

Abstract
A proline to serine substitution at position 56 in the gene encoding vesicle-associated membrane protein-associated protein B (VAPB) causes some dominantly inherited familial forms of motor neuron disease including amyotrophic lateral sclerosis (ALS) type-8. VAPB is an integral endoplasmic reticulum (ER) protein whose amino-terminus projects into the cytosol. Overexpression of ALS mutant VAPBP56S disrupts ER structure but the mechanisms by which it induces disease are not properly understood. Here we show that VAPB interacts with the outer mitochondrial membrane protein, protein tyrosine phosphatase-interacting protein 51 (PTPIP51). ER and mitochondria are both stores for intracellular calcium (Ca2+) and Ca2+ exchange between these organelles occurs at regions of ER that are closely apposed to mitochondria. These are termed mitochondria-associated membranes (MAM). We demonstrate that VAPB is a MAM protein and that loss of either VAPB or PTPIP51 perturbs uptake of Ca2+ by mitochondria following release from ER stores. Finally, we demonstrate that VAPBP56S has altered binding to PTPIP51 and increases Ca2+ uptake by mitochondria following release from ER stores. Damage to ER, mitochondria and Ca2+ homeostasis are all seen in ALS and we discuss the implications of our findings in this context.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is an adult onset neurodegenerative disease characterized by selective loss of motor neurons in the spinal cord, motor cortex and brain stem, which leads to progressive muscle atrophy and ultimately paralysis and death, typically within 3–5 years of onset. Most forms of ALS are sporadic but approximately 5% are inherited and mutations in a number of genes have now been shown to be causative for these familial forms (1,2).
A mutation in the gene encoding vesicle-associated membrane protein-associated protein B (VAPB) causes ALS type-8 and some other related forms of motor neuron disease including late onset spinal muscular atrophy (3). VAPB is an integral endoplasmic reticulum (ER) membrane protein. It contains an N-terminal domain homologous to the major sperm protein of nematode worms, a central coiled-coil region and a C-terminal transmembrane domain through which it is anchored in the ER membrane; the N-terminus of VAPB projects from the ER into the cytoplasm (4–9). The mutation that causes ALS type-8 involves a proline to serine substitution at position-56 (VAPBP56S) although a further mutation (VAPBT46I) has recently been identified in a single ALS patient and this too may cause ALS (3,10).
VAPBP56S induces the formation of abnormal ER-derived inclusions (3,8,9,11–13) but the mechanisms by which VAPBP56S induces disease are not clear and this is partly because the function of VAPB is not properly understood. VAPB has been implicated in a variety of processes including ER stress and the unfolded protein response (UPR), ER to Golgi transport and bouton formation at the neuromuscular junction (6,14–19). There is also evidence that implicates VAPB in microtubule organization (7,14,20) and finally, a cleaved and secreted VAPB fragment acts as a ligand for ephrin receptors (21). ER stress is linked to the pathogenesis of ALS (22,23) and several studies implicate VAPBP56S in abnormal UPR but again the mechanisms are unclear (6,10,11,18,19,21).
Here, we report that VAPB interacts with the outer mitochondrial membrane protein, protein tyrosine phosphatase-interacting protein 51 (PTPIP51). ER and mitochondria are both stores for calcium (Ca2+) and we demonstrate that the VAPB–PTPIP51 interaction impacts on intracellular Ca2+ handling and that VAPBP56S has altered properties in this function. Damage to Ca2+ homeostasis is seen in ALS (24,25) and as such, our results provide a novel function for VAPB that has relevance to ALS.
RESULTS
VAPB interacts with PTPIP51
We screened a human brain cDNA yeast two-hybrid library with the cytoplasmic domain of VAPB as ‘bait’ [i.e. VAPB lacking its carboxy-terminal transmembrane domain; amino acids (aa) 1–220]. We isolated four interacting clones that all encoded partial PTPIP51 sequences [aa36–470, aa84–470 (2 clones), aa75–175] (Fig. 1A). PTPIP51 is also known as family with sequence similarity 82 member A2 (FAM82A2), human cerebral protein-10 and regulator of microtubule dynamics protein-3 (RMD-3). PTPIP51 binds protein tyrosine phosphatase-1B and T-cell protein tyrosine phosphatase in yeast two-hybrid (26) and proximity ligation assays (27), but the functional significance of these interactions is unknown. RMD-3 is a putative homologue of PTPIP51 in Caenorhabditis elegans and another RMD family member, RMD-1 functions in chromosome segregation (28). However, in mammalian cells, PTPIP51 is a mitochondrial protein of unclear function that has been implicated in the regulation of cell morphology, motility and apoptosis and which contains an amino-terminal transmembrane domain and a central coiled-coil domain (29,30) (Fig. 1A). Both VAPB and PTPIP51 are ubiquitously expressed although their expression levels vary in different tissues (7,26,29).
Figure 1.

VAPB interacts with PTPIP51. (A) Domain structure of PTPIP51. CC, coiled coil domain; TM, transmembrane domain. The VAPB-interacting PTPIP51 clones identified by yeast two-hybrid are indicated. (B) Myc-VAPB and HA-PTPIP51 co-immunoprecipitate in transfected HEK293 cells. Myc-VAPB was immunoprecipitated with anti-myc antibody from cells transfected with myc-VAPB and/or HA-PTPIP51. Non-immune mouse antibody was used as control (Ctrl). The immune pellets were probed for myc-VAPB (Myc) and HA-PTPIP51 (HA) on immunoblots. The input levels of myc-VAPB and HA-PTPIP51 in the transfected cells are shown (input). (C) Endogenous PTPIP51 and VAPB co-immunoprecipitate in HEK293 cells. PTPIP51 was immunoprecipitated using rat anti-PTPIP51 antibody (PTPIP51 IP) and the immune pellet probed for VAPB and PTPIP51 on immunoblots with rabbit VAPB (#3504) and PTPIP51 (FAM82A2) antibodies, respectively; immunoprecipitation with pre-immune rat serum was used as a control (Ctrl IP). A sample of the input lysate is also shown. (D) Quantification of in situ proximity ligation assay results (mean ± SEM). Cells were probed with no primary antibodies (no Abs) (n = 10), with VAPB antibody only (VAPB) (n = 10), with PTPIP51 antibody only (PTPIP51) (n = 10) and with VAPB+PTPIP51 antibodies (n = 32) and the numbers of signals/cell determined. Representative VAPB+PTPIP51 labelling with corresponding phase contrast image are shown (scale bar, 20 μm).
To confirm the interaction between VAPB and PTPIP51, we first performed co-immunoprecipitation assays of transfected and endogenous proteins in HEK293 cells. Hemagglutinin (HA)-tagged PTPIP51 (HA-PTPIP51) co-immunoprecipitated with myc-tagged VAPB (myc-VAPB) from HA-PTPIP51 and myc-VAPB co-transfected cells but not HA-PTPIP51-only transfected cells and this interaction was not detected when the immunoprecipitating myc antibody was replaced with non-immune mouse antibody (Fig. 1B). Likewise, endogenous VAPB co-immunoprecipitated with endogenous PTPIP51 but this interaction was abrogated when pre-immune serum was used instead of immunoprecipitating PTPIP51 antibody (Fig. 1C). Both PTPIP51 and VAPB antibodies detected protein species of the correct molecular masses (Supplementary Material, Fig. S1).
We also monitored the endogenous VAPB and PTPIP51 interaction in cells using an in vivo, in situ proximity ligation assay (31). Here, conventionally fixed and permeabilized cultured CV1 cells were probed with anti-VAPB and anti-PTPIP51 primary antibodies. However, instead of fluorescently labelled secondary antibodies, antibodies coupled to different oligonucleotides (one for each of the two primary antibodies) were used as secondary antibodies. If the distance between two antibody-coupled oligonucleotides of different types is less than 50 nm, they can hybridize and serve as primers for rolling-circle amplification with fluorescent oligonucleotides. The resulting fluorescent signals can then be imaged using by fluorescence microscopy and correspond to interacting protein pairs. Using VAPB and PTPIP51 antibodies, we observed proximity signals in all cells examined (Fig. 1D), confirming the VAPB–PTPIP51 interaction in intact cells. As negative controls, we omitted either or both of the primary antibodies from the assays and these produced only very low numbers of proximity signals. Together these data identify PTPIP51 as a novel VAPB binding protein.
PTPIP51 is an amino-terminal anchored outer mitochondrial membrane protein
Although PTPIP51 is a mitochondrial protein (29), its precise sub-mitochondrial localization and membrane orientation are not known. Our discovery that PTPIP51 interacts with VAPB, an ER protein whose amino-terminus project into the cytosol, suggests that PTPIP51 is an outer mitochondrial membrane protein whose carboxy-terminus projects into the cytosol. To begin to test this possibility, we first confirmed the mitochondrial localization of PTPIP51 and the role of its amino-terminal transmembrane domain. Endogenous PTPIP51 co-localized with the known outer mitochondrial membrane protein translocase of outer membrane-20 (TOM20) (Fig. 2A). Likewise, transfected HA-PTPIP51 also localized to mitochondria (Fig. 2B). However, HA-PTPIP51 lacking its amino-terminal transmembrane domain (HA-PTPIP51ΔTM) displayed diffuse labelling with no clear mitochondrial localization whereas an amino-terminal PTPIP51 domain containing the transmembrane domain fused to EGFP [EGFP-PTPIP51(1–72)] localized to mitochondria (Fig. 2B). Thus, sequences encompassing the amino-terminal transmembrane domain are responsible for targeting PTPIP51 to mitochondria. These findings are in agreement with a previous report (29).
