

Abstract
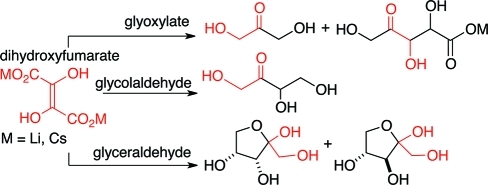
In the context of a “glyoxylate scenario” of primordial metabolism,1 the reactions of dihydroxyfumarate (DHF) with reactive small molecule aldehydes (e.g., glyoxylate, formaldehyde, glycolaldehyde, and glyceraldehyde) in water were investigated and shown to form dihydroxyacetone, tetrulose, and the two pentuloses, with almost quantitative conversion. The practically clean and selective formation of ketoses in these reactions, with no detectable admixture of aldoses, stands in stark contrast to the formose reaction, where a complex mixture of linear and branched aldoses and ketoses are produced. These results suggest that the reaction of DHF with aldehydes could constitute a reasonable pathway for the formation of carbohydrates and allow for alternative potential prebiotic scenarios to the formose reaction to be considered.
Introduction
In a recent review entitled “The Search for Chemistry of Life’s Origin”, Eschenmoser has propounded a “glyoxylate scenario”,1 wherein glyoxylate (1) and its formal dimer, dihydroxyfumarate (DHF, 2), are proposed to be the central starting materials of a chemical constitution of a possible primordial metabolism, serving as source molecules for the formation of biogenic molecules (such as sugars, α-amino acids, pyrimidines, and constituents of the citric acid cycle), by reactions deemed to be compatible with the constraints of prebiotic chemistry.1a An important component of the proposal was the possibility that prebiotic chemistry might have to consider, besides the classical formose reaction,2 additional pathways to carbohydrates, starting not from formaldehyde, but rather from “aquo-oligomers of carbon monoxide”, such as glyoxylate and dihydroxyfumarate. The proposal put forward, specifically, that reactions of DHF with glyoxylate, glycolaldehyde, and glyceraldehyde could lead to the formation of the corresponding sugars. If this were to be demonstrated experimentally, then it would bolster this segment of the “glyoxylate scenario” proposal. This paper describes results of exploratory investigations with the aim of examining some of the chemical and potentially prebiotic aspects of this proposal.
When we considered what was already known regarding the reactions involving glyoxylate or DHF in aqueous environment, literature experimental precedents were unpropitious. Only rare cases involving glyoxylic acid (or its salts) as a partner in aldol reaction, especially in mixed-aqueous medium, had been documented.3 Moreover, for DHF,4 only two major reaction pathways in aqueous medium had been identified previously: (a) the decarboxylative conversion to glycolaldehyde5,6 and (b) the oxidative transformation to dioxosuccinic acid,7 leading to the perception that DHF (and its salts) in water are “unstable”; in addition, DHF and its Na+, K+, and NH4+ salts are sparingly soluble in water. Of these documented observations, the decarboxylative conversion of DHF to glycolaldehyde and the (in)solubility of DHF (and its salts) in aqueous medium are expected to play a central role in the context of the “glyoxylate scenario”. For example, the sparing solubility of DHF (and its salts) in water could act as protecting factor against decomposition, while ensuring a slow and steady source of the starting material, while the decarboxylation of DHF, en route to glycolaldehyde, is expected to produce various reactive intermediates that would be able to take part in the “glyoxylate scenario”, while also acting as “driver” of the reaction.
At the beginning of our study of the aldolization chemistry of DHF in the presence of glyoxylate, we also investigated the behavior of water-soluble lithium,5c cesium, and magnesium salts of DHF alone, and observed that DHF is capable of undergoing aldolization either with itself (dimerization) or with its decarboxylated intermediate (α-carboxy-glycolaldehyde). The product is a single threo-diastereomer of pentulosonic acid 3 (Scheme 1) formed in quantitative yields; we recently have reported this result.8
Scheme 1. Condensation Reaction between 0.4 M (Li or Cs Salts of) DHF (2) and 0.4 M Sodium Glyoxylate (1) Leading to the Formation of Dihydroxyacetone (4) and a (∼1:1) Diastereomeric Mixture of Pentulosonic Acid (3a + 3b) as Deciphered by Monitoring with 13C NMR Spectroscopy.

