

Abstract
Background:
Infection with human papillomavirus (HPV) is a critical factor in the development of cervical cancer. Smoking is an additional risk factor. Tobacco smoke carcinogens, such as benzo[a]pyrene (B[a]P), and their cytochrome P450-related metabolites are present in significantly higher levels in the cervical mucus of women smokers than in nonsmokers. We determined the metabolism and P450 expression of B[a]P-treated human keratinocytes infected with HPV-16 or -18.
Materials and Methods:
Monolayer cultures of uninfected primary human foreskin keratinocytes, human vaginal and cervical keratinocytes carrying episomal genomes of HPV-16 and -18, respectively, and invasive cervical carcinoma cell lines carrying either HPV-16 or -18 genomes integrated into the host DNA, were incubated with 0.1 μM [3H]B[a]P. The resulting oxidative metabolites were analyzed and quantified by radioflow high-performance liquid chromatography. Additionally, all cell lines were incubated with unlabeled 0.1 μM B[a]P for Western blot analysis of cytochrome P450 1A1 and 1B1.
Results:
Significant enhancement in levels of both detoxification and activation metabolites was found in incubations with all types of HPV-infected cells compared with control incubations (P < 0.05). The highest capacity to metabolize B[a]P was observed with cells containing integrated HPV-18 genomes. Induction of cytochrome 1B1 was observed in HPV-16 and -18 integrated, and in HPV-16 episomal cell types.
Conclusions:
Both viral genotype and genomic status in the host cell affect B[a]P metabolism and cytochrome P450 1B1 expression. An increase of DNA-damaging metabolites might result from exposure of HPV-infected women to cigarette smoke carcinogens.
Keywords: Benzo[a]pyrene metabolism, benzo[a]pyrene, cervical cancer, cigarette smoke carcinogen, cytochrome P450 1A1, cytochrome P450 1B1, human papillomavirus
BACKGROUND
Cervical cancer is the second most prevalent cancer type in females and ranks fifth in cancer-related deaths for women worldwide.[1,2] Human papillomavirus (HPV) infection is associated with more than 90% of all human cervical cancers and is an established etiological factor in the development of this disease.[3] Over 100 HPV genotypes have been identified, and they are classified as either high (e.g., HPV types -16 and -18) or low risk (e.g., HPV types -6 and -11). HPV morphogenesis is intimately connected with host cell and tissue differentiation.[4] From initial infection to the morphogenesis of new virions, the HPV genome is present as an episome in the host cell. In high-grade cervical lesions, the HPV DNA is integrated into the host genome and viral replication ceases.[4] HPV genome integration marks the end of the viral replication cycle and is a critical step in the development of cervical cancer.[5]
Most HPV infections clear spontaneously.[6] Consequently, interest in tobacco use, a secondary factor that might promote cervical carcinogenesis in HPV-infected women, has grown. Cigarette smoking has been linked to an increased risk for cervical cancer of up to three-fold in HPV-positive women smokers compared with nonsmokers.[7] Over 4000 compounds have been identified in tobacco and tobacco smoke, and more than 60 of these are established carcinogens.[8] Of these carcinogens, polycyclic aromatic hydrocarbons (PAH), including the ubiquitous environmental carcinogen benzo[a]pyrene (B[a]P), are among the most toxic and carcinogenic.[9] Topical application of B[a]P to the cervix induces squamous cell carcinoma in mice and hamsters.[10,11] B[a]P treatment of cells infected with the high-risk HPVs -31, -16 and -18 increases viral morphogenesis in organotypic raft cultures derived from a cervical intraepithelial neoplasia type I cell line.[12] An increase in viral load is thought to be important for the persistence of HPV infection. Persistent infection is considered by many to be necessary for progression from initial infection to malignancy.[6]
As with many chemical carcinogens, B[a]P requires metabolic activation in order to exert its carcinogenic effect. The cytochrome P450 group of enzymes, including cytochromes 1A1 and 1B1 (CYP1A1 and CYP1B1), contribute to the formation of B[a]P metabolites, including both activation and detoxification products [Figure 1].[13–15] Detoxification metabolites derived from B[a]P include trans-9,10-dihydroxy-9,10-dihydro-benzo[a]pyrene (B[a]P-9,10-diol) and 3-hydroxy-benzo[a]pyrene (3-OH-B[a]P). Activation metabolites include the trans-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene (B[a]P-7,8-diol) as well as r-7,t-8,9,c-10-tetrahydroxy-7,8,9,10-tetrahydro-benzo[a]pyrene (trans,anti-B[a]P-tetraol).[13–15] The trans,anti-B[a]P-tetraol is used as an indication of anti-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydro-benzo[a]pyrene (anti-BPDE) formation. This metabolite is the ultimate carcinogen of B[a]P, but is too reactive to be identified in cellular incubations.[14]
Figure 1.
