

Abstract
Studies of amyloid disease-associated proteins in aqueous solutions containing 2,2,2-trifluoroethanol (TFE) have shown that the formation of structural intermediates is often correlated with enhanced protein aggregation. Here, enhanced green fluorescent protein (EGFP) is used as a model protein system to investigate the causal relationship between TFE-induced structural transitions and aggregation. Using circular dichroism spectroscopy, light scattering measurements, and transmission electron microscopy imaging, we demonstrate that population of a partially α-helical, monomeric intermediate is roughly correlated with the growth of β-sheet-rich, flexible fibrils for acid-denatured EGFP. By fitting our circular dichroism data to a model in which TFE-water mixtures are assumed to be ideal solutions, we show that increasing entropic costs of protein solvation in TFE-water mixtures may both cause the population of the intermediate state and increase aggregate production. Tertiary structure and electrostatic repulsion also impede aggregation. We conclude that initiation of EGFP aggregation in TFE likely involves overcoming of multiple protective factors, rather than stabilization of aggregation-prone structural elements.
Introduction
Numerous human diseases, including Alzheimer's disease, Parkinson's disease, and type II diabetes, are associated with protein aggregation (1). Biophysical investigations of aggregation processes frequently employ chemical cosolvents to reduce experimental variability, simulate cellular conditions, or induce the formation of atypical aggregate structures. The fluorinated alcohol 2,2,2-trifluoroethanol (TFE) is one of the most commonly used cosolvents in these studies (2).
The effects of TFE on protein structure have been studied for decades, but the physical interactions underlying TFE-induced aggregation enhancement are not well understood (2,3). Disordered proteins and peptides generally undergo a gradual coil-to-helix transition as TFE is added to a solution, reaching their maximally helical state by ∼30–40% TFE. Additionally, TFE can denature globular proteins, typically leading to the formation of nonnative α-helical structure. When present at intermediate (∼10–40% v/v) concentrations, TFE often induces or increases the aggregation of both globular and disordered proteins. For globular proteins, tertiary structure disruption often, but not always, precedes aggregation.
Many researchers have hypothesized that fluorinated alcohols promote protein aggregation by stabilizing fibrillogenic structural intermediates. Helical intermediates are frequently associated with aggregation for disordered and denatured proteins (4–9). However, short (5–6-mer) peptides, which should not be capable of forming significant helical structure, also show enhanced fibril formation in ∼7–10% TFE (10). Moreover, β-sheet-rich species have been detected before aggregation for some proteins (11–13). Also, multiple conformational states can lead to similar aggregates for some proteins (14), and TFE-induced aggregation can even occur in the absence of significant tertiary structure disruption (15,16). It is possible that various structural intermediates promote protein aggregation via multiple mechanisms. Alternatively, some solvent-dependent effect may cause reduced protein solubility, with structural transitions being coincidental.
The observation that fluoroalcohol molecules form clusters in water has led to the suggestion that aggregation might result from increased effective hydrophobicity of a protein bound to a cluster of relatively hydrophobic alcohol molecules (17,18). However, protein structural transitions and aggregation can occur at fluoroalcohol concentrations lower than those required for cluster formation (19). The decrease in the dielectric constant of alcohol-water mixtures has also been linked to the formation of H-bonded structures, including structural intermediates and β-sheet-rich aggregates, although polarity alone cannot account for the differences in behavior between fluorinated and simple alcohols (20). Therefore, relationships between bulk properties of TFE-water mixtures and protein structural transitions remain uncertain.
In this article, we investigate TFE-induced structural rearrangements and aggregation using enhanced green fluorescent protein (EGFP) as a model system. Near physiological pH, EGFP possesses β-can tertiary structure, whereas low and high pH conditions denature the protein, leading to readily detectable loss of green fluorescence (21). Thus, EGFP is a nearly ideal system for examining the roles of tertiary versus secondary structure in TFE-induced conformational rearrangements. By combining circular dichroism (CD) spectroscopy with thermodynamic modeling, we show that changes in the chemical potential of solvent molecules may drive the observed structural transitions. We conclude that increased entropic costs of protein solvation may promote EGFP aggregation at moderate [TFE].
Materials and Methods
Materials and solutions
Enhanced GFP (GFPmut1), derived from wild-type Aequorea victoria GFP with substitutions F64L and S65T (22), was synthesized by Dr. Cynthia Kinsland and the Cornell University Life Sciences Core Laboratories Center Protein Production Facility. Details of the expression can be found in the Supporting Material.
Acros Organics brand 99.8% pure 2,2,2-trifluoroethanol was purchased from Fisher Scientific (Pittsburgh, PA). Our pH measurements refer to aqueous samples; we did not correct our measurements to account for TFE effects on ionization constants of protein, water, or buffer components. These changes are likely to be small at low pH but can be significant at higher pH (23,24). Samples labeled pH 2.4 contained 10 mM phosphoric acid and pH 7.5 samples contained 10 mM sodium phosphate buffer. Additional solution conditions are specified in the figure legends.
Light scattering
Scattering experiments were performed using a QuantaMaster fluorescence spectrofluorometer (Photon Technology International, Birmingham, NJ) using 600-nm light to avoid EGFP absorption. The scattering angle was 90° and the same quartz cuvette was used to obtain all the data. Before measurement, 50 μM EGFP samples, along with baseline solutions containing identical ingredients excepting the protein, were maintained under quiescent conditions for 24 ± 2 h in a 37°C incubator. The sample signals were normalized to the baseline signals.
CD spectroscopy
Far-UV CD measurements were performed using an Aviv 400 CD spectrometer (Aviv Biomedical, Lakewood, NJ). Data were collected at 1-nm intervals using a 1-nm bandwidth and a scan speed of 1 s per nm. Buffer-only baseline samples were measured and subtracted from the protein spectra and noise was reduced via a smoothing routine in the instrument software. The protein samples were first mixed with acid or buffer salts, and then TFE was added immediately before the measurements. However, for pH 3–5 samples, which were aggregation-prone, the protein was diluted into water, and the buffer salts and TFE were added together before the measurements.
Transmission electron microscopy imaging
A 37°C incubator was used to maintain solutions in quiescent conditions, whereas agitated samples were incubated in an orbital shaker operating at 200 RPM and 37°C. Sodium azide (0.02% w/v) was added to solutions incubated for longer than 24 h. To prevent grid damage, samples containing >15% TFE or large amounts of aggregates were diluted with water or buffer immediately before analysis. Images were obtained as described in Anderson et al. (4).