13C NMR chemical shifts (δ, ppm) are shown in the Cs series.
Reaction of DHF with Glyoxylate
We began by investigating the reaction between glyoxylate (a “carboxylated formaldehyde”) and DHF, because of their proposed generational relationship1 and used 13C NMR spectroscopy extensively to monitor and characterize the reactions. In our preliminary experiments, we observed the existence of a rapid condensation reaction between lithium or cesium salts of DHF and sodium or cesium glyoxylate (0.3–0.4 M, 1:1 equiv, room temperature, pH ≈ 7–8).9 The dimerization of DHF8 was suppressed in the presence of glyoxylate (no signals corresponding to the DHF dimer8 could be observed in the 13C NMR spectrum). The absorbance of DHF decreased about 7 times faster (at rt) in the presence of 1 equiv of glyoxylate, compared with DHF alone under identical conditions (as monitored by UV spectroscopy at 289 nm, Figure S9 in Supporting Information).
Monitoring of the reaction mixture (both at 4 °C and rt) by 13C NMR spectroscopy revealed the formation of a discrete keto acid adduct 5 formed from the initial condensation reaction of DHF with glyoxylate (Scheme 1). This intermediate 5 decarboxylated (losing the β-carboxyl group) leading to the formation of a symmetrical intermediate dicarboxy-dihydroxyacetone6. The loss of the second β-carboxyl group (from either position in 6) gave rise to monocarboxyl dihydroxyacetone 7, which on further decarboxylation gave rise to the final product dihydroxyacetone (DHA, 4); it was of interest to note that no glyceraldehyde could be detected. However, apart from 4, a diastereomeric mixture (∼1:1) of pentulosonic acid (3a + 3b) was also formed (Figure 1); this was not anticipated, since only one equivalent of glyoxylate was employed (which should have been consumed in the first condensation step with DHF to give 5). No branched products were detected.
Figure 1.

13C NMR (150 MHz, H2O/D2O) of the reaction mixture from Cs2DHF with cesium glyoxylate (0.5 M, 1:1) after 20 h at rt, showing the 1:1 mixture of pentulosonic acid diastereomers (3a + 3b) and dihydroxyacetone (4).
Time elapsed 13C NMR monitoring divulged not only the formation and demise of intermediates 5, 6, and 7 but also that they have markedly different stabilities and different rates of decarboxylation, which are highly temperature dependent. The initially formed adduct 5 was stable for about 30–60 min at 4 °C, while it disappears within minutes at rt decarboxylating to afford intermediate 6. This dicarboxy-dihydroxyacetone intermediate 6 was stable for about 12–24 h at 4 °C and for about 4–6 h at rt, decarboxylating to intermediate 7. The monocarboxyl dihydroxyacetone intermediate 7 was stable for a longer time (24–36 h at 4 °C or for about 12 h at rt) en route to the final products. Thus, there was an intervening period where both intermediates 6 and 7 were present at 4 °C as the major species (Figure S10, Supporting Information). The change in the counterion (from Li+ to Cs+) did not seem to make any difference either in the pathway or the final outcome of this reaction, except that the rates of the reaction in the presence of Cs+ seemed to be slower compared with those in the presence of Li+.
The unexpected formation of diastereomeric mixture of pentulosonic acid (3a + 3b) could be rationalized if an additional molecule of glyoxylate would react with intermediate 6 or 7 or the final product 4. Careful monitoring by 13C NMR revealed that the intensity of the signals for DHF or glyoxylate were very small after the initial mixing period and were not observed during further progress of the reaction.10 For example, the major species observed (at 24 h at 4 °C or at 20 min at rt) were the dicarboxyl-DHA and DHA (6 and 7). However, as time progressed at room temperature, signals attributed to DHA (4) and diastereomeric mixtures of pentulosonic acid (3a + 3b) appeared, with none of the signals corresponding to DHF or glyoxylate detectable. These observations seem to negate the notion that the reaction of another molecule of glyoxylate reacting with 6, 7, or 4 could constitute a major pathway for the formation of 3.11 In order to clarify the pathway by which diastereomeric mixture of pentulosonic acid is formed, we used doubly 13C-labeled glyoxylate to study the reaction with DHF. The persistence of even minute amounts of glyoxylate and the intermediates that are formed when it reacts further should be revealed by measurements on the sensitive cryo-probe 13C NMR instrument. Moreover, if glyoxylate reacted with the dicarboxyl-DHA (6), monocarboxyl-DHA (7), or DHA (4), this pathway should eventually give rise to a pentulosonic acid skeleton where the 13C labeled carbons would end up at three consecutive carbon positions (Scheme 2).
Scheme 2. The Reaction of 0.4 M DHF with 1 Equiv of Doubly 13C-Labeled Glyoxylate (as Followed by 13C NMR Spectroscopy).