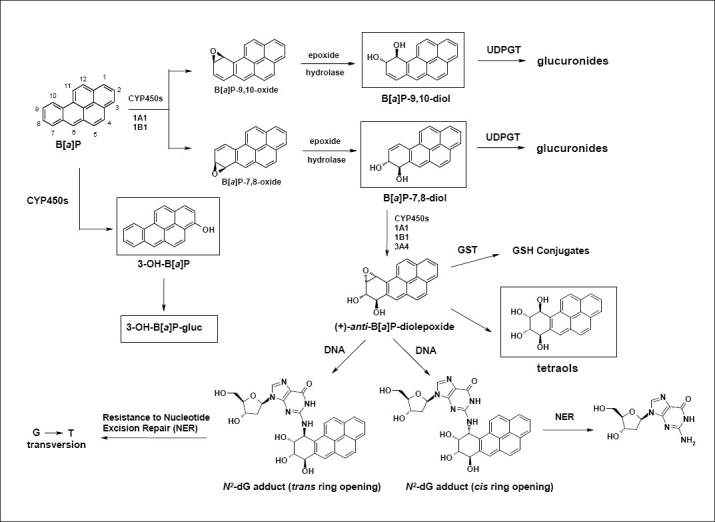
Metabolic oxidation of benzo[a]pyrene. Metabolites identified in this study are depicted in boxes
Both CYP1A1 and CYP1B1 have been found in human uterine tissue, including the cervix.[16,17] An increase of CYP1A1 was found by Farin et al. in human cervical cells immortalized by HPV-16 compared with normal cervical cells.[18] We have previously demonstrated the presence of B[a]P, B[a]P-9,10-diol, 3-OH-B[a]P and trans,anti-B[a]P-tetraol in the cervical mucus of both smokers and nonsmokers. Additionally we found significantly higher levels of anti-BPDE adducts in DNA isolated from the cervical epithelial tissue of smokers compared with the adduct levels in nonsmokers.[19] Melikian et al. found that B[a]P treatment of human HPV-16 immortalized human cervical cells resulted in significantly higher BPDE-deoxyguanosine levels when compared with B[a]P-treated normal cervical cells.[20]
The increases in viral load and in the levels of DNA adducts found in the above mentioned studies suggest that B[a]P exposure may be an important secondary factor for the development of cervical carcinoma. In the current study, we have investigated the effects of HPV infection on B[a]P metabolism and cytochrome P450 expression in cells infected with either HPV-16 or -18 as episomes or integrated into the host genome.
MATERIALS AND METHODS
Chemicals
Unlabelled B[a]P was purchased from Aldrich Chemical Co., Milwaukee, WI, USA. [3H]B[a]P, specific activity = 83.0 Ci/mmol, was obtained from Amersham Life Science, Buckinghamshire, England. The following chemicals were purchased from the National Cancer Institute's Chemical Carcinogen Reference Standard Repository at the Midwest Research Institute, Kansas City, MO, USA: trans-4,5-dihydroxy-4,5-dihydro-benzo[a]pyrene (B[a]P-4,5-diol), B[a]P-7,8-diol, B[a]P-9,10-diol and 3-OH-B[a]P. trans,anti-B[a]P-tetraol was synthesized as previously described.[21,22]
Metabolism of B[a]P by human cells
The HPV-16 and HPV-18 infected human keratinocyte cell lines were isolated from high-grade lesion human cervical biopsy samples as previously described.[23] In these cells, the respective HPV DNA is integrated into the host genome. The HPV-16 cell line containing episomal HPV DNA was laboratory derived and generated by electroporation of the HPV DNA into human vaginal keratinocytes derived from a surgical sample using protocols previously reported.[24] The HPV-18 cell line was derived in a similar manner from human cervical keratinocytes.[25] All the HPV-positive lines were maintained in a monolayer culture with E Medium containing 5% fetal bovine serum in the presence of mitomycin C-treated J2 feeder cells.[23] Primary foreskin keratinocytes (HPV negative) were derived from newborn foreskin via trypsin digestion at 37°C.[26] These cells were maintained in monolayer cultures without feeder cells, with 154 Medium supplemented with antibiotics and human keratinocyte growth supplement (Cascade Biologics, Portland, OR, USA). Cells were grown to approximately 80% confluence, trypsinized and plated at a density of 1 million cells in 100-mm plates in E Media without addition of mitomycin C-treated J2 feeder cells. After plating, cells were incubated between 10 and 12 h, at which time [3H]B[a]P diluted with unlabeled B[a]P in DMSO was added to obtain a concentration of 0.1 μM (specific activity = 20 Ci/mmol). The media was collected following a 24 h incubation at 37°C. All incubations were performed in duplicate.