Results
Secondary and tertiary structure of EGFP in TFE
Fig. 1, A and B, shows CD spectra of 0.3 μM EGFP in the presence of 0–60% TFE. We show spectra that are consistent with monomeric or mostly monomeric protein, based on comparisons of 0.3 μM and 3 μM samples and the observation that the curves do not change significantly during the ∼20 min (per sample) experimental duration (see Fig. S1 in the Supporting Material).
Figure 1.
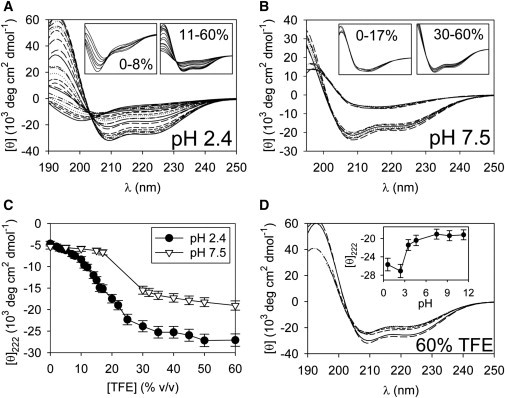
TFE-induced secondary structural transitions for EGFP. (A and B) Far-UV CD spectra for 0.3 μM EGFP in 0–60% TFE at 37°C. (Insets) Selected spectra from the main plots, and the inset x axes' ranges are the same as the main plot ranges. (A) Data for EGFP at pH 2.4. The TFE concentrations for spectra with increasing negative ellipticity at 222 nm are 0, 2, 3, 4, 5, 7, 8, 10, 11, 12, 14, 15, 16, 17, 20, 22, 25, 30, 35, 40, 45, 60, and 50% TFE. (Insets) Spectra that share isodichroics. (Left y-axis range) −18–5 × 103 deg cm2 dmol−1. (Right y-axis range) −37–65 × 103 deg cm2 dmol−1. (B) Data for EGFP at pH 7.5. The TFE concentrations for spectra with increasing negative ellipticity at 222 nm are 0, 5, 10, 15, 17, 30, 32, 35, 40, 45, 50, and 60% TFE. (Left inset) Low-TFE spectra that are nearly invariant. (Right inset) High-TFE spectra that share an isodichroic point. (Left y-axis range) −9–18 × 103 deg cm2 dmol−1. (Right y-axis range) −26–26 × 103 deg cm2 dmol−1. (C) The mean residue ellipticity at 222 nm versus [TFE] for the spectra in A and B. (Error bars) Experimental uncertainties. (D) The CD spectra of EGFP in 60% TFE at various pH. The solution conditions for spectra with increasing negative ellipticity at 222 nm are 10 mM sodium phosphate (pH 7.5), 2 mM NaOH (pH 11.3), 10 mM borax (pH 9.3), 10 mM citrate-phosphate buffer (pH 4.6), 10 mM citrate-phosphate buffer (pH 3.5), 0.25 N sulfuric acid (pH 0.6), and 10 mM phosphoric acid (pH 2.4). (Inset) Ellipticity at 222 nm (in the same units as the main plot) as a function of pH for these samples. (Error bars) Experimental uncertainties.
The pH 2.4, 0% TFE spectrum (Fig. 1 A) features the negative peak near 200 nm that is characteristic of polyproline type II (PPII) or statistical coil structure (25). As [TFE] increases, the ellipticity near 222 nm becomes larger-negative and the 200-nm peak becomes less prominent. At high TFE, the curves show the double minima at 208 and 222 that are expected for α-helical secondary structure; these TFE-induced helical states likely contain little to no tertiary structure (3).
Isodichroic points are apparent for two subsets of the spectra (Fig. 1 A, insets). The wavelength positions of these points are ∼209 nm for the 0–8% TFE samples and ∼203 nm for the 11–60% TFE samples. We also verify that EGFP is nonfluorescent at low pH in the presence of 0–60% TFE (see Fig. S2 A), as expected for denatured protein.
For EGFP in pH 7.5 solutions, the low (0–17%) TFE curves (Fig. 1 B, left inset) are consistent with the expected signal from β-can structure (26). Above 30% TFE, the spectra appear α-helical and an isodichroic point occurs near 203 nm (Fig. 1 B, right inset). We verify that EGFP remains fluorescent in pH 7.5 solutions immediately after heating to 37°C for [TFE] <≈20% (see Fig. S2 A). In contrast, rapid loss of fluorescence is observed for ≥30% TFE, pH 7.5 samples. Note that we omit the ∼20–28% TFE, pH 7.5 spectra from Fig. 1 B because EGFP unfolds and oligomerizes during the experimental time frame in these conditions (see Fig. S1 and Fig. S3).
Plots of the ellipticity at 222 nm versus [TFE] (Fig. 1 C) reveal that the low-pH, high-TFE signal is relatively strong; the estimated number of helical residues (27) is ∼50% higher in the acidic, 60% TFE solution compared to pH 7.5 conditions (see Table S1 in the Supporting Material). Examination of additional solution conditions reveals that the ellipticity shift occurs at pH ∼3.5 (Fig. 1 D), which coincides roughly with the pKa of acidic residues, although TFE may modify buffer and protein ionization constants (23,24). It is unlikely that an α-helix can propagate through the EGFP chromophore, which involves a covalent bond between Thr-65 and Gly-67 backbone groups. At pH 2.4, ∼160 residues are estimated to be helical (see Table S1). Therefore, structure formation probably does not involve only the portion of the protein N-terminal to the chromophore. The C-terminal portion may form a continuous helix, or multiple short segments could form in various parts of the protein. Thus, the pH-dependent changes might involve either a change in helix lengths or in the number of helical segments. Electrostatic attraction between oppositely charged residues may tend to favor compact structures, rather than extended helices, near neutral pH (28,29). However, higher-resolution experiments are necessary to determine the origin of the pH dependence of the helicity.
Reconstruction of the I-state spectrum and estimation of state populations
A transition-diagram (30) plot of the pH 2.4 CD data shows two linear segments (Fig. 2). The existence of two isodichroics in the CD spectra (Fig. 1 A) and two linear segments in the transition diagram indicate that EGFP is probably sampling at least three secondary structural conformations, which appear to include a low-TFE, PPII-like state (U), a high-TFE, highly helical state (HH), and an intermediate conformation (I).
Figure 2.