Figure 2.

PTPIP51 is an integral OMM protein. (A) PTPIP51 localizes to mitochondria. Endogenous PTPIP51 and the mitochondrial protein TOM20 were visualized in CV1 cells by immunofluorescence microscopy. Scale bar, 20 μm. (B) Sequences spanning the amino-terminal transmembrane domain of PTPIP5 are involved in targeting PTPIP51 to mitochondria. CV1 cells were co-transfected with either full-length HA-PTPIP51 and AcGFP-Mito (a), or HA-PTPIP51 lacking its amino-terminal transmembrane domain (HA-PTPIP51▵TM) and AcGFP-Mito (b), or the amino-terminal 72 amino acids of PTPIP51 (which contain the transmembrane domain) fused to EGFP (EGFP-PTPIP51TM) (c). In (a) and (b), HA-PTPIP51 and HA-PTPIP51▵TM were detected by immunostaining with anti-HA antibody and mitochondria with AcGFP-mito. In (c), PTPIP51TM was detected with the EGFP tag and mitochondria by immunostaining with antibody to MnSOD. Full-length HA-PTPIP51 and EGFP-PTPIP51TM both localized to mitochondria whereas HA-PTPIP51▵TM labelling was cytosolic. Scale bar, 20 μm. (C–E) PTPIP51 is an outer mitochondrial membrane protein. (C) Outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM) plus mitochondrial matrix fractions (matrix/IMM) were prepared and probed for PTPIP51, the OMM markers Miro1, VDAC and TOM20, the integral IMM protein TIM23 and mitochondrial matrix protein HSP60. (D) Mitochondria were treated with proteinase K or alkaline as indicated; control treatments involved incubation in appropriate buffer (Ctrl). After treatment, samples were probed for PTPIP51, HSP60 (mitochondrial matrix), TIM23 (IMM), TOM20 (OMM) and cytochrome c (peripheral IMM) on immunoblots. Input shows untreated mitochondria (left panel). (E) Isolated mitochondria labelled with MitoTracker GreenFM were immunostained for PTPIP51 and the mitochondrial matrix, inner and outer mitochondrial membrane proteins TOM20, TIM23, COXIV, MnSOD and HSP60. Antibodies were detected with Alexa633-conjugated secondary antibody and the numbers of mitochondria showing Alexa633 signal were quantified from confocal images. As a negative control, mitochondria were incubated with secondary antibody only (CTRL). Bar graph shows percentages of mitochondria labelled with the different antibodies. Also shown are representative images mitochondria in control (CTRL), PTPIP51 and TOM20 labelled samples (image width = 5 μm).
We next determined the sub-mitochondrial localization and membrane orientation of PTPIP51 using three different assays. First, we separated outer from inner mitochondrial membranes and the mitochondrial matrix by digitonin treatment of HEK293 cell mitochondria (32) and probed these fractions for PTPIP51 on immunoblots. As controls, we probed for well-characterized outer mitochondrial membrane proteins TOM20, voltage-dependent anion channel (VDAC) and mitochondrial Rho GTPase 1 (Miro1), the inner mitochondrial membrane proteins translocase of inner membrane-23 (TIM23) and the mitochondrial matrix protein heat shock protein 60 (HSP60). The known mitochondrial proteins were present in the correct fractions and PTPIP51 was exclusively in the outer mitochondrial membrane fraction (Fig. 2C).
Secondly, we treated mitochondria with proteinase K or alkaline. Membrane-anchored outer mitochondrial membrane proteins exposed to the cytosol are degraded by proteinase K but are resistant to alkaline (33). Proteinase K but not alkaline treatment removed PTPIP51 signal on immunoblots and this was identical to TOM20 that was used as a positive control (Fig. 2D). The loss of PTPIP51 signal was not due to disintegration of mitochondria because the matrix protein HSP60 and two inner mitochondrial membrane proteins, TIM23 and cytochrome c, resisted the proteinase K treatment. Alkaline extraction was effective because the peripheral inner mitochondrial membrane protein cytochrome c was removed after treatment (Fig. 2D).
Thirdly, we immunostained freshly purified mitochondria in solution using an antibody raised against PTPIP51 that lacked the amino-terminal transmembrane domain; only PTPIP51 exposed to the cytosol can interact with the antibody in this assay. As a positive control, we labelled mitochondria with an antibody for the outer mitochondrial membrane protein TOM20, and as negative controls we labelled mitochondria with antibodies for the inner mitochondrial membrane proteins TIM23 and cytochrome oxidase subunit IV (COXIV), or the matrix proteins HSP60 and Mn-superoxide dismutase (MnSOD). Most mitochondria were labelled with PTPIP51 and positive control TOM20 antibodies but the negative control antibodies stained only few mitochondria (Fig. 2E). These data support the notion that PTPIP51 is an integral outer mitochondrial membrane protein that projects into the cytosol and complement the immunoprecipitation and proximity ligation assays, which demonstrate that VAPB and PTPIP51 are binding partners.
VAPB is present in mitochondria-associated membranes (MAM) and modulating PTPIP51 expression alters the association of VAPB with mitochondria
Mitochondria and ER are closely associated with between 5 and 20% of the mitochondrial surface being closely apposed to ER membrane domains (34). These ER domains are termed mitochondria-associated membranes (MAM) and can be purified by Percoll gradient centrifugation (34–36). Our finding that VAPB interacts with PTPIP51 suggests that VAPB is present in MAM. To test this possibility, we prepared MAM, mitochondria and ER from HEK293 cells and probed for VAPB on immunoblots. As controls, the samples were also probed for PTPIP51 and HSP60 as mitochondrial markers, fatty-acid-coenzyme A ligase long-chain 4 (FACL4) as a MAM marker (37,38) and protein disulfide-isomerase (PDI) as a general ER marker. All markers were enriched in their respective fractions and a prominent signal for VAPB was obtained in the MAM fraction (Fig. 3A). We also compared the localization of endogenous VAPB with that of the type III inositol 1,4,5-trisphosphate receptor (IP3R3), an ER protein enriched in MAM (38), and mitochondria by confocal laser scanning microscopy. A proportion of VAPB and IP3R3 were present as punctate structures that aligned with mitochondria (Fig. 3B).
Figure 3.

VAPB is a MAM protein. (A) MAM, mitochondria (Mito) and ER fractions were prepared from HEK293 cells and equal protein amounts from each fraction probed on immunoblots for PTPIP51 and HSP60 (mitochondrial proteins) FACL4 (MAM enriched protein), PDI (ER protein) and VAPB. (B) A proportion of VAPB colocalizes with IP3R3 and mitochondria. CV1 cells were immunostained for VAPB, IP3R3 (MAM enriched protein) and HSP60 (mitochondrial protein). Some punctate VAPB and IP3R3 staining aligned with mitochondria (Merge). Zoomed cellular areas are indicated [zoom (a), and zoom (b)]. Colocalized VAPB and IP3R3 labelling along mitochondria is indicated with arrowheads. Scale bar, 20 μm; zoom (a), 4 μm.
We next tested whether expression of PTPIP51 influences the association of VAPB with mitochondria. To do so, we modulated PTPIP51 expression and monitored on immunoblots, the presence of VAPB in a biochemical fraction that contains both mitochondria and MAM but not ER. PTPIP51 expression was increased by transfection of HA-PTPIP51 and reduced by use of PTPIP51 siRNAs. Two different PTPIP51 siRNAs (PTPIP51#10 and PTPIP51#11) were used and both achieved knockdowns of over 90% without affecting VAPB expression (Fig. 4C). Transfection of HA-PTPIP51 increased whereas siRNA knockdown decreased the amount of endogenous VAPB that was associated with mitochondria (Fig. 4A and B). We also monitored how overexpression of PTPIP51 influenced the cellular distribution of VAPB in relation to mitochondria by immunofluorescence microscopy. In non-transfected control cells, VAPB displayed a vesicular cytosolic pattern consistent with its known localization to ER. However, transfection of HA-PTPIP51 induced a noticeable redistribution of VAPB such that it appeared more closely aligned with mitochondria (Fig. 4D). Thus, although VAPB is not exclusively localized to MAM, a significant proportion is present in this ER compartment and modulating PTPIP51 expression influences the amount of VAPB that is associated with mitochondria.
Figure 4.

