

Abstract
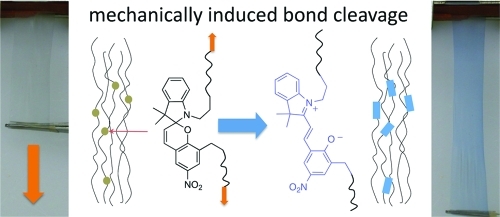
A photochromic polymer exhibiting mechanochromic behavior is prepared by means of ring-opening polymerization (ROP) of ε-caprolactone by utilizing a difunctional indolinospiropyran as an initiator. The configuration of having the photochromic initiating species within the polymer backbone leads to a mechanochromic effect with deformation of polymer films leading to ring-opening of the spiro C−O bond to form the colored merocyanine. Active stress monitoring by dynamic mechanical analysis (DMA) in tension mode was used to probe the effects of UV irradiation on polymer films held under constant strain. Irradiation with UV light induces a negative change in the polymer stress of approximately 50 kPa. Finally, a model of the mechanochromic effect was performed using density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. A sharp increase in the relative molecular energy and the absorption wavelength as well as a drastic decrease in the spiro-oxygen atom charge occurred at a molecular elongation of >39%.
Keywords: mechanochromic, photochromic, spiropyran, ROP, DFT
Introduction
The incorporation of functional molecules (liquid crystals, multiple hydrogen-bonding groups, multidentate ligands, etc.) into polymer systems can impart dynamic material properties useful for the development of novel sensor and actuator devices. Advances in computational simulations and advanced synthetic techniques have led to rapid progress in the study of smart materials, or compounds that respond to external stimuli by design. These materials are generally tailored for specific changes in environmental conditions such as thermal gradients (1), mechanical deformation (2), electrical field fluctuation (3,4), and photoirradiation (5). Typically, an observable response (i.e., actuation or chromic shift) occurs through a dynamic shift in a bulk material property. Integrating photochromic molecules into a polymer matrix is one method of preparing materials with controllable photochemical properties. The formulation and polymerization type, binding interactions, and overall configuration of the molecular scaffolding are generally tailored to a directed application. Indolinospriopyrans (spiropyran or SP) are well-characterized photochromes that have been utilized extensively in sensor and actuator research through covalently binding or blending these materials into polymeric substrates (6−9).
The photochromic response observed in spiropyrans is a result of a reversible photochemical rearrangement that occurs upon exposure to UV light. Photoexcitation of an SP with UV light leads to cleavage of the C−O spiro bond and ring-opening, resulting in an electronic rearrangement and the formation of a highly conjugated zwitterionic merocyanine (MC) that is responsible for an observed bathochromic shift (10,11). The merocyanine form is highly polarized and imparts changes in solubility, opens up chelation possibilities, and can cause aggregation of dye molecules in non-polar environments. The ease of control of the reversible SP rearrangement, and the various driving forces resulting from it, has led to a number of scientific applications including molecular motors (12), chemical sensing (13), optoelectronic switching (14), and controlled polymer micelle formation (15).
Photoexcitation is typically the principal method to promote the spiropyran to merocyanine conversion, but this transformation can also be influenced by changes in solvent polarity (solvatochromism) (16) as well as by mechanical deformation (17). Potisek et al. reported on findings of mechanical induced ring-opening of a polymer bound spiropyran that occurred during ultrasonication of SP-polymer solutions (18). More recently they have pursued characterization of bulk mechanochromic responses of polymer backbone integrated spiropyrans in elastomeric poly(methacrylate) (19). Active monitoring of stresses induced in polymer samples under applied loads was performed with fluorescence spectroscopy to determine the strain rates that induce the spiro-merocyanine transformation. The high molar extinction coefficient of the SP allows changes in stress to be observed at very low loadings. This characteristic is ideal considering that a single photochromic molecule is used to initiate polymerization, and therefore only one molecule is integrated per polymer chain (not accounting for dimerization chain termination events in the case of radical acrylate polymerization).