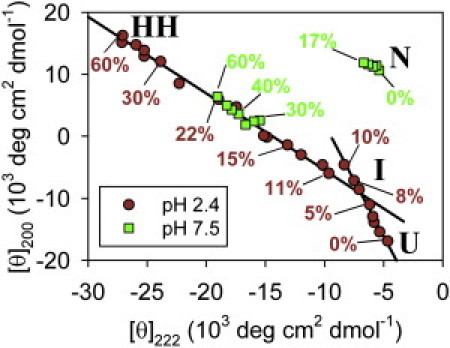
Transition diagram constructed from the CD spectra in Fig. 1, A and B. The plot shows the ellipticity at 200 vs. 222 nm. Some points are labeled with their [TFE], and the labels are color-coded to show pH. (Solid lines) Fits to the pH 2.4 points derived from spectra that share isodichroics (Fig. 1A, insets).
Points derived from 0–17% TFE, pH 7.5 curves (Fig. 1 B) cluster in a region of the transition diagram that corresponds to the native β-can fold (N in Fig. 2). EGFP remains fluorescent in these solution conditions (see Fig. S2 A); therefore the slight spectral differences for these samples likely involve changes in loop regions, rather than the core β-can. In contrast, the higher-TFE, pH 7.5 points lie along the I ↔ HH transition line and reflect loss of tertiary structure in favor of nonnative helical conformations.
Following the procedure from Anderson et al. (4), we use principal component analysis (PCA) to obtain information about the conformations contributing to our CD spectra (see the Supporting Material). We find that acid-denatured EGFP samples three structural states within the resolution of our measurements (see Fig. S4 A), as might be expected given that the spectra contain two isodichroics (Fig. 1 A) and that the corresponding points lie along two lines in the transition diagram (Fig. 2).
We also use PCA to infer the CD spectrum of the I state (see the Supporting Material). The resulting curve (Fig. 3) features weak double minima, which are suggestive of partial α-helical structure. Deconvolutions via several algorithms also predict an increase in helicity for the I state compared to the 0% TFE conformation (see Table S2). Furthermore, the reconstructed EGFP I state curve is very similar to the TFE-induced intermediate state spectrum for human wild-type αS, which may be partly helical (4).
Figure 3.
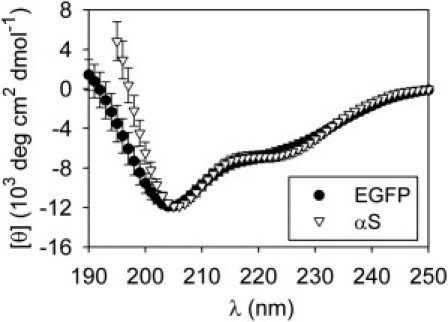
Reconstructed I-state spectrum for EGFP. The PCA-inferred EGFP curve is compared to the αS data from Anderson et al. (4). (Error bars) Uncertainties in the PCA estimates.
By fitting our CD data to a linear combination of the inferred I-state spectrum (Fig. 3) and the measured 0% and 60% TFE curves (Fig. 1 A), we can obtain an estimate of the state populations over the full TFE range (Fig. 4 A, symbols; see Fig. S5 for more information about the fits). We find that the EGFP I state is maximally populated near 8% TFE. In contrast, fits to the data in Anderson et al. (4) suggest that peak I-state population for αS occurs at ∼17% TFE (Fig. 4 B, symbols). Although the αS structural transitions were measured at 25°C and pH 7.5, whereas our EGFP data reflect 37°C and pH 2.4 conditions, we subsequently found that the TFE concentration at which the αS intermediate is maximally populated is not significantly affected by temperature variations or low pH conditions (V. L. Anderson, W. W. Webb, and D. Eliezer, unpublished data). Therefore, differences in solution conditions do not appear to be primarily responsible for the state population differences between the two proteins.
Figure 4.
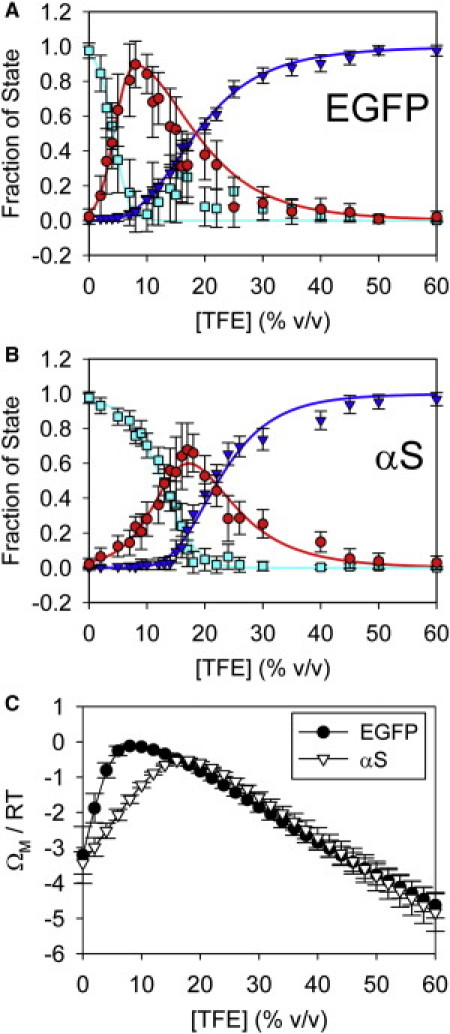
State populations and model fits for αS and EGFP. (A and B) State populations (symbols) for (A) acid-denatured EGFP and (B) αS, which were calculated by fitting the experimental spectra to a linear combination of the U (cyan), I (red), and HH (dark blue) state spectra. The error bars reflect estimated uncertainties due to noise in the experimental and I state spectra (see the Supporting Material). The EGFP populations were calculated from the data in Fig. 1A, and the αS populations were calculated from the data in Anderson et al. (4). The lines show fits of the populations to our desolvation model (Eq. 3). (C) The average monomer grand potentials versus [TFE] for EGFP and αS. The error bars reflect standard errors resulting from the desolvation model fits.
A desolvation model for TFE-induced secondary structural transitions
We construct a very simple model for the structural changes of denatured and disordered proteins in TFE-water mixtures. We briefly summarize the model here, and a more detailed derivation can be found in the Supporting Material.
Our desolvation model assumes that changes in the water and TFE chemical potentials perturb the grand potentials of the U and HH states. Therefore, this model predicts the relative populations of hydrated, TFE-solvated, and desolvated conformations, but does not account for the structural details of these states.
