

Abstract
Humulus lupulus is commonly known as hops, a member of the family moraceae. Currently many projects are underway leading to the accumulation of voluminous genomic and expressed sequence tag sequences in public databases. The genetically characterized domains in these databases are limited due to non-availability of reliable molecular markers. The large data of EST sequences are available in hops. The simple sequence repeat markers extracted from EST data are used as molecular markers for genetic characterization, in the present study. 25,495 EST sequences were examined and assembled to get full-length sequences. Maximum frequency distribution was shown by mononucleotide SSR motifs i.e. 60.44% in contig and 62.16% in singleton where as minimum frequency are observed for hexanucleotide SSR in contig (0.09%) and pentanucleotide SSR in singletons (0.12%). Maximum trinucleotide motifs code for Glutamic acid (GAA) while AT/TA were the most frequent repeat of dinucleotide SSRs. Flanking primer pairs were designed in-silico for the SSR containing sequences. Functional categorization of SSRs containing sequences was done through gene ontology terms like biological process, cellular component and molecular function.
Keywords: Humulus lupulus, expressed sequence tag, molecular markers, simple sequence repeats
Background
Hop (Humulus lupulus) is a medicinal plant, but its major profitable use is in flavoring of beer. This plant is dioecious (2n = 2x = 20) with two heteromorphic sex chromosomes, X and Y [1, 2, 3]. The reproductive mode affects many aspects of breeding and crop management such as male and female reproductive organs are dimorphic [4], families are highly heterozygous, phenotypically variable and breeding while cultivar developments are accomplished by single mating followed by selection and fixation of favorable genotypes by various means of asexual reproduction. SSR discovery in hops has relied on the hybridization-based screening of genomic libraries by means of artificial repetitive sequences and sequencing of isolated clones in order to build up locus-specific primers previously [5]. However, high-throughput sequencing results engender information on thousands of expressed sequence tags (ESTs) [6].
Microsatellite or Simple Sequence Repeats (SSR) or Short tandem repeats (STR) are 1-6 bp tandemly repeated motifs present in both coding and non-coding regions of prokaryotic and eukaryotic genome. The prominent frequencies of length polymorphism associated with microsatellites provide the basis for development of a marker system that has extensive application in genetic research including studies of genetic variation, linkage mapping, gene tagging and evolution [7]. SSRs are used extensively as molecular markers because of their multiallelic nature, co-dominant inheritance and relative abundance. The foremost annoyance of SSRs as markers has been their time consuming development in laboratory. However, with fast-paced boost of nucleic acid in recent years, it became realistic to screen for microsatellites in database for numerous plant species. Variations in SSR regions originate mostly from errors during the replication process, frequently DNA polymerase slippage. These errors generate base pair insertions or deletions respectively. [8, 9]. We have mined SSRs from EST of Humulus lupulus to get the SSR polymorphism. ESTs are short and single pass sequences read from mRNA (cDNA) [10]. This represents a snapshot of genes expressed in a given tissue. The use of EST or cDNA-based SSRs has been reported for several species including grape [11], sugarcane [12], durum wheat [13], rye [14], medicinal plant like basil [15] and Periwinkle [16]. There are various SSR identification softwares such as MISA, SSR Finder, SSRIT, TRF, TROLL and sputnik. We used MISA [17] to identify SSR. Different types of SSRs and their percentage distributions were examined. The forward and reverse primer pairs were designed from the flanking ends of SSRs. The functional annotation of these SSR containing sequence was done.
Methodology
Sequence data source:
There are 25,495 ESTs of Humulus lupulus present in dbEST at NCBI. The retrieved sequences were isolated from different plant tissues like leaves, stem, root, etc. There is a chance of occurrence of redundancy in the EST sequences. In order to remove the redundancy, CAP3 assembler [18, 19] was used for sequence assembly. The resulting non-redundant sequences are contigs and singletons.
Microsatellite Identification:
SSR were detected using MIcroSAtellite identification tool (MISA) written in the Perl scripting language [17, 20]. This tool analyzes microsatellite repeats in FASTA formatted contig and singlet files. EST derived SSRs were considered to contain repeat motifs ranging in length from 1 to 6 bp. The minimum numbers of repeats were 10 for mononucleotides, 6 for dinucleotides and 5 for trinucleotides, tetranucleotides, pentanucleotides and hexanucleotides. The analysis of SSRs was done based on their types (mono to hexanucleotides), number of repeats, percentage frequency of occurrences of each SSR motif and their distribution in the sequence.