13C-labeled glyoxylate contains 1.5% oxalic acid and 3% glycolic acid. 13C NMR chemical shifts (δ, ppm) are as indicated; d = doublet (J ≈ 45 Hz), s = singlet.
Reaction of doubly 13C-labeled glyoxylate with unlabeled DHF (0.4 M each, 4 °C and rt) was investigated. The 13C NMR spectrum of the reaction did not show a triplet or doublet of doublet (resulting from a 13C–13C–13C-type coupling) in the signals arising from the final pentulosonic acid; all 13C-enriched signals were either a singlet or doublet. Therefore, this observation ruled out the pathway involving 6a, 7a, or 4a reacting with glyoxylate en route to pentulosonic acid.11 These observations, however, pointed to a possible alternative route, where the initially formed adduct 5, after decarboxylation, gives rise to a putative enolate intermediate 2-ene-dioate that could react faster (compared with DHF), selectively at C(2)-position, with glyoxylate to give a β-keto acid adduct,12 which then undergoes stepwise decarboxylation affording the observed 13C-label distributionin pentulosonic acid (Scheme 2). Any remaining DHF would react with itself to produce (unlabeled) pentulosonic acid.8
The keto- and CHOH (α to the keto group) of the pentulosonic acid 3 do not appear in the (enriched) 13C NMR spectrum indicating that these carbon atoms are unlabeled and must originate from the DHF. The CO2—, CH2OH, and the CHOH (β to the keto group) of the pentulosonic acid contain the 13C label and, therefore, must originate from the doubly 13C-labeled glyoxylate. One of the CH2OH groups of DHA 4 is 13C-labeled, while the other CH2OH and the keto groups are not. This indicates that the 13C-labeled CH2OH must have come from glyoxylate, while the unlabeled CH2OH and the keto groups have their origins in DHF. These observations once again reaffirm that DHF salts in water are equivalent to an α-hydroxy acetyl anion umpolung synthon.8
When the same reaction of DHF and doubly 13C-labeled glyoxylate was carried out at lower concentration (0.04 M), the reaction proceeded identically (Scheme 3) up to the stage of formation of a mixture of 3 and 4. At this lower concentration, a newer reaction pathway emerged: pentulosonic acid 3 was found to undergo a ketose–aldose transformation and to exist in equilibrium with xyluornic acid (major, 8a) along with traces of riburonic acid (minor, 8b). The identification of 8a and 8b was made by comparison with 13C NMR chemical shifts of known compounds.13 This observation suggests that for reactions at lower concentrations, there may be pathways that are able to shift the equilibrium toward the thermodynamically less favored aldoses; in this specific case, the aldoses may be stabilized as a cyclic hemiacetal.
Scheme 3. Reaction of DHF and 13C-Labeled Glyoxylate under Dilute (0.04 M) Conditions, with 13C NMR Chemical Shifts (δ, ppm).