In the ideal solution approximation, the chemical potentials of water and TFE (μW and μT, respectively) are equal to the pure substance chemical potentials plus terms related to the mixture entropy, so that (31)
| (1) |
where xw and xT are the mole fractions of water and TFE (xw + xT = 1), T is the temperature in degrees Kelvin, and R is the ideal gas constant. The expressions in Eq. 1 imply that protein-water and protein-TFE interactions are entropically costly in TFE-water mixtures; therefore, the potentials of water and/or TFE solvated states will increase at moderate [TFE].
We consider the dilute-protein limit, so that the properties of the bulk solvent are not affected by protein solvation. We further assume that the 0% TFE, U state corresponds to a hydrated conformation in which some protein region interacts with nW water molecules, the I state is a desolvated conformation in which the region is buried or solvent-shielded, and the HH state is TFE-solvated so that the region interacts with nT TFE molecules. Also, we set the arbitrary zero-point potential to correspond to the I-state energy. Then, the grand canonical partition function (Z) for our system is
| (2) |
where eU (eHH) is the grand potential of state U (HH) in pure water (TFE) (see the Supporting Material).
Using Eq. 2, we calculate the fractions of the U, I, and HH states (fU, fI, and fHH, respectively):
| (3) |
We fit our estimated state populations to Eq. 3, converting between vol % TFE and xT (Fig. 4, A and B, lines). The expressions in Eq. 3 fit both our EGFP data and the previously obtained αS data (4) fairly well, and we obtain similar reasonable fits for additional αS variants (see Fig. S6 and Table S3). Table 1 shows the parameters resulting from the fits.
Table 1.
Parameters resulting from fits of the EGFP and αS state populations
| EGFP | αS | |
|---|---|---|
| nW | 286 ± 85 | 92 ± 10 |
| nT | 2.84 ± 0.20 | 3.27 ± 0.36 |
| eU/RT | −3.17 ± 0.84 | −3.40 ± 0.33 |
| eHH/RT | −8.32 ± 0.62 | −9.2 ± 1.0 |
In Fig. 4 C, we show the model prediction for the average monomer protein grand potential (ΩM; see Eq. S12 in the Supporting Material). Note that ΩM = 0 corresponds to 100% population of the I state. The range of TFE concentrations over which the monomer protein is destabilized (ΩM is high) is predicted to be wider for EGFP than for αS.
Aggregate production versus [TFE]
Fig. 5 shows light scattering data, reflecting relative amounts of aggregate production, for 50 μM EGFP samples incubated for 24 h at 37°C. At pH 2.4 with no added salt, aggregation is minimal over the entire 0–60% TFE range. However, the addition of 75 mM NaCl leads to aggregation enhancement for samples containing ∼7.5–30% TFE.
Figure 5.
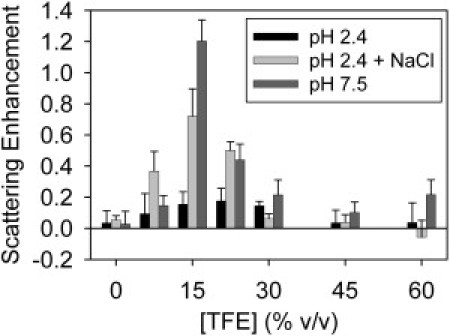
EGFP aggregation as a function of [TFE]. The plot shows the scattering signal from 50 μM EGFP at pH 2.4 (solid bars), pH 7.5 (dark-shaded bars), and pH 2.4 with 75 mM NaCl (light-shaded bars). The samples were incubated for 24 ± 2 h at 37°C in the presence of 0, 7.5, 15, 22.5, 30, 45, and 60% TFE. (Error bars) Standard deviations of measurements of three identical samples and baseline uncertainties.
At pH 7.5, aggregation is low for 0–7.5% TFE samples, increases sharply at 15% TFE, and then decreases at higher [TFE]. Fluorescence loss occurs above 15% TFE for pH 7.5 samples (see Fig. S2 A); therefore, EGFP aggregation appears to require tertiary structure loss. However, very high TFE conditions stabilize monomeric protein.
Structural features of EGFP aggregates
CD spectra for EGFP samples identical to those examined in Fig. 5 reveal single minima near 216 nm, suggesting the presence of β-sheet structure (Fig. 6). For 50 μM EGFP in pH 7.5 solutions containing 15–35% TFE (Fig. 6 B), the magnitude of the CD spectra near 216 nm is ∼2× larger than the native protein value (Fig. 1 B), suggesting that aggregates are β-structured but not natively folded. The ∼15–25% TFE samples also appear cloudy-white after 24 h. We verify that removal of large aggregates from 15% TFE samples similar to those examined in Fig. 6 results in CD signal loss, indicating that large aggregates contribute to the observed spectra (see Fig. S7).
Figure 6.
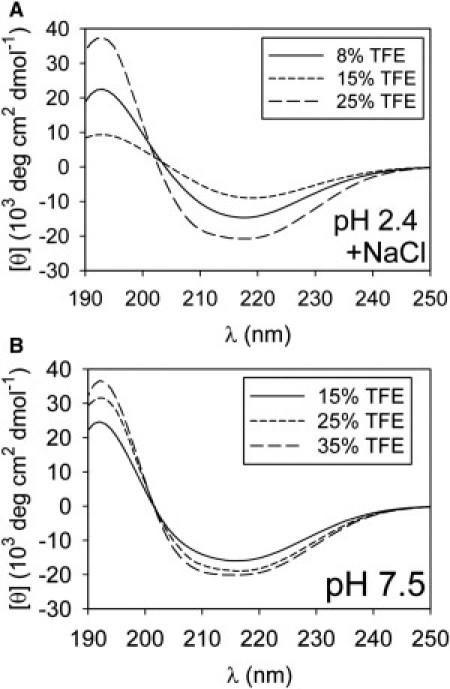
CD spectra of EGFP in aggregation-promoting conditions. 50 μM EGFP samples were incubated for 24 ± 2 h at 37°C in various [TFE]. (A) Spectra for pH 2.4 solutions containing 75 mM NaCl. (B) Spectra for pH 7.5 samples.
Even at low (∼μM) protein concentrations, pH 7.5, 15–25% TFE samples appear to be aggregation-prone, whereas pH 2.4 samples in low salt and/or reduced protein concentrations show CD spectra that are similar to the monomer curves (see Fig. S8).
