

Abstract
Human kinetochores are transcriptionally active, producing very low levels of transcripts of the underlying alpha-satellite DNA. However, it is not known whether kinetochores can tolerate acetylated chromatin and the levels of transcription that are characteristic of housekeeping genes, or whether kinetochore-associated ‘centrochromatin’, despite being transcribed at a low level, is essentially a form of repressive chromatin. Here, we have engineered two types of acetylated chromatin within the centromere of a synthetic human artificial chromosome. Tethering a minimal NF-κB p65 activation domain within kinetochore-associated chromatin produced chromatin with high levels of histone H3 acetylated on lysine 9 (H3K9ac) and an ~10-fold elevation in transcript levels, but had no substantial effect on kinetochore assembly or function. By contrast, tethering the herpes virus VP16 activation domain produced similar modifications in the chromatin but resulted in an ~150-fold elevation in transcripts, approaching the level of transcription of an endogenous housekeeping gene. This rapidly inactivated kinetochores, causing a loss of assembled CENP-A and blocking further CENP-A assembly. Our data reveal that functional centromeres in vivo show a remarkable plasticity – kinetochores tolerate profound changes to their chromatin environment, but appear to be critically sensitive to the level of centromeric transcription.
Key words: Centromere, CENP-A, Chromatin, Epigenetics, Human artificial chromosome, Kinetochore
Introduction
The centromere is a specialized chromatin domain that directs the assembly of functional kinetochores during mitosis (Carroll and Straight, 2006; Cheeseman and Desai, 2008; Maiato et al., 2004). In humans, endogenous centromeres typically form on chromosome-specific higher-order ‘alphoid’ DNA arrays, which are composed of 171 bp alpha-satellite monomer units that are tandemly arranged in a directional head-to-tail fashion (Choo et al., 1991; Mitchell et al., 1985; Schueler and Sullivan, 2006; Willard and Waye, 1987). Independent of this sequence preference, specific deposition of the centromeric histone H3 variant CENP-A (Earnshaw and Rothfield, 1985) is thought to form the basis for an ‘epigenetic’ maintenance of centromere identity (Allshire and Karpen, 2008; Okamoto et al., 2007; Vafa and Sullivan, 1997; Warburton et al., 1997). The epigenetic control of centromere activity is strikingly illustrated by the inactivation of centromeres on dicentric chromosomes (Earnshaw and Migeon, 1985; Earnshaw et al., 1989; Merry et al., 1985; Sugata et al., 2000; Sullivan and Schwartz, 1995) and by rare neocentromeres that recruit CENP-A and assemble fully functional kinetochore structures on non-alphoid DNA (Alonso et al., 2007; Depinet et al., 1997; du Sart et al., 1997; Saffery et al., 2000; Warburton et al., 1997).
Besides CENP-A, active centromeres display a specific profile of post-translational modifications on their associated histone H3-containing nucleosomes (Bergmann et al., 2011; Lam et al., 2006; Ribeiro et al., 2010; Sullivan and Karpen, 2004). This special chromatin, which has been termedx ‘centrochromatin’ (Sullivan and Karpen, 2004), suggests a functional link between the local chromatin environment and kinetochore function. Indeed, previous work from our laboratories indicates that the local chromatin state is crucial for the maintenance of a functional kinetochore structure (Bergmann et al., 2011; Cardinale et al., 2009; Nakano et al., 2008; Nakano et al., 2003; Nakashima et al., 2005).
The generation of an artificial chromosome carrying a synthetic, higher-order alpha-satellite repeat array with a tet operator (tetO) sequence in every other alphoid monomer has been described previously (Nakano et al., 2008). This human artificial chromosome (HAC) carrying the alphoid array with the tetO sequence replacing every second CENP-B box (hereafter referred to as the alphoidtetO HAC) exhibits a mitotic stability that is comparable to that of native chromosomes in cultured human cells (Nakano et al., 2008). We have since successfully used the alphoidtetO HAC to manipulate the chromatin environment underlying its functional kinetochore by the specific targeting of tet repressor (tetR) fusion proteins (Cardinale et al., 2009; Nakano et al., 2008). Using this approach, we recently showed that H3 dimethylated on lysine 4 (H3K4me2) is required for the long-term maintenance of the kinetochore (Bergmann et al., 2011).
Our previous work contradicted the classical view that vertebrate kinetochores are transcriptionally inactive by showing that either a histone modification linked to transcriptional elongation or that transcriptional elongation itself is required for the maintenance of kinetochore chromatin in human cells (Bergmann et al., 2011). Here, we have explored the impact of seeding two types of ‘open’ chromatin within the HAC centromere on the maintenance of its kinetochore structure and function. Our studies reveal that human centromeres are remarkably tolerant of an open chromatin structure. Thus, even centromeres in which the histone H3 is highly acetylated on lysine 9 remain capable of promoting functional kinetochore assembly. However, when acetylated chromatin is coupled with high levels of transcription, kinetochores are rapidly disassembled.
Results
Tethering of p65 and VP16 to the alphoidtetO HAC centromere promotes the formation of two distinct types of ‘open’ chromatin
In order to begin to distinguish between the role of chromatin modifications and transcription in kinetochore structure and function, we generated two tetR fusion constructs as tools for engineering distinct types of ‘open’ chromatin at the functional alphoidtetO HAC centromere (Fig. 1A). For the purpose of this study, we operationally define ‘open’ chromatin as chromatin that contains substantial levels of H3K9ac and produces significant levels of transcripts. As shown in Fig. 1A, the alphoidtetO HAC consists of ~6000 synthetic alphoid dimer repeats plus ~30 copies of a vector backbone carrying a blasticidin S resistance (BSr) cassette for selection. Each dimer contains tetO and CENP-B box sequences in the first and second monomers, respectively.
Fig. 1.

Tethering of p65 and VP16 into the alphoidtetO HAC kinetochore creates two distinct types of ‘open’ chromatin. (A) Schematic drawings of the alphoidtetO DNA array and tetR fusion constructs expressed in HeLa 1C7 cells. The synthetic monomer was based on a published consensus sequence for alphoid DNA (Choo et al., 1991). (B) Real-time RT-PCR analysis of non-transfected 1C7 cells stably carrying the alphoidtetO HAC and 2 days after transfecting constructs expressing tetR-EYFP (tetR), tetR–EYFP–p65 (p65) or tetR–EYFP–VP16 (VP16). We selected for transfected cells expressing the tethered p65 and VP16 constructs using an IRES-Puro vector by puromycin selection. Expression levels of the alphoidtetO array (tetO) and the chromosome 21 centromere (chr. 21) were normalized to those of β-actin. AlphoidtetO RNA levels in untransfected cells were arbitrarily set as 1.0. Data represent the mean and s.e.m. of three or more independent experiments. (C) ChIP analysis of nontransfected 1C7 cells or cells transfected as in B using an antibody against the elongating form of RNA polymerase II. Transfections with tetR–EYFP–VP16 were performed either in the absence of or in the presence (+Dox) of 1 μg/ml doxycycline, which prevents the binding of tetR to the alphoidtetO array. RNA polymerase II association with alphoidtetO (tetO), chromosome 21 alphoid (chr. 21) DNA, the blasticidin S resistance (BSr) marker in the BAC vector and the endogenous PABPC1 gene was determined. Values were normalized to the RNA polymerase II occupancy at the endogenous PP1A housekeeping gene locus. Data represent the mean and s.e.m. of two or more independent experiments.
