

Abstract
Toxoplasma gondii is an obligate intracellular apicomplexan parasite that can infect a wide range of warm-blooded animals including humans. In humans and other intermediate hosts, toxoplasma develops into chronic infection that cannot be eliminated by host’s immune response or by currently used drugs. In most cases, chronic infections are largely asymptomatic unless the host becomes immune compromised. Thus, toxoplasma is a global health problem and the situation has become more precarious due to the advent of HIV infections and poor toleration of drugs used to treat toxoplasma infection, having severe side effects and also resistance have been developed to the current generation of drugs. The emergence of these drug resistant varieties of T. gondii has led to a search for novel drug targets. We have performed a comparative analysis of metabolic pathways of the host Homo sapiens and the pathogen T. gondii. The enzymes in the unique pathways of T. gondii, which do not show similarity to any protein from the host, represent attractive potential drug targets. We have listed out 11 such potential drug targets which are playing some important work in more than one pathway. Out of these, one important target is Glutamate dehydrogenase enzyme; it plays crucial part in oxidation reduction, metabolic process and amino acid metabolic process. As this is also present in the targets of tropical diseases of TDR (Tropical disease related Drug) target database and no PDB and MODBASE 3D structural model is available, homology models for Glutamate dehydrogenase enzyme were generated using MODELLER9v6. The model was further explored for the molecular dynamics simulation study with GROMACS, virtual screening and docking studies with suitable inhibitors against the NCI diversity subset molecules from ZINC database, by using AutoDock-Vina. The best ten docking solutions were selected (ZINC01690699, ZINC17465979, ZINC17465983, ZINC18141294_03, ZINC05462670, ZINC01572309, ZINC18055497_01, ZINC18141294, ZINC05462674 and ZINC13152284_01). Further the Complexes were analyzed through LIGPLOT. On the basis of Complex scoring and binding ability it is deciphered that these NCI diversity set II compounds, specifically ZINC01690699 (as it has minimum energy score and one of the highest number of interactions with the active site residue), could be promising inhibitors for T. gondii using Glutamate dehydrogenase as Drug target.
Keywords: Homology modeling, Molecular dynamics, Docking, Metabolic Pathway Analysis, Glutamate dehydrogenase, Toxoplasma gondii, Structural biology, Drug targets, KEGG
Background
Toxoplasma gondii is an obligate intracellular Apicomplexan parasite that can infect a wide range of warmblooded animals including humans [1]. Toxoplasma gondii was initially discovered by accident, in 1908, by a scientist, Charles Nicolle, who was working in North Africa and searching for a reservoir of Leishmania in a native rodent, Ctenodactylus gundi. The gundis live in the foothills and mountains of Southern Tunisia and were commonly used to study Leishmania at the Pasteur Institute in Tunis. The name Toxoplasma means ‘arc form’ in Greek and was named according to the crescentshaped morphology of the tachyzoite and bradyzoite stages of the organism observed by the scientists. At about the same time, Alfonso Splendore working in Sao Paulo discovered a similar parasite in rabbits. This pathogen is one of the most common in humans due to many contributing factors that include: (1) its complex life cycle allows it to be transmitted both sexually via felid fecal matter and asexually via carnivorism. (2) Toxoplasma has an extremely wide host cell tropism that includes most nucleated cells. (3) In humans and other intermediate hosts, Toxoplasma develops into a chronic infection that cannot be eliminated by the host’s immune response or by currently used drugs. In most cases, chronic infections are largely asymptomatic unless the host becomes immune compromised. Together, these and other properties have allowed Toxoplasma to achieve infection rates that range from ~23% in the USA [2] to 50-70% in France [3].
There are two major reasons that new drugs are needed to treat Toxoplasma infections. First, the drugs currently used to treat Toxoplasma infections are poorly tolerated, have severe side effects, and cannot act against bradyzoites [4]. Second, there are reports that Toxoplasma is developing resistance to the current generation of drugs [5, 6]. How resistance to these drugs has developed is not known but is critical to understand because it will lead to improved drug design and will increase our understanding of the biological functions of these drug targets. One way to understanding mechanisms of resistance is to compare the transcriptional profiles of wild-type and resistant parasites grown in the absence or presence of the drug. Such studies in bacterial resistance have demonstrated that pathogen responses to antibiotics are multifactorial and complex [7]. Whether the same will be true in Toxoplasma is unclear, but data from these types of experiments will likely impact new anti- Toxoplasma drug design.