Gene Ontology Classification:
SSR-ESTs sequences with significant matches to protein entries of Swiss Prot-Uniprot KB database were functionally classified. Characterization of SSR-ESTs performed through gene ontology terms using Amigo [21, 22]. The ontology classification was done in terms of their biological process, molecular function and cellular component. This characterization has been based on analyzed SSR repeats.
Marker development:
Primer pairs for the SSR containing sequences were designed using BatchPrimer3 software for developing microsatellite markers [23]. The microsatellites containing contigs and singletons were used for designing primers pairs. Forward and reverse primer pairs were designed for marker development.
Discussion
ESTs are often represented by redundant cDNA sequences making them difficult to analyze effectively for SSRs. To eliminate the redundancy in sequences CAP3 program was used. The identification of overlapping sequences to generate contig and singleton sequences was done. 88.84% of ESTs forming contigs indicate that the majority of the ESTs have overlapping sequences while only 27.47% sequences were unique and have no corresponding overlapping sequences. The reduction of redundancy was found to be a sizeable proportion that has reduced 61.39% that means that number of ESTs prior to SSR analysis Table 1 (see supplementary material). The study of occurrence of different types of SSR repeats revealed that percentage distribution of mononucleotide SSRs is 60.44% in contigs and 62.16% in singletons followed by dinucleotide SSRs, 21.48% in contig and 19.9% in singleton (Figure 1a, Figure 2a).
Figure 1.
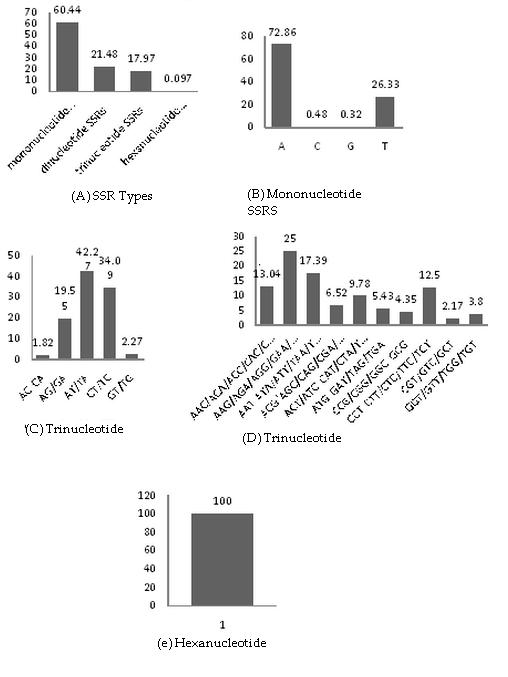
(A) Percentage distribution of different SSRs; (B) Percentage distribution of mononucleotide SSRs; (C) Percentage distribution of drinucleotide; (D) Percentage distribution of rinucleotide SSRs; (E) Percentage distribution of hexanucleotide SSRs
Figure 2.
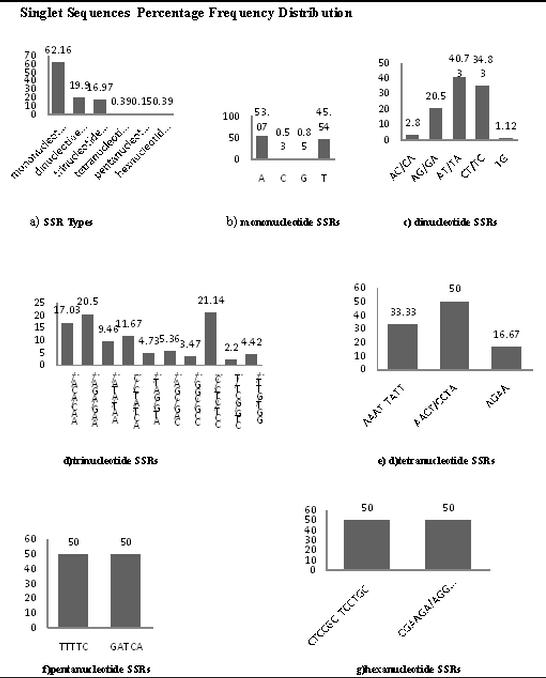
(a) Percentage distribution of different SSRs; (b) Percentage distribution of mononucleotide SSRs; (c) Percentage distribution of drinucleotide SSRs; (d) Percentage distribution of trinucleotide SSRs; (e) Percentage distribution of tetranucleotide SSRs; (f) Percentage distribution of pentanucleotide SSRs; (g) Percentage distribution of hexanucleotide SSRs.