We generated a fusion construct linking VP16 to TetR labeled with enhanced yellow fluorescent protein (TetR–EYFP–VP16) that resembles the widely used tTA transcription factor, but contains an EYFP tag that is inserted between the tetracycline repressor and the C-terminal herpes virus VP16 trans-activation domain. This domain promotes the opening of the local chromatin structure and results in a substantial increase in local RNA polymerase activity (Carpenter et al., 2005). TetR–EYFP–p65 is an analogous construct that contains a minimal C-terminal trans-activation domain derived from the NF-κB subunit p65. This domain induces an open chromatin environment but is less efficient at promoting transcriptional activity (Carpenter et al., 2005).
To determine the impact of these two chimeric proteins on transcription at the alphoidtetO HAC centromere, we transiently expressed the two constructs in human 1C7 cells stably carrying a single copy of the alphoidtetO HAC (Cardinale et al., 2009). Tethering of the tetR–EYFP control construct caused a mild (2.5-fold) increase in transcription of the HAC centromere, as determined by quantitative real-time RT-PCR using primers that were specific for the alphoidtetO repeat (Fig. 1B). By contrast, tethering of the VP16 fusion construct promoted a dramatic (150-fold) increase in local transcription (Fig. 1B). TetR–EYFP–p65 caused a lesser, but still significant (P=0.01, 10-fold) increase in alphoidtetO transcription (Fig. 1B). Importantly, none of these constructs affected transcription of the native chromosome 21 alpha-satellite DNA. This confirms the specificity of this type of epigenetic engineering, where effects are exerted only at sites of tetR:tetO binding within the HAC centromere.
Consistent with the relatively mild effect on transcript levels observed, chromatin immunoprecipitation (ChIP) analysis of cells expressing tetR–EYFP–p65 revealed only a small and statistically insignificant increase (P=0.23) of the very low levels of elongating RNA polymerase II (phosphorylated at serine 2 of the C-terminal hepta-peptide repeat) associated with the alphoidtetO centromere (Fig. 1C). By contrast, tethering of the tetR–EYFP–VP16 construct caused a significant (P=0.02) increase in RNA polymerase II occupancy. This increase was not observed in control-transfected cells grown in the presence of doxycycline, which prevents the binding of tetR to the alphoidtetO array (Fig. 1C). This confirms that transcriptional induction through VP16 is a direct consequence of local binding of the fusion construct. Importantly, levels of elongating RNA polymerase on the alphoidtetO array induced by tethering of VP16 were within the physiologically relevant range, remaining below those detected within the body of the actively transcribed housekeeping gene encoding poly-A binding protein-C1 (PABPC1) (Fig. 1C) (Blobel et al., 2009).
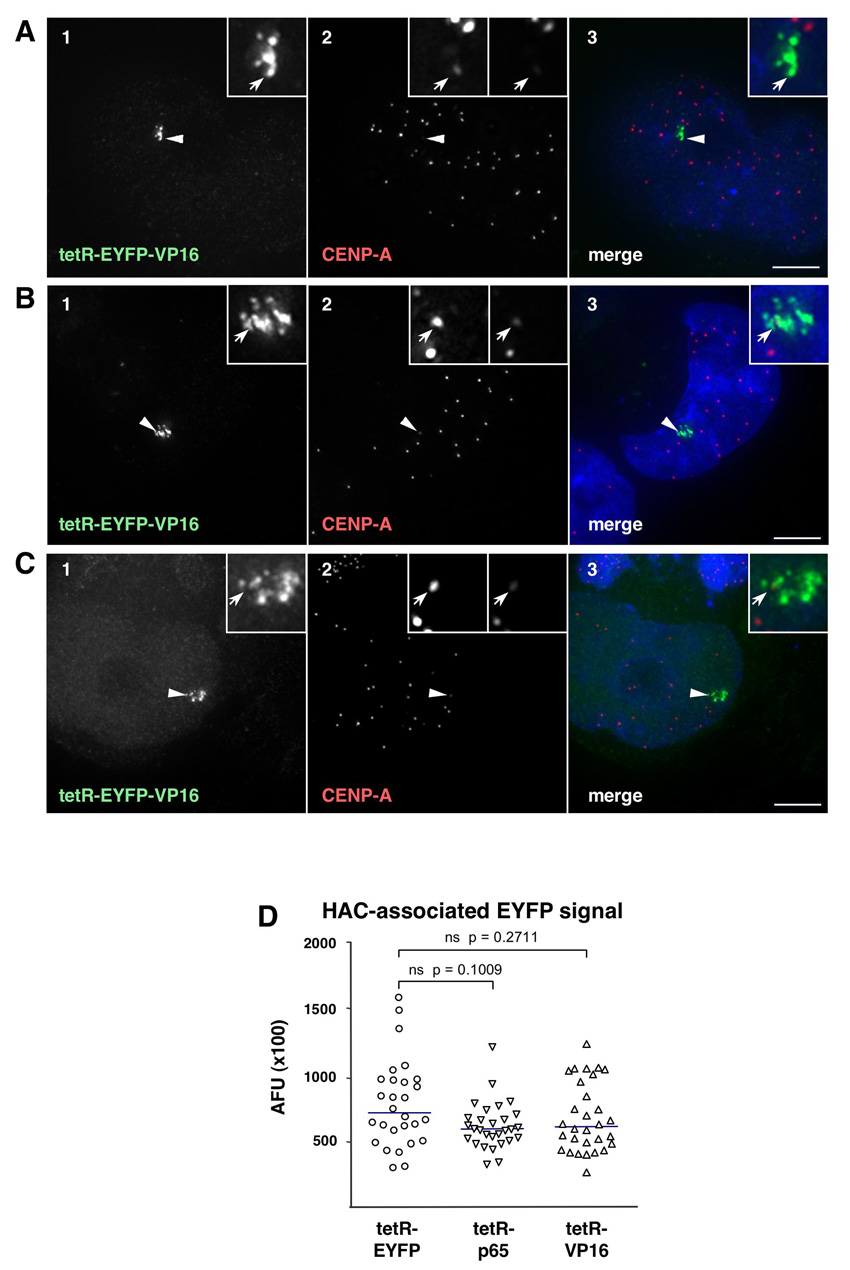
Tethering of either the tetR–EYFP–p65 or tetR–EYFP–VP16 fusion constructs resulted in a pronounced increase in the levels of H3K9ac in the underlying HAC chromatin that were readily detected by indirect immunofluorescence (Fig. 2A–C). In the control experiments, tethering of tetR–EYFP produced no such effect. This increase in H3K9ac was specific, as staining for H4K8ac and H4K16ac was comparable at HACs targeted by p65 and VP16 and did not show an appreciable increase over HACs targeted by tetR–EYFP (data not shown). The quantification of both HAC-associated H3K9ac staining (Fig. 2D) and levels of H3K9ac on the alphoidtetO array by ChIP (Fig. 2E) demonstrated that both p65 and VP16 induced a comparable increase in this modification at the HAC centromere.