Over the last decade, complete genome sequences of several pathogens have been determined, and many more such projects are currently under way. While, these data potentially contain all the determinants of host-pathogen interactions and possible drug targets, Development of effective therapies for intracellular eukaryotic pathogens is a serious challenge, given the protected location of these pathogens and the similarity of their biology to that of the host [8]. Genomics approach can be applied to evaluate the suitability of potential targets using two criteria, i.e. “essentiality” and “selectivity” [9]. The target must be essential for the growth, replication, viability or survival of the microorganism, i.e. encoded by genes critical for pathogenic lifestages. The microbial target for treatment should not have any well-conserved homolog in the host, in order to address cytotoxicity issues. This can help to avoid expensive deadends when a lead target is identified and investigated in great detail only to discover at a later stage that all its inhibitors are invariably toxic to the host. Genes that are conserved in different genomes often turn out to be essential [10]. A gene is deemed to be essential if the cell cannot tolerate its inactivation by mutation, and its status is confirmed using conditional lethal mutants. Identifying cellular processes that are unique to the parasite is therefore a crucial step towards defining appropriate drug targets. Detection of genes that are non-homologous to human genes, and are essential for the survival of the pathogen represents a promising means of identifying novel drug targets [9].
We have performed a comparative analysis of metabolic pathways of the host Homo sapiens and the pathogen T. gondii. Enzymes from the biochemical pathways of T. gondii from the KEGG metabolic pathway database were compared with proteins from the host H. sapiens, by performing a BLASTp search against the non-redundant database restricted to the H. sapiens subset. Enzymes, which do not show similarity to any of the host proteins, below the threshold, were filtered out as potential drug targets [11]. The T. gondii genes for the identified enzymes were also retrieved for BLASTP search to identify any homologous protein in humans. We have listed out such potential drug targets which are playing some important work in more than one pathway. Out of these, one important target is Glutamate dehydrogenase enzyme, which is having several activities like catalytic activity, oxidoreductase activity and binding activity. Glutamate dehydrogenase is also involved in oxidation process, metabolic process and amino acid metabolic process. During metabolic pathways analysis it was found that, it is a common enzyme in three metabolic pathways Alanine, Aspartate and Glutamate Metabolism, Arginine and Proline Metabolism and Nitrogen Metabolism. As this is also present in the targets of tropical diseases of TDR (Tropical disease related Drug) target database and no PDB and MODBASE 3D structural model is available. Thus, as a case study, we have built homology models and validated by using various online servers. The model was further explored for the molecular dynamics simulation study, virtual screening and molecular docking studies with suitable inhibitors. The three dimensional model of glutamate dehydrogenase presented here would be helpful in guiding both enzymatic studies as well as design of specific inhibitors. A complete drug target identification using metabolic pathways analysis and molecular modeling, docking studies workflow is followed in this work and given in Figure 1.
Figure 1.
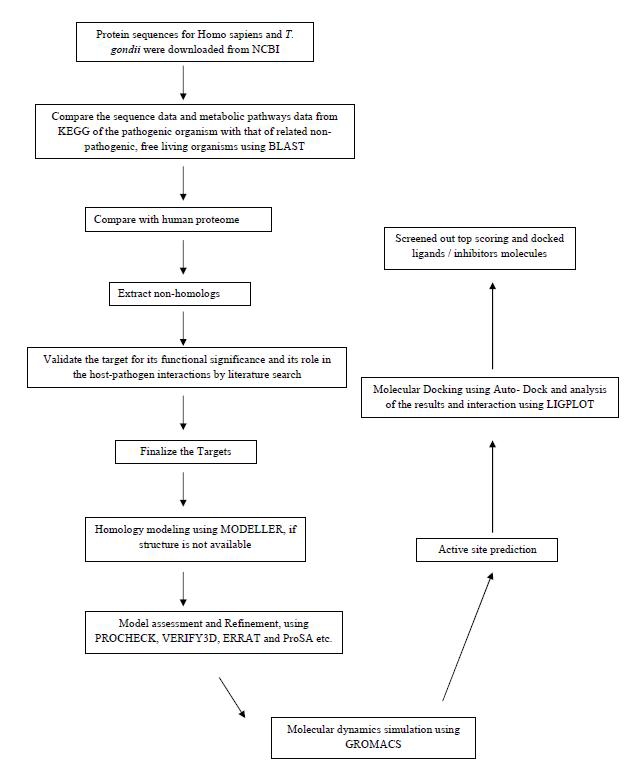
A workflow for complete drug target identification using metabolic pathways analysis and molecular modeling, docking studies.
