-packed in a cake layer:
| (4) |
where a is the particle radius, τ is the surface shear stress, φ0 is the initial bulk volume fraction, and μ0 is the solution viscosity. The critical permeation flux cr is
| (5) |
where φmax is the maximum packing density on the surface, assumed to be ∼0.6 for hard spheres. This is a conservative estimate that may underestimate the packing density of deformable cells (35). Nevertheless, the empty space between cells in a close-packed layer (∼40%) is still considerably larger than the membrane porosity (∼10%), making it unlikely that a single close-packed layer of cells would block a significant number of pores. This calculation thus assumes that the presence of ∼10 μm cells does not affect the fluidic resistance of the membrane or associated transverse flux.
Phase diagrams for the critical distance xcr as a function of axial distance x and initial cell volume fraction φ0 at varying permeation fluxes are shown in Fig. 6 and Fig. S5. To prevent caking, the critical distance xcr should exceed the device length L at any point along the length of the channel so that the particle fraction on the porous surface never reaches its maximum cell packing density, φmax. In general, the critical distance xcr decreases with increasing initial cell volume fraction φ0, because a smaller enhancement in concentration is required to reach the maximum. In the case of minimal permeation flux (A = 10%), slow advection to the surface means that caking will only occur at relatively high bulk volume fractions. For instance, for φ0 = 0.4 ∼ 2 · 106 cells/mL, φmax is reached at xcr = 3 cm (Fig. 6 A). Experimentally, operating in this regime leads to a visible buildup of white blood cells on the reaction surface, despite the lack of specific cell-surface interactions. A further increase to φ0 = 0.45 ∼ 2.5 · 106 cells/mL, corresponds to xcr essentially at the entrance, which is not a usable condition. In general, operating at this minimal flux regime (A = 10%) is both inefficient and unselective because 90% of the cells never reach the surface, but those that do are not subjected to sufficient shear to remove white blood cells while retaining cancer cells.
Figure 6.
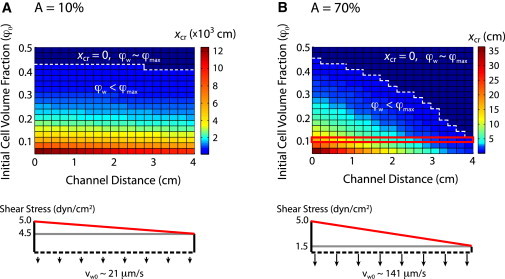
Phase diagram of the critical distance (xcr) where the volume fraction of cells reaches the maximum close packing (φw ∼ φmax ∼ 0.6) as a function of the initial volume fraction φo and channel location. (A) Low permeation flux (A = 10%). (B) High permeation flux (A = 70%). At a critical value of initial volume fraction, the maximum close packing is reached along the length of the channel, causing excess cell buildup (caking) and hindering cell capture (white dotted line). Devices were operated in the optimum regime (φo = 0.1, Qin = 6 mL/h, A = 70%) to maximize throughput without excess cell buildup (red line).
At the higher permeation flux described previously (A = 70%) and φ0 = 0.4, xcr = 0.8 cm, corresponding to caking within the device (Fig. 6 B). Instead, at φ0 = 0.1, corresponding to ∼500,000 cells/mL, the critical length xcr = 5 cm is larger than the length of the channel (L = 4 cm) and caking should not occur within the device. Experimentally, device operation was optimal under these conditions, and efficient cell transport, selective capture, and minimal accumulation of white blood cells were achieved at relatively high flow rates.
Discussion
Conventional platforms based on adhesion-based cell capture on solid surfaces exhibit highly diminished capture efficiency at elevated flow rates due to two coupled mechanisms. First, the rapid advection of cells through the device limits the transport of cells to the capture surface. As a result, only a small fraction of the total cells in a sample actually reach the surface. Second, cells near the surface experience strong shear stresses that cause rapid rolling and translation across the surface. The limited interaction between cellular receptors and surface ligands hinders the formation of strong adhesive bonds, and any transient bonds that do form are more likely to dissociate at elevated forces. Although the first limitation can be addressed by increasing the capture surface area or enhancing mixing, the second limitation is more difficult to overcome, because the shear stress scales directly with the flow rate near a solid surface. Given this limiting flow rate, one can still increase the overall throughput by scaling up to multiple parallel channels, but this approach soon becomes impractical for device manufacture and readout.