Fig. 2.

Tethering of VP16 and p65 mediates HAC centromere hyper-acetylation. (A–C) Immunofluorescence (IF) analysis of 1C7 cells 2 days after transfection with the indicated tetR fusion constructs. Cells were co-stained with antibodies against acetylated H3K9 (H3K9ac, green, panel 2) and CENP-A (red, panel 3). Arrowheads depict the HAC, as determined by the EYFP signal (blue, panel 1). Merged images (panel 4) represent the overlay of EYFP signals with antibody and 4′,6-diamidino-2-phenylindole (DAPI) staining (light grey). Scale bars: 5 μm. (D) Fluorescence signals of HAC-associated H3K9ac staining in individual cells transfected as in A–C were quantified and plotted as arbitrary fluorescence units (AFU). Solid lines indicate the median. (E) ChIP analysis as in Fig. 1C of 1C7 cells transfected with tetR–EYFP, tetR–EYFP–p65 (p65) or tetR–EYFP–VP16 (VP16), using an antibody against H3K9ac. Values are normalized to the chromosome 21 centromere locus and represent the mean and s.e.m. of three independent experiments. (F) Quantification of HAC decondensation in cells transfected as in A–C. The HAC-bound EYFP area was measured in maximum-intensity projections of cells expressing the indicated fusion constructs. Sold lines indicate the median.
It was previously reported that targeting of either VP16 or p65 caused dramatic decondensation of non-centromeric reporter arrays (Carpenter et al., 2005). We also observed some decondensation of the HAC when targeted by p65 or VP16 (Fig. 2F). A fraction of HACs targeted by the VP16 fusion construct showed substantial large-scale unfolding within the interphase nucleus (Fig. 2F; also compare Fig. 2C and supplementary material Fig. S1A–C). Strikingly, the associated CENP-A staining in those cases typically remained compacted (supplementary material Fig. S1A–C), indicating resistance of the CENP-A domain to large-scale chromatin unfolding in vivo. This is consistent with the increased resistance of CENP-A chromatin to low-salt-induced unfolding observed in our previous studies (Ribeiro et al., 2009). It remains to be determined whether this difference can be ascribed to a higher rigidity for CENP-A nucleosomes, as revealed by deuterium exchange experiments (Black et al., 2004; Black et al., 2007).
Together, these data demonstrate that the TetR–EYFP–p65 and TetR–EYFP–VP16 fusion constructs are useful as tools to specifically engineer acetylated chromatin with low or high RNA polymerase levels, respectively, within a functional kinetochore.
Acetylated chromatin is compatible with the integrity of the CENP-A domain and kinetochore function
HACs targeted by TetR–EYFP–VP16 suffered a dramatic loss of CENP-A staining within two days of transfection (Fig. 2C). Remarkably, CENP-A staining remained normal at HACs tethered with tetR–EYFP–p65 over this same time period, despite fully overlapping with robust staining for the ectopically induced H3K9 acetylation (Fig. 2B). As expected from previous studies (Cardinale et al., 2009; Nakano et al., 2008), in parallel control experiments, interphase HACs targeted by tetR–EYFP retained robust CENP-A staining for at least 2 days after transfection (Fig. 2A). These results do not arise from differences in the expression of the chimeric proteins, because steady-state levels of the three fusion proteins bound to the HAC centromere were similar, as determined by quantification of the respective EYFP signals (supplementary material Fig. S1D).
Quantification of the HAC-associated CENP-A protein by either quantitative fluorescence microscopy or ChIP confirmed that tetR–EYFP–p65 did not negatively affect the levels of CENP-A at the HAC centromere (Fig. 3A,B). By contrast, both methods of measurement confirmed a pronounced reduction of CENP-A at the HAC centromere in cells expressing tetR–EYFP–VP16 (Fig. 3A,B). As with the levels of active RNA polymerase, loss of CENP-A was crucially dependent on binding of the VP16 fusion construct to the alphoidtetO array and did not occur in cells expressing this chimeric protein in the presence of doxycycline (Fig. 3B). As a further control, the negative effect of VP16 was specific for CENP-A, and the levels of canonical histone H3 were not affected (Fig. 3C).
Fig. 3.
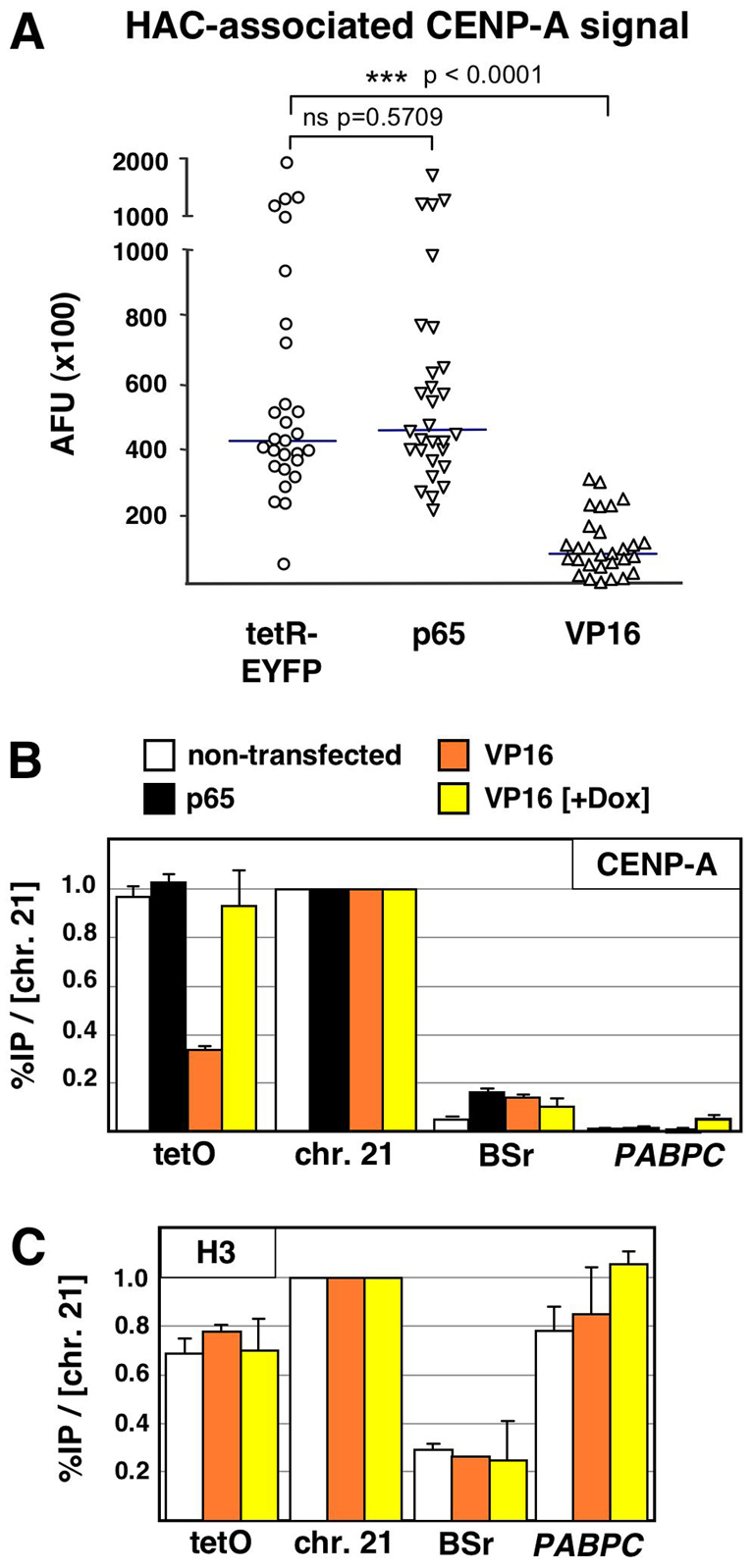
Tethering of VP16, but not p65, causes a specific loss of CENP-A from the alphoidtetO HAC centromere. (A) 1C7 cells were transfected with the indicated tetR fusion constructs and fixed after 2 days. CENP-A immunofluorescence staining associated with the HAC was quantified. Solid lines indicate the median. (B,C) ChIP analysis as in Fig. 1C, using antibodies against CENP-A (B) or canonical histone H3 (C). ns, no significant difference.