The following report describes a similar mechanochromic response observed in a polymer prepared from an indolinospiropyran diol utilized as an initiator in the ring-opening polymerization (ROP) of poly(ε-caprolactone) (PCL). Real-time stress monitoring during plastic deformation can communicate valuable information about material weaknesses and possibly signal component failure, an important application when relying on plastic components in mechanical systems. The mechanochromic response of this material was observed during an investigation into light activated responses in shape-memory polymers (SMP). In the present work the labile C−O spiro bond of an SP acts as a weak link along the plane of the polymer backbone in which it is embedded. We have carried out quantum chemical calculations of the chromic response of PCL subject to an applied mechanical stress. These calculations allow us to support the experimental observation that the color change in PCL as a function of external load is controlled by cleavage of the weak C−O spiro bond at the molecular level. The mechanochromic effect for these materials can be summarized as follows: an induced strain causes alignment of polymer strands in the bulk material which results in stress induced cleavage at the spiro C−O bond. An electronic reconfiguration to the zwitterionic merocyanine gives rise to the observable color change that has previously been correlated to the SP → MC conversion. Consequently, the color change occurs just before failure in a polymer sample, with the most intense chromic shift being observed immediately following material rupture. This report describes stress monitoring in a non-elastomeric material in which polymer chain alignment is retained after sample preparation. Because of retention of the polymer chain configuration, the present material is sensitive to relatively minor deformations.
Results and Discussion
Synthesis and Characterization of Photochromic Poly(ε-caprolactone)
Photochromic indolinospiropyran 5 was readily prepared by a procedure adapted from the work of Raymo et al, Scheme 1(20). Indolium bromide 1 (prepared according to reference) was converted to indole 2 by grinding in a mortar and pestle with potassium hydroxide in open air until a fine paste was obtained. This green chemistry approach resulted in near quantitative yield of indole 2 after extraction of the paste. Hydrolysis of 3-chloromethyl-5-nitrosalicylaldehyde gave methylhydroxy substituted salicylaldehyde 4 in good yield after recrystallization from water. Spiropyran 5 was prepared by condensation of 2 and 4 in refluxing 50% aqueous ethanol, which gave the product as a precipitate. The photochrome was recrystallized from acetonitrile, which accounts for a majority of the material loss; however, X-ray quality crystals were obtained and the solved X-ray crystal structure showed the compound to be present in the merocyanine form as a dihydrate (21). The di-hydroxy-functionalized photochrome was then utilized as a double-ended initiator in the ring-opening polymerization of ε-caprolactone. To a solution containing photochrome 5 in ε-caprolactone (dried over 3 Å MS and filtered before use) was added a catalytic amount of stannous octoate and heated. Small aliquots were removed from the solution periodically to determine molecular weight conversion by size exclusion chromatography (SEC). The polymerization was determined to be complete after 24 h. The resulting polymeric material was purified by repeated precipitation into methanol, dried under a vacuum, and analyzed by SEC and proton NMR chain end analysis to determine the molecular weight distribution.
Scheme 1. Preparation of Photochromic Spiropyran Diol and Subsequent Polymerization of ε-Caprolactone.

(i) KOH, grind open in air 5−10 min, >99%. (ii) 6M NaOH, acetone/water reflux, recryst. water, 81%. (iii) 50:50 ethanol:water reflux, 5 h, recryst. 70% CH3CN (aq), 50%. (iv) ε-caprolactone, toluene, cat. stannous octoate, reflux 24 h.
Figure 1 displays the polymerization evolution of PCL by SEC analysis over a period of 24 h. The final number average molecular weight (Mn) of the obtained polymer was 42 kDa with a polydispersity index of 1.45. Figure 2 shows the assignment of the proton NMR spectrum to chain end groups and the incorporated spiropyran initiator. Utilizing integration of the chain end group protons (assigned to δ) and the protons present in the backbone of the polymer (assigned to ε), the number of monomeric repeat units (n) was determined to be 150, for a total of 300 monomeric repeat units per spiropyran initiator. The estimated average molecular weight based on this data is 34 kDa, in close approximation with the molecular weight determination by SEC.
Figure 1.

Time-resolved SEC analysis from polymerization of 6.
Figure 2.

Proton NMR assignment of polymer backbone protons (labeled with Greek characters) of 6. An inset of the expanded region from 3.2−8.0 ppm (empty regions cut out for clarity) is blown-up to display the protons associated with the polymer chain end group (labeled δ) and those of the initiating spiropyran (labeled with Roman characters).