Among mononucleotide repeats, polyA/polyT repeats were predominant while polyC/polyG repeats were rare (Figure 1b, Figure 2b). A-T repeat motifs are the most abundant type of SSRs in plants [22]. All dinucleotide repeat combinations excluding homomeric dinucleotides can be grouped into classes namely, (AG)n, (AT)n, (AC)n, (GT)n and (TC)n. It is evident that AT/TA dinucleotide repeats were more frequent followed by CT/TC and AG/GA (Figure 1c, Figure 2c).
Among 10 unique trinucleotide repeat classes, AAG/AGA/AGG/GAA/GAG/GGA (Contig-25% and singlet - 20.50) was the most frequent. The lowest frequency of trinucleotides was observed with TGC/TCG/GTC/CGT/CTG (contig-2.17% and singlet-2.2%) (Figure 1d, Figure 2d). No tetranucleotide, pentanucleotide microsatellite was observed in contig sequences however these can be seen in singleton sequences Frequency of AACT/CCTA (50%) was maximum followed by AAAT/TATT and least frequency depicted in AGAA (Figure 2e). Only two pentanucleotides repeats were found there contribution was 50% (Figure 2f).In hexanucleotide SSR motif CCGCCT depicted in contig and in singlet CTCCGC/TCCTGC and CGAAGA/AGGAGC both are seen with frequency of 50% (Figure 2g).
Codon Repetitions:
The trinucleotide SSRs are triplet codon that code for a particular amino acid. It was observed that out of all triplet codons of contig sequences, GAA (encoding Glutamic Acid) repetitions are predominant followed by AAG (encoding Lysine) and CAC (Histidine), while in singleton sequences CAC is predominated followed by GAA. The triplet codon forms an open reading frame (ORF) translated to proteins (Figure 3).
Figure 3.

Frequency of distribution of contig triplet codon (A) Repetition of contig sequence codon; (B) Repetition of singlet sequence codon.
Amino acid distribution:
The trinucleotide microsatellite codes for 21 types of amino acids, which includes stop codon. It was observed that out of all coded amino acid in contigs sequences Asparagine and Glutamic acid demonstrated the highest percentage of occurrence followed by Isoleucine and Leucine (Figure 4a). Where as in Singleton sequences Serine demonstrated the highest percentage followed by Histidine (Figure 4b).
Figure 4.
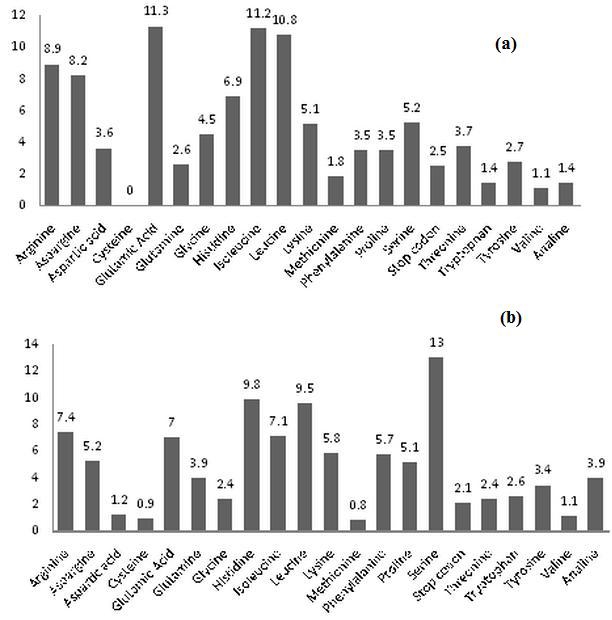
(a) Percentage distribution of amino acids of contig sequences; (b) percentage distribution of amino acids of singlet sequences.
In Humulus lupulus serine occurs in Serine/threonine-protein kinase and is involved in protein phosphorylation, positive regulation of DNA and Serine acetyltransferase is involved in cellular response to sulfate starvation. In contigs sequences valine showed the least occurrence and in singlets methionine and Cysteine were lowest in frequency.