High-performance liquid chromatography analysis of B[a]P metabolites
B[a]P metabolites were identified based on comparison of elution times of the radioactive peaks with those of unlabeled synthetic standards monitored by UV detection (230 or 254 nm) and quantified by high-performance liquid chromatography (HPLC) interfaced with a radioflow detector. The system was composed of an HP 1050 automatic injector (Agilent Technologies, Wilmington, DE, USA), a Waters 600 Multisolvent Delivery System (Waters, Milford, MA, USA), a Shimadzu SPD 10A UV detector (Shimadzu Scientific Instruments, Columbia, MD, USA), an IN/US β-RAM radioactivity detector (IN/US Systems, Tampa, FL, USA) and a Phenomenex Synergi MAX-RP column (250 mm x 4.6 mm, 4 μ; Phenomenex, Torrance, CA, USA). Solvent A was 20 mM sodium phosphate, pH 7.0, while solvent B was 95% methanol/5% water. The elution program (1 ml/min) was a modification of the one employed by Staretz et al.[27] Initial conditions were 10% B, followed by increases to 40% B in 15 min, 55% B in 10 min, 70% B in 20 min, 80% B in 15 min, a 5-min hold at 80% B and then an increase to 100% B in 5 min. The final conditions were held for 17 min before returning to the initial conditions. Before injection, all samples were centrifuged at 13,000 rpm for 5 min and 350 μl was removed and placed in a vial containing the following unlabeled standards in 5 μl DMSO: trans,anti-B[a]P-tetraol, B[a]P-9,10-diol, B[a]P-7,8-diol, and, in some samples, B[a]P-4,5-diol. The injection volume was 250 μl. All samples were analyzed twice. The results for each metabolite are expressed as percent of total radioactivity. Statistical analyses were performed using the Student's t-test.
β-Glucuronidase assay
The assay mixture consisted of 100–150 μl of sample, 200 μl of 75 mM potassium phosphate (KP) buffer, pH 6.8, containing 0.1% BSA, 25 μl phenolphthalein glucuronide (3 mM in water) and 20 μl β-glucuronidase (540 U/ml KP buffer, pH 6.8, with 0.1% BSA, Type IX-A from E. coli; Sigma-Aldrich, St. Louis, MO, USA) heated overnight at 37°C. HPLC analysis of these incubations was accomplished as described for the metabolism study, except that a Rainin C18 Microsorb MV column (5 μ, 250 mm x 1.6 mm; Varian Inc., Lake Forest, CA, USA) was used.
Sulfatase assay
One hundred microliters of media from an incubation with the HPV-16 episomal cell line was incubated overnight at 37°C with 300 μl 10 mM Tris, pH 7.1, 40 μl p-nitrophenyl sulfate (0.12 M in water) and 20 μl sulfatase (0.5 U/ml 10 mM Tris, pH 7.1, Type V from Limpets, Sigma-Aldrich). The sample was analyzed as described for the β-glucuronidase assay.
Western blot analysis
Protein extracts were prepared as described by Meyers.[28] A total of 60 μg of protein for each sample was loaded onto a 7.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel for separation of either CYP1A1 or CYP1B1. Following transfer to a nitrocellulose membrane, the proteins were incubated with a 1:2000 dilution of either CYP1A1 or CYP1B1 antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) overnight. After washing, the blots were incubated with anti-mouse horseradish peroxidase-labeled secondary antibody (Amersham Life Science) and the proteins detected utilizing a chemiluminescence reagent (Amersham Life Science) according to the manufacturer's instructions. Actin was analyzed using an 8% SDS-polyacrylamide gel and a primary antibody at 1:10000 dilution (Santa-Cruz). The bands of interest were quantified using UVP VisionWorks LS Image Acquisition Software (version 6.3.3, UVP Inc., Upland, CA, USA).
RESULTS
B[a]P metabolism
Figure 2a, shows the elution profile of synthetic standards monitored by UV detection. Figure 2b–f, are representative HPLC traces of B[a]P incubations with the different cell types. Referring to panel F, three radioactive peaks, by virtue of their co-elution with synthetic standards, were separately identified as trans,anti-B[a]P-tetraol (peak 2), B[a]P-9,10-diol (peak 3) and B[a]P-7,8-diol (peak 6). Upon treatment with β-glucuronidase, peak 4 co-eluted with the synthetic standard of 3-OH-B[a]P (data not shown) and was thereby identified as 3-OH-B[a]P-glucuronide (3-OH-[BaP]-gluc). Peak 1, following β-glucuronidase hydrolysis, shifted to a later retention time (data not shown). The retention time of unknown peak 5 did not change upon treatment with either β-glucuronidase or sulfatase. The identities of these two peaks (unknown glucuronide and unknown, respectively) remain undetermined.
Figure 2.