With the intention of maximizing dihydroxyacetone formation, we explored the role of lowering the pH and the role of divalent metals (such as Mg2+, Cu2+, and Zn2+). After some exploration,14 we found that the reaction pathways in Scheme 2 could be influenced by simply acidifying the reaction mixture after the elapse of a certain amount of time. For example, it was observed that after 10 min at rt, 13C NMR shows predominantly the presence of the dicarboxyl-DHA intermediate (6). Therefore, acidification at this stage should induce decarboxylation leading to the formation of DHA (4). Accordingly, acidification of the reaction mixture after 10 min at rt resulted in the predominant formation of 4 (as evidence by 13C NMR, Figure S14, Supporting Information) with little or no pentulosonic acid formed.15 No peaks corresponding to glyceraldehyde could be observed. In one case, after workup, dihydroxyacetone was isolated (as the 2,4-dinitrophenylhydrazone derivative) in only about 20% yield, pointing to the dichotomy of the low isolated yields against the apparent high conversion efficiency in this reaction.
The formation of dihydroxyacetone in the reaction of DHF with glyoxylate (formally a carboxylated formaldehyde) encouraged us to further explore the reactions of DHF with other α-hydroxy aldehydes (glycolaldehyde, glyceraldehyde), formaldehyde, and ketones (dihydroxyacetone and acetone). Reactions were monitored by 13C NMR: intermediates and end products were identified based on 13C and 1H NMR data (and where available by comparison, and spiking, with authentic samples). We used high enough concentrations (ca. 0.1 M and upward) in the reaction to ensure detection by 13C NMR spectroscopy would be possible within a reasonable amount of time.
Reaction of DHF with Formaldehyde
The “glyoxylate scenario” calls for bypassing formaldehyde, since glyoxylate itself is the potential alternative source molecule.1However, we did explore the reaction of DHF with formaldehyde purely from a chemical viewpoint.
The reactions of Li2DHF (0.8 M) with aqueous solution of formaldehyde (formalin, 37 wt % in water containing 10–15% MeOH as stabilizer) at rt and 4 °C was examined (Scheme 4); pH of the reaction mixtures ranged from 8 to 9. The course of the reaction (as monitored by 13C NMR) showed the formation of the initial keto adduct 9, which is transformed to different end products, by two different pathways depending on the amount of formaldehyde present:
-
(a)
When one equivalent of formaldehyde was present, intermediate 9 decarboxylated to yield monocarboxylated-DHA 7, which further decarboxylated to yield DHA (4), quantitatively; 13C NMR of the reaction mixture showed the presence of HCO3– (and MeOH from formalin) as the only other byproduct. During the course of the reaction, peaks (with low intensity) attributable to DHF-dimer formation could be observed; however, as the reaction progressed, only signals for 4 were observed. This suggested that there could be a minor equilibration between the initial keto-adduct 9 with its starting materials, which with progress of time is channeled toward DHA (4) formation, by subsequent decarboxylations.
-
(b)
When more than one equivalent of formaldehyde was present, intermediate 9 (after decarboxylation, via the putative 2-ene-diolate, by analogy with earlier observation) reacted further with another molecule of formaldehyde to give adduct 10, which then lost a molecule of carbon dioxide to form tetrulose 11 with complete conversion. It is interesting to note here that further reaction with formaldehyde, under the reaction conditions, gave rise to a linear, and not a branched, sugar derivative. This points once again to the strong preference of the (putative) ene-diol intermediate derived from the decarboxylation of adduct 9 to react exclusively as nucleophile at the C(2), and not C(3), carbon leading to β-keto acid intermediate 10.
Scheme 4. Reaction of DHF with Formaldehyde Leads to Formation of Dihydroxyacetone or Tetrulose As Determined by 13C NMR Spectroscopy.

Acidification leads to the formation of (hydrated) 2,3-diketobutanol (12). 13C NMR chemical shifts (δ, ppm) are indicated.
When a reaction mixture containing DHF and 2 equiv of formaldehyde was acidified after 30 min at rt (to pH ≈ 2–3) and left overnight at rt, a methyl diketone derivative 12 (presumably from elimination of water from intermediate 10)16 was formed as essentially the sole product in the reaction mixture. The constitution of diketo 12 was identified by 13C NMR spectral analysis.
Reaction of DHF with Glycolaldehyde
Reaction of 1.0 equiv (0.1–0.5 M) of Li2DHF with 1.0 equiv of glycolaldehyde (pH ≈ 8–9, at 4 °C and rt) led to the clean formation of tetrulose as the sole product (Scheme 5). 13C NMR monitoring revealed that the reaction proceeded via a five-membered cyclic intermediate 13, which experienced stepwise decarboxylations (presumably via the putative linear form) to yield tetrulose 11 over a period of 24–48 h. For the reaction conducted at 4 °C, 13C NMR spectral data indicated that two diastereomers of the cyclic intermediate 13 are formed. Based on the differences in the disappearance of the signal intensity of each species over time, it was concluded that one diastereomer of the cyclic intermediate displayed the tendency to decarboxylate faster than the other diastereomer.
Scheme 5. Reaction Pathway of 1.0 Equiv of DHF with 1.0 Equiv of Glycolaldehyde Leading to the Formation of Tetrulose As Discerned by 13C NMR Spectroscopy.