Methodology
Identification of Unique Enzymes as Drug Targets:
Metabolic pathway information was obtained from the pathway database Kyoto Encyclopedia of Genes and Genomes. Enzyme commission numbers (EC) of the pathogen Toxoplasma gondii and the host H. sapiens were extracted from the KEGG database. Pathways unique to Toxoplasma gondii were filtered out. Out of total 84 metabolic pathways in humans 55 metabolic pathways were present in T. gondii. Out of these 55 metabolic pathways, 29 were such which contain unique enzymes which are only present in T. gondii and absent in humans (Table 1, see supplementary material). These are the pathways that do not appear in the host (H. sapiens) but are present in the pathogen. We further identified unique enzymes among shared pathways under carbohydrate metabolism, energy metabolism, lipid metabolism, nucleotide metabolism, amino acid metabolism, glycan biosynthesis and metabolism of co-factors and vitamins were obtained from the KEGG database. A total of 48 enzymes that are present in Toxoplasma gondii but absent in H. sapiens were obtained and their corresponding protein sequences were retrieved from the KEGG database. Out of these 48 enzymes, 11 enzymes were commonly present in more than one pathway (Table 2, see supplementary material) The protein sequences for these 48 unique enzymes were retrieved and were subjected to BLAST search against human protein sequences database at an expectation E-value cutoff of 10-2 to identify nonhomologous genes in Toxoplasma gondii. Removing enzymes from the pathogen that share a similarity with the host protein ensures that the targets have nothing in common with the host proteins, thereby, eliminating undesired host protein-drug interactions [12]. The above search resulted in 48 enzymes that had “no hits” in BLAST search. Thirty of these 48 “no hits” belonged to the unique pathways set.
Comparative Homology Modeling:
Out of the 11 enzymes, commonly present in more than one pathway, we have built a homology model of one of the potential drug target, Glutamate dehydrogenase enzyme (Gene I.D.: TGME49_093180 and EC No. is EC: 1.4.1.4.
Template Selection and Model Building:
Template selection is a critical step in homology modeling. The amino acid sequence of NADP-specific glutamate dehydrogenase of Toxoplasma gondii ME49 (target) (XP_002370120.1) was retrieved from the protein database of National Center of Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/). The three-dimensional structure of the protein was not yet available in Protein Data Bank and Modbase Database. Also there is no expression information available for this gene; hence the present exercise of developing the 3D model of the glutamate dehydrogenase of Toxoplasma gondii was undertaken. BLASTP search was performed against Brookhaven Protein Data Bank (PDB) with the default parameters to find suitable templates for homology modeling.
The academic version of MODELLER9v6 (http//:www.salilab.org/modeller), was used for 3D structure generation based on the information obtained from sequence alignment. The MODELLER software employs probability density functions (PDFs) as the spatial restraints rather than energy. The 3D model of a protein is obtained by optimization of the molecular pdf such that the model violates the input restraints as possible. The molecular pdf was derived as a combination of pdfs restraining individual spatial features of the whole molecule.
Model Assessment and Refinement:
Out of 6 models generated, 5 by MODELLER and one by ModWeb server, the one with the best G-score of PROCHECK and with the best VERIFY3D profile was subjected to energy minimization. VERIFY3D (a structure evaluation server) was used to check the residue profiles of the obtained threedimensional models. In order to assess the stereo-chemical qualities of the three dimensional models, PROCHECK analysis was performed. Quality evaluation of the model for the environment profile was also predicted using ERRAT structure evaluation server. The final refined model was evaluated for its atomic contacts using the Whatif program to identify bad packing of side chain atoms or unusual residue contacts. The predicted model was also analyzed for the recognition of errors in the three dimensional structure by using the ProSA web server. This model was further subjected for identification of active site and docking study.