The analysis of data revealed that the majority of amino acids were polar in nature, both in contig (63.57%) and singleton sequences (61.47%) and frequency of non polar contigs and singleton sequences are 37.38% ,36.87% respectively (Figure 5a, Figure 6a). Similarly, frequency of occurance of aliphatic amino acids in contig and singleton sequences 27.7% and 23.01% were more than aromatic amino acids in contig and singleton (14.68% and 20.8%) (Figure 5b, Figure 6b). The distribution study of chemical nature of amino acids gives an insight that neutral amino acid occurred more frequently than with 53.24 in contigs sequences and 56.73 in singleton sequences in comparison with basic amino acids (contig-22.28%,singleton-23.01%) and acidic amino acids (contigs-15.21%, singleton-8.83%) (Figure 5c, Figure 6c).
Figure 5.

(a) Percentage frequency of polar & non-polar amino acids; (b) Percentage frequency of hydrop hilic & hydrophobic amino acids; (C) Percentage frequency of aromatic & aliphatic amino acids
Figure 6.
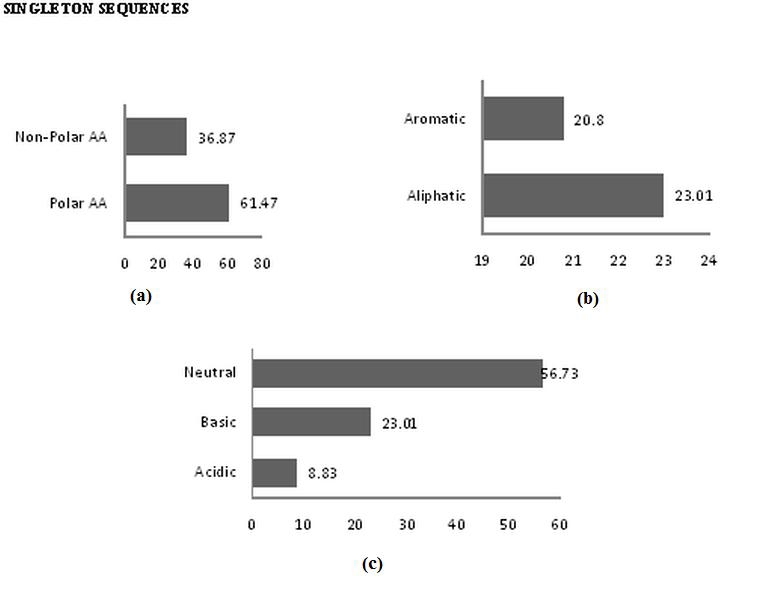
a) Percentage frequency of polar & non-polar amino acids; b) Percentage frequency of hydrophilic & hydrophilic amino acids; c) Percentage frequency of aromatic & aliphatic amino acids.
Gene Ontology Classification:
Gene ontology based functional annotation of SSR- ESTs was performed through BLASTx using NCBI database. BLAST best hit were retained meeting the following criteria: E-value < 1e-4, and similarity >=70%. The most significant matches for the SSRESTs with unique SSR motif were considered. Out of 2651 unique SSR-ESTs, 835 had significant matches to proteins. Functional annotation of these SSR-ESTs was performed using Amigo. Gene ontology for the corresponding SSR's was determined on the basis of sequence, domain and motif similarity Table 2 and Table 3 (see supplementary material). A biological process is a series of events accompolished by one or more ordered assemblies of molecular functions. In a gamut of biological process corresponding to SSR-ESTs, the most frequent was Ribosome Biogenesis (32SSR-EST) followed by translation, protein folding, embroyo development in seed dormancy,ATP synthesis coupled proton transport, brassinosteroid biosynthetic process, regulation of transcription, DNA-dependent, protein phosphorylation, Photosynthesis.Molecular Function describes activities, such as catalytic or binding activities,that occur at the molecular level.In a gamut of molecular function, the most frequent was Structural constitue of ribosome (95 SSR-ESTs), protein binding, DNA binding, ATP binding, electron carrier activity, hydrogen ion transporting, ATP synthase activity, rotational mechanism, metal ion binding, lipid binding, sequence specific DNA binding. A cellular component is a component of a cell, but with the provision that it is a part of some larger object; this may be an anatomical structure or a gene product group. In a gamut of cellular components housing putative proteins,the most frequent was chloroplast (151SSRESTs) followed by cytosol (94 SSR-ESTs), Cell wall (66 SSRESTs), Cytoplasm, Nucleus, Plasma Membrane.