Consistent with the near-complete loss of detectable CENP-A at the HAC centromere in cells expressing tetR–EYFP–VP16 for 2 days, the majority of HACs in these cells suffered from mitotic defects. These included misalignment in metaphase and segregation defects of HAC sister chromatids in anaphase (Fig. 4C). By contrast, tethering of p65 did not affect HAC mitotic behaviour – the HACs in all cells that were examined displayed normal mitotic phenotypes (Fig. 4B). In the control experiments, HACs targeted by tetR–EYFP displayed a normal mitotic kinetochore function, aligning on the metaphase plate under tension, and with sister chromatids segregating normally in subsequent anaphase (Fig. 4A).
Fig. 4.

Tethering of p65 is compatible with HAC kinetochore function. (A–C) Cells expressing the indicated tetR fusion proteins were fixed and stained 2 days after transfection. HAC sister chromatids (arrowheads) were analyzed in metaphase (panel 1) and late anaphase or telophase (panel 2). The percentages of HACs with mitotic defects, determined as unaligned HACs in metaphase cells (e.g. C1) or mis-segregated HAC sister chromatids (e.g. C2) are indicated (n=53, 44 and 42 for tetR–EYFP, tetR–EYFP–p65 and tetR–EYFP–VP16, respectively). Scale bar: 5 μm.
This functional analysis was supported by staining for CENP-C – a crucial kinetochore protein, which links the inner and outer kinetochore components (Saitoh et al., 1992; Tomkiel et al., 1994; Gascoigne et al., 2011; Przewloka et al., 2011; Screpanti et al., 2011). CENP-C was severely depleted from the HAC kinetochore following the tethering of tetR–EYFP–VP16, but could be detected at HAC kinetochores in the presence of tetR–EYFP–p65 and the control, tetR–EYFP (Fig. 5).
Fig. 5.

Tethered VP16, but not p65, removes CENP-C from the HAC kinetochore. (A–C) Cells expressing the indicated tetR fusion proteins were fixed and stained 2 days after transfection. tetR–EYFP fusion protein (green, panel 1), CENP-C (red, panel 2) and CENP-A (blue, panel 3). Merged images (panel 4) represent the overlay of EYFP signal, antibody and DAPI staining (light grey). Arrows depict the HAC. Scale bars: 5 μm.
Combined, these data demonstrate that tethering tetR–EYFP–p65 in the kinetochore does not interfere with the integrity of centromeric CENP-A chromatin, kinetochore structure or function of the HAC, despite the formation of an open chromatin environment and a substantial upregulation of local transcriptional activity. By contrast, despite producing an apparently similar open chromatin environment, tethering of tetR–EYFP–VP16 profoundly disrupted the maintenance of CENP-A levels and kinetochore function. This difference is likely to arise as a result of the much more robust induction of transcription by tetR–EYFP–VP16.
VP16, but not p65, abrogates centromeric loading of CENP-A
The rapid decrease in CENP-A levels observed after targeting of tetR–EYFP–VP16 into the kinetochore could be due either to an active disassembly of the kinetochore chromatin or to the inhibition of CENP-A incorporation, as previously observed following the removal of H3K4me2 from the kinetochore (Bergmann et al., 2011). To look specifically at incorporation of CENP-A synthesized de novo, we conducted a quench–pulse-chase experiment using SNAP-tagged CENP-A (Jansen et al., 2007). Cells were co-transfected with constructs expressing SNAP-tagged CENP-A along with either tetR–EYFP as a control, tetR–EYFP–VP16 or tetR–EYFP–p65, and subsequently arrested in S phase with thymidine (Fig. 6A). We then irreversibly quenched existing CENP-A–SNAP molecules by using the nonfluorescent BG substrate. Following the release of cells from the thymidine block, we assessed the incorporation of newly synthesized CENP-A–SNAP at centromeres in G1 by labelling with the fluorescent tetramethylrhodamine (TMR)-Star substrate. A prominent TMR-Star signal was generally associated with the HAC in cells expressing tetR–EYFP (Fig. 6B,E). By contrast, tethering of VP16 virtually completely abolished the accumulation of newly synthesized CENP-A at the HAC (Fig. 6D,E). Therefore, the rapid loss of centromeric CENP-A at the HAC is at least in part due to a profound defect in CENP-A loading.
Fig. 6.

VP16, but not p65 tethering, negatively affects CENP-A loading at the HAC centromere. (A) Schematic diagram of quench–pulse-chase experiments in cells co-transfected with SNAP-tagged CENP-A and either of the tetR fusion constructs to determine the centromeric loading of newly synthesized CENP-A. (B–D) Cells expressing the indicated tetR fusion construct (green, panel 1) and pulse-labelled with TMR-Star (red, panel 2). Merged images (panel 3) represent the overlay of EYFP signal with TMR and DAPI staining (blue). Arrowheads depict the HAC. Scale bar: 5 μm. (E) Quantification of HAC-associated TMR-Star signal in cells as in B–D. AFU values are plotted relative to the average TMR-Star signal measured at endogenous centromeres. Solid lines indicate the median. ns, no significant difference.
Consistent with the maintenance of interphase CENP-A staining at the HAC, p65 had no negative impact on CENP-A loading when compared with tetR–EYFP (Fig. 6C,E). Indeed, tethering of tetR–EYFP–p65 appeared to augment the amount of newly deposited CENP-A mildly relative to that observed with tethered tetR–EYFP, although this effect was statistically not significant (Fig. 6E).
The rapid loss of CENP-A following the tethering of VP16 strongly suggested that tethered tetR–EYFP–VP16 could actively evict CENP-A from centromeric nucleosomes. Cells virtually lacking any detectable CENP-A signal at the HAC could be observed within two cell cycles of transfection (see Fig. 2C). A loading defect, such as that identified in Fig. 6 would only produce a passive loss of CENP-A, with levels of CENP-A decreasing by a factor of two at each mitosis. Thus, at 2 days post-transfection, these cells would be expected to retain 25% of their initial CENP-A chromatin. We tested this hypothesis for both exogenous and endogenous CENP-A, using two different protocols.