In this work, we have demonstrated a fluid-permeable capture surface that overcomes both of these limitations, enabling excellent capture efficiency and selectivity at flow rates 20-fold higher than those achieved by a comparable device with a solid surface. Remarkably, by controlling the fluid permeation through the membrane, we were able to divert streamlines into the membrane even at high flow rates. On a cellular scale, the transverse flux has an additional advantage in that the transverse flow through the membrane significantly decreases the axial fluid flow near the surface. Thus, despite high overall flow rates, cells near the surface experience a considerably diminished shear as well as a braking suction force. In contrast, these reduced surface shear conditions can only be achieved near a solid surface at substantially lower flow rates. One can independently vary these transport and cell-surface mechanisms by adjusting the overall flow rate or the ratio of transverse to axial flux, respectively, thereby optimizing the device operation.
Another phenomenon that occurs with fluid-permeable surfaces is a linear decrease in the shear stress along the length of the channel, which is reminiscent of microfluidic devices that use a Hele-Shaw channel geometry (with solid surfaces) (36). As a result, the density of captured cells increases with distance in the channel, because cells are more likely to remain arrested at lower shear. This trend is the opposite of what is often observed on solid surfaces, where the shear is constant and the density of captured cells decreases exponentially along the length of the device (12). In both cases, the spatial distribution of captured cells reflects how the target subpopulation interacts specifically with an antibody-functionalized surface at a particular shear rate. An intriguing possibility is that additional biophysical information, such as the variation in receptor expression levels, may be encoded in this distribution. For instance, circulating tumor cells in a clinical sample may exhibit much greater heterogeneity than a spiked cell line (5), leading to a different distribution of captured cells in the channel. These effects could be explored with the use of improved surface chemistries (37) as well as spatial patterning schemes such as stripes (38), gradients (39), or discrete regions of different capture antibodies (40).
In addition to selective cell capture, microfluidic platforms incorporating porous surfaces may be useful as a well-controlled in vitro assay to elucidate cell trafficking behaviors in vivo. Indeed, a variety of cell types undergo rolling and arrest in vivo to enable separation from a highly heterogeneous background population (41). For example, leukocytes are selectively recruited from the circulation to home in on sites of inflammation (42), and mesenchymal stem cells participate in organogenesis, wound-healing, and natural cell turnover (43). Similar strategies may be utilized by circulating tumor cells before extravasation and metastatic colonization take place (44,45). Many of these behaviors are associated with a porous vasculature, such as capillaries in the bone marrow and discontinuous fenestrated sinusoid cells in the liver (46). This vascular permeability can be enhanced during acute and chronic inflammation as well as in cancer (47). Although cell rolling and adhesion are frequently observed along sinusoids in vivo (48), previous in vitro assays have only used solid surfaces to elucidate the biochemical interactions that occur between cellular receptors and surface ligands.
By using porous surfaces with slightly larger pores, we may be able to delve deeper into the multistep trafficking cascade under biomimetic conditions. In particular, this geometry is highly reminiscent of the transwell/Boyden chamber assay for chemotaxis studies (49). After capturing cells, one could reconfigure the flow conditions in the microfluidic device to minimize transverse permeation while achieving identical continuous flows in the top and bottom chambers. If the bottom chamber were loaded with a chemoattractant solution, a stable gradient would be formed through the membrane, promoting cell migration through the pores. A further step could be to culture a layer of endothelial cells on the porous surface instead of simply patterning ligands (50–52). By combining biomimetic features with precisely controlled microfluidic flows, we may be able to recapitulate complex biological behaviors in vitro, replicating the dynamics of the leaky vasculature within tumor microenvironments.