With the aim of evaluating the stability and folding, conformational changes and getting insights into the natural dynamics on different timescale of protein in solution, 10 nanoseconds (ns) molecular dynamics simulation were performed. The molecular dynamics (MD) simulations of modeled glutamate dehydrogenase protein were carried out with the GROMACS 4.0.6 software package by employing GROMOS 96 force field and the flexible SPC (Simple Point Charge) water model. The initial structure was immersed in a periodic water box of cubic shape (0.5 nm thick). Electrostatic energy was calculated using the particle mesh Ewald method. Cutoff distance for the calculation of the coulomb and van der Waals interaction was 1.0 Ǻ. After energy minimization using a steepest descent for 1000 steps, the system was subjected to equilibration at 300k and normal pressure for 100 ps under the conditions of position restraints for heavy atoms. LINCS constraints were performed for all bonds, keeping the whole protein molecule fixed and allowing only the water molecule to move to equilibrate with respect to the protein structure. The system was coupled to the external bath by the Berendsen pressure and temperature coupling. The final MD calculations were performed for 10 ns under the same conditions except that the position restraints were removed. The results were analyzed using the standard software provided by the GROMACS package. An average structure was refined further using a steepest descent energy minimization.
Binding Site Prediction:
Binding site was characterized by CASTp and PASS and compared by using the information of binding sites. By comparing prediction of above two algorithms, best active site was selected.
Molecular Docking:
The docking of glutamate dehydrogenase was performed, against the NCI subset II molecules retrieved from ZINC database by using Autodock- vina (http://vina.scripps.edu/), where 1,364 molecules from the NCI diversity subset II (http://zinc.docking.org/index.shtml) were screened. The Python scripts in MGL tools package were used to analyze the docking results.
Analyzing the Docking Results:
The search for the best ways to fit ligand molecules from the NCI diversity subset II, into glutamate dehydrogenase modeled structure, using Autodock- vina resulted in docking files that contained detailed records of docking. These log files were read into ADT (Auto Dock Tool) to analyze the results of docking. The similarity of docked structures was measured by computing the root mean square deviation (RMSD) between the coordinates of the atoms and creating clustering of the conformations based on the RMSD values. In most cases the first cluster was also the largest cluster found. The lowest binding energy conformation in the first cluster was considered as the most favorable docking pose. Binding energies that are reported represent the sum of the total intermolecular energy, total internal energy and torsional free energy minus the energy of the unbound system. The top ten ligands were selected based on the energy score after virtual screening Table 3 (see supplementary material) and were further analyzed by the program LIGPLOT. The Ligplot represents the hydrogen and hydrophobic interactions between ligand and active site residues.
Discussion
Metabolic pathways analysis:
A total of 48 enzymes that are found present in Toxoplasma gondii but absent in H. sapiens were listed out as potential drug targets. Out of these 48 enzymes, some are already reported in literature and often used as target in treatment of toxoplasmosis e.g.
Template search and Model Building:
Based on the maximum identity with high score and lower evalue crystal structure of Glutamate dehydrogenase (NADP+) of Plasmodium falciparum (PDB code: 2bma_A) was selected as template. The sequence identity and similarity between the target and template are 58% and 73%, respectively. E- Value was 0.0. The sequence alignment of glutamase dehydrogenase of Toxoplasma gondii and Plasmodium falciparum was carried out using the CLUSTALW (http://www.ebi.ac.uk/clustalw/) program.