Modulating PTPIP51 expression influences VAPB association with mitochondria. (A and B) Overexpression or siRNA knockdown of PTPIP51 influences the amount of endogenous VAPB present in a mitochondria plus MAM but not ER containing fraction. Mitochondria plus MAM fractions were prepared from HEK293 cells transfected with control empty vector (Ctrl) or HA-PTPIP51 (A), or control siRNA (Ctrl) or PTPIP51 siRNAs (B). Samples were probed for HA-PTPIP51 (using anti HA) in (A) or endogenous PTPIP51 in (B), and VAPB and COXIV as a mitochondrial marker. (C) shows siRNA knockdown of PTPIP51 in cells transfected with control or PTPIP51 (PTPIP51#10, PTPIP51#11) siRNAs. Samples were also probed for VAPB and tubulin as indicated. (D) Overexpression of PTPIP51 increases the localization of endogenous VAPB with mitochondria. CV1 cells were co-transfected with DsRed-Mito (to label mitochondria) and either control empty vector (CRTL) or HA-PTPIP51 (+HA-PTPIP51). Cells were then immunostained for VAPB and HA-PTPIP51 (using HA antibody). Representative confocal images show DeRed-Mito (mitochondria), VAPB and HA-PTPIP51. Scale bar, 20 μm.
Depletion of VAPB and PTPIP51 disturbs Ca2+ handling
Both ER and mitochondria are stores for intracellular Ca2+. Moreover, the ER–mitochondria interface and MAM play a crucial role in Ca2+ exchange between these organelles; disruption of ER–mitochondria connections perturbs Ca2+ uptake by mitochondria following release from ER stores (34,36,37,39). To assess if VAPB or PTPIP51 affect Ca2+ homeostasis, we suppressed VAPB or PTPIP51 expression using siRNAs and monitored cytosolic Ca2+ levels ([Ca2+]c) after induction of inositol 1,4,5-trisphosphate receptor (IP3R)-mediated Ca2+ release from ER stores. Again two different siRNAs each for PTPIP51 and VAPB were used and all achieved knockdowns of over 90%; siRNA loss of PTPIP51 did not influence expression of VAPB and loss of VAPB did not influence expression of PTPIP51 (Figs 4C and 5A). For these experiments, we used HEK293 cells transfected with the M3 muscarinic-ACh-receptor (M3R) and triggered physiological IP3R-mediated Ca2+ release from ER stores by application of the M3R agonist oxotremorine-M (Oxo-M). [Ca2+]c was measured with the Ca2+ indicator dye Fluo4-AM and was calculated as relative fluorescence compared to baseline fluorescence at the beginning of the measurement (F/F0). In VAPB or PTPIP51 siRNA-treated cells, the peak Oxo-M-induced [Ca2+]c reached significantly higher levels compared to control siRNA-treated cells (Fig. 5B). Mitochondria rapidly take up Ca2+ released from ER stores (34,36,37,39). Thus, the increased peak [Ca2+]c observed in these cells might be caused by reduction and/or delay in mitochondrial Ca2+ uptake. We therefore measured mitochondrial Ca2+ levels ([Ca2+]m) under the same conditions using Rhod2-AM. Depletion of VAPB or PTPIP51 reduced the peak [Ca2+]m after IP3R-mediated Ca2+ release from ER stores (Fig. 5C). The magnitude of this reduction was similar to that previously reported in mitofusin 2 knockout cells; mitofusin 2 is known to connect and to regulate Ca2+ exchange between ER and mitochondria (37). Finally, we analysed the time lag between the time points where the peak [Ca2+]c and [Ca2+]m were reached in Fluo4/Rhod2 co-loaded cells. Both VAPB and PTPIP51 knockdown caused a significant delay in mitochondrial Ca2+ uptake (Fig. 5D). Thus, siRNA-mediated knockdown of either VAPB or PTPIP51 increased peak [Ca2+]c following IP3R-mediated Ca2+ release from ER stores and this was associated with a reduction and delay in mitochondrial Ca2+ uptake.
Figure 5.

siRNA knockdown of VAPB or PTPIP51 disturbs Ca2+ handling. (A) shows siRNA knockdown of VAPB without influencing PTPIP51 expression. HEK293 cells were transfected control siRNA (CTRL) or two different VAPB siRNAs (VAPB#05, VAPB#07) and the samples then probed on immunoblots for VAPB and PTPIP51 as indicated. (B and C) siRNA loss of either VAPB or PTPIP51 increases cytosolic (B) and decreases mitochondrial (C) Ca2+ levels upon Oxo-M induced Ca2+ release from ER stores. HEK293 cells were transfected with M3R and control (CTRL), VAPB#05, VAPB#07, PTPIP51#10 or PTPIP51#11 siRNAs and treated with Oxo-M. Representative traces of Fluo4 and Rhod2 fluorescence are shown on the left and normalized peak values are shown on the right. Fluo4 and Rhod2 fluorescence show a transient increase in cytosolic (B) and mitochondrial (C) Ca2+ levels upon Oxo-M induced Ca2+ release from ER stores. However, compared to control, VAPB and PTPIP51 siRNAs increased the peak [Ca2+]c levels (B, right; mean ± SEM; CTRL, n = 158 cells; VAPB#05, n = 255; VAPB#07, n = 108; PTPIP51#10, n = 149; PTPIP51#11, n = 184), and decreased the peak [Ca2+]m levels (C, right; control siRNA, n = 64; VAPB#05, n = 49; VAPB#07, n = 55; PTPIP51#10, n = 34; PTPIP51#11, n = 64). (D) siRNA knockdown of VAPB or PTPIP51 increases the time lag between peak [Ca2+]c and [Ca2+]m after Oxo-M induced release of Ca2+ from ER stores. The time lag between peak [Ca2+]c and [Ca2+]m after Oxo-M induced release of Ca2+ from ER stores was measured in HEK293 cells-treated with control (CTRL), VAPB#07 or PTPIP51#10 siRNAs as indicated. Representative traces show [Ca2+]c (full line) and [Ca2+]m (dashed line) in cells treated with control or VAPB siRNAs (left), or control or PTPIP51 siRNAs (middle). Bar chart shows time lag between peak [Ca2+]c and [Ca2+]m (mean ± SEM; control siRNA, n = 59; VAPB#07, n = 33; PTPIP51#10, n = 20).
VAPBP56S displays increased binding to PTPIP51 in immunoprecipitation assays and accumulates in MAM
VAPBP56S causes familial ALS type-8 and disrupts smooth ER (3,6,8,9,19,40). We therefore examined whether VAPBP56S had altered binding to PTPIP51 and distribution in MAM. First, we analysed the binding of VAPBP56S to PTPIP51 in immunoprecipitation assays. We co-transfected HEK293 cells with HA-PTPIP51 and myc-VAPB or HA-PTPIP51 and myc-VAPBP56S and compared the amounts of bound proteins following immunoprecipitation of either myc-VAPB/myc-VAPBP56S or HA-PTPIP51. Compared to myc-VAPB, myc-VAPBP56S bound approximately 3–4-fold more PTPIP51 in these assays (Fig. 6A).
Figure 6.

Compared to VAPB, VAPBP56S levels associated with PTPIP51 are elevated and VAPBP56S levels are increased in MAM. (A) VAPBP56S binds more PTPIP51 than VAPB in immunoprecipitation assays. HEK293 cells were co-transfected with either HA-PTPIP51 and myc-VAPB, or HA-PTPIP51 and myc-VAPBP56S. myc-VAPB/VAPBP56S or HA-PTPIP51 were immunoprecipitated using anti-myc or anti-HA antibodies and bound HA-PTPIP51/myc-VAPB then detected on immunoblots. Control immunoprecipitations (Ctrl) were performed with non-immune antibody. Upper panel shows myc-VAPB/VAPBP56S immunoprecipitations; middle panel shows HA-PTPIP51 immunoprecipitations. Lower panel shows input levels of transfected proteins. Bar chart shows relative amounts of co-immunoprecipitated PTPIP51 (upper) and VAPB (middle) following densitometric quantification of signals. **P < 0.01, n = 3, t-test. Error bars are mean ± SEM. (B) Compared to VAPB, VAPBP56S levels associated with mitochondria are increased. Mitochondria containing MAM were purified from HEK293 cells transfected with empty vector (Ctrl), myc-VAPB (VAPB) or myc-VAPBP56S (P56S). The samples were then probed on immunoblots for myc-VAPB/VAPBP56S using anti-myc antibody, tubulin and COXIV (as a mitochondrial marker). Bar chart shows relative amounts of myc-VAPB and myc-VAPBP56S following densitometric quantification of signals. *P< 0.05, n = 3, t-test. Error bars are mean ± SEM. (C) Compared to VAPB, VAPBP56S levels are increased in MAM and decreased in non-MAM ER. HEK293 cells transfected as in (B) were fractionated into MAM, pure mitochondria and ER. The samples were then probed on immunoblots for myc-VAPB, myc-VAPBP56S, IP3R3 (MAM marker), COXIV (mitochondria marker) and PDI (ER marker). Total shows immunoblot of the total cell lysates (before fractionation); equal loading was confirmed by immunoblot for tubulin.