UV−visible Characterization of Photochromic Poly(ε-caprolactone)
Photochromic behavior, imparted to the polymer from the initiating spiropyran, was clearly observed upon irradiating samples with 365 nm UV light. The photoexcitation of a spiropyran to its merocyanine form (depending on environmental conditions) is a short-lived response as the zwitterionic merocyanine equilibrates back to the more thermally stable spiro form (22). This transformation can be monitored via spectroscopic techniques to determine the rate at which this process proceeds. A study of this transformation was probed by measuring the absorption decay of 5 and 6 in ethyl acetate solutions (4.93 × 10−5 M) (23) saturated with the visibly active merocyanine via UV (365 nm) irradiation for 5 min. The absorption at λmax (580 nm) was monitored over time and followed an exponential trend according to eq 1(24) The rate of decay (k1) was found by solving eq 1 with respect to time and plotting ln((A − A∞)/(Ao − A∞)) to produce the time-dependent kinetic plot in Figure 3. The slope of a linear approximation to the displayed curves was used to determine the value of the rate of decoloration, kd. The rate of absorption decay observed in the polymer bound merocyanine was somewhat slower, k1 = −1.70 × 10−2 s−1, when compared to the rate obtained for small molecule 5, k1 = −4.08 × 10−2 s−1. The decreased rate is attributed to steric interaction of polymer chains inhibiting ring closure back to the spiro form.
| 1 |
The decoloration of a thin film of polymer 6 (0.02 mm) was monitored under the same conditions as previously stated to determine the decoloration rate of spiropyran in a solid polymer system. The rate, k1, determined to be −5.6 × 10−4 s−1, is a dramatic decrease when compared to the rate of decoloration of solvated material. This increase is attributed to inhibited molecular motion in a solid matrix (25). The results from this experiment can be found in the Supporting Information.
Figure 3.

Kinetic decoloration study of 5 (◻) and 6 (○) in ethyl acetate.
Mechanochromic and Photomechanical Phenomena in PCL 6
In addition to the photochromic behavior, a mechanochromic effect was observed at the edges of polymer samples when scrapped or deformed. To investigate this phenomena, we pressed a sample of the precipitated polymer at a temperature above its melting point (70 °C) into a thin sheet (film thickness was between 0.1−0.2 mm). Heat-pressed samples required adequate cooling time (1 h) at room temperature prior to further processing, otherwise drawn samples would fracture before alignment was achieved. Differential scanning calorimetry of the formed material was used to determine a crystallinity of 42% (Figure S2 in the Supporting Information). Rectangular film strips cut from the sheet were heated to 50 °C by immersion into a pre-heated temperature controlled water bath for 30 s. Immediately upon removal, the sample was pulled by hand on either end to affect alignment of polymer chains (film thickness after alignment was 0.02 −0.04 mm). It should be noted that the polymer chain alignment step is necessary before a mechanochromic response can be observed in this polymer system, the chromic response of samples pulled to failure before alignment is very minor by comparison. Alignment increases the number of spiro to merocyanine transformations by imparting a direct mechanical force to the weakest bond. Prior to alignment, deforming a sample results in material failure with a mechanochromic change being observed only at the edges where failure occurs. Conversely, deformation of aligned polymer films results in a noticeable color change. The color change of this material can be observed upon direct deformation from an aligned state; a video demonstration can be found in the Supporting Information. We believe the nearly immediate color change arising from deformation occurs in response to a strain held on the spiropyran via the alignment process. Films deformed to failure result in even more drastic chromic effects, with color gradients observed near fracture points. Figure 4 exhibits a chromic shift in an aligned polymer film with increasing load, ultimately ending with a significant color change seen just after sample failure. Control PCL films non-covalently dyed with a comparable percentage of photochromic spiropyran 5, and subjected to the above alignment process did not display any mechanochromic behavior upon deformation. We can conclude that this phenomenon is in fact a result from embedding the spiropyran via a covalent attachment.
Figure 4.

Photo series displaying mechanochromism in PCL film 6 as it is deformed to failure.
The effect of mechanically induced stress on thin films of 6 to produce a color change prompted an evaluation of the opposing relationship: UV light irradiation induced spiropyran to merocyanine transition on stress in polymer films. Testing of polymer stress under different light conditions was accomplished with a TA Instruments Q800 dynamic mechanical analyzer with a black box erected to fit over the sample holding clamps. A hole was cut in the middle face of the box to insert a UV (365 nm) LED irradiation source. Samples were affixed to the film tension clamp holders and equilibrated in the dark at room temperature until a flat baseline was achieved on the DMA (roughly 5 min). A constant strain rate of 1% was applied, and after 5 min the UV light source was turned on. A significant decrease in stress was immediately observed (Figure 5). After 2 min of irradiation, the light source was turned off resulting in increased stress. Two minute UV on/off intervals were repeated multiple times to demonstrate a controlled cyclic response to light irradiation. The decrease in stress during UV exposure is attributed to lengthening of the polymer sample through C−O bond cleavage and ring-opening in the spiropyran, resulting in decreased stress. Conversely, stress recovery takes place during the lamp off cycle, a response which corresponds to contraction of polymer films and occurs as the merocyanine undergoes ring closure to the more constrained spiro form. Similar results have been observed with polymer films dyed with photochromic compounds (26). Temperature fluctuations from the light source we not observed during irradiation cycling, and a temperature versus time plot included in Figure 5 shows a nearly isothermal process over this time frame. Under appropriate conditions in an optimized matrix, this light-induced stress relaxation could be utilized as a trigger for a macroscale actuation event.