Model Assessment and Refinement:
All the six glutamate dehydrogenase models obtained were validated by using the SAVS and PROSA server. Ramachandran plots were drawn and the structures were analyzed by PROCHECK, a well-known protein structure checking program. By comparison of the results for all the models second model was judged as best among six (Figure 2). In the case of second model, it was found that the phi/psi angles of 91.3% of the residues fell in the most favored regions, 8.0% of the residues fell in the additional allowed regions, 0.7% fell in the generously allowed regions, and only 0.0 % of the residues fell in the disallowed regions. All these findings suggest a stereo-chemically very good model. The overall PROCHECK G factor for the homology-modeled structure was 0.00. The energetic architecture as predicted by PROSA score was negative (9.62) for the modeled protein. The value is close to that of template (9.60), which indicates the reliability of the model. The final structure was validated by an ERRAT graph. The quality factor of 77.106 indicated good quality, as scores of >50 are acceptable for a reasonable model. High quality of model is also confirmed from VERIFY 3D server as 76.00% of residues of modeled protein showed a score higher than 0.02 thus the model showed satisfactory 3D-1D scores for all the residues in the sequence. Results of WHAT_CHECK also indicate about the correctness of the modeled structure. These results indicate that the obtained structure has reasonably good quality. This model was used for virtual screening of the potential inhibitors for glutamate dehydrogenase.
Figure 2.

Ribbon representation of modeled glutamate dehydrogenase protein. The a-helices & β -sheets are shown as helices and ribbons respectively & rest are shown as loops. (Pymol Model)
Molecular Dynamics Simulation:
The predicted Model of glutamate dehydrogenase has shown good accuracy of the structure. But in order to use this model for virtual screening predicted model should have stable molecular dynamic behavior. The molecular dynamic stability has been established by performing the molecular dynamic simulation study using GROMACS 4.0.6 software package. The root mean square deviation (RMSD) during the simulation was increasing in the beginning but after 220 ps it becomes almost constant for rest of the duration of the simulation. This suggests that the glutamate dehydrogenase model has very less RMSD for the backbone and it also has less flexibility, indicating that model has a stable dynamic behavior structure. Molecular dynamic simulation study showed that the energy of the molecule was found to be constant throughout the time period of simulation. This suggests that the molecule has a stable structure as required for the virtual screening and drug designing. The root mean square (RMS) fluctuations were very less. Most the atoms were free from the RMS fluctuations. Very few atoms have shown RMS fluctuation at C and N terminal due to the loop region. This suggests the glutamate dehydrogenase model has an accurate and stable structure which can be used for virtual screening. The simulation studies also indicated that radius of gyration was increasing in the beginning but after 400 ps it decreases upto 420 ps and finally became almost constant for rest of the duration of the simulation. This suggests that the glutamate dehydrogenase model has a compact structure which provides the required stability to the molecule.
Active Site Identification:
After getting the final model, the possible binding sites of modeled structure were searched using the CASTp server and PASS program. Ten possible binding sites were obtained. These sites were compared with active site of the template and it was found that pocket1 is highly conserved with the template. Since the glutamate dehydrogenase protein of Toxoplasma gondii and Plasmodium falciparum are well conserved in both sequence and structure, it is predicted that their biological function may be identical. From the structure-structure comparison of template and final refined models of glutamate dehydrogenase using SPDBV program, it was found that the residues in the site1 Phe294, Pro295, Lys292, Gly293, Gly275, Ala272, Leu296, Ala276, Lys277, Arg417 and Asp421 are highly conserved within the active site of template. In this study, site1 is chosen as the binding site to dock with the NCI diversity set molecules.
Virtual screening:
The top ten ligand molecules having minimum energy were screened out as the possible inhibitors for glutamate dehydrogenase [19] Table 3 (see supplementary material). All selected molecules were having energy score as follows: Ligand ZINC01690699 is having minimum energy score 9.1 Kcal/Mol. Ligand ZINC17465979 are having 8.1 Kcal/Mol. Four ligands, ZINC17465983, ZINC18141294_03, ZINC05462670 and ZINC01572309 are having energy score 7.9 Kcal/Mol, Four ligands, ZINC18055497_01, ZINC18141294, ZINC05462674 and ZINC13152284_01 are having energy score 7.8. With the help of the Ligplot study we have selected ZINC01690699 as the possible inhibitor lead molecule, as it has minimum energy score and one of the highest number of interactions with the active site residue, it has 09 hydrophobic and 2 hydrogen interactions. Figure 3 (a) to (e), represent interactions of the top five ligands drawn by ligplot according to the energy score.
Figure 3.