Fluid-permeable surfaces represent a powerful and versatile approach for specific analyte capture because they overcome fundamental limitations associated with interfacial effects near solid surfaces (53,54). This work elucidates the physical mechanisms that govern both transport and cell-surface interactions in these conditions, and thus establishes engineering design rules for future devices. It is expected that the overall throughput can be further increased by scaling up the device architecture with additional demultiplexed channels. This might yield an additional order-of-magnitude increase in throughput, building on the demonstrated 20-fold enhancement in effective flow rate (relative to comparable devices). The ability to process hundreds of milliliters of blood efficiently may lead to transformative approaches for point-of-care diagnostics and personalized medicine, such as the capture of extremely rare antigen-specific T-cells or fetal cells.
Acknowledgments
We thank O. Hurtado for assistance with fabrication, B. Hassell for help with image processing, A. Maheshwari for providing cells, A. A. Yanik for performing scanning electron microscopy, and K. T. Kotz for useful discussions. We also thank J. Voldman for insightful comments on the manuscript.
This work was supported by the National Institute of Biomedical Imaging and Bioengineering under grant P41-EB002503 (BioMEMS Resource Center). I.Y.W. is the Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2065-10).
Supporting Material
References
- 1.Toner M., Irimia D. Blood-on-a-chip. Annu. Rev. Biomed. Eng. 2005;7:77–103. doi: 10.1146/annurev.bioeng.7.011205.135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Didar T.F., Tabrizian M. Adhesion based detection, sorting and enrichment of cells in microfluidic Lab-on-Chip devices. Lab Chip. 2010;10:3043–3053. doi: 10.1039/c0lc00130a. [DOI] [PubMed] [Google Scholar]
- 3.Pratt E.D., Huang C., Kirby B.J. Rare cell capture in microfluidic devices. Chem. Eng. Sci. 2011;66:1508–1522. doi: 10.1016/j.ces.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wintrobe M.M., Greer J.P. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia: 2009. Wintrobe's Clinical Hematology. [Google Scholar]
- 5.Yu M., Stott S., Haber D.A. Circulating tumor cells: approaches to isolation and characterization. J. Cell Biol. 2011;192:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gossett D.R., Weaver W.M., Di Carlo D. Label-free cell separation and sorting in microfluidic systems. Anal. Bioanal. Chem. 2010;397:3249–3267. doi: 10.1007/s00216-010-3721-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonner W.A., Hulett H.R., Herzenberg L.A. Fluorescence activated cell sorting. Rev. Sci. Instrum. 1972;43:404–409. doi: 10.1063/1.1685647. [DOI] [PubMed] [Google Scholar]
- 8.Miltenyi S., Müller W., Radbruch A. High gradient magnetic cell separation with MACS. Cytometry. 1990;11:231–238. doi: 10.1002/cyto.990110203. [DOI] [PubMed] [Google Scholar]
- 9.Kotz K.T., Xiao W., Toner M., Inflammation and the Host Response to Injury Collaborative Research Program Clinical microfluidics for neutrophil genomics and proteomics. Nat. Med. 2010;16:1042–1047. doi: 10.1038/nm.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sekine K., Revzin A., Toner M. Panning of multiple subsets of leukocytes on antibody-decorated poly(ethylene) glycol-coated glass slides. J. Immunol. Methods. 2006;313:96–109. doi: 10.1016/j.jim.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 11.Murthy S.K., Sin A., Toner M. Effect of flow and surface conditions on human lymphocyte isolation using microfluidic chambers. Langmuir. 2004;20:11649–11655. doi: 10.1021/la048047b. [DOI] [PubMed] [Google Scholar]
- 12.Cheng X., Irimia D., Toner M. A microfluidic device for practical label-free CD4(+) T cell counting of HIV-infected subjects. Lab Chip. 2007;7:170–178. doi: 10.1039/b612966h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plouffe B.D., Brown M.A., Murthy S.K. Controlled capture and release of cardiac fibroblasts using peptide-functionalized alginate gels in microfluidic channels. Lab Chip. 2009;9:1507–1510. doi: 10.1039/b823523f. [DOI] [PubMed] [Google Scholar]