Figure 5.

Plot of stress monitoring over time (+) of a polymer film held under constant strain (1%) while cycling UV irradiation (365 nm) at 2 min intervals. The temperature measured by the DMA over this time scale is plotted on the second y-axis (•).
Model Studies of Mechanochromic Transformation
To understand the mechanochromic properties of the prepared spiropyran embedded PCL as a function of the applied stress, we have carried out a series of density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. Because the applied stress on poly(ε-caprolactone) creates a transition from the spiropyran form to the charge-separated zwitterionic form, it is necessary to choose a functional which accurately captures this charge separation on the potential energy surface. To this end, all DFT calculations were performed using a long-range corrected BLYP functional, LC-BLYP (27−29) which incorporates an exact −1/r exchange energy dependence at large interelectronic distances. For the LC-BLYP calculations in this work, we specifically used a range separation parameter of μ = 0.31 Bohr−1, which we previously found was optimal (compared to other hybrid functionals) for simultaneously describing both absorption and fluorescence charge-transfer excitations in large oligothiophenes (30). Davis et al. also carried out modeling studies on their polymer systems and computed force-activity data based on DFT calculations (19).
The initial equilibrium geometry for a single spiropyran unit tethered with pendant poly(ε-caprolactone) polymer chains, shown in Figure 6, was first optimized at the LC-BLYP/6-31G(d,p) level of theory. From this equilibrium spiropyran geometry, the effect of an applied stress was obtained by gradually increasing the distance between the two terminal methyl carbon atoms (cf. Figure 6) while optimizing all other internal coordinates to minimize the total energy. We evaluated the relaxed geometries and energies in intervals of 0.1 Å up to a final molecular elongation of 60%. Single-point LC-BLYP properties with a diffuse 6-31+G(d,p) basis set were subsequently performed at each of the LC-BLYP/6-31G(d,p) relaxed geometries.
Figure 6.

Equilibrium molecular geometry of poly(ε-caprolactone) integrated spiropyran 5 in the spiro form. The effect of an applied stress was calculated by gradually increasing the distance between the methyl units (denoted by arrows) while optimizing all other internal coordinates.
The relaxed ground-state energies as a function of molecular elongation are shown in Figure 7a, and the corresponding Mulliken charge of the oxygen atom in the C−O spiro bond is displayed in Figure 7b. As seen in Figure 7a, the relative energy increases rapidly along the reaction path, and we observe a distinct rupture of the C−O spiro bond at 39% elongation (31). After the poly(ε-caprolactone) has been stretched past the 39% transition point, the energy sharply decreases from 2.7 eV to 1.6 eV as the spiropyran has relaxed into the charge-separated zwitterionic merocyanine form. As a result, the energies for elongation lengths greater than 39% are associated with stretching molecular bonds along the backbone of the zwitterionic configuration. The electronic charge of the oxygen atom (Figure 7b) in the C−O spiro bond also reveals a distinct transition between the spiropyran and zwitterionic forms as the polymer is stretched. The negative charge of the oxygen atom slowly decreases along the reaction path and sharply decreases by a factor of 2 at 39% elongation. This trend can be associated with increasing polarization of the C−O bond as the internuclear distance increases. The DFT calculations indicate that the C−O spiro bond is weakest, and as the polymer is stretched, the C−O bond must begin to break, leading to a more negative electron distribution for the O atom in the charge-separated zwitterionic form.
Figure 7.

(a) Relaxed potential energy of poly(ε-caprolactone) as a function of molecular elongation. A sharp transition between spiropyran and zwitterionic forms occurs at 39% elongation length. (b) Mulliken charge of the oxygen atom in the C−O spiro bond. The electronic charge of the oxygen atom decreases by a factor of 2 at the 39% transition point. The electronic properties for a and b were obtained at the LC-BLYP/6-31+G(d,p) level of theory at LC-BLYP/6-31G(d,p) relaxed geometries.