The use of a SNAP-tag pulse–chase protocol clearly showed that tethered tetR–EYFP–VP16 causes a rapid loss of incorporated CENP-A from the HAC kinetochore (Fig. 7A,B). HeLa 1C7 cells stably expressing CENP-A–SNAP were given a pulse of TMR-Star to label all kinetochores. Two hours after washing out the label, the cells were transfected with constructs expressing tetR–EYFP, tetR–EYFP–p65 or tetR–EYFP–VP16. Following a 24-hour chase, the relative labelling at the HAC kinetochore was compared with that of ten endogenous kinetochores that were selected at random. Indeed, tethering of tetR–VP16 caused a dramatic and highly significant (P<0.0007) drop in CENP-A levels at the HAC kinetochore (Fig. 7B). By contrast, tethering of tetR–p65 to the HAC centromere caused only a slight and insignificant (P=0.2) loss of CENP-A in this experiment.
Fig. 7.

VP16 causes an increased rate of loss of CENP-A from the HAC centromere. (A) Schematic diagram of pulse-chase experiments in cells stably expressing SNAP-tagged CENP-A and transfected with the various tetR fusion constructs. (B) Quantification of HAC-associated TMR-Star signal in cells plotted relative to the average TMR-Star signal measured at ten endogenous centromeres. Solid lines indicate the median. (C–E) Cells expressing the indicated tetR fusion construct (green, panel 1) and pulse-labelled with TMR-Star (red, panel 2). Merged images (panel 3) represent the overlay of EYFP signal with TMR Star and DAPI staining (blue). Arrowheads depict the HAC. Scale bar: 5 μm.
In a complementary approach to look at endogenous rather than transgenic CENP-A, we assessed CENP-A levels before the first mitosis following transfection with tetR fusion proteins (supplementary material Fig. S2A). This protocol should examine loss rather than assembly defects, as the progression through mitotic exit is required to trigger loading of CENP-A (Jansen et al., 2007). We assessed CENP-A staining 19 hours after expressing either tetR–EYFP or tetR–EYFP–VP16 in the presence of nocodazole. Late G2 cells were selected for analysis, using the large nuclei and frequent CENP-A doublets of endogenous centromeres as criteria (supplementary material Fig. S2A). Quantification revealed a substantial decrease in CENP-A staining at VP16-targeted HACs compared with those targeted by tetR–EYFP, which is consistent with an active loss of CENP-A (supplementary material Fig. S2B). We independently confirmed this finding by arresting cells with colcemid 16 hours after transfection. These cells would also be in their first cell cycle after transfection, and by definition, would not yet have gone through mitotic exit (the time during which CENP-A loading defects would be observed). Robust CENP-A signals were typically associated with both kinetochores of mitotic HACs targeted by tetR–EYFP (supplementary material Fig. S2C). By contrast, HACs in cells expressing the VP16 construct frequently displayed reduced CENP-A staining (supplementary material Fig. S2C).
Combined, these findings demonstrate that acetylation of kinetochore chromatin and a mild stimulation of transcription by tethered tetR–EYFP–p65 does not interfere with either the loading or the maintenance of centromeric CENP-A. However, tethering of tetR–EYFP–VP16, which produces a similar chromatin environment but is accompanied by much higher levels of transcription, is incompatible with both the loading of newly synthesized CENP-A molecules and the retention of established CENP-A.
Discussion
Centromere identity is maintained epigenetically through the local deposition of CENP-A and at least in part through the associated chromatin context (Allshire and Karpen, 2008; Black and Bassett, 2008; Nakano et al., 2003; Nakashima et al., 2005; Okada et al., 2007). Classically, centromeres were assumed to be composed of heterochromatin. However, the groundbreaking study of Sullivan and Karpen revealed that the kinetochore-specific histone CENP-A resides in a specialized chromatin environment, which has been termed ‘centrochromatin’ (Sullivan and Karpen, 2004). A more recent study has shown that centrochromatin has a histone modification pattern resembling that found in the downstream body of transcribed genes (Bergmann et al., 2011). Indeed, previous studies have detected transcription within rice centromeres (Nagaki et al., 2004), human neocentromeres (Saffery et al., 2003) and also within the alphoid array of a derivative of the synthetic HAC used in the present study (Iida et al., 2010). The chromatin profile at active kinetochores resembles the ‘yellow’ chromatin described in a recent paper (Filion et al., 2010). This chromatin type is characteristic of genes with broad expression patterns (‘housekeeping’ genes). We note, however, that the level of transcripts observed here in human alpha-satellite DNA was 200–300 times lower than that observed for RNA polymerase-II-transcribed housekeeping genes in euchromatin.
Our experimental system enables us to engineer changes directly in the chromatin of a stable artificial chromosome (the alphoidtetO HAC), whose centromeric DNA contains an array of alpha-satellite repeats containing tetracycline operators (Nakano et al., 2008). Using this system, we previously found that the specific removal of the histone mark H3K4me2 from kinetochore chromatin resulted in a concomitant decrease in the level of transcription of this chromatin in human cells (Bergmann et al., 2011). This was accompanied by the impaired loading of new kinetochore histone CENP-A during mitotic exit. The net result was a gradual loss of kinetochore identity. Those results were consistent with the results from two previous studies in which we had shown that the nucleation of heterochromatin within the kinetochore causes a dramatic loss of kinetochore proteins, with subsequent inactivation of the kinetochore (Cardinale et al., 2009; Nakano et al., 2008).
One important question raised by the previous studies was whether it is the chromatin environment, the transcription or both that are important for CENP-A loading and kinetochore maintenance. The results of the present study are consistent with the level of transcription being a crucial factor in kinetochore structure and function.
We were surprised to find that kinetochores are tolerant of seeding of domains that are highly enriched in H3K9ac overlapping the CENP-A domain. Previous characterisation of centrochromatin had shown that levels of H3K9ac are low in this region (Bergmann et al., 2011; Sullivan and Karpen, 2004), and, at least in chicken kinetochores, we had been able to detect H3K9me3 interspersed within the overall domain of CENP-A binding by using super-resolution microscopy (Ribeiro et al., 2010). Following the binding of p65, which contains the 30 amino acid transcription activation domain of NF-κB subunit p65, kinetochore-associated alpha-satellite DNA underwent an ~10-fold increase in transcription, although importantly the transcription levels remained far below those observed at a control euchromatic locus. Indeed, tethering of tetR–EYFP–p65, despite causing a substantial increase in kinetochore-associated H3K9ac, appeared to have no negative functional consequences for kinetochore structure, function or propagation.
Kinetochores were not nearly so tolerant of targeting by the potent transcriptional activator VP16. VP16 produced changes in the chromatin environment resembling those seen with p65 – H3K9ac was induced to a similar level – however, transcript levels increased ~150 fold. TetR-EYFP-VP16 targeting virtually completely abrogates the loading of newly synthesized CENP-A at the alphoidtetO centromere. Furthermore, VP16 mediates active and selective displacement of pre-established CENP-A. The impact of VP16 on the maintenance of CENP-A chromatin is thus even more pronounced than that of the heterochromatin-seeding transcriptional repressors tTS or KAP1 (Cardinale et al., 2009; Nakano et al., 2008).