- 14.Plouffe B.D., Kniazeva T., Sales V.L. Development of microfluidics as endothelial progenitor cell capture technology for cardiovascular tissue engineering and diagnostic medicine. FASEB J. 2009;23:3309–3314. doi: 10.1096/fj.09-130260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narasipura S.D., Wojciechowski J.C., King M.R. Purification of CD45+ hematopoietic cells directly from human bone marrow using a flow-based P-selectin-coated microtube. Am. J. Hematol. 2008;83:627–629. doi: 10.1002/ajh.21204. [DOI] [PubMed] [Google Scholar]
- 16.Guo K.-T., SchAfer R., Wendel H.P. A new technique for the isolation and surface immobilization of mesenchymal stem cells from whole bone marrow using high-specific DNA aptamers. Stem Cells. 2006;24:2220–2231. doi: 10.1634/stemcells.2006-0015. [DOI] [PubMed] [Google Scholar]
- 17.Nagrath S., Sequist L.V., Toner M. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stott S.L., Hsu C.-H., Toner M. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA. 2010;107:18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S., Liu K., Tseng H.R. Highly efficient capture of circulating tumor cells by using nanostructured silicon substrates with integrated chaotic micromixers. Angew. Chem. Int. Ed. Engl. 2011;50:3084–3088. doi: 10.1002/anie.201005853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S., Wang H., Tseng H.R. Three-dimensional nanostructured substrates toward efficient capture of circulating tumor cells. Angew. Chem. Int. Ed. Engl. 2009;48:8970–8973. doi: 10.1002/anie.200901668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gleghorn J.P., Pratt E.D., Kirby B.J. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip. 2010;10:27–29. doi: 10.1039/b917959c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips J.A., Xu Y., Tan W. Enrichment of cancer cells using aptamers immobilized on a microfluidic channel. Anal. Chem. 2009;81:1033–1039. doi: 10.1021/ac802092j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone H.A., Stroock A.D., Ajdari A. Engineering flows in small devices. Annu. Rev. Fluid Mech. 2004;36:381–411. [Google Scholar]
- 24.Squires T.M., Quake S.R. Microfluidics: fluid physics at the nanoliter scale. Rev. Mod. Phys. 2005;77:977–1026. [Google Scholar]
- 25.Gervais T., Jensen K.F. Mass transport and surface reactions in microfluidic systems. Chem. Eng. Sci. 2006;61:1102–1121. [Google Scholar]
- 26.Squires T.M., Messinger R.J., Manalis S.R. Making it stick: convection, reaction and diffusion in surface-based biosensors. Nat. Biotechnol. 2008;26:417–426. doi: 10.1038/nbt1388. [DOI] [PubMed] [Google Scholar]
- 27.Liu J., Williams B.A., Quake S. Enhanced signals and fast nucleic acid hybridization by microfluidic chaotic mixing. Angew. Chem. Int. Ed. Engl. 2006;45:3618–3623. doi: 10.1002/anie.200503830. [DOI] [PubMed] [Google Scholar]
- 28.Hammer D.A., Lauffenburger D.A. A dynamical model for receptor-mediated cell adhesion to surfaces. Biophys. J. 1987;52:475–487. doi: 10.1016/S0006-3495(87)83236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammer D.A., Tirrell M. Biological adhesion at interfaces. Annu. Rev. Mater. Sci. 1996;26:651–691. [Google Scholar]
- 30.Chueh B.H., Huh D., Takayama S. Leakage-free bonding of porous membranes into layered microfluidic array systems. Anal. Chem. 2007;79:3504–3508. doi: 10.1021/ac062118p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suye S., Kumon Y., Ishigaki A. Immobilization of glucose oxidase on poly-(L-lysine)-modified polycarbonate membrane. Biotechnol. Appl. Biochem. 1998;27:245–248. [Google Scholar]
- 32.Davis J.A., Inglis D.W., Austin R.H. Deterministic hydrodynamics: taking blood apart. Proc. Natl. Acad. Sci. USA. 2006;103:14779–14784. doi: 10.1073/pnas.0605967103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munn L.L., Melder R.J., Jain R.K. Analysis of cell flux in the parallel plate flow chamber: implications for cell capture studies. Biophys. J. 1994;67:889–895. doi: 10.1016/S0006-3495(94)80550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldman A.J., Cox R.G., Brenner H. Slow viscous motion of a sphere parallel to a plane wall–II Couette flow. Chem. Eng. Sci. 1967;22:653–660. [Google Scholar]