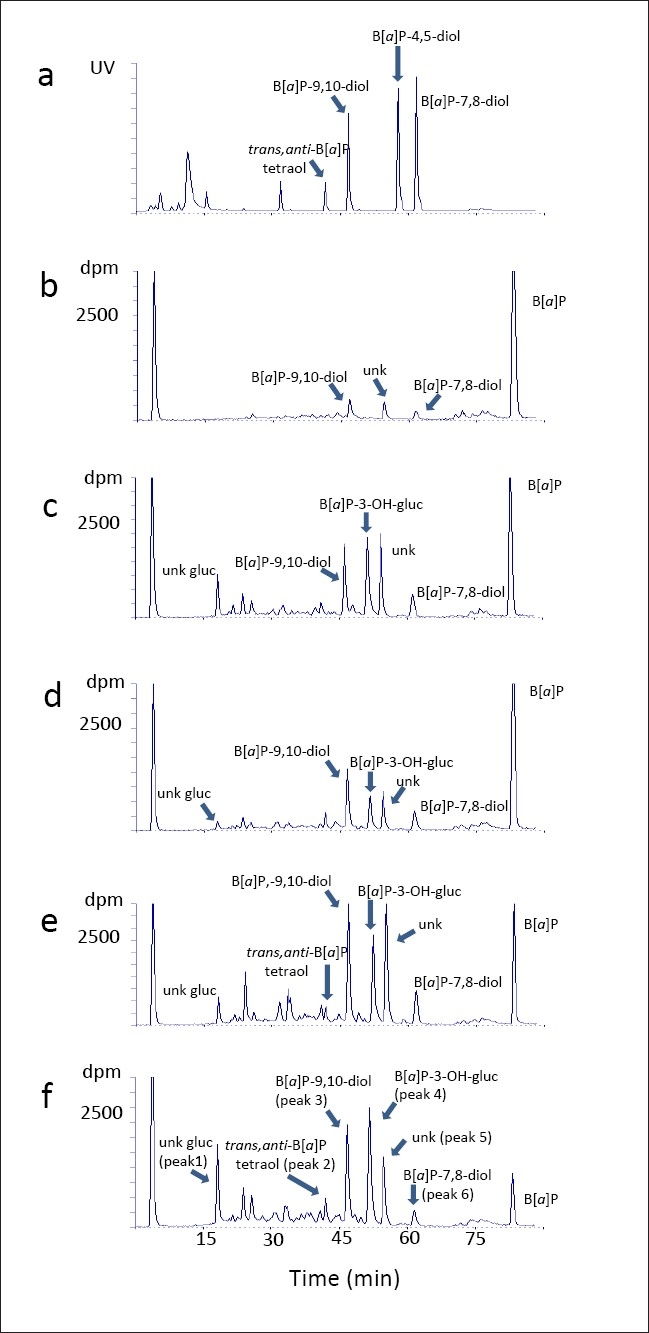
High-performance liquid chromatography elution profiles of [3H]benzo[a]pyrene metabolites. (a) synthetic standards. (b) uninfected primary human foreskin keratinocytes. (c) human vaginal keratinocytes infected with episomal human papillomavirus (HPV-16). (d) human cervical keratinocytes infected with episomal HPV-18. (e) invasive cervical carcinoma keratinocytes with HPV-16 integrated into the genome. (f) invasive cervical carcinoma keratinocytes with HPV-18 integrated into the genome. Arrows have been used to clarify the positions of closely eluting peaks in the chromatograph
The metabolism results are displayed graphically in Figure 3 and the percent metabolism results are shown in Table 1. The lowest total metabolism of B[a]P was found in HPV-negative primary keratinocytes from newborn foreskin (5.7% of total radioactivity). The presence of high-risk HPV-16 or -18, whether integrated into the genome or present as an episome in keratinocytes, clearly increased the overall metabolism of B[a]P (P < 0.05). The highest overall metabolism was found in incubations with HPV-18 and -16 integrated into the genome (31.5% and 27.1%, respectively), followed by cells infected with episomal HPV-16 and -18 (24.4% and 16.8%, respectively).
Figure 3.
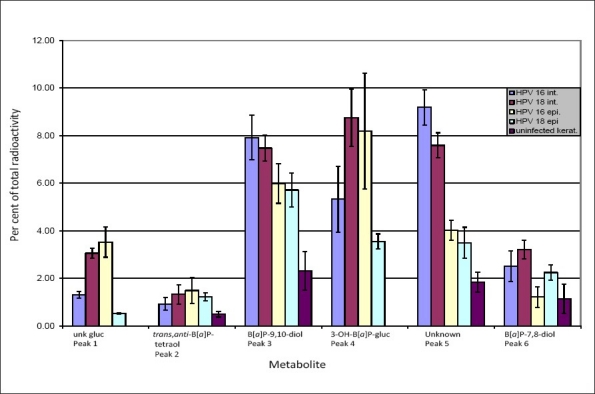
Levels of benzo[a]pyrene (B[a]P) metabolites, expressed as a percent of total radioactivity, from cell cultures after exposure for 24 h with 0.1 μM [3H]B[a]P
Table 1.
Metabolism of B[a]P in HPV infected cells and in uninfected control cells
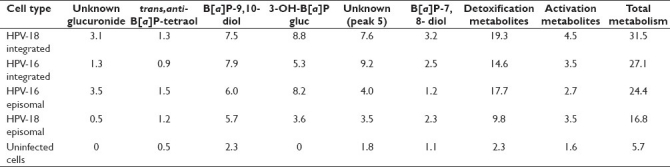
Both detoxification and activation metabolites were present at significantly higher levels in HPV-infected cells compared with uninfected control cells. For the two metabolites representing B[a]P activation, the trans,anti-B[a]P-tetraol and the B[a]P-7,8-diol, the combined metabolism was greatest in HPV-18 integrated cells, followed by HPV-18 episomal and HPV-16 integrated, HPV-16 episomal and, lastly, uninfected keratinocytes (4.5%, 3.5%, 3.5%, 2.7% and 1.6%, respectively). The combined metabolism of the three likely detoxification metabolites, the glucuronide of unknown structure, the B[a]P-9,10-diol and the 3-OH-B[a]P-gluc was highest in HPV-18 integrated infected cells, followed by HPV-16 episomal, HPV-16 integrated, HPV-18 episomal and uninfected keratinocytes (19.3%, 17.7%, 14.6%, 9.8% and 2.3%, respectively).