13C NMR chemical shifts (δ, ppm) are shown.
At low temperatures (4 °C), an equilibrium between cyclic adduct 13 and the starting materials (DHF and glycolaldehyde) seems to exist as suggested by the presence of signals assignable to DHF and glycolaldehyde, which eventually with progress of time is funneled off into tetrulose formation. Such a phenomenon was not observed at room temperature and higher concentrations, presumably (also) due to higher rates of decarboxylation, which would siphon off intermediate 13 in an irreversible manner. Once again, no branched sugar products were observed.
Conducting the reaction in D2O (instead of H2O) led to the formation of the corresponding α-CD2 derivative of tetrulose, confirming (a) the decarboxylation-followed-by-protonation (deuteration) steps of the proposed mechanistic pathway and (b) that DHF acted as the hydroxyl-ketone anion equivalent8 in attacking the carbonyl group of the glycolaldehyde.
The identity of the end product, tetrulose 11 (Figure 2), was confirmed by comparison of the 13C NMR spectral data with those of commercially available erythrulose. The 13C NMR spectrum of the reaction mixture indicated the quantitative conversion to tetrulose 11 with no other organic products discernible by 13C NMR. Attempts to isolate pure tetrulose 11 were discontinued after it became clear that the process of isolation and purification led to decomposition and further reaction of tetrulose under the isolation conditions.
Figure 2.

13C NMR spectrum (150 MHz, drops of D2O) of the reaction mixture of 0.1 M Li2DHF + 0.1 M glycolaldehyde (1:1) at rt, after 20 h. Peak at 160.95 ppm is HCO3–.
Reaction of DHF with Glyceraldehyde
The reaction of DHF with dl-glyceraldehyde was investigated most intensively, because it held the promise of a straightforward route to the five-carbon sugars. Based on the reactions with other aldehydes, DHF was expected to act as the hydroxyacetyl umpolung synthon, attacking the carbonyl group of glyceraldehyde to afford the pentoses/pentuloses.
Exploratory experiments were conducted with 0.4 M Li2DHF with 1.0 equiv of dl-glyceraldehyde at 4 °C (pH ≈ 8–9). Monitoring the reaction with 13C NMR spectroscopy suggested that the reaction pathway is complex in its details; for example, the reaction seemed to proceed via at least three different (five- and six-membered) cyclic intermediates, all of which with time were converted to end products, thought to be a mixture of the isomers of pentuloses/pentoses. At least one of the end products was preliminarily identified as ribulose by comparison with an authentic sample based on 13C NMR spectral data.17a ESI-mass spectral data of the crude reaction mixture showed peak(s) corresponding to a pentose/pentulose.
Encouraged by these preliminary results, we began a systematic investigation of the reaction of Li2DHF (0.33 M, 1.0 equiv) withdl-glyceraldehyde (1.0 equiv) mixed at 4 °C (pH ≈ 8–9); then, reaction conditions were varied (pH, divalent metal ions, temperature, and concentration; see Table S1, Supporting Information, for details), and all reactions were monitored by 13C NMR spectroscopy (Scheme 6). The results from these explorations pointed out the following:
-
(a)
The reaction proceeded through the formation of two five- and one six-membered cyclic adducts, which lost CO2 in succession, presumably via the corresponding linear intermediates that are not visible in the 13C NMR spectra.
-
(b)
The 13C NMR spectral data of the final (crude) reaction mixtures indicated that xylulose was formed as the major product with ribulose as the minor product; the two pentuloses were present as α- and β-anomers and the open keto form. Identification and assignments were based on comparison with literature values,17 and comparison and spiking with authentic materials.
-
(c)
The six-membered cyclic intermediate identified as 16-p was quite stable over period of time (for 7 days) at 4 °C, or even at rt for 24 h, at near neutral pH. We were even able to identify this cyclic adduct in the mass spectrum (Figure S61, Supporting Information). Under acidic pH (∼1–2), this pyranosyl intermediate 16-p was found to be remarkably stable (even after 2 days at rt); the low pH seems to prevent the formation of pentuloses from this cyclic intermediate, by impeding the conversion of 16-p to the open α-keto acid form. Presumably at this low pH, the open (linear) form of the α-keto acid is highly destabilized (by the conversion of the less electron-withdrawing carboxylate moiety to the more electron-withdrawing carboxylic acid moiety), and the compound alleviates this problem by transforming itself to the cyclic acetal.
-
(d)
Decarboxylation was accelerated by mild acidification or by the presence of divalent metals. The addition of Lewis acids (such as MgCl2 and ZnCl2) hastened the formation of pentuloses from the (furanosyl- and pyranosyl-) intermediates and seemed to do it in a more controlled fashion than simple acidification. ZnCl2 seemed to offer the cleanest conversion to the products within the shortest amount of time.
-
(e)
When conducted at 4 °C, the reaction proceeded more slowly with corresponding delay in the rate of appearance and disappearance of the intermediates; however, the nature of the final products and their distribution remained the same compared with the reaction at rt.
Scheme 6. Formation of Pentuloses from the Reaction of DHF with Glyceraldehyde by the Reaction Pathway As Inferred by Monitoring with 13C NMR Spectroscopy.