Together with our recent findings (Bergmann et al., 2011) as well as our previous work (Nakashima et al., 2005; Okada et al., 2007), this reinforces the notion that a careful balance of transcriptionally permissive chromatin is essential for the maintenance of functional kinetochores. It is unlikely that VP16 disrupts the kinetochore by directly bombarding it with large numbers of transcribing polymerases, as is the case for conditional centromeres in budding yeast (Hill and Bloom, 1987). The level of RNA polymerase II induced by tetR–EYFP–VP16 remained at only about half of that of a typical euchromatic gene. It is known that RNA polymerase can interact with factors transiently destabilizing nucleosomes to promote elongation through chromatin (Selth et al., 2010; Workman, 2006). Therefore, excessive centromeric polymerase activity might cause specific loss of CENP-A as a consequence of a limiting pool of available CENP-A or as a result of its association with histone chaperones, such as HJURP (Dunleavy et al., 2009; Foltz et al., 2009).
Our study reveals a remarkable degree of plasticity in the chromatin environment of active kinetochores in vivo in human cells. Given the historic assumptions about the links between centromeres and heterochromatin, it might seem surprising that kinetochore chromatin appears to be more tolerant of an ‘open’ state, defined by high levels of acetylation and partial decondensation, than a ‘closed’ and/or repressive state. However, it has been known for a number of years that the Mis18 complex, which is associated with CENP-A loading, is associated with acetyltransferase activity (Fujita et al., 2007). Furthermore, recent work from our laboratories has revealed that H3K9 appears to function as an acetyl/methyl switch, regulating CENP-A loading by HJURP (Ohzeki et al., unpublished). What appears to be most crucial for kinetochore maintenance is the level of transcripts being produced from the underlying alpha-satellite DNA. Important questions for future studies include whether the transcripts act as structural elements at the kinetochore or whether it is the process of transcription that is important.
Materials and Methods
Generation of tetR fusion constructs
The coding sequence of the VP16 acidic activation domain was amplified by PCR from pTet-On (Clontech) using VP16-RE_F (5′-AATAGGATCCTCCGCGTACAGCCG-3′) and VP16-RE_R (5′-ATAAGGATCCCTACCCACCGTACTCG-3′). The coding sequence of the p65 C-terminal acidic activation domain was amplified from NYE108 (Carpenter et al., 2005) by using p65-RE_F (5′-TCCGGAGCGTCGACCCCGGGG-3′) and p65-RE_R (5′-TCCGGATTAGGAGCTGATCTGACTCAGC-3′). The PCR products were subcloned into pGEM-T Easy vectors (Promega). The 0.4 kb BamHI containing the VP16 coding sequence was ligated into the BamHI site of tYIP (Cardinale et al., 2009) to generate tYIP–VP16, expressing tetR–EYFP–VP16 from a cytomegalovirus (CMV) promoter and conferring resistance to puromycin through an IRES motif. Analogously, the 0.1 kb BspEI fragment containing the p65 coding sequence was ligated into the BspEI site of tYIP to generate tYIP–p65. Final constructs were verified by DNA sequencing.
Cell culture
HT1080-derived AB2.2.18.21 cells carrying the alphoidtetO HAC and 1C7 cells (a fusion of AB2.2.18.21 with HeLa cells) (Cardinale et al., 2009) were maintained in RPMI Medium 1640 (with l-Glutamine) (Invitrogen), supplemented with 10% FBS (Invitrogen), 100 U/ml penicillin G and 100 μg/ml streptomycin sulfate (Invitrogen). Blasticidin S (Invitrogen) was added to a final concentration of 2–4 μg/ml. Cells were grown at 37°C in 5% CO2 in a humidified atmosphere.
Transfections were routinely performed in six-well plates using Fugene 6 (Roche), according to the manufacturer's instructions. Transfection complexes containing 3 μl Fugene 6 reagent and 1 μg plasmid DNA were prepared in 100 μl OptiMEM (Invitrogen). Transfections for ChIP were performed in 10-cm dishes using five volumes of the transfection complex. Where desired, transfected cells were selected for by the addition of 2–5 μg/ml puromycin (Sigma).
Immunostaining, cytological analysis and fluorescence signal quantification
Indirect immunofluorescence staining of cells fixed in 4% paraformaldehyde (PFA) in PBS was performed using standard protocols. The following antibodies were used: mouse anti-CENP-A (AN1); rabbit anti-CENP-A (Valdivia et al., 1998); rabbit anti-CENP-C (R554) and rabbit anti-H3K9ac (R607). Fluorophore-conjugated secondary antibodies were purchased from Jackson Labs.
Microscopic images were typically acquired on a DeltaVision Core system (Applied Precision) using an inverted Olympus IX-71 stand, with an Olympus UPlanSApo 100× oil-immersion objective [numerical aperture (NA) 1.4] and a 250W Xenon light source. Camera (Photometrix Cool Snap HQ), shutter and stage were controlled through SoftWorx (Applied Precision). Z-series were collected with a spacing of 0.2 μm. Image stacks were subsequently deconvolved in SoftWorx. For display purposes, the maximum-intensity projections are shown.
For quantification of CENP-A staining in single-stained cells, a custom-written macro was used in ImagePro software (Media Cybernetics), details of which are available upon request. In brief, HAC-associated EYFP and TexasRed signals were determined for each HAC-containing z section within a circular region of interest that was nine pixels in diameter. For each section, the average nuclear background for each channel was determined within three regions of interest of the same size and was subtracted from the specific signal.
SNAP-tag quench–pulse-chase experiments
Cells were co-transfected as described above, with 0.8 μg of the relevant tetR fusion construct and 0.2 μg of the pCenA–SNAP–IP construct (Bergmann et al, 2011). 16 hours after transfection, thymidine was added to a final concentration of 2 mM for 10 hours. Existing CENP-A–SNAP was blocked for 30 minutes using BG-block (New England Biolabs) according to the manufacturer's instructions. BG-block and thymidine were washed out and cells were incubated for a further 18 hours in the presence of deoxycytidine. Newly synthesized CENP-A–SNAP was fluorescently labelled using the TMR-Star substrate (New England Biolabs) for 20 minutes according to the manufacturer's instructions. Following the wash-out of unbound substrate, cells were fixed after 1 hour in 4% PFA and analyzed by fluorescence microscopy.
Quantification of HAC-associated TMR signal was performed on maximum-intensity projections in SoftWorx within a circular region of interest with a nine-pixel diameter. The average TMR signal at endogenous centromeres was determined using a custom-written macro in ImagePro. For HAC and endogenous centromeres, the average nuclear background signals determined within an equal-sized region of interest were subtracted.
Chromatin immunoprecipitation analysis
ChIP from cells crosslinked in 1% formaldehyde and subsequent real-time PCR analysis were performed as described in detail previously (Bergmann et al., 2011). Monoclonal antibodies for ChIP were described previously (Kimura et al., 2008), with the exception of mouse antibody against RNA Polymerase II Ser2p (PC26B5) and mouse antibody against histone H3 (Abcam, ab10799).