Finally, to evaluate the photochromic properties of polymer 6 as a function of the applied stress, we carried out TDDFT calculations for each of the constrained geometries along the reaction path. In our TDDFT calculations, the lowest 10 singlet electronic excitations and oscillator strengths were calculated at the LC-BLYP/6-31+G(d,p) level of theory. In Figure 8, we plot the absorption wavelength having the highest oscillator strength (among the 10 lowest excitations) for each constrained geometry. For molecular elongations less than 39%, the poly(ε-caprolactone) spiropyran unit does not absorb strongly in the visible spectrum and has a maximum absorbance at 250 nm (oscillator strength ∼0.3). However, as the weak C−O spiro bond is placed under stress, a large molecular dipole (9.6 D) begins to emerge, and the polymer develops a strong absorption at 450 nm (oscillator strength ∼0.5) for elongations past the 39% transition point. We should also mention that these TDDFT calculations were performed in vacuo and do not take into account the effect of neighboring interactions between adjacent spiropyran polymer chains. The result of these neighboring interactions would increase the effective dielectric constant of the individual molecular unit and would, therefore, increase the absorption wavelengths relative to our TDDFT calculations performed in vacuo. Nevertheless, the TDDFT calculations reproduce and confirm the experimental observation of a significant photochromic shift as the spiropyran within 6 converts from the neutral spiro to the zwitterionic form as the polymer is placed under load.
Figure 8.
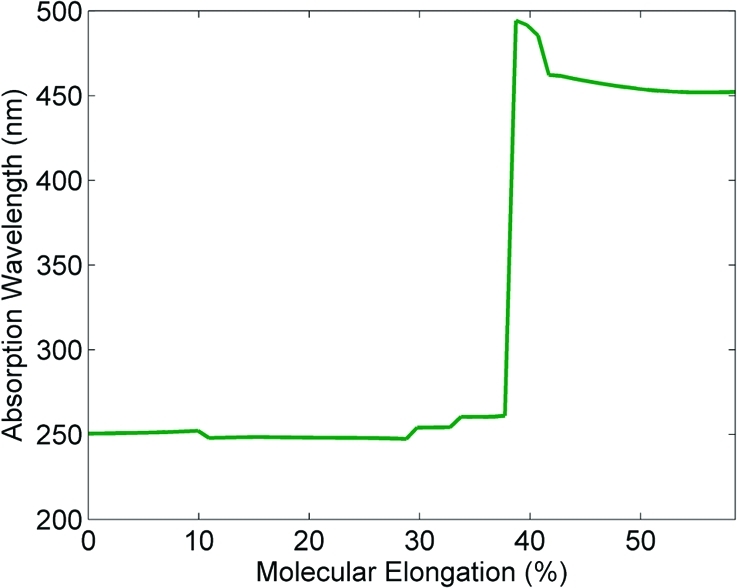
Absorption wavelength of poly(ε-caprolactone) as a function of molecular elongation. The absorption abruptly changes from the ultraviolet region (<39% elongation) to a strong absorption in the visible spectrum (>39% elongation). The absorption profile was obtained from TDDFT calculations at the LC-BLYP/6-31+G(d,p) level of theory.
Conclusion
A photochromic poly(ε-caprolactone) polymer was prepared by ring-opening polymerization with a di-functional spiropyran. The resulting polymer was characterized by standard techniques and found to respond to mechanically induced stress events and UV irradiation. It was found that deformation of thin polymer films results in bond cleavage at the weak C−O bond of the integrated spiropyran initiator. Light induced stress relaxation was demonstrated by irradiating polymer films while holding samples at a constant strain. Both phenomena are a direct result of incorporating a photochromic spiropyran diol within the polymer backbone, a configuration that is readily attained via ROP with the initiating molecule. Through both experimental and theoretical studies, this work illustrates that an induced stress (even for small loads) can lead to a significant mechanochromic response in bulk polymer materials. The change in photochromic response associated with an external stress in these materials may serve as a spectroscopic indicator for other types of mechanical stimuli. We expect that molecular engineering of functional groups within the spiropyran can induce sensitive detection in other regions of the optical spectrum.