Transmission electron microscopy (TEM) imaging reveals that the TFE-induced aggregates are thin, fibrillar structures, which often clump together (Fig. 7, A–C, and see Fig. S9). Rigid fibrils were sometimes observed after extended incubations (Fig. 7 D, and see Fig. S9).
Figure 7.
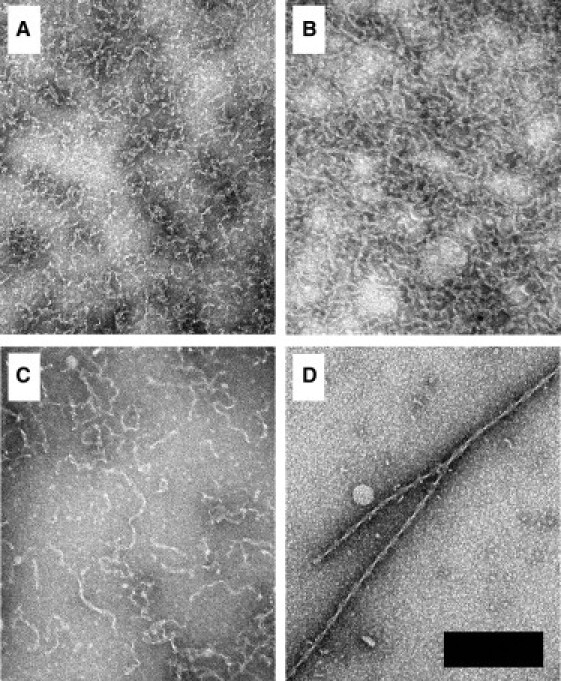
TEM images of EGFP aggregates. The scale bar is 200-nm wide and all images are shown at the same magnification. All solutions were incubated at 37°C. Unless otherwise specified, the samples were incubated for 24 ± 2 h. (A) Flexible fibrils grown from 50 μM EGFP at pH 2.4 with 15% TFE and 75 mM NaCl. (B) Flexible fibrils grown from 50 μM EGFP at pH 7.5 with 15% TFE. (C) Similar to panel B, except the solution contained 45% TFE. (D) Rigid fibrils grown from 50 μM EGFP after seven weeks with shaking at pH 2.4 in the absence of TFE or NaCl.
Discussion
We have demonstrated that TFE-induced EGFP aggregation requires tertiary structure disruption and is associated with reduced PPII-like and/or α-helical secondary structure. Using a simplified model, we showed that changes in solvent component chemical potentials may drive protein structural transitions. The free energy of the TFE-water mixture decreases at intermediate [TFE]. Consequently, it becomes costly to move solvent molecules from the bulk solvent into the protein solvation layer. The elevated entropic costs of protein solvation at moderate [TFE] may result in reduced population of solvated states in favor of solvent-shielded monomeric or oligomeric structures.
Entropic costs of protein solvation may drive secondary structural transitions in TFE-water mixtures
For both acid-denatured EGFP (Fig. 1 A) and disordered αS (4), the presence of small amounts (<≈10–15%) of TFE causes a decrease in the magnitude of the negative peak near 200 nm in the CD spectra. This peak is thought to reflect a conformational bias toward PPII structure in the ensemble of disordered conformations (25). Water-protein H-bonding may enable the formation of sterically favorable PPII structure (32); therefore, the loss of the 200-nm CD peak is consistent with the reduction of protein hydration that is predicted by our model (Eq. 3).
The inferred CD spectrum of the EGFP I state appears partially α-helical (Fig. 3, and see Table S2). Partial helical structure has been observed before TFE-induced aggregation for many additional disordered or denatured proteins and peptides (4,5,7,9). If our desolvation model is correct, the intermediate structure may be a relatively compact, solvent-avoiding conformation containing partial secondary structure. However, additional experiments are necessary to test this hypothesis.
Our >∼30% TFE CD spectra (Fig. 1, A and B), which appear highly α-helical, are suggestive of preferential solvation of EGFP by TFE. Both experimental (33,34) and theoretical (35) studies have found that TFE binds to or coats proteins when present at medium-to-high (>≈30% v/v) concentrations. Preferential solvation of proteins by TFE stabilizes α-helical structure, possibly as a result of chaotropic interactions or the low dielectric constant of TFE compared to water (36,37).
In the desolvation model, preferential TFE solvation is partly a consequence of favorable protein-TFE interactions (i.e., negative eHH values in Table 1). Interestingly, the interactions that determine eHH in our model are expected to be the same interactions that are responsible for protein solubilization and structure in pure TFE, and the fact that eHH is larger-negative than eU may reflect the high solubility of many proteins in neat TFE compared to water (38,39). Moreover, it is less entropically unfavorable to move a smaller number of molecules from the bulk solvent into the protein solvation shell; therefore the relatively small nT values compared to nW values (Table 1) tend to favor TFE, rather than water, solvation. Notably, for both EGFP and αS, the nT/nW ratios (Table 1) are larger than might be expected given the relative molecular sizes. However, changes in protein solvation are associated with changes in protein conformation; therefore, solvent-accessible surface areas may be different for the U and HH states.
Interestingly, nT, eU, and eHH are similar for EGFP and αS. The major difference between the fit results for the two proteins is the number of water molecules predicted to be bound in the U state. The value nW is approximately three times larger for EGFP than for αS. Both proteins contain a comparable fraction of nonpolar amino acids (38% for αS and 37% for EGFP when the numbers of Ala, Ile, Leu, Met, Phe, Tyr, and Trp residues are counted). However, αS contains a large proportion of Ala residues, whereas EGFP has more bulky nonpolar residues. Consequently, more water molecules should be required to form a solvation shell around denatured EGFP compared to αS. In addition, the sequence context of nonpolar residues in EGFP may favor water exclusion, given that EGFP adopts a globular fold near neutral pH.
The relative helix induction capabilities of some alcohols, hexafluoro-2-propanol > TFE > isopropanol > ethanol > methanol (18), appears to follow the order of the sizes of the molecules. Our model predicts that smaller nT values will reduce the entropic costs of alcohol solvation, leading to HH-state population at relatively low alcohol concentrations. Thus, if fewer larger molecules are needed to solvate a protein, it may be expected that larger alcohols may be better helix inducers. However, the strengths of alcohol-water interactions (eHH) should also influence helix formation, and the structural details of the high-alcohol states will be affected by the specific properties of the alcohols.