Since PTPIP51 influences the amount of VAPB that is associated with mitochondria (Fig. 4), this increase in binding of VAPBP56S to PTPIP51 might lead to increased levels of VAPBP56S in MAM. To test this possibility, we prepared mitochondria and associated MAM but not ER from cells transfected with myc-VAPB or myc-VAPBP56S and determined the amount of VAPB in this fraction. Compared to myc-VAPB, myc-VAPBP56S levels were increased almost 2-fold in the mitochondria+MAM fraction (Fig. 6B). To further characterize this enrichment, we also compared the levels of myc-VAPB and myc-VAPBP56S in purified ER, mitochondria and MAM. Compared to myc-VAPB, myc-VAPBP56S levels were elevated in MAM and correspondingly decreased in non-MAM ER; myc-VAPB and myc-VAPBP56S were not detected in pure mitochondria (Fig. 6C). This increase in VAPBP56S in MAM was not due to altered fractionation properties of ER, as the levels of PDI in MAM did not change upon expression of VAPBP56S. Thus, VAPBP56S binds more PTPIP51 than VAPB in immunoprecipitation assays and VAPBP56S levels are elevated in MAM.
VAPBP56S induces clustering of mitochondria
The increased interaction of VAPBP56S with PTPIP51 suggests that VAPBP56S may influence mitochondria in some fashion. We therefore monitored mitochondrial distribution by immunostaining in cells transfected with either myc-VAPB or myc-VAPBP56S. Transfected VAPB was detected using the myc-tag and mitochondria visualized by staining with anti-MnSOD. Mitochondria in cells transfected with myc-VAPB were distributed throughout the cytoplasm in a pattern not noticeably different to that seen in empty vector-transfected cells (Fig. 7A and B). By contrast, transfection of myc-VAPBP56S induced perinuclear clustering of mitochondria. This was particularly noticeable in cells expressing higher levels of myc-VAPBP56S (as determined by fluorescent signal intensity) (Fig. 7C and D). This clustering of mitochondria was most marked in perinuclear regions containing VAPBP56S aggregates.
Figure 7.
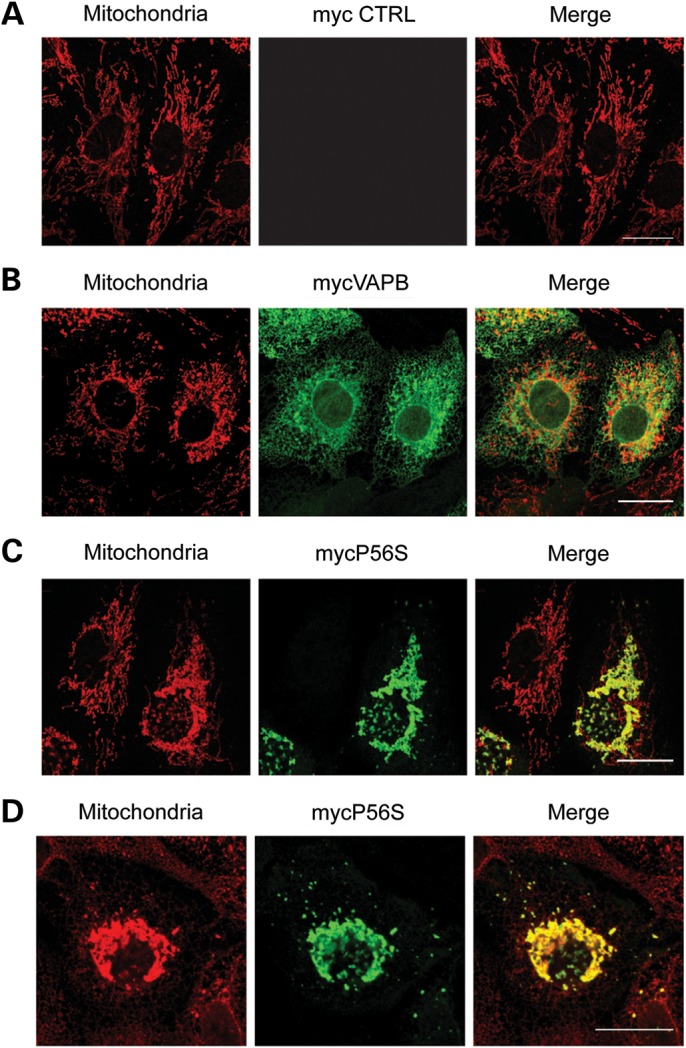
VAPBP56S induces clustering of mitochondria. CV1 cells transfected with empty vector (A), myc-VAPB (B) or myc-VAPBP56S (C and D) were immunostained for mitochondria (red) and VAPB/VAPBP56S (green) using MnSOD and myc antibodies, respectively. Representative CLSM images are shown. Scale bar = 20 μm.
VAPBP56S perturbs Ca2+ handling
Our findings that loss of VAPB and PTPIP51 disrupts Ca2+ handling following Ca2+ release from ER stores and that VAPBP56S has increased binding to PTPIP51 suggest that VAPBP56S may influence Ca2+ handling. To test this, we monitored [Ca2+]m and [Ca2+]c upon IP3R-mediated Ca2+ release from ER stores in HEK293 cells co-transfected with M3R and control vector, myc-VAPB or myc-VAPBP56S. Transfection of myc-VAPBP56S but not myc-VAPB significantly increased peak [Ca2+]m and decreased [Ca2+]c compared to control (Fig. 8A and B). To gain insight into the role of PTPIP51 in this effect, we reduced PTPIP51 expression using siRNAs and again monitored [Ca2+]m upon IP3R-mediated Ca2+ release from ER stores. Loss of PTPIP51 abrogated the effect of VAPBP56S on elevation of [Ca2+]m (Fig. 8C). Thus, expression of VAPBP56S but not VAPB disrupts mitochondrial Ca2+ handling and this effect is associated with its binding partner PTPIP51.
Figure 8.
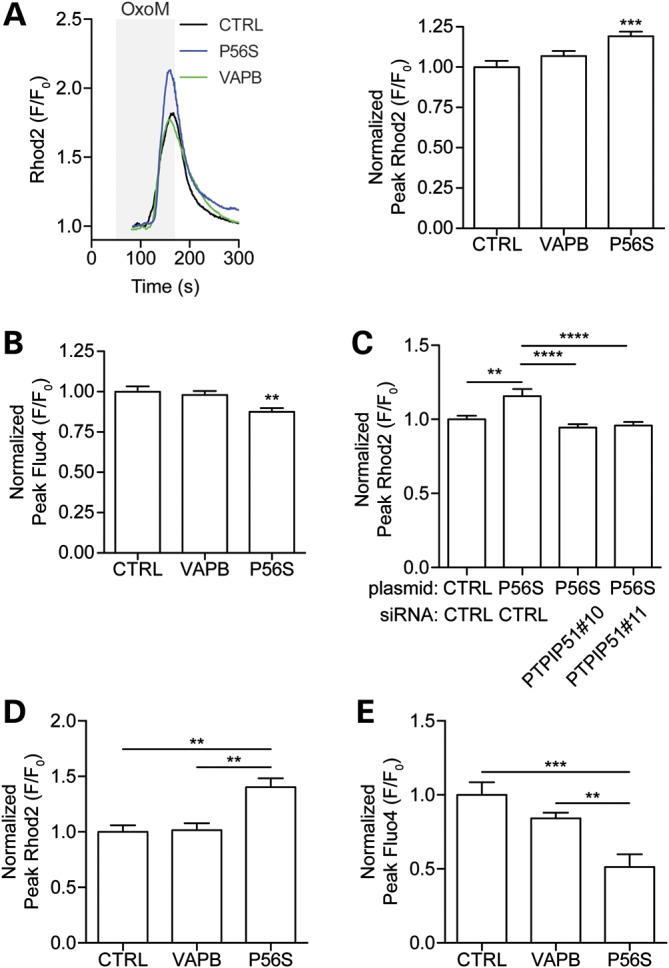
VAPBP56S perturbs Ca2+ handling. (A) VAPBP56S increases the peak [Ca2+]m following Ca2+ release from ER stores. HEK293 cells were co-transfected with M3R and either empty vector (CTRL), myc-VAPB (VAPB) or myc-VAPBP56S (P56S). Peak [Ca2+]m levels were then determined following application of Oxo-M to induce Ca2+ release from ER stores. Representative traces of Rhod2 fluorescence are shown on the left and normalized peak values are shown on the right (mean ± SEM; CTRL, n = 45; VAPB, n = 46, VAPBP56S, n = 79). (B) VAPBP56S decreases the peak [Ca2+]c following Ca2+ release from ER stores. HEK293 cells were co-transfected with M3R and either empty vector (CTRL), myc-VAPB (VAPB) or myc-VAPBP56S (P56S). Peak [Ca2+]c levels were then determined following application of Oxo-M to induce Ca2+ release from ER stores (mean ± SEM; CTRL, n = 47; VAPB, n = 82, VAPBP56S, n = 67). (C) siRNA loss of PTPIP51 abrogates the VAPBP56S induced increase in peak [Ca2+]m following Ca2+ release from ER stores. HEK293 cells treated with control (CTRL) or PTPIP51 siRNAs were co-transfected with M3R and either empty vector (CTRL), or myc-VAPBP56S (P56S). Peak [Ca2+]m were then determined following application of Oxo-M to induce Ca2+ release from ER stores. Values are mean ± SEM; CTRL siRNA + CTRL, n = 43; CTRL siRNA+VAPBP56S, n = 28; PTPIP51#10 siRNA+VAPBP56S, n = 39; PTPIP51#11 siRNA+VAPBP56S, n = 40. (D) VAPBP56S increases the peak [Ca2+]m following depolarization of rat cortical neurons. Neurons were transfected with either CFP (CTRL), CFP-VAPB (VAPB) or CFP-VAPBP56S (P56S) and [Ca2+]m determined using Rhod2 after transient influx of Ca2+ by a 2 min application of 50 mm KCl to depolarize the neurons (mean ± SEM; CTRL, n = 6; VAPB, n = 10; P56S, n = 4). (E) VAPBP56S decreases the peak [Ca2+]c following depolarization of rat cortical neurons. Neurons were transfected with either CFP (CTRL), CFP-VAPB (VAPB) or CFP-VAPBP56S (P56S) and [Ca2+]c determined using Fluo4 after transient influx of Ca2+ by a 2 min application of 50 mm KCl to depolarize the neurons (mean ± SEM; CTRL, n = 4; VAPB, n = 9; P56S, n = 5).