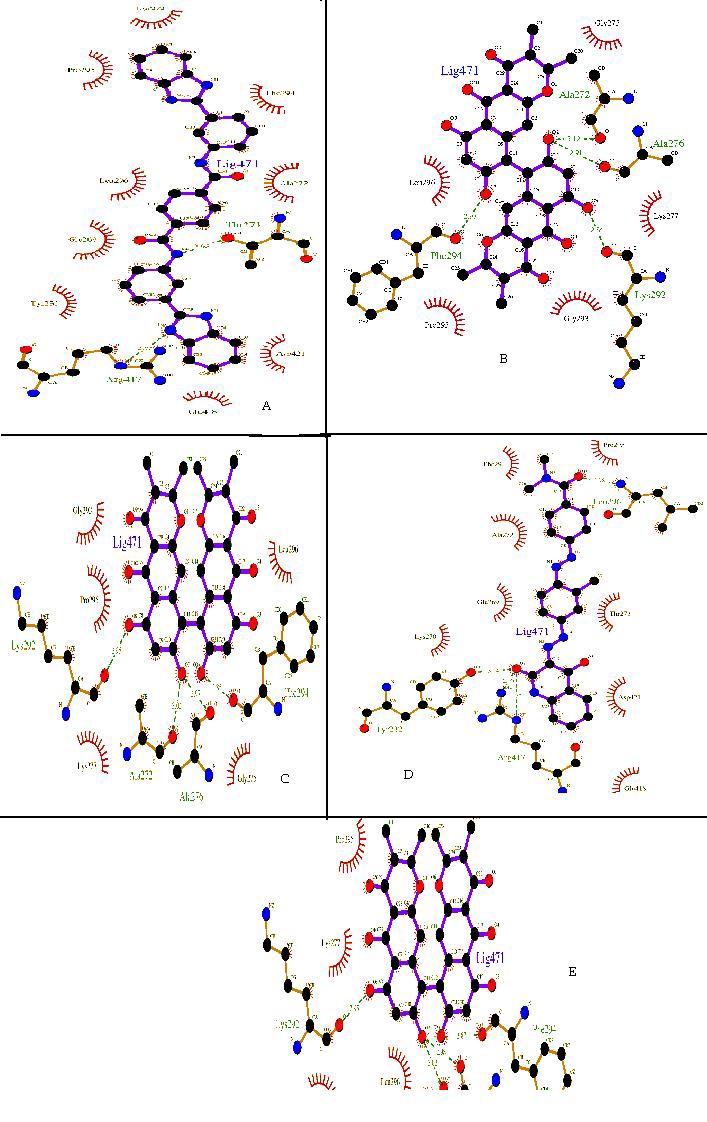
Ligplot showing the protein-ligand interactions of top five ligands, based on energy score (hydrogen bonding and hydrophobic), generated by Ligplot program. (A) ZINC01690699; (B) ZINC17465979; (C) ZINC17465983; (D) ZINC18141294_03 and (E) ZINC05462670.
Correlation coefficient analysis was performed between energy score calculated for all selected ligands molecules; log P value and molecular weight Table 3 (see supplementary material). The correlation coefficient is a statistical calculation that is used to examine the relationship between two sets of value. The Correlation coefficient between energy score and log P value was 0.101603358 and between energy score and molecular weight was 0.236816724. These values suggest a clear negative correlation between energy score of the ligands and their molecular weight and log P value. Thus the energy score of the ligands is independent of their molecular weight and log P value. The value of the correlation coefficient tells us about the strength and the nature of the relationship. Correlation coefficient values can range between +1.00 to 1.00. The negative correlation suggests that energy score may depend on interactions or the conformation of ligands and active site residues [20].
It was found the inhibitor ZINC01690699 has minimum energy score which reveals higher binding affinity towards the glutamate dehydrogenase and the inhibitor was also showing one of the best interactions with residues of the active site. The other important drug like properties like molecular weight and logP value were also found within the limits of drug like molecules. For the wet laboratory validation of present study, samples of T. gondii can be collected from the pathological laboratories and the molecule ZINC01690699 can be procured from the Pubchem database. Growth inhibition of T. gondii by inhibitor ZINC01690699 can be established by the qualitative and quantities microbiological techniques. Thus, the three dimensional model of glutamate dehydrogenase presented here would be helpful in guiding both enzymatic studies as well as design of specific inhibitors.