Oligonucleotide primer pairs for tetO alphoid DNA and the BSr gene in the HAC, as well as for alphoid DNA of human chromosome 21, have been described previously (Cardinale et al., 2009), with the exception of the following primer pairs: PABPC-10_F (5′-CTAGCATCCGTGGGCCAAGAG-3′) and PABPC-10_R (5′-CTCTTCCCCAACCCCAGCAAAAT-3′) for the +10 kb locus of the PABPC1 gene (Blobel et al., 2009); PP1A-3_F (5′-AGTGTGGGGAAGAAAGGAGCCG-3′) and PP1A-3_R (5′-CTAAAAGAGACAGTGTTGGTCAGGC-3′) for the +3kb locus of the PP1A gene.
Real-time RT-PCR
RNA extraction with TRIzol (Invitrogen), RT-PCR using random hexamer primers, real-time PCR analysis and the oligonucleotide primers were described in detail previously (Bergmann et al, 2011). On each plate and for each oligonucleotide primer used, a standard curve was prepared from a serial dilution of corresponding genomic DNA.
Supplementary Material
Acknowledgements
We thank Andrew Belmont for providing the construct carrying the p65 cDNA.
Footnotes
Funding
J.H.B. was funded by a PhD studentship from The Wellcome Trust [grant number 080458]. This work was supported by the intramural research program of the National Institutes of Health, the National Cancer Institute, the Center for Cancer Research to V.L.; a grant-in-aid from the Ministry of Education, Sports, Culture, Sports, Science and Technology (MEXT) [grant number 23247030 to H.M.]; and the Genome Network project and grants-in-aid from the MEXT of Japan [grant number 22370063 to H.K.]. Work in the W.C.E. lab is funded by The Wellcome Trust, of which he is a Principal Research Fellow [grant number 073915]. The Wellcome Trust Centre for Cell Biology is supported by grant number 092076. Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.090639/-/DC1
References
- Allshire R. C., Karpen G. H. (2008). Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat. Rev. Genet. 9, 923-937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A., Fritz B., Hasson D., Abrusan G., Cheung F., Yoda K., Radlwimmer B., Ladurner A. G., Warburton P. E. (2007). Co-localization of CENP-C and CENP-H to discontinuous domains of CENP-A chromatin at human neocentromeres. Genome Biol. 8, R148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann J. H., Rodriguez M. G., Martins N. M., Kimura H., Kelly D. A., Masumoto H., Larionov V., Jansen L. E., Earnshaw W. C. (2011). Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J. 30, 328-340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black B. E., Bassett E. A. (2008). The histone variant CENP-A and centromere specification. Curr. Opin. Cell Biol. 20, 91-100 [DOI] [PubMed] [Google Scholar]
- Black B. E., Foltz D. R., Chakravarthy S., Luger K., Woods V. L., Jr, Cleveland D. W. (2004). Structural determinants for generating centromeric chromatin. Nature 430, 578-882 [DOI] [PubMed] [Google Scholar]
- Black B. E., Jansen L. E., Maddox P. S., Foltz D. R., Desai A. B., Shah J. V., Cleveland D. W. (2007). Centromere identity maintained by nucleosomes assembled with histone H3 containing the CENP-A targeting domain. Mol. Cell 25, 309-322 [DOI] [PubMed] [Google Scholar]
- Blobel G. A., Kadauke S., Wang E., Lau A. W., Zuber J., Chou M. M., Vakoc C. R. (2009). A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol. Cell 36, 970-983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale S., Bergmann J. H., Kelly D., Nakano M., Valdivia M. M., Kimura H., Masumoto H., Larionov V., Earnshaw W. C. (2009). Hierarchical inactivation of a synthetic human kinetochore by a chromatin modifier. Mol. Biol. Cell 20, 4194-4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter A. E., Memedula S., Plutz M. J., Belmont A. S. (2005). Common effects of acidic activators on large-scale chromatin structure and transcription. Mol. Cell. Biol. 25, 958-968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C. W., Straight A. F. (2006). Centromere formation: from epigenetics to self-assembly. Trends Cell Biol. 16, 70-78 [DOI] [PubMed] [Google Scholar]
- Cheeseman I. M., Desai A. (2008). Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 9, 33-46 [DOI] [PubMed] [Google Scholar]
- Choo K. H., Vissel B., Nagy A., Earle E., Kalitsis P. (1991). A survey of the genomic distribution of alpha satellite DNA on all human chromosomes, and derivation of a new consensus sequence. Nucleic Acids Res. 19, 1179-1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depinet T. W., Zackowski J. L., Earnshaw W. C., Kaffe S., Sekhon G. S., Stallard R., Sullivan B. A., Vance G. H., Van Dyke D. L., Willard H. F., et al. (1997). Characterization of neo-centromeres in marker chromosomes lacking detectable alpha-satellite DNA. Hum. Mol. Genet. 6, 1195-1204 [DOI] [PubMed] [Google Scholar]
- du Sart D., Cancilla M. R., Earle E., Mao J. I., Saffery R., Tainton K. M., Kalitsis P., Martyn J., Barry A. E., Choo K. H. (1997). A functional neo-centromere formed through activation of a latent human centromere and consisting of non-alpha-satellite DNA. Nat. Genet. 16, 144-153 [DOI] [PubMed] [Google Scholar]
- Dunleavy E. M., Roche D., Tagami H., Lacoste N., Ray-Gallet D., Nakamura Y., Daigo Y., Nakatani Y., Almouzni-Pettinotti G. (2009). HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137, 485-497 [DOI] [PubMed] [Google Scholar]
- Earnshaw W. C., Migeon B. (1985). Three related centromere proteins are absent from the inactive centromere of a stable isodicentric chromosome. Chromosoma 92, 290-296 [DOI] [PubMed] [Google Scholar]
- Earnshaw W. C., Rothfield N. (1985). Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma 91, 313-321 [DOI] [PubMed] [Google Scholar]
- Earnshaw W. C., Ratrie H., Stetten G. (1989). Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma 98, 1-12 [DOI] [PubMed] [Google Scholar]
- Filion G. J., van Bemmel J. G., Braunschweig U., Talhout W., Kind J., Ward L. D., Brugman W., de Castro I. J., Kerkhoven R. M., Bussemaker H. J., et al. (2010). Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 143, 212-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz D. R., Jansen L. E., Bailey A. O., Yates J. R., 3rd, Bassett E. A., Wood S., Black B. E., Cleveland D. W. (2009). Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 137, 472-484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y., Hayashi T., Kiyomitsu T., Toyoda Y., Kokubu A., Obuse C., Yanagida M. (2007). Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev. Cell 12, 17-30 [DOI] [PubMed] [Google Scholar]