Upon comparing B[a]P metabolism of the two different HPV-18 infected cell types, we found significantly higher levels (P < 0. 01) of the unknown glucuronide (3.1% vs. 0.5%), the B[a]P-9,10-diol (7.5% vs. 5.7%), 3-OH-B[a]P-gluc (8.8% vs. 3.6%), the unknown metabolite (peak five, 7.6% vs. 3.5%) and the B[a]P-7,8-diol (3.2% vs. 2.3%) in incubations with cells containing integrated HPV-18. There was no significant difference in the level of trans,anti-B[a]P-tetraol between these cell types.
In incubations of cells containing integrated HPV-16, significantly higher conversion of B[a]P to the B[a]P-9,10-diol (7.9% vs. 6.0%, P < 0.05), the unknown peak five (9.2% vs. 4.0%, P < 0.004) and to B[a]P-7,8-diol (2.5% vs. 1.2%, P < 0.02) was observed as compared with cells carrying episomal HPV-16. Metabolism to the unknown glucuronide (1.3% vs. 3.5%) and to trans,anti-B[a]P-tetraol (0.9% vs. 1.5%) was significantly lower (P < 0.02) than in cell incubations with episomal HPV-16. The level of 3-OH-B[a]P-gluc was also lower in cells with integrated HPV-16 (5.3% vs. 8.2%, respectively), although the difference was not statistically significant (P = 0.1).
Western blot analysis
The results of the Western blot analysis for CYP1A1 and CYP1B1 are shown in Figure 4. Constitutive levels of CYP1A1 were low in both episomal (lane 3) and integrated HPV-16 (lane 5) cells compared with the other cell types. Upon B[a]P treatment, expression of this enzyme increased two-fold in cells infected with integrated HPV-16 (lanes 5 and 6). Little change in enzyme expression from the constitutive levels was found in the other B[a]P-treated cell types. A high constitutive level of CYP1A1, which appeared to decrease upon B[a]P treatment (by 25%), was apparent in the control cells (lanes 1 and 2). Except for cells infected with episomal HPV-16 (lane 4), expression of this enzyme appeared roughly equal in all cell types following B[a]P treatment.
Figure 4.
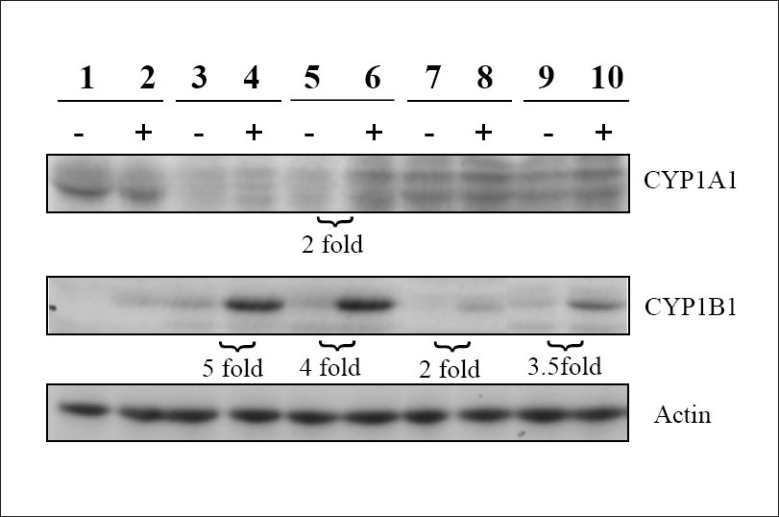
Western blot analysis of cell extracts. Lanes 1 and 2: uninfected primary foreskin keratinocytes. Lanes 3 and 4: cells infected with episomal human papillomavirus (HPV-16) DNA. Lanes 5 and 6: integrated HPV-16 DNA. Lanes 7 and 8: episomal HPV-18 DNA. Lanes 9 and 10: integrated HPV-18 DNA. The minus sign signifies untreated cells and the plus sign signifies treatment with 0.1 μM benzo[a]pyrene
CYP1B1 constitutive levels were low in all cell types analyzed. Treatment with 0.1 μM B[a]P resulted in increases of five-fold and four-fold in cells infected with episomal (lanes 3 and 4) and integrated HPV-16 (lanes 5 and 6), respectively. Following B[a]P treatment, cells infected with HPV-18 integrated into the genome showed a 3.5-fold increase of CYP1B1 expression (lanes 9 and 10), while an increase of two-fold was seen in cells infected with episomal HPV-18 (lanes 7 and 8). Constitutive expression of CYP1B1 was not detectable in control cells, but expression of this enzyme was apparent following B[a]P treatment (lanes 1 and 2).