Finally, we monitored the effect of VAPBP56S on calcium handling in neurons. Cultured rat cortical neurons were transfected with CFP, CFP-VAPB or CFP-VAPBP56S and [Ca2+]m and [Ca2+]c determined following depolarization of the neurons to induce a transient increase in intracellular Ca2+. VAPBP56S caused an increase in peak [Ca2+]m and a decrease in peak [Ca2+]c compared to VAPB or CFP controls (Fig. 8D and E). Thus VAPBP56S also disturbs Ca2+ handling in neurons.
DISCUSSION
Both ER and mitochondria are important stores for intracellular Ca2+ and exchange of Ca2+ between these organelles impacts upon a number of physiological processes. To facilitate this exchange, up to 20% of the mitochondrial surface is closely apposed to ER membranes (34,36). These ER domains associated with mitochondria are termed MAM (41–43). Here, we report that the integral ER protein VAPB interacts with the outer mitochondrial membrane protein PTPIP51 and that a proportion of VAPB is present in MAM. We also present evidence that both VAPB and PTPIP51 are involved in the regulation of intracellular Ca2+ homeostasis. In particular, siRNA loss of VAPB or PTPIP51 reduced [Ca2+]m following release of Ca2+ from ER stores. Interestingly, the magnitude of this reduction (approximately 86% of control) was very similar to that reported previously in mitofusin 2 knockout cells (approximately 82% of control); mitofusin 2 is a known mediator of ER–mitochondria connections that regulates Ca2+ exchange between these organelles (37). Finally, we demonstrate that VAPBP56S, which causes familial ALS type-8, has altered binding to PTPIP51 and disturbs Ca2+ handling.
The mechanisms by which VAPBP56S induce disease are not known although several studies implicate a role for ER stress and the UPR in this process. However, the precise details are controversial. In some reports, expression of VAPB induces UPR and VAPBP56S inhibits this process (6,10,18). In another, expression of VAPB and VAPBP56S both inhibit UPR but VAPBP56S is more potent in this effect (19). Finally, other studies have shown that VAPB and/or VAPBP56S can induce ER stress and UPR (11,21). Whatever the precise details, recent studies have shown that the UPR can influence Ca2+ release from ER stores (44). VAPBP56S perturbation of the ER structure and/or UPR may therefore impact on this process. Interestingly, mutants of Cu/Zn superoxide dismutase-1 (SOD1) that cause familial ALS type-1 have also been implicated in ER stress (22) and elevated Ca2+ levels are also seen in mutant SOD1 expressing cells (24). Despite this, mutant SOD1 induced alterations to mitochondria morphology and cell toxicity can be independent of any [Ca2+]m increases (24,45). By contrast, others have recently demonstrated that VAPBP56S toxicity in motor neurons is linked to defects in Ca2+ handling (11). Clearly, further studies on this topic will help resolve these issues.
Damage to both mitochondria and ER are seen in ALS (reviewed in 23,46,47). Thus mutant ALS SOD1 selectively associates with mitochondria and perturbs mitochondrial function (48–53). Likewise, TDP-43 that is present in ubiquitinated inclusions in ALS and which when mutated can also cause familial ALS (54–56) has been linked to mitochondrial dysfunction (57,58). In apparent contrast, both mutant SOD1 and TDP-43 mis-metabolism have been linked to ER stress (22,59) and VAPB functions in ER stress and the UPR (6,10,11,18,19). Our findings that VAPB is a MAM protein that interacts with PTPIP51 suggest that such damage to mitochondria and ER in ALS may involve the ER–mitochondrial axis. Indeed, one known molecular target for damage by ALS mutant SOD1 is the VDAC (45). VDAC is located in the outer mitochondrial membrane and interacts with IP3Rs in ER via glucose-regulated protein 75 (60,61). Also, mutations in mitofusin 2 cause forms of Charcot-Marie-Tooth disease (62) and mitofusin 2 has recently been shown to tether ER to mitochondria and regulate Ca2+ exchange between these organelles (37). Future studies aimed at examining the role of MAM and the ER/mitochondria axis in ALS may thus be warranted.
MATERIALS AND METHODS
Plasmids
Amino-terminal myc-tagged VAPB (myc-VAPB) was generated by polymerase chain reaction (PCR) from a human VAPB clone purchased from Origene and cloned into pCI-Neo (Promega). Myc-VAPBP56S was generated from myc-VAPB with a QuikChange mutagenesis kit (Stratagene). CFP-VAPB and CFP-VAPBP56S were produced by cloning into pECFP-C1 (Clontech). Carboxy-terminal HA-tagged PTPIP51 (HA-PTPIP51) was generated by PCR from a full-length human PTPIP51 cDNA clone (National Institute of Technology and Evaluation, Tokyo, Japan) and cloned into pCI-Neo.
Primer sequences were—myc-VAPB: ATCGGTCGACGCCACCATGGAGCAGAAACTCATCTCTGAAGAGGATCTGATGGCGAAGGTGGAGCAGGTCCTGAGCCTCGAGCCGCAG and ATCGGTCGACCTACAAGGCAATCTTCCCAATAATTACACCAACG; myc-VAPBP56S mutagenic primer: ACCACGTAGGTACTGTGTGAGGTCCAACAGCGGAATCATCGATGC, HA-PTPIP51: ATCGGAATTCGCCACCATGTCTAGACTGGGAGCCCTGGGTGGTGCCCGTG and CGATGTCGACTTAAGCGTAATCTGGAACATCGTATGGGTAGTCTCGTAAAATGACTTCCAGTTCTTCCAGGTCCTTCTGGATAGC.
pDsRed-mito and pAcGFP1-mito were from Clontech and muscarinic-ACh-receptor type 3 was a gift of Dr E. Seward, University of Sheffield, UK.
Antibodies
VAPB antibodies were raised in rat (sk83) and rabbit (#3504) and PTPIP51 antibodies in rat (skr9) by immunization with glutathione S-transferase (GST)-VAPB(1–220) and GST-PTPIP51(36–470), respectively. These antibodies detected single species of the correct molecular mass by immunoblotting (Supplementary Material, Fig. S1). Primary antibodies used were: rabbit anti-PTPIP51 (FAM82A2) and rabbit anti-Miro1 (RHOT1) (both from Atlas Antibodies); mouse anti-TIM23 and mouse anti-TOM20 (BD Transduction Laboratories), mouse anti-myc (9B11), rabbit anti-cytochrome c, rabbit anti-COXIV and rabbit anti-VDAC (Cell Signaling Technology); rabbit anti-HA, mouse anti-tubulin (DM1A) and mouse anti-HSP60 (Sigma); rabbit anti-myc (Upstate Biotechnology); rabbit anti-MnSOD (Stressgen); mouse anti-PDI (RL77, Affinity BioReagents); rabbit anti-IP3R3 (Millipore); rabbit anti-ACLS4 (FACL4; Santa Cruz). Secondary antibodies were horseradish peroxidase-coupled goat anti-mouse, anti-rabbit and anti-rat Ig (GE Healthcare or Dako), Alexa fluorophore (488, 546, 633)-coupled goat anti-mouse, anti-rabbit and anti-rat IgG (Invitrogen), Cy3 or Cy5-coupled donkey anti-mouse, anti-rabbit and anti-rat IgG (Jackson ImmunoResearch).
Yeast two-hybrid screen
Library screens were performed using a pre-transformed human brain cDNA library (Clontech) with VAPB ‘bait’ lacking it carboxy-terminal transmembrane domain (VAPB1–220) in pY1 according to the manufacturer's instructions. Following mating yeast underwent tryp−/leu−/his− selection and vigorously growing clones were subjected to β-galactosidase assays. Prey plasmids were rescued by transformation into Escherichia coli and positive plasmids identified by co-transformation back into yeast either alone or with VAPB(1–220) bait as described elsewhere (63).