Primer designing:
The primer designing have been done for PCR amplification of the desired microsatellites using BatchPrimer3.0 software. Out of 829 SSR-ESTs with significant matches, primers were designed for 268 SSR-EST contigs and 373 SSR-EST singletons. It was observed that forward and reverse primer pairs were obtained from mononucleotide SSRs (264) followed by trinucleotide (226). Hence a total of 641 SSR primer pairs were designed.
Conclusion
Microsatellites serve for divergent roles in the field of plant genomics.EST database provide a valuable resource for the development of microsatellite markers, which are associated with transcribed genes. Simple Sequence Repeats are an important class of molecular markers for genomics and plant breeding applications due to their abundance, hyper variability, and suitability for high-throughput analysis, high polymorphism and transportability. Development of SSR markers from the EST database saves both cost and time, once sufficient amounts of EST sequence are available. Computational Approaches have been used here to mine ever increasing EST sequences in public databases. The publicly available collections of 25,495 expressed sequence tags (ESTs) from Humulus lupulus have been assembled and clustered using CAP3 assembly program. Assembly of EST sequences resulted in 9844 non-redundant EST sequences which were reported to have 2955 EST-SSRs. Among all the percentage frequency of mono-nucleotide SSRs is maximum and hexanucleotide has minimum frequency. Functional annotation of 2651 SSR-EST was performed and 835 have significant matches.829 SSR-ESTs were subject for primer designing which yielded a total of 641 primer set for Humulus lupulus that can be applied in studies of genetic variation, linkage mapping and comparative genomics.
Supplementary material
Acknowledgments
First Author is thankful to University Grants Commission, New Delhi for a research fellowship. The help of Sanchita Gupta, Research Scholar, CIMAP, at various stages of manuscript preparation is also acknowledged.
Footnotes
Citation:Singh et al, Bioinformation 8(3): 114-122 (2012)
References
- 1.Winge, et al. Compt Rend Trav Lab Carlsberg. 1923;15:1. [Google Scholar]
- 2.Derenne, et al. Bull Inst Agro de Gembloux. 1954;22:18. [Google Scholar]
- 3.Jacobsen, et al. Hereditas. 1957;43:357. [Google Scholar]
- 4.KW Carter, et al. J Fla Med Assoc. 1983;70:774. [Google Scholar]
- 5.Queller, et al. Trends Ecol Evol. 1993;8:285. doi: 10.1016/0169-5347(93)90256-O. [DOI] [PubMed] [Google Scholar]
- 6.Nagel, et al. Plant Cell. 2008;20:186. doi: 10.1105/tpc.107.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.RV Kantety, et al. Plant Molecular Biology. 2002;48:501. doi: 10.1023/a:1014875206165. [DOI] [PubMed] [Google Scholar]
- 8.R Ravi, Iyer The journal of biological chemistry. 2000;2174 doi: 10.1074/jbc.275.3.2174. [DOI] [PubMed] [Google Scholar]
- 9.Victoria, et al. BMC Plant Biology. 2011;11:15. doi: 10.1186/1471-2229-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MD Adams, et al. Science. 1991;252:1651. [Google Scholar]
- 11.KD Scott, et al. Theor Appl Genet. 2000;100:723. [Google Scholar]
- 12.G Cordeiro, et al. Plant Sci. 2001;160:1115. doi: 10.1016/s0168-9452(01)00365-x. [DOI] [PubMed] [Google Scholar]
- 13.I Eujayl, et al. Theor Appl Genet. 2002;104:399. doi: 10.1007/s001220100738. [DOI] [PubMed] [Google Scholar]
- 14.B Hackauf, et al. Plant Breed. 2002;121:17. [Google Scholar]
- 15.S Gupta, et al. Plant Omics J. 2010;3:121. [Google Scholar]
- 16.RK Joshi, et al. Bioinformation. 2011;5:378. [Google Scholar]
- 17.T Thiel, et al. Theor Appl Genet. 2003;106:411. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 18. http://www.genome.clemson.edu/resources/online_tools/cap3.
- 19.OI Ramalakshmi, et al. Bioinformation. 2010;5:240. [Google Scholar]
- 20. http://pgrc.ipk-gatersleben.de/misa/misa.html.
- 21.S Carbon, et al. Bioinformatics. 2009;25:288. [Google Scholar]
- 22.Sanchita, et al. Bioinformation. 2010;5:113. [Google Scholar]
- 23. http://probes.pw.usda.gov/cgibin/batchprimer3/batchprimer3.cgi.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.