DISCUSSION
B[a]P metabolism
Cigarette smoking increases the risk of developing cervical cancer in women infected with high-risk HPV.[6] B[a]P, a carcinogen and a tobacco smoke constituent, and the trans,anti-B[a]P-tetraol, a metabolic product of enzyme activation of B[a]P, have been detected in the cervical mucus of smokers.[19] In the same study, BPDE adducts from DNA isolated from cervical tissue were also significantly higher in smokers compared with nonsmokers. Both CYP1A1 and CYP1B1, enzymes involved in the formation of both activation and detoxification metabolites of B[a]P, have been found in human uterine tissue, including the cervix.[16,17] An increase of CYP1A1 was found by Farin et al. in human cervical cells immortalized by HPV-16 compared with normal cervical cells.[18] To the best of our knowledge, however, the impact of the type and genomic status of HPV infection on the metabolism of B[a]P has not been investigated. In this study, we attempted to address this issue.
The presence of high-risk HPV-16 or -18, whether integrated into the genome or present as an episome in keratinocytes, substantially increased the overall metabolism of B[a]P. Both detoxification and activation metabolites were present at significantly higher levels in HPV-infected cells compared with uninfected control cells. Overall metabolism was roughly comparable in the HPV-18 and -16 integrated and HPV-16 episomal cell types, but lower in the HPV-18 episomal cells. In the latter, however, the level of activation metabolites was comparable to that of the other cell types. There was a clear difference in B[a]P metabolism between HPV-18 integrated and HPV-18 episomal cell types. Excluding the trans,anti-B[a]P-tetraol, levels of all metabolites were significantly higher in the HPV-18 integrated cells. The differences in metabolism were less pronounced between the two types of HPV-16 infected cell types. Levels of both the glucuronides and the trans,anti-B[a]P-tetraol were higher in the HPV-16 episomal cells while levels of the 9.10- and 7,8-B[a]P-diols and of the metabolite of unknown identity were higher in the HPV-16 integrated cells. High levels of 3-OH-B[a]P-gluc and a glucuronide of unknown identity were found in all infected cell types. Neither 3-OH-B[a]P-gluc nor unconjugated 3-OH-B[a]P was seen in control cells. The absence of glucuronidation in these cells, therefore, may simply reflect an inability of this cell type, under these conditions, to form the necessary metabolites that lend themselves to conjugation.
Western blot analysis
The enzymes CYP1A1 and CYP1B1 were selected for analysis as they have been shown to be more active toward B[a]P than other cytochromes, such as 1A2.[29] We found that constitutive levels of CYP1A1 were much lower in both types of HPV-16 infected cells than in the other cell types. Treatment with B[a]P increased expression of this enzyme only in HPV-16 integrated cells. Following this treatment, expression of CYP1A1 was roughly equivalent in all cell types except for HPV-16 episomal cells. CYP1B1 constitutive levels were low in all cell types. The strongest increases in expression occurred in HPV-16 episomal and integrated cells, followed by HPV-18 integrated cells. The weakest increases in expression occurred in HPV-18 episomal and in normal uninfected control cells. These protein expression results for both CYP1A1 and CYP1B1 are consistent with the result of Wen, in which B[a]P induced CYP1B1 protein expression to a greater extent than CYP1A1 in human oral epithelial cells.[29] Tsuji, however, reported the opposite result in human bronchial epithelial cells.[30] It is therefore possible that changes in enzyme expression upon B[a]P treatment will vary depending on the cell type. In addition, our results suggest that the type of HPV infection may modulate the extent of enzyme expression, as we found that increases in CYP1B1 were greater in HPV-16 infected cells than in HPV-18 infected cells [Figure 4].
Based on the metabolism data from this study, it is uncertain as to what role CYP1A1 might play in B[a]P metabolism in these cell types. CYP1A1 metabolizes B[a]P to 3-OH-B[a]P.[15] Despite constitutive expression of CYP1A1, we saw no evidence of 3-OH-B[a]P formation (free or as the glucuronide) in control cells. Additionally, after B[a]P treatment, the lowest apparent CYP1A1 expression [Figure 4, lane 4] was seen in HPV-16 episomal cells. Yet, the 3-OH-B[a]P-gluc level in these cells was approximately 2.3-times the level seen in HPV-18 episomal cells [Figure 3], which appeared to have the highest CYP1A1 levels [Figure 4, lane 8] following B[a]P treatment. These data suggest that this enzyme is either inactive or may be inhibited by some factor present in these incubations.
The extent to which CYP1B1 participates in B[a]P metabolism in these incubations is also unclear. Similar to CYP1A1, CYP1B1 also metabolizes B[a]P to 3-OH-B[a]P.[15] Following B[a]P treatment, CYP1B1 expression is low in control cells [Figure 4, lane 2] and, correspondingly, no 3-OH-B[a]P was seen in these incubations (discussed previously). In infected cells, the lowest 3-OH-B[a]P level (determined as the glucuronide) was found in HPV-18 episomal cells [Figure 3]. This corresponds to the relatively low enzyme expression seen in these cell types [Figure 4, lane 8]. However, the levels of 3-OH-B[a]P-gluc in the other infected cell types [Figure 3] do not correspond to CYP1B1 expression. This is apparent when comparing HPV-18 and -16 integrated enzyme expression [Figure 4, lanes 10 and 6] with the corresponding levels of 3-OH-B[a]P-gluc [Figure 3]. Protein expression is clearly lower in HPV-18 infected cells; however, in these cells, the levels of 3-OH-B[a]P-gluc are the highest of all the cell types. Regarding the activation metabolites, B[a]P-7,8-diol and the trans,anti-B[a]P-tetraol, the relative order of enzyme expression [Figure 4] following B[a]P treatment (with the associated percent metabolism in parentheses) is: HPV-16 integrated, lane 6 (3.5%) ≈ HPV-16 episomal, lane 4 (2.7%) > HPV-18 integrated, lane 10 (4.5%) > HPV-18 episomal, lane 8 (3.5%) > uninfected cells, lane 2 (1.1%). Aside from the fact that the lowest expression of CYP1B1 (normal cells) corresponds to the lowest level of activation metabolism, no clear pattern of CYP1B1 activity on B[a]P activation emerges from these data. The metabolism results for both these enzymes, combined with the results for the protein expression of CYP1A1 and CYP1B1, suggest that, under the conditions of this study, other enzymes may be involved in the metabolism of B[a]P in these cell types. Clarification of the enzymes responsible for B[a]P metabolism in HPV-infected cells as well as identification of the unknown metabolites found in this study and determination of the levels of DNA adducts in B[a]P-treated cells remain the goals for future work.