- Gascoigne K. E., Takeuchi K., Suzuki A., Hori T., Fukagawa T., Cheeseman I. M. (2011). Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell 145, 410-422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A., Bloom K. (1987). Genetic manipulation of centromere function. Mol. Cell. Biol. 7, 2397-2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida Y., Kim J. H., Kazuki Y., Hoshiya H., Takiguchi M., Hayashi M., Erliandri I., Lee H. S., Samoshkin A., Masumoto H., et al. (2010). Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res. 17, 293-301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen L. E., Black B. E., Foltz D. R., Cleveland D. W. (2007). Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol. 176, 795-805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H., Hayashi-Takanaka Y., Goto Y., Takizawa N., Nozaki N. (2008). The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct. Funct. 33, 61-73 [DOI] [PubMed] [Google Scholar]
- Lam A. L., Boivin C. D., Bonney C. F., Rudd M. K., Sullivan B. A. (2006). Human centromeric chromatin is a dynamic chromosomal domain that can spread over noncentromeric DNA. Proc. Natl. Acad. Sci. USA 103, 4186-4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiato H., Deluca J., Salmon E. D., Earnshaw W. C. (2004). The dynamic kinetochore-microtubule interface. J. Cell Sci. 117, 5461-5477 [DOI] [PubMed] [Google Scholar]
- Merry D. W., Pathak S., Hsu T. C., Brinkley B. R. (1985). Anti-kinetochore antibodies: use as probes for inactive centromeres. Am. J. Hum. Genet. 37, 425-430 [PMC free article] [PubMed] [Google Scholar]
- Mitchell A. R., Gosden J. R., Miller D. A. (1985). A cloned sequence, p82H, of the alphoid repeated DNA family found at the centromeres of all human chromosomes. Chromosoma 92, 369-377 [DOI] [PubMed] [Google Scholar]
- Nagaki K., Cheng Z., Ouyang S., Talbert P. B., Kim M., Jones K. M., Henikoff S., Buell C. R., Jiang J. (2004). Sequencing of a rice centromere uncovers active genes. Nat. Genet. 36, 138-145 [DOI] [PubMed] [Google Scholar]
- Nakano M., Okamoto Y., Ohzeki J., Masumoto H. (2003). Epigenetic assembly of centromeric chromatin at ectopic alpha-satellite sites on human chromosomes. J. Cell Sci. 116, 4021-4034 [DOI] [PubMed] [Google Scholar]
- Nakano M., Cardinale S., Noskov V. N., Gassmann R., Vagnarelli P., Kandels-Lewis S., Larionov V., Earnshaw W. C., Masumoto H. (2008). Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev. Cell 14, 507-522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima H., Nakano M., Ohnishi R., Hiraoka Y., Kaneda Y., Sugino A., Masumoto H. (2005). Assembly of additional heterochromatin distinct from centromere-kinetochore chromatin is required for de novo formation of human artificial chromosome. J. Cell Sci. 118, 5885-5898 [DOI] [PubMed] [Google Scholar]
- Okada T., Ohzeki J., Nakano M., Yoda K., Brinkley W. R., Larionov V., Masumoto H. (2007). CENP-B controls centromere formation depending on the chromatin context. Cell 131, 1287-1300 [DOI] [PubMed] [Google Scholar]
- Okamoto Y., Nakano M., Ohzeki J., Larionov V., Masumoto H. (2007). A minimal CENP-A core is required for nucleation and maintenance of a functional human centromere. EMBO J. 26, 1279-1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przewloka M. R., Venkei Z., Bolanos-Garcia V. M., Debski J., Dadlez M., Glover D. M. (2011). CENP-C is a structural platform for kinetochore assembly. Curr. Biol. 21, 399-405 [DOI] [PubMed] [Google Scholar]
- Ribeiro S. A., Gatlin J. C., Dong Y., Joglekar A., Cameron L., Hudson D. F., Farr C. J., McEwen B. F., Salmon E. D., Earnshaw W. C., et al. (2009). Condensin regulates the stiffness of vertebrate centromeres. Mol. Biol. Cell 20, 2371-2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro S. A., Vagnarelli P., Dong Y., Hori T., McEwen B. F., Fukagawa T., Flors C., Earnshaw W. C. (2010). A super-resolution map of the vertebrate kinetochore. Proc. Natl. Acad. Sci. USA 107, 10484-10489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffery R., Irvine D. V., Griffiths B., Kalitsis P., Wordeman L., Choo K. H. (2000). Human centromeres and neocentromeres show identical distribution patterns of >20 functionally important kinetochore-associated proteins. Hum. Mol. Genet. 9, 175-185 [DOI] [PubMed] [Google Scholar]
- Saffery R., Sumer H., Hassan S., Wong L. H., Craig J. M., Todokoro K., Anderson M., Stafford A., Choo K. H. (2003). Transcription within a functional human centromere. Mol. Cell 12, 509-516 [DOI] [PubMed] [Google Scholar]
- Saitoh H., Tomkiel J. E., Cooke C. A., Ratrie H. R., Maurer M., Rothfield N. F., Earnshaw W. C. (1992). CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell 70, 115-125 [DOI] [PubMed] [Google Scholar]
- Schueler M. G., Sullivan B. A. (2006). Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genomics Hum. Genet. 7, 301-313 [DOI] [PubMed] [Google Scholar]
- Screpanti E., De Antoni A., Alushin G. M., Petrovic A., Melis T., Nogales E., Musacchio A. (2011). Direct binding of Cenp-C to the Mis12 complex joins the inner and outer kinetochore. Curr. Biol. 21, 391-398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selth L. A., Sigurdsson S., Svejstrup J. Q. (2010). Transcript Elongation by RNA Polymerase II. Annu. Rev. Biochem. 79, 271-293 [DOI] [PubMed] [Google Scholar]
- Sugata N., Li S., Earnshaw W. C., Yen T. J., Yoda K., Masumoto H., Munekata E., Warburton P. E., Todokoro K. (2000). Human CENP-H multimers colocalize with CENP-A and CENP-C at active centromere–kinetochore complexes. Hum. Mol. Genet. 9, 2919-2926 [DOI] [PubMed] [Google Scholar]
- Sullivan B. A., Schwartz S. (1995). Identification of centromeric antigens in dicentric Robertsonian translocations: CENP-C and CENP-E are necessary components of functional centromeres. Hum. Mol. Genet. 4, 2189-2197 [DOI] [PubMed] [Google Scholar]
- Sullivan B. A., Karpen G. H. (2004). Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 11, 1076-1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkiel J. E., Cooke C. A., Saitoh H., Bernat R. L., Earnshaw W. C. (1994). CENP-C is required for maintaining proper kinetochore size and for a timely transition to anaphase. J. Cell Biol. 125, 531-545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafa O., Sullivan K. F. (1997). Chromatin containing CENP-A and a-satellite DNA is a major component of the inner kinetochore plate. Curr. Biol. 7, 897-900 [DOI] [PubMed] [Google Scholar]
- Valdivia M. M., Figueroa J., Iglesias C., Ortiz M. (1998). A novel centromere monospecific serum to a human autoepitope on the histone H3-like protein CENP-A. FEBS Lett. 422, 5-9 [DOI] [PubMed] [Google Scholar]
- Warburton P. E., Cooke C., Bourassa S., Vafa O., Sullivan B. A., Stetten G., Gimelli G., Warburton D., Tyler-Smith C., Sullivan K. F., et al. (1997). Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Curr. Biol. 7, 901-904 [DOI] [PubMed] [Google Scholar]
- Willard H. F., Waye J. S. (1987). Hierarchical order in chromosome-specific human alpha satellite DNA. Trends Genet. 3, 192-198 [Google Scholar]
- Workman J. L. (2006). Nucleosome displacement in transcription. Genes Dev. 20, 2009-2017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}