Cell culture and plasmid transfection
HEK293 and CV1 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) containing 4.5 g/l glucose, 10% fetal bovine serum (Sera Laboratories), 2 mm l-glutamine (Invitrogen), and 1 mm sodium pyruvate (Sigma) and were transfected with Exgen500 according to the manufacturer's instructions (Fermentas). HEK293 cells were used for biochemical and Ca2+ measurement studies because of the high transfection efficiencies that are achieved in this cell type. CV1 cells were used for immunostaining experiments because they are particularly suitable for studying intracellular structure due to their large size and highly spread morphology. All cells were used in experiments 16–24 h post-transfection.
Cortical neurons were isolated from embryonic day 18 rat embryos and cultured on glass coverslips coated with poly-l-lysine in 6 or 12-well plates in neurobasal medium supplemented with B27 supplement (Invitrogen), 100 IU/ml penicillin, 100 μg/ml streptomycin and 2 mm l-glutamine. Neurons were cultured for 5 days and then transfected using a calcium phosphate Profection kit (Promega) as previously described (64). Neurons were used in experiments 48 h post-transfection.
siRNA sequences and transfection
VAPB and PTPIP51 siRNAs were from Dharmacon or Invitrogen. Non-targeting control siRNA was from Dharmacon. siRNA sequences were: VAPB#05: UGUUACAGCCUUUCGAUUA, VAPB#07: GCUCUUGGCUCUGGUGGUU, PTPIP51#10: CCUUAGACCUUGCUGAGAUUU, and PTPIP51#11: GAAGCUAGAUGGUGGAUGAUU. HEK293 cells were siRNA transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The cells were used for experiments 4 days post-transfection.
Immunoprecipitation
Cells were harvested in ice-cold lysis buffer [50 mm Tris–HCl pH 7.4, 150 mm NaCl, 1 mm ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100 and complete protease inhibitors (Roche)], and lysed for 30 min on ice. The lysate was cleared by centrifugation at 100 000g for 30 min at 4°C and precleared with protein G sepharose beads (Sigma), followed by incubation with antibodies for 16 h at 4°C. Antibody was captured with protein G sepharose for 2 h at 4°C. The immune pellets were washed with ice-cold lysis buffer and analysed by SDS–PAGE and immunoblotting.
Immunostaining
Immunostaining was performed as described previously (65). CV1 cells on glass coverslips were fixed with 3.7% formaldehyde in phosphate buffered saline (PBS) for 20 min at room temperature. After washing with PBS, residual formaldehyde was quenched by incubation with 50 mm NH4Cl in PBS for 15 min at room temperature, followed by a second round of washing with PBS. Subsequently, the cells were permeabilized with 0.2% Triton X-100 in PBS for 3 min. Alternatively, cells were fixed and permeabilized with −20°C MeOH for 20 min (Figs 3B and 7), and MeOH was removed by washing with PBS.
After fixing, the cells were incubated with PBS containing 0.2% fish gelatin (PBS/F) for 30 min at room temperature and then with the primary antibody in PBS/F for 1 h. After washing with PBS/F, the cells were incubated with secondary antibody in PBS/F for 45 min at room temperature. After a final wash, the samples were mounted in Moviol (Calbiochem) containing 1% DABCO (Sigma). Immunostaining of isolated mitochondria was performed as described previously (66).
In situ proximity ligation assays
In situ proximity ligation assays were performed with rabbit VAPB and rat PTPIP51 antibodies using a Duolink kit following the manufacturer's protocol (Olink Bioscience). Proximity signals were quantified using the Particle Analysis function of ImageJ.
Microscopy
Confocal images were recorded with an LSM510Meta confocal microscope equipped with a 63×/1.4NA Plan-Apochromat objective using a pinhole size of one Airy unit (Carl Zeiss). Conventional immunofluorescence microscopy was done with a Leica DM5000B microscope equipped with 63×/1.25NA and 40×/0.75NA HCX-PL-FLUOTAR objectives and appropriate filtersets (Leica).
Cell fractionation
ER, MAM and mitochondria were prepared from HEK293 cells as described elsewhere (35). Briefly, cells were harvested and washed by centrifugation at 13 000g for 30 s once with PBS and once with isolation buffer (250 mm mannitol, 5 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.4, 0.5 mm ethylene glycol tetraacetic acid (EGTA), 0.1% bovine serum albumin (BSA) and complete protease inhibitors) at 4°C. The cell pellet was resuspended in isolation buffer and homogenized using a teflon/glass dounce homogenizer (100 strokes; Kontes Pestle 19; Kimble Chase). The homogenate was centrifuged twice at 600g for 5 min to remove nuclei and unbroken cells. The MAM-enriched mitochondrial fraction was pelleted by centrifugation at 10 300g for 10 min. The supernatants were centrifuged at 100 000g for 30 min to pellet the ER/microsomes. To separate MAM and mitochondria, the MAM-enriched mitochondrial pellet was resuspended in isolation buffer and layered on top of a self-forming 30% Percoll gradient (225 mm mannitol, 1 mm EGTA, 0.05% BSA, 30% Percoll, 25 mm Na-HEPES pH 7.4). After centrifugation at 95 000g for 30 min, a dense band containing the mitochondria was recovered at the bottom of the gradient; the MAM-containing band was retrieved above the mitochondrial band. To remove residual Percoll, the mitochondrial band was diluted in isolation medium and mitochondria were washed twice by centrifugation at 6300g for 10 min. The MAM band was diluted with isolation buffer and centrifuged once at 6300g for 10 min to remove contaminating mitochondria. MAM was pelleted from the resulting supernatant by centrifugation at 100 000g for 1 h. All final organelle pellets were resuspended and lysed in radio-immunoprecipitation assay (RIPA) buffer (50 mm Tris–HCl pH 6.8, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, complete protease inhibitors) and the protein concentrations determined by Bradford assay (Bio-Rad Laboratories).
For sub-fractionation of mitochondria, mitochondria were isolated from HEK293 cells on a discontinuous sucrose gradient as described elsewhere (67). Proteinase K and alkaline treatments were essentially as described previously (33). For proteinase K treatment, 0.5 μl 2.5 mg/ml proteinase K was added to 100 μl mitochondria in isolation buffer (10 mm K-HEPES pH 7.4, 220 mm mannitol, 70 mm sucrose, 1 mm EDTA, 1 mm dithiothreitol). After 10 min incubation, 1 μl 200 μm phenylmethanesulfonylfluoride was added to inhibit proteinase K and the mitochondria were pelleted by centrifugation at 10 300g for 10 min and washed once with isolation buffer. For alkaline extraction, mitochondria were resuspended in 100 μm Na2CO3 pH 11.5, and incubated for 30 min on ice. To remove the alkaline solution, mitochondria were pelleted by centrifugation at 10 300g for 10 min and washed once with isolation buffer.
Mitochondria were sub-fractionated into outer mitochondrial membrane and inner mitochondrial membrane/matrix fractions as described previously (32). Briefly, purified mitochondria were incubated with 3 mg/ml digitonin (Calbiochem) in 250 mm sucrose, 10 mm K-MOPS pH 7.2 for 20 min at 4°C. After incubation, an inner mitochondrial membrane/matrix pellet was recovered by centrifugation (9500g, 15 min, 4°C). An outer mitochondrial membrane pellet was recovered from the supernatant by centrifugation at 100 000g, for 1 h at 4°C. Both pellets were resuspended in isolation buffer and loaded on top of sucrose gradients (51.3, 37.4 and 23.2% sucrose in 10 mm KH2PO4 pH 7). Pure inner mitochondrial membrane/matrix and outer mitochondrial membrane fractions were recovered and concentrated by centrifugation (100 000g, 1 h, 4°C). All fractions were solubilized in RIPA buffer and protein concentrations determined by Bradford assay.
SDS–PAGE and immunoblotting
Protein samples were separated by SDS–PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell) by wet electroblotting (BioRad). After transfer, membranes were blocked with Tris–HCl-buffered saline (TBS) containing 5% milk, and 0.1% Tween-20 for 1 h at room temperature, and then incubated with primary antibodies in blocking buffer for 1 h at room temperature. After washing with wash buffer (TBS, 0.1% Tween-20), the membranes were incubated with secondary antibodies in wash buffer for 1 h at room temperature. After washing, the membranes were processed for chemiluminescent detection with the Pierce SuperSignal West Pico Chemiluminescent Substrate System according to the manufacturer's instructions. Signal intensities on immunoblots were quantified with ImageJ after scanning with an Epson Precision V700 Photo scanner as described (68).
Ca2+ measurements
HEK293 cells and cortical neurons were loaded with 2 μm Fluo4-AM and/or Rhod2-AM dye (Invitrogen) in external solution (145 mm NaCl, 2 mm KCl, 5 mm NaHCO3, 1 mm MgCl2, 2.5 mm CaCl2, 10 mm glucose, 10 mm Na-HEPES pH 7.25) containing 0.02% Pluronic-F27 (Invitrogen) for 15 min at 37°C, followed by washing in external solution for 15 min at 37°C. Fluo4 and Rhod2 fluorescence was timelapse recorded (1 s interval) at 37°C with MetaMorph (Molecular Dynamics) on an Axiovert S100 microscope (Zeiss) equipped with YFP (Fluo4) and DsRed (Rhod2) filtersets (Chroma Technology), a 40×/1.3NA Plan-Neofluar objective (Zeiss) and a Photometrics Cascade-II 512B EMCCD. The cells were kept under constant perfusion with external solution (0.5 ml/min). IP3R-mediated Ca2+ release from ER stores was triggered by application of 100 μm Oxo-M for 2 min. Neurons were depolarized by application of 50 mm KCl for 2 min. Ca2+ levels were calculated as relative Fluo4 or Rhod2 fluorescence compared to baseline fluorescence at the start of the measurement.