CONCLUSION
HPV infection clearly influences the metabolic capabilities of the different cell types studied. We have demonstrated that cells infected with HPV are capable of generating high levels of both detoxification metabolites and increased levels of B[a]P metabolites that are known to damage DNA as compared with controls. CYP1B1 expression is increased in HPV-16 infected cells, although its role in B[a]P metabolism remains uncertain. At present, therefore, it is unclear which enzymes are responsible for this increase in metabolism. Despite this ambiguity, the authors believe that cigarette smoking is likely to result in increased exposure of the cervical epithelium to potentially mutagenic metabolites of this carcinogen and, consequently, be a factor in the development of cervical cancer.
AUTHORS’ CONTRIBUTIONS
NT carried out the analyses of metabolites, participated in the study design and helped draft the manuscript. SA carried out the cell incubations, the Western blot analysis, participated in the study design and helped draft the manuscript. KEB participated in the study design and helped draft the manuscript. JK synthesized metabolite standards. SGA synthesized metabolite standards. JG carried out the cell incubations. CM helped design the study and provided expertise on incubations using HPV. BP conceived the study and drafted the manuscript.
AUTHOR'S PROFILE
Neil Trushin, Department of Pharmacology, Penn State Cancer Institute, CH76, 500 University Drive, Hershey, PA 17033
Dr. Samina Alam, Department of Microbiology and Immunology, Penn State College of Medicine, H107, 500 University Drive, Hershey, PA 17033
Dr. Craig Meyers, Department of Microbiology and Immunology, Penn State College of Medicine, H107, 500 University Drive, Hershey, PA 17033
Dr. Bogdan Prokopczyk, Department of Pharmacology, Penn State Cancer Institute, CH76, 500 University Drive, Hershey, PA 17033
ACKNOWLEDGMENTS
The authors would like to thank Dr. Arun Sharma for his assistance in the preparation of [Figure 1], and Dr. Raghu Sinha and Indu Sinha for their assistance in the preparation of Figure 4.
REFERENCES
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: GLOBOCAN 2000. Int J Cancer. 2001;94:153–6. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 3.Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–27. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 4.Frattini MG, Lim HB, Laimins LA. In vitro synthesis of oncongenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc Natl Acad Sci U S A. 1996;93:3062–7. doi: 10.1073/pnas.93.7.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond) 2006;110:525–41. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 6.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 7.Castellsague X, Bosch FX, Munoz N. Environmental co-factors in HPV carcinogenesis. Virus Res. 2002;89:191–9. doi: 10.1016/s0168-1702(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 8.Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: A controversial issue.A tribute to Ernst L. Wynder. Chem Res Toxicol. 2001;14:767–90. doi: 10.1021/tx000260u. [DOI] [PubMed] [Google Scholar]
- 9.Hecht SS. Cigarette smoking: cancer risks, carcinogens, and mechanisms. Langenbecks Arch Surg. 2006;391:603–13. doi: 10.1007/s00423-006-0111-z. [DOI] [PubMed] [Google Scholar]
- 10.Koprowska I, Bogacz J. A cyto-pathologic study of tobacco tar-induced lesions of uterine cervix of mouse. J Natl Cancer Inst. 1959;23:1–19. [PubMed] [Google Scholar]
- 11.Chu EW, Herrold KM, Wood TA., Jr Cytopathological changes of the uterine cervix of Syrian hamsters after painting with DMBA, benzo(a)pyrene, and tobacco tar. Acta Cytol. 1962;6:376–84. [PubMed] [Google Scholar]
- 12.Alam S, Conway MJ, Chen HS, Meyers C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J Virol. 2008;82:1053–8. doi: 10.1128/JVI.01813-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thakker DR, Yagi H, Levin W, Wood AW, Conney AH, Jerina DM. Polycyclic Aromatic-Hydrocarbons: metabolic activation to ultimate carcinogens. In: Anders MW, editor. Bioactivation of Foreign Compounds. New York: Academic Press; 1985. pp. 177–242. [Google Scholar]