- 35.Romero C.A., Davis R.H. Global model of crossflow microfiltration based on hydrodynamic particle diffusion. J. Membr. Sci. 1988;39:157–185. [Google Scholar]
- 36.Usami S., Chen H.H., Skalak R. Design and construction of a linear shear stress flow chamber. Ann. Biomed. Eng. 1993;21:77–83. doi: 10.1007/BF02368167. [DOI] [PubMed] [Google Scholar]
- 37.Hong S., Lee D., Karp J.M. Covalent immobilization of p-selectin enhances cell rolling. Langmuir. 2007;23:12261–12268. doi: 10.1021/la7014397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karnik R., Hong S., Langer R. Nanomechanical control of cell rolling in two dimensions through surface patterning of receptors. Nano Lett. 2008;8:1153–1158. doi: 10.1021/nl073322a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herman C.T., Potts G.K., Bailey R.C. Probing dynamic cell-substrate interactions using photochemically generated surface-immobilized gradients: application to selectin-mediated leukocyte rolling. Integr Biol (Camb) 2011;3:779–791. doi: 10.1039/c0ib00151a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myung J.H., Launiere C.A., Hong S. Enhanced tumor cell isolation by a biomimetic combination of E-selectin and anti-EpCAM: implications for the effective separation of circulating tumor cells (CTCs) Langmuir. 2010;26:8589–8596. doi: 10.1021/la904678p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McEver R.P., Zhu C. Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 2010;26:363–396. doi: 10.1146/annurev.cellbio.042308.113238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luster A.D., Alon R., von Andrian U.H. Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 43.Laird D.J., von Andrian U.H., Wagers A.J. Stem cell trafficking in tissue development, growth, and disease. Cell. 2008;132:612–630. doi: 10.1016/j.cell.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 44.Wirtz D., Konstantopoulos K., Searson P.C. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shibue T., Weinberg R.A. Metastatic colonization: settlement, adaptation and propagation of tumor cells in a foreign tissue environment. Semin. Cancer Biol. 2011;21:99–106. doi: 10.1016/j.semcancer.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Jain R.K. Determinants of tumor blood flow: a review. Cancer Res. 1988;48:2641–2658. [PubMed] [Google Scholar]
- 47.Nagy J.A., Benjamin L., Dvorak H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mazo I.B., Gutierrez-Ramos J.C., von Andrian U.H. Hematopoietic progenitor cell rolling in bone marrow microvessels: parallel contributions by endothelial selectins and vascular cell adhesion molecule 1. J. Exp. Med. 1998;188:465–474. doi: 10.1084/jem.188.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boyden S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962;115:453–466. doi: 10.1084/jem.115.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huh D., Matthews B.D., Ingber D.E. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Srigunapalan S., Lam C., Simmons C.A. A microfluidic membrane device to mimic critical components of the vascular microenvironment. Biomicrofluidics. 2011;5:13409. doi: 10.1063/1.3530598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song J.W., Cavnar S.P., Takayama S. Microfluidic endothelium for studying the intravascular adhesion of metastatic breast cancer cells. PLoS ONE. 2009;4:e5756. doi: 10.1371/journal.pone.0005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yanik A.A., Huang M., Altug H. Integrated nanoplasmonic-nanofluidic biosensors with targeted delivery of analytes. Appl. Phys. Lett. 2010;96:021101. [Google Scholar]
- 54.Chen G.D., Fachin F., Toner M. Nanoporous elements in microfluidics for multiscale manipulation of bioparticles. Small. 2011;7:1061–1067. doi: 10.1002/smll.201002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.