Statistics
All experiments were repeated at least three times. Statistical analysis was performed with Prism 5.0d (GraphPad Software). Unless stated otherwise, statistical significance was determined by one-way ANOVA followed by Bonferroni's multiple comparison test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Medical Research Council (MRC; http://www.mrc.ac.uk/index.htm) (G0501573 to C.C.J.M. and C.E.S.), by the Wellcome Trust (http://www.wellcome.ac.uk/) (078662 to C.C.J.M.), the Motor Neurone Disease Association (MNDA; http://www.mndassociation.org/) (Miller6231 to C.C.J.M.) and the European Union 7th Framework Programme for RTD (http://ec.europa.eu/research/fp7) (Project MitoTarget—Grant Agreement HEALTH-F2-2008-223388 to C.C.J.M. and K.J.D.V.). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Supplementary Material
ACKNOWLEDGEMENTS
We thank C. Bauer, K. Brady, A. Brain and E. Gray for technical assistance, and E. Seward for providing reagents.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lill C.M., Abel O., Bertram L., Al-Chalabi A. Keeping up with genetic discoveries in amyotrophic lateral sclerosis: The ALSoD and ALSGene databases. Amyotroph. Lateral Scler. 2011;12:238–249. doi: 10.3109/17482968.2011.584629. doi:10.3109/17482968.2011.584629. [DOI] [PubMed] [Google Scholar]
- 2.Byrne S., Walsh C., Lynch C., Bede P., Elamin M., Kenna K., McLaughlin R., Hardiman O. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry. 2011;82:623–627. doi: 10.1136/jnnp.2010.224501. doi:10.1136/jnnp.2010.224501. [DOI] [PubMed] [Google Scholar]
- 3.Nishimura A.L., Mitne-Neto M., Silva H.C., Richieri-Costa A., Middleton S., Cascio D., Kok F., Oliveira J.R., Gillingwater T., Webb J., et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004;75:822–831. doi: 10.1086/425287. doi:10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kagiwada S., Hosaka K., Murata M., Nikawa J., Takatsuki A. The Saccharomyces cerevisiae SCS2 gene product, a homolog of a synaptobrevin-associated protein, is an integral membrane protein of the endoplasmic reticulum and is required for inositol metabolism. J. Bacteriol. 1998;180:1700–1708. doi: 10.1128/jb.180.7.1700-1708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaiser S.E., Brickner J.H., Reilein A.R., Fenn T.D., Walter P., Brunger A.T. Structural basis of FFAT motif-mediated ER targeting. Structure. 2005;13:1035–1045. doi: 10.1016/j.str.2005.04.010. doi:10.1016/j.str.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Kanekura K., Nishimoto I., Aiso S., Matsuoka M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8) J. Biol. Chem. 2006;28:30223–30232. doi: 10.1074/jbc.M605049200. doi:10.1074/jbc.M605049200. [DOI] [PubMed] [Google Scholar]
- 7.Skehel P.A., Fabian-Fine R., Kandel E.R. Mouse VAP33 is associated with the endoplasmic reticulum and microtubules. Proc. Natl Acad. Sci. USA. 2000;97:1101–1106. doi: 10.1073/pnas.97.3.1101. doi:10.1073/pnas.97.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teuling E., Ahmed S., Haasdijk E., Demmers J., Steinmetz M.O., Akhmanova A., Jaarsma D., Hoogenraad C.C. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J. Neurosci. 2007;27:9801–9815. doi: 10.1523/JNEUROSCI.2661-07.2007. doi:10.1523/JNEUROSCI.2661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fasana E., Fossati M., Ruggiano A., Brambillasca S., Hoogenraad C.C., Navone F., Francolini M., Borgese N. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 2010;24:1419–1430. doi: 10.1096/fj.09-147850. doi:10.1096/fj.09-147850. [DOI] [PubMed] [Google Scholar]
- 10.Chen H.J., Anagnostou G., Chai A., Withers J., Morris A., Adhikaree J., Pennetta G., de Belleroche J.S. Characterisation of the properties of a novel mutation in VAPB in familial ALS. J. Biol. Chem. 2010;285:40266–40281. doi: 10.1074/jbc.M110.161398. doi:10.1074/jbc.M110.161398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langou K., Moumen A., Pellegrino C., Aebischer J., Medina I., Aebischer P., Raoul C. AAV-mediated expression of wildtype and ALS-linked mutant VAPB selectively triggers death of motoneurons through a Ca-dependent ER-associated pathway. J. Neurochem. 2010;114:795–809. doi: 10.1111/j.1471-4159.2010.06806.x. doi:10.1111/j.1471-4159.2010.06806.x. [DOI] [PubMed] [Google Scholar]
- 12.Kim S., Leal S.S., Ben Halevy D., Gomes C.M., Lev S. Structural requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-associated VAP-B(P56S) neurotoxicity. J. Biol. Chem. 2010;285:13839–13849. doi: 10.1074/jbc.M109.097345. doi:10.1074/jbc.M109.097345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tudor E.L., Galtrey C.M., Perkinton M.S., Lau K.F., De Vos K.J., Mitchell J.C., Ackerley S., Hortobagyi T., Vamos E., Leigh P.N., et al. Amyotrophic lateral sclerosis mutant VAPB transgenic mice develop TDP-43 pathology. Neuroscience. 2010;167:774–785. doi: 10.1016/j.neuroscience.2010.02.035. doi:10.1016/j.neuroscience.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 14.Pennetta G., Hiesinger P., Fabian-Fine R., Meinertzhagen I., Bellen H. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. doi: 10.1016/s0896-6273(02)00769-9. doi:10.1016/S0896-6273(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 15.Ratnaparkhi A., Lawless G.M., Schweizer F.E., Golshani P., Jackson G.R. A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE. 2008;3:e2334. doi: 10.1371/journal.pone.0002334. doi:10.1371/journal.pone.0002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prosser D.C., Tran D., Gougeon P.Y., Verly C., Ngsee J.K. FFAT rescues VAPA-mediated inhibition of ER-to-Golgi transport and VAPB-mediated ER aggregation. J. Cell Sci. 2008;121:3052–3061. doi: 10.1242/jcs.028696. doi:10.1242/jcs.028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peretti D., Dahan N., Shimoni E., Hirschberg K., Lev S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP Proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell. 2008;19:3871–3884. doi: 10.1091/mbc.E08-05-0498. doi:10.1091/mbc.E08-05-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki H., Kanekura K., Levine T.P., Kohno K., Olkkonen V.M., Aiso S., Matsuoka M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 2009;108:973–985. doi: 10.1111/j.1471-4159.2008.05857.x. doi:10.1111/j.1471-4159.2008.05857.x. [DOI] [PubMed] [Google Scholar]
- 19.Gkogkas C., Middleton S., Kremer A.M., Wardrope C., Hannah M., Gillingwater T.H., Skehel P. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 2008;17:1517–1526. doi: 10.1093/hmg/ddn040. doi:10.1093/hmg/ddn040. [DOI] [PubMed] [Google Scholar]
- 20.Amarilio R., Ramachandran S., Sabanay H., Lev S. Differential regulation of endoplasmic reticulum structure through VAP-Nir protein interaction. J. Biol. Chem. 2005;280:5934–5944. doi: 10.1074/jbc.M409566200. doi:10.1074/jbc.M409566200. [DOI] [PubMed] [Google Scholar]
- 21.Tsuda H., Han S.M., Yang Y., Tong C., Lin Y.Q., Mohan K., Haueter C., Zoghbi A., Harati Y., Kwan J., et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008;133:963–977. doi: 10.1016/j.cell.2008.04.039. doi:10.1016/j.cell.2008.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saxena S., Cabuy E., Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. doi:10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 23.Kanekura K., Suzuki H., Aiso S., Matsuoka M. ER Stress and unfolded protein response in amyotrophic lateral sclerosis. Mol. Neurobiol. 2009;39:81–89. doi: 10.1007/s12035-009-8054-3. doi:10.1007/s12035-009-8054-3. [DOI] [PubMed] [Google Scholar]
- 24.Tradewell M.L., Cooper L.A., Minotti S., Durham H.D. Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 2011;42:265–275. doi: 10.1016/j.nbd.2011.01.016. doi:10.1016/j.nbd.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Grosskreutz J., Van Den Bosch L., Keller B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium. 2010;47:165–174. doi: 10.1016/j.ceca.2009.12.002. doi:10.1016/j.ceca.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Stenzinger A., Kajosch T., Tag C., Porsche A., Welte I., Hofer H.W., Steger K., Wimmer M. The novel protein PTPIP51 exhibits tissue- and cell-specific expression. Histochem. Cell Biol. 2005;123:19–28. doi: 10.1007/s00418-004-0732-7. doi:10.1007/s00418-004-0732-7. [DOI] [PubMed] [Google Scholar]
- 27.Brobeil A., Graf M., Oeschger S., Steger K., Wimmer M. PTPIP51-a myeloid lineage specific protein interacts with PTP1B in neutrophil granulocytes. Blood Cells Mol. Dis. 2010;45:159–168. doi: 10.1016/j.bcmd.2010.04.009. doi:10.1016/j.bcmd.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Oishi K., Okano H., Sawa H. RMD-1, a novel microtubule-associated protein, functions in chromosome segregation in Caenorhabditis elegans. J. Cell Biol. 2007;179:1149–1162. doi: 10.1083/jcb.200705108. doi:10.1083/jcb.200705108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lv B.F., Yu C.F., Chen Y.Y., Lu Y., Guo J.H., Song Q.S., Ma D.L., Shi T.P., Wang L. Protein tyrosine phosphatase interacting protein 51 (PTPIP51) is a novel mitochondria protein with an N-terminal mitochondrial targeting sequence and induces apoptosis. Apoptosis. 2006;11:1489–1501. doi: 10.1007/s10495-006-8882-9. doi:10.1007/s10495-006-8882-9. [DOI] [PubMed] [Google Scholar]