- 14.Kim JH, Stansbury KH, Walker NJ, Trush MA, Strickland PT, Sutter TR. Metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-diol by human cytochrome P450 1B1. Carcinogenesis. 1998;19:1847–53. doi: 10.1093/carcin/19.10.1847. [DOI] [PubMed] [Google Scholar]
- 15.Bauer E, Guo ZY, Ueng YF, Bell LC, Zeldin D, Guengerich FP. Oxidation of benzo[a]pyrene by recombinant human cytochrome P450 enzymes. Chem Res Toxicol. 1995;8:136–42. doi: 10.1021/tx00043a018. [DOI] [PubMed] [Google Scholar]
- 16.Vadlamuri SV, Glover DD, Turner T, Sarkar MA. Regiospecific expression of cytochrome P4501A1 and 1B1 in human uterine tissue. Cancer Lett. 1998;122:143–50. doi: 10.1016/s0304-3835(97)00382-0. [DOI] [PubMed] [Google Scholar]
- 17.Muskhelishvili L, Thompson PA, Kusewitt DF, Wang C, Kadlubar FF. In situ hybridization and immunohistochemical analysis of cytochrome P450 1B1 expression in human normal tissues. J Histochem Cytochem. 2001;49:229–36. doi: 10.1177/002215540104900210. [DOI] [PubMed] [Google Scholar]
- 18.Farin FM, Bigler LG, Oda D, Mcdougall JK, Omiecinski CJ. Expression of cytochrome P450 and microsomal epoxide hydrolase in cervical and oral epithelial cells immortalized by human papillomavirus type 16 E6/E7 genes. Carcinogenesis. 1995;16:1391–401. doi: 10.1093/carcin/16.6.1391. [DOI] [PubMed] [Google Scholar]
- 19.Melikian AA, Sun P, Prokopczyk B, El-Bayoumy K, Hoffmann D, Wang X, et al. Identification of benzo[a]pyrene metabolites in cervical mucus and DNA adducts in cervical tissues in humans by gas chromatography-mass spectrometry. Cancer Lett. 1999;146:127–34. doi: 10.1016/s0304-3835(99)00203-7. [DOI] [PubMed] [Google Scholar]
- 20.Melikian AA, Wang X, Waggoner S, Hoffmann D, El-Bayoumy K. Comparative response of normal and of human papillomavirus-16 immortalized human epithelial cervical cells to benzo[a]pyrene. Oncol Rep. 1999;6:1371–6. doi: 10.3892/or.6.6.1371. [DOI] [PubMed] [Google Scholar]
- 21.Yagi H, Thakker DR, Hernandez O, Koreeda M, Jerina DM. Synthesis and reactions of highly mutagenic 7,8-diol 9,10-epoxides of carcinogen benzo[a]pyrene. J Am Chem Soc. 1977;99:1604–11. doi: 10.1021/ja00447a053. [DOI] [PubMed] [Google Scholar]
- 22.Whalen DL, Ross AM, Yagi H, Karle JM, Jerina DM. Stereoelectronic factors in solvolysis of bay region diol epoxides of polycyclic aromatic hydrocarbons. J Am Chem Soc. 1978;100:5218–21. [Google Scholar]
- 23.Ozbun MA, Meyers C. Transforming growth factor beta1 induces differentiation in human papillomavirus-positive keratinocytes. J Virol. 1996;70:5437–46. doi: 10.1128/jvi.70.8.5437-5446.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyers C, Mayer TJ, Ozbun MA. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J Virol. 1997;71:7381–86. doi: 10.1128/jvi.71.10.7381-7386.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bedell MA, Hudson JB, Golub TR, Turyk ME, Hosken M, Wilbanks GD, et al. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J Virol. 1991;65:2254–60. doi: 10.1128/jvi.65.5.2254-2260.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruesch MN, Stubenrauch F, Laimins LA. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin-10. J Virol. 1998;72:5016–24. doi: 10.1128/jvi.72.6.5016-5024.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staretz ME, Murphy SE, Patten CJ, Nunes MG, Koehl W, Amin S, et al. Comparative metabolism of the tobacco-related carcinogens benzo[a]pyrene, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol, and N’- nitrosonornicotine in human hepatic microsomes. Drug Metab Dispos. 1997;25:154–62. [PubMed] [Google Scholar]
- 28.Meyers C, Alam S, Mane M, Hermonatt PL. Altered biology of adeno-associated virus type 2 and human papillomavirus during dual infection of natural host tissue. Virology. 2001;287:30–9. doi: 10.1006/viro.2001.0968. [DOI] [PubMed] [Google Scholar]
- 29.Wen X, Walle T. Preferential induction of CYP1B1 by benzo[a]pyrene in human oral epithelial cells: impact on DNA adduct formation and prevention by polyphenols. Carcinogenesis. 2005;26:1774–81. doi: 10.1093/carcin/bgi127. [DOI] [PubMed] [Google Scholar]
- 30.Tsuji PA, Walle T. Inhibition of benzo[a]pyrene-activating enzymes and DNA binding in human bronchial epithelial BEAS-2B cells by methoxylated flavonoids. Carcinogenesis. 2006;27:1570–85. doi: 10.1093/carcin/bgi358. [DOI] [PubMed] [Google Scholar]