Experimental Section
Experimental Methods
500 MHz 1H and 125 MHz 13C NMR spectra were recorded on a Varian Inova Unity NMR in CDCl3 (unless otherwise stated). Mass spectrometry was performed on a Waters LCT Premier XE using an electrospray (ESI) ionization probe coupled with a time-of-flight (TOF) detector. Samples were dissolved in acetonitrile and directly infused into the probe from an attached syringe pump (Hamilton 500 μL syringe). UV−visible analysis was performed on a Shimadzu UV-2501PC spectrophotometer in ethyl acetate solution in 1 cm path length quartz cells. Fourier-transform infrared (FT-IR) spectroscopy was performed on a Varian Scimitar Series 800 using a Pike ATR attachment. Size-exclusion chromatography (SEC) was conducted on an Agilent 1100 LC system with tetrahydrofuran as the mobile phase at a flow rate of 1 mL/min with a Agilent PLgel 5 μm Mixed-C 300 × 7.5 mm column. Dynamic mechanical analysis (DMA) experiments were performed on a TA Instruments Q800 using a black box fabricated from steel shims and a Nichia 365 nm UV LED flashlight to irradiate samples. Differential scanning calorimetry (DSC) was performed on a Mettler Toledo DSC822e heating from room temperature to 90 °C at a rate of 1° per min in a 40 μL aluminum pan with a blank pan for reference. Melting points are uncorrected. Solvents were purified with a VAC closed loop recirculation module solvent filtration/purification system. Reagents 1,3,3-trimethyl-2-methyleneindoline (Aldrich, 97%), 2-bromoethanol (Aldrich, 95%), 3-chloromethyl-5-nitrosalicylaldehyde (TCI America, 96%), ε-caprolactone (Acros, 99%), and tin(II) 2-ethylhexanoate (NuSil Silicone Technologies) were used as received.
Synthetic Procedures
3-Hydroxymethyl-5-nitrosalicylaldehyde (4)
3-Chloromethyl-5-nitrosalicylaldehyde (5.742 g, 26.63 mmol) was dissolved in 30 mL of acetone and added to a 100 mL round bottom flask containing deionized water (10 mL) and a magnetic stir bar. The solution was heated to reflux with stirring for 20 min. Sodium hydroxide solution (4.45 mL of a 6 M solution) was added drop wise over a period of 10 minutes. The reaction mixture was stirred and refluxed for 3 h then cooled to room temperature. The resulting solution was filtered and chilled to precipitate the product. The material was recrystallized from water to obtain 3.913 g (75% yield) of light green crystals. MP = 202−203 °C. IR (ATR): 3265 (br), 3081, 1654, 1628, 1522, 1444, 1352, 1322, 1067 cm−1. 1H NMR (500 MHz, CDCl3) ∂: 11.93 (br s, 1 H, phenol), 10.24 (s, 1 H, −C(O)H), 8.72 (d, 1 H, J = 3 Hz, Ar−H ortho CH2OH), 8.62 (m, 1 H, Ar−H para CH2OH), 4.82 (s, 2 H, −CH2OH), 2.84 (br s, 1 H, CH2OH).
9,9,9a-Trimethyl-2,3,9,9a-tetrahydro-oxazolo[3,2-a]indole (2)
The preparation for the title compound was adapted from an existing procedure of Raymo et al (20). Anhydrous potassium hydroxide (315 mg, 5.62 mmol) was ground in a mortar and pestle to a fine powder. 1-(2-hydroxyethyl)-2,3,3-trimethyl-3H-indolium bromide (1.00 g, 3.51 mmol) was added and the mixture ground until a fine paste was achieved and the color changed from purple to yellow. The paste was extracted with petroleum ether, and the extracts dried over anhydrous sodium sulfate, filtered, concentrated, then dried in vacuo to obtain 712 mg (>99% yield) of a yellow oil. The structure was confirmed by IR and 1H NMR analysis. IR (ATR): 2964, 1607, 1479, 1293, 1021, 745 cm−1. 1H NMR (500 MHz, CDCl3) ∂: 7.07 (m, 2 H, Ar−H m-NR2), 6.84 (m, 1 H, Ar−H p-NR2), 6.76 (m, 1 H, Ar−H o-NR2), 3.76 (m, 2 H, −NCH2CH2O−), 3.53 (m, 1 H, −NCH2CH2O−), 3.37 (m, 1 H, −NCH2CH2O−), 1.37 (s, 3 H, C(N)(O)CH3), 1.31 (s, 3 H, CH3), 1.23 (s, 3 H, CH3).