Our desolvation model assumes that mixing entropy alters the chemical potentials of TFE and water, leading to structural transitions. We do not take into account changes in the dielectric constant or viscosity of the bulk solvent, which may also affect the energies of the various states. Despite its simplicity, our model can reproduce many key features of our data. However, it is possible that corrections to account for solvent properties will be needed to refine the model. Measurements of protein structures and solubility in various alcohols could help to assess the validity of the ideal solution approximation and determine the primary factors responsible for the high helix induction capabilities of fluoroalcohols.
Desolvation may initiate protein aggregation at moderate [TFE]
Enhanced aggregation of acid-denatured EGFP in 75 mM NaCl (Fig. 5) occurs for TFE ranges that are roughly similar to those in which the I-state population is high in 0 mM NaCl solutions (Fig. 4 A). Note that we compare EGFP monomer structures at 0 mM NaCl with aggregate production at 75 mM NaCl, although the protein conformations may not be identical in the different solution conditions. We are unable to measure monomeric EGFP spectra for ∼0–20% TFE, 75 mM NaCl solutions because of aggregation (see Fig. S10). However, the high-TFE spectra are qualitatively similar in the presence and absence of NaCl, and EGFP may sample a similar partially α-helical intermediate state in both conditions. Partially helical states appear to be more aggregation-prone than highly helical states at for acid-denatured EGFP at 75 mM NaCl, as well as for high TFE, pH 7.5 samples (Fig. 5). A similar correlation between the formation of partial α-helical intermediates and aggregation has been observed for many additional disordered and denatured proteins in fluoroalcohols (4–9).
Our model (Eq. 3) predicts that I-state population and aggregation enhancement both may result from a solvent entropy-driven increase in the grand potentials of solvated conformations. At moderate [TFE], the average potential of the monomer protein increases (Fig. 4 C), favoring aggregated conformations. However, the amount of aggregate produced will also depend on energies of oligomeric and fibrillar species, which may vary with [TFE]. Therefore, peak aggregation may not exactly coincide with maximal destabilization of monomeric protein.
For acid-denatured EGFP in 75 mM NaCl, we find that the peak scattering signal may be slightly shifted rightward to ∼15% TFE, compared to the ∼8% TFE I-state peak (Fig. 4 B), although the differences are within the measurement uncertainties. The presence of NaCl may affect the state populations, or the relatively low dielectric constant of TFE compared to water may shift aggregation peaks rightward as a result of favorable H-bond formation (20).
Decreases in the free energy of the bulk solvent are expected to favor the formation of solvent-shielded and oligomeric structures (40). In addition, desolvation is consistent with previous reports that hydration impedes amyloid aggregation (41–44). Therefore, we believe that the simplicity of our model, combined with the fact that it offers a straightforward explanation for both the structural changes and the aggregation enhancement observed for proteins in TFE, make it a reasonable initial analysis. However, we do not directly measure protein-solvent interactions, and the fact that the desolvation model fits our data does not prove that it is correct. Moreover, because changes in solvent properties and protein conformation are likely to occur in parallel, it may be difficult to definitively separate correlation from causation in aggregation reactions. Experiments and simulations that directly address the hydration status of proteins might help verify the primary factors driving aggregation in the presence of TFE (41,43).
Multiple protective factors impede EGFP aggregation
Previous studies have identified electrostatic repulsion and globular structures as natural protective mechanisms, which likely evolved to prevent abnormal protein aggregation (45). Hydration is also an important stabilizing interaction for disordered proteins (46). Our data suggest that several types of protective interactions impede EGFP aggregation.
We find that aggregation of acid-denatured EGFP is minimal in low ionic strength solutions (Fig. 5). The net charge per EGFP molecule is predicted to be +36e pH 2.4, compared to −7.7e at pH 7.5 (47); therefore electrostatic repulsion likely opposes aggregation of the acid-denatured protein. The addition of 75 mM NaCl enables significant aggregation in solutions containing ∼7.5–30% TFE, indicating that electrostatic screening can overcome these protective repulsive forces.
For EGFP in pH 7.5 solutions, aggregation jumps sharply at 15% TFE (Fig. 5), where tertiary structure disruption occurs (see Fig. S2 A). However, increased α-helical structure at higher TFE (Fig. 1) is associated with reduced fibrilization. Therefore both intact tertiary structure and TFE-protein interactions may impede EGFP fibrilization at neutral pH.
Our model predicts that increased entropic costs of protein solvation overcome protective protein-solvent interactions, leading to monomer destabilization at intermediate [TFE] (Fig. 4 C). Moreover, if a modification could be made to a protein that would prevent the formation of the I state while leaving the potentials of other states unchanged, the ensemble-average monomer potential would increase in the TFE-water mixture (see Fig. S11). Therefore, the I state may be somewhat protective, in the sense that adoption of solvent-shielded intermediate conformations could reduce aggregate production.
Conclusion
The secondary structural transitions for acid-denatured EGFP in TFE are qualitatively similar to those previously observed for αS (4) and are consistent with dehydration leading to loss of PPII structure at low-to-moderate TFE. At high TFE, favorable TFE-protein interactions induce the formation of α-helical structure and inhibit EGFP aggregation. We propose that monomer destabilization initiates fibrilization at intermediate [TFE]. However, intact tertiary structure and electrostatic repulsion impede aggregation. Hence, TFE-induced aggregation likely involves overcoming multiple protective factors, rather than the stabilization of aggregation-prone conformations.
Acknowledgments
The authors thank Prof. David Piston and Dr. Gert-Jan Kremers at Vanderbilt University for their generous gift of GFP variant vectors. We also are grateful to Mark Williams for help with manuscript preparation, and to Prof. D. Eliezer, Prof. J. Sethna, Prof. L. Nicholson, J. Grazul, and Y. Zhang for helpful discussions.