Conclusion
Our approach of comparative metabolic pathway analysis and molecular docking analysis resulted in the identification of potential drug targets. For the first time, the availability of complete genome sequences of many bacterial species is facilitating many novel computational approaches. The complete definition of all gene products by gene identification tools exemplified here is just the first step. The data presented here demonstrates that stepwise prioritization of genome open reading frames using simple biological criteria can be an effective way of rapidly reducing the number of genes of interest to an experimentally manageable number. This process is an efficient way for enriching potential target genes, and for identifying those that are critical for normal cell function. The generation of a comprehensive essential gene list will allow an accelerated genetic dissection of traits such as metabolic flexibility and inherent drug resistance that render Toxoplasma gondii such a tenacious pathogen. Such a strategy will enable us to locate critical pathways and steps in pathogenesis; to target these steps by designing new drugs; and to inhibit the infectious agent of interest with new antimicrobial agents. Hence, in present study, it can be concluded that the molecule ZINC01690699 1-N, 4-N-bis [3-(1H-benzimidazol-2-yl) phenyl] benzene-1, 4-dicarboxamide has the potential to inhibit the growth of Toxoplasma gondii and can act as remedy for the treatment of Toxoplasma gondii infection.
Supplementary material
Acknowledgments
The authors are grateful to the Sam Higginbottom Institute of Agriculture, Technology & Sciences, Deemed to be University, Allahabad for providing the facilities and support to complete the present research work.
Footnotes
Citation:Gautam et al, Bioinformation 8(3): 134-141 (2012)
References
- 1.JG Montova, O Liesenfeld. Lancet. 2004;363:1965. [Google Scholar]
- 2.JL Jones, et al. Am J Epidemiol. 2001;154:357. doi: 10.1093/aje/154.4.357. [DOI] [PubMed] [Google Scholar]
- 3.AM Tenter, et al. Int J Parasitol. 2000;30:1217. doi: 10.1016/s0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.R Mccabe. Cambridge University Press; 2001. pp. 319–359. [Google Scholar]
- 5.H Baatz, et al. Ocul Immunol Inflamm. 2006;14:185. doi: 10.1080/09273940600659740. [DOI] [PubMed] [Google Scholar]
- 6.TV Aspinall, et al. J Infect Dis. 2002;185:1637. doi: 10.1086/340577. [DOI] [PubMed] [Google Scholar]
- 7.MD Brazas, RE Hancock. Drug Discov Today. 2005;10:1245. doi: 10.1016/S1359-6446(05)03566-X. [DOI] [PubMed] [Google Scholar]
- 8.L Miesel, et al. Nat Rev Genet. 2003;4:442. doi: 10.1038/nrg1086. [DOI] [PubMed] [Google Scholar]
- 9.KR Sakharkar, et al. In Silico Biol. 2004;4:355. [PubMed] [Google Scholar]
- 10.K Kobayashi, et al. Proc. Natl. Acad. Sci. USA. 2003;100:4678. [Google Scholar]
- 11.Singh, et al. Elixir Bio Phy. 2011;32 [Google Scholar]
- 12.Singh, et al. Bioinformation. 2011;6:31. doi: 10.6026/97320630006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.BI Schweitzer, et al. FASEB J. 1990;4:2441. [Google Scholar]
- 14.LC Chio, SF Queener. Antimicrob. Agents Chemother. 1993;37:1914. doi: 10.1128/aac.37.9.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.G Dorsey, et al. Am J Trop Med Hyg. 2004;71:758. [Google Scholar]
- 16.BR Ogutu, et al. Trop Med Int Health. 2005;10:484. doi: 10.1111/j.1365-3156.2005.01415.x. [DOI] [PubMed] [Google Scholar]
- 17.SA Omar, et al. Exp Parasitol. 2005;110:73. doi: 10.1016/j.exppara.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 18.F Seeber. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:99. doi: 10.2174/1568008033340261. [DOI] [PubMed] [Google Scholar]
- 19.AU Khan, et al. Bioinformation. 2011;5:331. [Google Scholar]
- 20.R Farmer, et al. Bioinformation. 2010;4:290. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.