2-(8-(Hydroxymethyl)-3′,3′-dimethyl-6-nitrospiro[chromene-2,2′-indolin]-1′-yl)ethanol (5)
The preparation for the title compound was adapted from an existing literature procedure of Raymo et al. (20). 3-Hydroxymethyl-5-nitrosalicylaldehyde (576 mg, 2.92 mmol) was dissolved in 10 mL of ethanol in a 50 mL round bottom flask. Water (10 mL) was added followed by addition of 2 (712 mg, 3.50 mmol). The reaction mixture was heated to reflux while stirring and monitored by TLC (7:3 ethyl acetate:petroleum ether, Rf = 0.73). The reaction was determined to be complete after a period of 5 h. During the course of the reaction, an iridescent precipitate formed on the sides of the flask. The mixture was allowed to cool to room temperature and then concentrated on a rotary evaporator to remove ethanol. Water (10 mL) was added and the mixture was brought to a boil. The solution was filtered and this procedure then repeated. The product was washed with benzene to remove unreacted starting materials. The resulting brown powder was directly recrystallized from roughly 70% acetonitrile (aq) solution to give 607 mg (49.6% yield) of dark red iridescent crystals. The structure was determined by X-ray crystallography to exist in the zwitterionic merocyanine form as a dihydrate (21). MP = 154−155 °C. IR (ATR): 3388 (br), 2964, 1704, 1607, 1519, 1484, 1457, 1337, 1270 cm−1. 1H NMR (500 MHz, acetone-d6) ∂: [8.22 & 8.04] (d, 2H, J = 3 Hz, Ar−H o-NO2), 7.17 (d, 1 H, J = 10.5 Hz, ArCH=CH−), 7.12 (m, 2 H, Ar−H m-NR2), 6.81 (t, 1 H, J = 7 Hz, Ar−H p-NR2), 6.69 (d, 1 H, J = 7.5 Hz, Ar o-NR2), 6.07 (d, 1 H, J = 10.5 Hz, −CH=CHAr), 4.43 (m, 2 H, ArCH2OH), [3.80, 3.73, & 3.65] (m, 2 H, NCH2CH2OH), [3.40 & 3.31] (m, 2 H, NCH2CH2OH), [1.27 & 1.18] (s, 3 H, C(CH3)2). HRMS (neg mode) exact mass calcd for [M − 1]− C21H21N2O5, 381.1450; found, 381.1497.
Polymerization of ε-Caprolactone from Indolinospiropyran Diol 5 (6)
A solution of photochromic diol 5 (191 mg, 0.5 mmol) in ε-caprolactone (20 g, 175.22 mmol) was prepared in an oven-dried flask. To the solution was added 3 Å molecular sieves and the mixture was dried for 48 h. The solution was filtered into a 100 mL round bottom flask using anhydrous toluene (20 mL) to aid in transferring the complete contents to the reaction vessel. Stannous octoate (69 mg, 0.17 mmol) in 1 mL of toluene was added via syringe. The reaction mixture was heated to reflux and aliquots were removed periodically and analyzed by SEC. Between 6 and 8 h, the reaction became extremely viscous and magnetic stirring became impracticable. After 24 h of heating, the reaction was cooled to room temperature. Toluene (∼10 mL) was added and the vessel sonicated to reduce the viscosity to a pouring consistency. The contents were then poured into methanol chilled in an ice bath to precipitate the polymer. The precipitation procedure was repeated two more times and the polymer filtered, washed with methanol, air dried, and then further dried under a vacuum to give 14.837 g (73% gravimetric conversion) of a beige colored powder. IR: 2944, 1721, 1365, 1293, 1240, 1175 cm−1. SEC: Mn = 49 kDa, Mw = 60 kDa, PDI = 1.45.
Acknowledgments
This project is supported by the Laboratory Directed Research and Development program at Sandia National Laboratories, a multi-program laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy under Contract DE-AC04-94-AL85000.