This research made use of the Hudson Mesoscale Processing facility of the Cornell Center for Materials Research with support from the National Science Foundation Materials Research Science and Engineering Centers program (DMR 1120296 and DMR 0520404). Funding was provided by grants from the National Institutes of Health (5 R21 AG026650) and the National Science Foundation (Science and Technology Centers program under agreement No. ECS-9876771).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Otzen D.E. Amyloid formation in surfactants and alcohols: membrane mimetics or structural switchers? Curr. Protein Pept. Sci. 2010;11:355–371. doi: 10.2174/138920310791330622. [DOI] [PubMed] [Google Scholar]
- 3.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 4.Anderson V.L., Ramlall T.F., Eliezer D. Identification of a helical intermediate in trifluoroethanol-induced α-synuclein aggregation. Proc. Natl. Acad. Sci. USA. 2010;107:18850–18855. doi: 10.1073/pnas.1012336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fezoui Y., Teplow D.B. Kinetic studies of amyloid β-protein fibril assembly. Differential effects of α-helix stabilization. J. Biol. Chem. 2002;277:36948–36954. doi: 10.1074/jbc.M204168200. [DOI] [PubMed] [Google Scholar]
- 6.Zerovnik E., Skarabot M., Staniforth R.A. Amyloid fibril formation by human stefin B: influence of pH and TFE on fibril growth and morphology. Amyloid. 2007;14:237–247. doi: 10.1080/13506120701461137. [DOI] [PubMed] [Google Scholar]
- 7.Sen P., Ahmad B., Khan R.H. 2,2,2-Trifluroethanol induces simultaneous increase in α-helicity and aggregation in alkaline unfolded state of bovine serum albumin. Int. J. Biol. Macromol. 2010;46:250–254. doi: 10.1016/j.ijbiomac.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Williamson J.A., Loria J.P., Miranker A.D. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J. Mol. Biol. 2009;393:383–396. doi: 10.1016/j.jmb.2009.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu W., Prausnitz J.M., Blanch H.W. Amyloid fibril formation by peptide LYS (11–36) in aqueous trifluoroethanol. Biomacromolecules. 2004;5:1818–1823. doi: 10.1021/bm049841e. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhary N., Singh S., Nagaraj R. Morphology of self-assembled structures formed by short peptides from the amyloidogenic protein τ depends on the solvent in which the peptides are dissolved. J. Pept. Sci. 2009;15:675–684. doi: 10.1002/psc.1172. [DOI] [PubMed] [Google Scholar]
- 11.Srisailam S., Kumar T.K.S., Yu C. Amyloid-like fibril formation in an all β-barrel protein. Partially structured intermediate state(s) is a precursor for fibril formation. J. Biol. Chem. 2003;278:17701–17709. doi: 10.1074/jbc.M300336200. [DOI] [PubMed] [Google Scholar]
- 12.Pallarès I., Vendrell J., Ventura S. Amyloid fibril formation by a partially structured intermediate state of α-chymotrypsin. J. Mol. Biol. 2004;342:321–331. doi: 10.1016/j.jmb.2004.06.089. [DOI] [PubMed] [Google Scholar]
- 13.Lim K.H., Le Y.T., Kenney J.M. Characterization of amyloidogenic intermediate states through a combined use of CD and NMR spectroscopy. Biophys. Chem. 2010;151:155–159. doi: 10.1016/j.bpc.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Calamai M., Chiti F., Dobson C.M. Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophys. J. 2005;89:4201–4210. doi: 10.1529/biophysj.105.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plakoutsi G., Taddei N., Chiti F. Aggregation of the acylphosphatase from Sulfolobus solfataricus: the folded and partially unfolded states can both be precursors for amyloid formation. J. Biol. Chem. 2004;279:14111–14119. doi: 10.1074/jbc.M312961200. [DOI] [PubMed] [Google Scholar]
- 16.Soldi G., Bemporad F., Chiti F. Amyloid formation of a protein in the absence of initial unfolding and destabilization of the native state. Biophys. J. 2005;89:4234–4244. doi: 10.1529/biophysj.105.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanagi K., Ashizaki M., Goto Y. Hexafluoroisopropanol induces amyloid fibrils of islet amyloid polypeptide by enhancing both hydrophobic and electrostatic interactions. J. Biol. Chem. 2011;286:23959–23966. doi: 10.1074/jbc.M111.226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirota N., Mizuno K., Goto Y. Cooperative α-helix formation of β-lactoglobulin and melittin induced by hexafluoroisopropanol. Protein Sci. 1997;6:416–421. doi: 10.1002/pro.5560060218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gast K., Siemer A., Damaschun G. Fluoroalcohol-induced structural changes of proteins: some aspects of cosolvent-protein interactions. Eur. Biophys. J. 2001;30:273–283. doi: 10.1007/s002490100148. [DOI] [PubMed] [Google Scholar]
- 20.Munishkina L.A., Phelan C., Fink A.L. Conformational behavior and aggregation of α-synuclein in organic solvents: modeling the effects of membranes. Biochemistry. 2003;42:2720–2730. doi: 10.1021/bi027166s. [DOI] [PubMed] [Google Scholar]
- 21.Hsu S.T., Blaser G., Jackson S.E. The folding, stability and conformational dynamics of β-barrel fluorescent proteins. Chem. Soc. Rev. 2009;38:2951–2965. doi: 10.1039/b908170b. [DOI] [PubMed] [Google Scholar]
- 22.Cormack B.P., Valdivia R.H., Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173(1 Spec No):33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 23.Espinosa S., Bosch E., Valkó K. Change of mobile phase pH during gradient reversed-phase chromatography with 2,2,2-trifluoroethanol-water as mobile phase and its effect on the chromatographic hydrophobicity index determination. J. Chromatogr. A. 2002;954:77–87. doi: 10.1016/s0021-9673(02)00165-6. [DOI] [PubMed] [Google Scholar]
- 24.Zagorski M.G., Barrow C.J. NMR studies of amyloid β-peptides: proton assignments, secondary structure, and mechanism of an α-helix-β-sheet conversion for a homologous, 28-residue, N-terminal fragment. Biochemistry. 1992;31:5621–5631. doi: 10.1021/bi00139a028. [DOI] [PubMed] [Google Scholar]
- 25.Tiffany M.L., Krimm S. Circular dichroism of the “random” polypeptide chain. Biopolymers. 1969;8:347–359. [Google Scholar]
- 26.Visser N.V., Hink M.A., Visser A.J. Circular dichroism spectroscopy of fluorescent proteins. FEBS Lett. 2002;521:31–35. doi: 10.1016/s0014-5793(02)02808-9. [DOI] [PubMed] [Google Scholar]
- 27.Luo P., Baldwin R.L. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 28.Fan F., Mayo K.H. Effect of pH on the conformation and backbone dynamics of a 27-residue peptide in trifluoroethanol. An NMR and CD study. J. Biol. Chem. 1995;270:24693–24701. doi: 10.1074/jbc.270.42.24693. [DOI] [PubMed] [Google Scholar]