- 30.Yu C., Han W., Shi T., Lv B., He Q., Zhang Y., Li T., Song Q., Wang L., Ma D. PTPIP51, a novel 14-3-3 binding protein, regulates cell morphology and motility via Raf-ERK pathway. Cell Signal. 2008;20:2208–2220. doi: 10.1016/j.cellsig.2008.07.020. doi:10.1016/j.cellsig.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 31.Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. doi:10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 32.Benga G., Hodarnau A., Tilinca R., Porutiu D., Dancea S., Pop V., Wrigglesworth J. Fractionation of human liver mitochondria: enzymic and morphological characterization of the inner and outer membranes as compared to rat liver mitochondria. J. Cell Sci. 1979;35:417–429. doi: 10.1242/jcs.35.1.417. [DOI] [PubMed] [Google Scholar]
- 33.Ryan M.T., Voos W., Pfanner N. Assaying protein import into mitochondria. Methods Cell Biol. 2001;65:189–215. doi: 10.1016/s0091-679x(01)65012-x. doi:10.1016/S0091-679X(01)65012-X. [DOI] [PubMed] [Google Scholar]
- 34.Rizzuto R., Pinton P., Carrington W., Fay F.S., Fogarty K.E., Lifshitz L.M., Tuft R.A., Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. doi:10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 35.Vance J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- 36.Csordas G., Renken C., Varnai P., Walter L., Weaver D., Buttle K.F., Balla T., Mannella C.A., Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. doi:10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. doi:10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 38.Mendes C.C., Gomes D.A., Thompson M., Souto N.C., Goes T.S., Goes A.M., Rodrigues M.A., Gomez M.V., Nathanson M.H., Leite M.F. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 2005;280:40892–40900. doi: 10.1074/jbc.M506623200. doi:10.1074/jbc.M506623200. [DOI] [PubMed] [Google Scholar]
- 39.Csordas G., Hajnoczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochim. Biophys. Acta. 2009;1787:1352–1362. doi: 10.1016/j.bbabio.2009.06.004. doi:10.1016/j.bbabio.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chai A., Withers J., Koh Y.H., Parry K., Bao H., Zhang B., Budnik V., Pennetta G. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum. Mol. Genet. 2008;17:266–280. doi: 10.1093/hmg/ddm303. doi:10.1093/hmg/ddm303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rusinol A.E., Cui Z., Chen M.H., Vance J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994;269:27494–27502. [PubMed] [Google Scholar]
- 42.Simmen T., Lynes E.M., Gesson K., Thomas G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM) Biochim. Biophys. Acta. 2010;1798:1465–1473. doi: 10.1016/j.bbamem.2010.04.009. doi:10.1016/j.bbamem.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayashi T., Rizzuto R., Hajnoczky G., Su T.P. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. doi:10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li G., Mongillo M., Chin K.T., Harding H., Ron D., Marks A.R., Tabas I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009;186:783–792. doi: 10.1083/jcb.200904060. doi:10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Israelson A., Arbel N., Da Cruz S., Ilieva H., Yamanaka K., Shoshan-Barmatz V., Cleveland D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. doi:10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cozzolino M., Carri M.T. Mitochondrial dysfunction in ALS. Prog. Neurobiol. 2011 doi: 10.1016/j.pneurobio.2011.06.003. in press. [DOI] [PubMed] [Google Scholar]
- 47.Duffy L.M., Chapman A.L., Shaw P.J., Grierson A.J. The role of mitochondria in the pathogenesis of Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2011;37:336–352. doi: 10.1111/j.1365-2990.2011.01166.x. doi:10.1111/j.1365-2990.2011.01166.x. [DOI] [PubMed] [Google Scholar]
- 48.Higgins C.M., Jung C., Ding H., Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002;22:RC215. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferri A., Cozzolino M., Crosio C., Nencini M., Casciati A., Gralla E.B., Rotilio G., Valentine J.S., Carri M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl Acad. Sci. USA. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. doi:10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mattiazzi M., D'Aurelio M., Gajewski C.D., Martushova K., Kiaei M., Beal M.F., Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. doi:10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 51.Pasinelli P., Belford M.E., Lennon N., Bacskai B.J., Hyman B.T., Trotti D., Brown R.H., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. doi:10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 52.Liu J., Lillo C., Jonsson P.A., Velde C.V., Ward C.M., Miller T.M., Subramaniam J.R., Rothstein J.D., Marklund S., Andersen P.M., et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. doi:10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 53.De Vos K.J., Chapman A.L., Tennant M.E., Manser C., Tudor E.L., Lau K.F., Brownlees J., Ackerley S., Shaw P.J., McLoughlin D.M., et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. doi:10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. doi:10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 55.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. doi:10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 56.Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E., et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shan X., Chiang P.M., Price D.L., Wong P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl Acad. Sci. USA. 2010;107:16325–16330. doi: 10.1073/pnas.1003459107. doi:10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y.F., Gendron T.F., Zhang Y.J., Lin W.L., D'Alton S., Sheng H., Casey M.C., Tong J., Knight J., Yu X., et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010;30:10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. doi:10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki H., Lee K., Matsuoka M. TDP-43-induced death is associated with altered regulation of BIM and BCL-XL and attenuated by caspase-mediated TDP-43 cleavage. J. Biol. Chem. 2011;286:13171–13183. doi: 10.1074/jbc.M110.197483. doi:10.1074/jbc.M110.197483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szabadkai G., Bianchi K., Varnai P., De Stefani D., Wieckowski M.R., Cavagna D., Nagy A.I., Balla T., Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. doi:10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rapizzi E., Pinton P., Szabadkai G., Wieckowski M.R., Vandecasteele G., Baird G., Tuft R.A., Fogarty K.E., Rizzuto R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002;159:613–624. doi: 10.1083/jcb.200205091. doi:10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zuchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J., et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. doi:10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 63.McLoughlin D.M., Miller C.C.J. The intracellular cytoplasmic domain of the Alzheimer's disease amyloid precursor protein interacts with phosphotyrosine binding domain proteins in the yeast two-hybrid system. FEBS Lett. 1996;397:197–200. doi: 10.1016/s0014-5793(96)01128-3. doi:10.1016/S0014-5793(96)01128-3. [DOI] [PubMed] [Google Scholar]
- 64.Ackerley S., Grierson A.J., Brownlees J., Thornhill P., Anderton B.H., Leigh P.N., Shaw C.E., Miller C.C.J. Glutamate slows axonal transport of neurofilaments in transfected neurons. J. Cell Biol. 2000;150:165–175. doi: 10.1083/jcb.150.1.165. doi:10.1083/jcb.150.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Vos K.J., Allan V.J., Grierson A.J., Sheetz M.P. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr. Biol. 2005;15:678–683. doi: 10.1016/j.cub.2005.02.064. doi:10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 66.De Vos K.J., Sable J., Miller K.E., Sheetz M.P. Expression of phosphatidylinositol (4,5) bisphosphate-specific pleckstrin homology domains alters direction but not the level of axonal transport of mitochondria. Mol. Biol. Cell. 2003;14:3636–3649. doi: 10.1091/mbc.E02-10-0638. doi:10.1091/mbc.E02-10-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Vos K., Severin F., Van Herreweghe F., Vancompernolle K., Goossens V., Hyman A., Grooten J. Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J. Cell Biol. 2000;149:1207–1214. doi: 10.1083/jcb.149.6.1207. doi:10.1083/jcb.149.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vagnoni A., Rodriguez L., Manser C., De Vos K.J., Miller C.C.J. Phosphorylation of kinesin light chain-1 at serine-460 modulates binding and trafficking of calsyntenin-1. J. Cell Sci. 2011;124:1032–1042. doi: 10.1242/jcs.075168. doi:10.1242/jcs.075168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.