Supporting Information Available
Decoloration kinetics plot of a thin film of polymer 6 and DSC data (PDF); video of a polymer film stretched in tension demonstrating a color change with induced stress (QT). This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Behl M.; Lendlein A. Mater. Today 2007, 10, 20–28. [Google Scholar]
- Kim S. J.; Reneker D. H. Polym. Bull. 1993, 31, 367–374. [Google Scholar]
- Bauer S., Bauer F. In Piezoelectricity; Heywang W., Wersing W., Lubitz K., Eds.; Springer Series in Materials Science; Springer: Berlin, 2008; Vol. 114, pp 157−177. [Google Scholar]
- O’Halloran A.; O’Malley F.; McHugh P. J. Appl. Phys. 2008, 104, 071101−1. [Google Scholar]
- Koemer H.; White T. J.; Tabiryan N. V.; Bunning T. J.; Vaia R. A. Mater. Today 2008, 11, 34–42. [Google Scholar]
- Smets G.; DeBlauwe F. Pure Appl. Chem. 1974, 39, 225–238. [Google Scholar]
- Irie M. Adv. Polym. Sci. 1990, 94, 27–67. [Google Scholar]
- Tamai N.; Miyasaka H. Chem. Rev. 2000, 100, 1875–1890. [DOI] [PubMed] [Google Scholar]
- Ichimura K. In Organic Photochromic and Thermochromic Compounds; Crano J. C., Guglielmetti R. J., Eds.; Topics in Applied Chemistry; Springer: New York, 2002; Vol. 2, pp 9−63. [Google Scholar]
- Minkin V. I. Chem. Rev. 2004, 104, 2751–2776. [DOI] [PubMed] [Google Scholar]
- Ikeda T.; Mamiya J. I.; Yu Y. Angew. Chem., Int. Ed. 2007, 46, 506–528. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Chen D.; Li J.; Xu J.; Lv J.; Liu H.; Li Y. Org. Lett. 2007, 9, 3929–3932. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y.; Adachi K.; Itoh M.; Hirai T. Org. Lett. 2009, 11, 3482–3485. [DOI] [PubMed] [Google Scholar]
- Guo X.; Zhang D.; ad D. Zhu Y. Z. J. Org. Chem. 2003, 68, 5681–5687. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K. Angew. Chem., Int. Ed. 2007, 46, 2453. [DOI] [PubMed] [Google Scholar]
- Drummond C. J.; Furlong D. N. J. Chem. Soc., Faraday Trans 1990, 86 (21), 3613–3621. [Google Scholar]
- Tipkin D. S. Russ. J. Phys. Chem. 2001, 75, 1720–1722. [Google Scholar]
- Potisek S. L.; Davis D. A.; Sottos N. R.; White S. R.; Moore J. S. J. Am. Chem. Soc. 2007, 129, 13808–13809. [DOI] [PubMed] [Google Scholar]
- Davis D. A.; Hamilton A.; Yang J.; Cremar L. D.; Gough D. V.; Potisek S. L.; Ong M. T.; Braun P. V.; Martinez T. J.; White S. R.; Moore J. S.; Sottos N. R. Nature 2009, 459, 68–72. [DOI] [PubMed] [Google Scholar]
- Raymo F. M.; Giordani S. J. Am. Chem. Soc. 2001, 123, 4651–4652. [DOI] [PubMed] [Google Scholar]
- Rodriguez M. A.; O’Bryan G.; Andrzejewski W. J.; McElhanon J. R. Acta Crystallogr., Sect. E 2009, 65, 1906–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelson R. C. In Organic Photochromic and Thermochromic Compounds; Crano J. C., Guglielmetti R. J., Eds.; Topics in Applied Chemistry;Springer: New York, 2002; Vol. 1, p 11−83. [Google Scholar]
- The concentration of 6 in solution was diluted until the absorbance value at λ580 nearly matched with the absorbance of 5 in solution to ensure concentration-dependant variables were maintained consistently.
- See the Supporting Information inRaymo F. M.; Giordani S. J. Am. Chem. Soc. 2001, 123, 4651–4652. [DOI] [PubMed] [Google Scholar]
- Evans R. A.; Hanley T. L.; Skidmore M. A.; Davis T. P.; Such G. K.; Yee L. H.; Ball G. E.; Lewis D. A. Nat. Mater. 2005, 4, 249–253. [DOI] [PubMed] [Google Scholar]
- Blair H. S.; Pogue H. I. Polymer 1982, 23, 779–783. [Google Scholar]
- Iikura H.; Tsuneda T.; Yanai T.; Hirao K. J. Chem. Phys. 2001, 115, 3540. [Google Scholar]
- Tawada Y.; Tsuneda T.; Yanagisawa S.; Yanai T.; Hirao K. J. Chem. Phys. 2004, 120, 8425. [DOI] [PubMed] [Google Scholar]
- Wong B. M.; Cordaro J. G. J. Chem. Phys. 2008, 129, 214703. [DOI] [PubMed] [Google Scholar]
- Wong B. M.; Piacenza M.; Della Sala F. Phys. Chem. Chem. Phys. 2009, 11, 4498. [DOI] [PubMed] [Google Scholar]
- Color change in PCL films was observed experimentally between 35 and 40% elongation during deformation studies. Video of the film deformation can be found in the the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.