- 29.Valerio M., Porcelli F., Conti F. pH effects on the conformational preferences of amyloid β-peptide (1–40) in HFIP aqueous solution by NMR spectroscopy. ChemMedChem. 2008;3:833–843. doi: 10.1002/cmdc.200700324. [DOI] [PubMed] [Google Scholar]
- 30.Kuznetsova I.M., Turoverov K.K., Uversky V.N. Use of the phase diagram method to analyze the protein unfolding-refolding reactions: fishing out the “invisible” intermediates. J. Proteome Res. 2004;3:485–494. doi: 10.1021/pr034094y. [DOI] [PubMed] [Google Scholar]
- 31.Klotz I.M., Rosenberg R.M. Chemical Thermodynamics: Basic Theory and Methods. In: Benjamin W.A., editor. 3rd Ed. Menlo Park; CA: 1972. [Google Scholar]
- 32.Drozdov A.N., Grossfield A., Pappu R.V. Role of solvent in determining conformational preferences of alanine dipeptide in water. J. Am. Chem. Soc. 2004;126:2574–2581. doi: 10.1021/ja039051x. [DOI] [PubMed] [Google Scholar]
- 33.Chatterjee C., Gerig J.T. Interactions of trifluoroethanol with the Trp-cage peptide. Biopolymers. 2007;87:115–123. doi: 10.1002/bip.20796. [DOI] [PubMed] [Google Scholar]
- 34.Díaz M.D., Fioroni M., Berger S. Evidence of complete hydrophobic coating of bombesin by trifluoroethanol in aqueous solution: an NMR spectroscopic and molecular dynamics study. Chemistry. 2002;8:1663–1669. doi: 10.1002/1521-3765(20020402)8:7<1663::aid-chem1663>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 35.Fioroni M., Diaz M.D., Berger S. Solvation phenomena of a tetrapeptide in water/trifluoroethanol and water/ethanol mixtures: a diffusion NMR, intermolecular NOE, and molecular dynamics study. J. Am. Chem. Soc. 2002;124:7737–7744. doi: 10.1021/ja0259335. [DOI] [PubMed] [Google Scholar]
- 36.Walgers R., Lee T.C., Cammers-Goodwin A. An indirect chaotropic mechanism for the stabilization of helix conformation of peptides in aqueous trifluoroethanol and hexafluoro-2-propanol. J. Am. Chem. Soc. 1998;120:5073–5079. [Google Scholar]
- 37.Uversky V.N., Narizhneva N.V., Löber G. Conformational transitions provoked by organic solvents in β-lactoglobulin: can a molten globule-like intermediate be induced by the decrease in dielectric constant? Fold. Des. 1997;2:163–172. doi: 10.1016/s1359-0278(97)00023-0. [DOI] [PubMed] [Google Scholar]
- 38.Chin J.T., Wheeler S.L., Klibanov A.M. On protein solubility in organic solvent. Biotechnol. Bioeng. 1994;44:140–145. doi: 10.1002/bit.260440120. [DOI] [PubMed] [Google Scholar]
- 39.Malavolta L., Pinto M.R., Nakaie C.R. Interpretation of the dissolution of insoluble peptide sequences based on the acid-base properties of the solvent. Protein Sci. 2006;15:1476–1488. doi: 10.1110/ps.051956206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eggers D.K. A bulk water-dependent desolvation energy model for analyzing the effects of secondary solutes on biological equilibria. Biochemistry. 2011;50:2004–2012. doi: 10.1021/bi1017717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rauscher S., Baud S., Pomès R. Proline and glycine control protein self-organization into elastomeric or amyloid fibrils. Structure. 2006;14:1667–1676. doi: 10.1016/j.str.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Balbirnie M., Grothe R., Eisenberg D.S. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated β-sheet structure for amyloid. Proc. Natl. Acad. Sci. USA. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J., Yan Y.B. Oligomerization and aggregation of bovine pancreatic ribonuclease A: backbone hydration probed by infrared band-shift. Protein Pept. Lett. 2008;15:650–657. doi: 10.2174/092986608785133645. [DOI] [PubMed] [Google Scholar]
- 44.Mukherjee S., Chowdhury P., Gai F. Effect of dehydration on the aggregation kinetics of two amyloid peptides. J. Phys. Chem. B. 2009;113:531–535. doi: 10.1021/jp809817s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monsellier E., Chiti F. Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 2007;8:737–742. doi: 10.1038/sj.embor.7401034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 47.Putnam, C. D. 2006. Protein Calculator V.3.3. Web access 1 June 2011. <http://www.scripps.edu/∼cdputnam/protcalc.html>.
- 48.Anderson V.L., Webb W.W. Transmission electron microscopy characterization of fluorescently labeled amyloid β 1-40 and α-synuclein aggregates. BMC Biotechnol. 2011;11:125. doi: 10.1186/1472-6750-11-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrade M.A., Chacón P., Morán F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 50.Brahms S., Brahms J. Determination of protein secondary structure in solution by vacuum ultraviolet circular dichroism. J. Mol. Biol. 1980;138:149–178. doi: 10.1016/0022-2836(80)90282-x. [DOI] [PubMed] [Google Scholar]
- 51.Cattell R. The screen test for the number of factors. Multivariate Behav. Res. 1966;1:245–276. doi: 10.1207/s15327906mbr0102_10. [DOI] [PubMed] [Google Scholar]
- 52.Compton L.A., Johnson W.C., Jr. Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 1986;155:155–167. doi: 10.1016/0003-2697(86)90241-1. [DOI] [PubMed] [Google Scholar]
- 53.Enoki S., Saeki K., Kuwajima K. Acid denaturation and refolding of green fluorescent protein. Biochemistry. 2004;43:14238–14248. doi: 10.1021/bi048733+. [DOI] [PubMed] [Google Scholar]
- 54.Greenfield N.J. Methods to estimate the conformation of proteins and polypeptides from circular dichroism data. Anal. Biochem. 1996;235:1–10. doi: 10.1006/abio.1996.0084. [DOI] [PubMed] [Google Scholar]
- 55.Provencher S.W., Glöckner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 56.Spiess A.N., Neumeyer N. An evaluation of R2 as an inadequate measure for nonlinear models in pharmacological and biochemical research: a Monte Carlo approach. BMC Pharmacol. 2010;10:6. doi: 10.1186/1471-2210-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sreerama N., Venyaminov S.Y., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 2000;287:243–251. doi: 10.1006/abio.2000.4879. [DOI] [PubMed] [Google Scholar]
- 58.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 59.van Stokkum I.H., Spoelder H.J., Groen F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990;191:110–118. doi: 10.1016/0003-2697(90)90396-q. [DOI] [PubMed] [Google Scholar]
- 60.Whitmore L., Wallace B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32(Web Server issue):W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.