Chemical shifts (δ, ppm) are shown.
Nowhere during the course of monitoring did we observe the reappearance of signals for DHF or glyceraldehyde. This is in contrast to the observations made in the glycolaldehyde series, where we were able to observe the re-emergence of peaks attributable to glycolaldehyde and DHF after the formation of the five-membered ring intermediate 13 (Scheme 5). The existence of equilibrium between 13 and its starting materials versus the absence of it in a six-membered cyclic intermediate (16), perhaps, is the result of the dichotomy in the relative kinetic and thermodynamic lability of the five-membered ring over that of a six-membered ring structure.
Based on this extended investigation, we decided to use ZnCl2 and study, in detail, the pathway of the formation of pentuloses. To this end, we reacted a suspension of glyceraldehyde in water with Li2–DHF (0.33 M each) at 4 °C for 2 h (pH ≈ 10.7), which resulted in a pale yellow clear solution. Then, 2.0 equiv of ZnCl2 was added, which lowered the pH to 4.39. The reaction mixture was brought to rt and monitored by 13C NMR spectroscopy (final pH ≈ 5.6).
Once again, we could follow the reaction and identify and confirm the pyranosyl intermediate (16-p) and the end products based on 13C NMR chemical shifts and multiplicity (Scheme 6). After the completion of the reaction as judged by 13C NMR(Figure 3), the violet suspension was centrifuged, and the yellow supernatant was separated and concentrated in vacuo to afford a syrup. 1H NMR of this crude material was surprisingly clean (Figure 4) and showed the formation of the two pentuloses 17 and 18. Comparison with authentic samples of ribulose (17) and xylulose (18) allowed the assignment of the different forms of the two pentuloses. Integration of the relevant peaks allowed us to determine the ratio of the pentuloses formed: 78% of xylulose and 22% of ribulose. Spiking with authentic xylulose and ribulose confirmed the assignments.
Figure 3.

13C NMR (150 MHz, drops of D2O) of reaction mixture supernatant after overnight at rt (0.33 M Li2DHF + 0.33 M glyceraldehyde in degassed H2O + ZnCl2).
Figure 4.

1H NMR (600 MHz, D2O) of crude product (0.33 M Li2DHF + 0.33 M glyceraldehyde in degassed H2O + ZnCl2).
The 1H and 13C NMR spectral data of the crude reaction mixture indicates that there are very minor side products formed, implying near quantitative conversion of starting materials to products; no aldoses seem to be present. Yields of the crude mixture of pentuloses were estimated to be at least 60% (calculated based on addition of a known amount of sodium benzoate as internal standard to the crude reaction mixture). We have not attempted to purify the pentuloses produced in these reactions since the crude mixture itself was reasonably clean by 1H NMR (Figure 5).
Figure 5.

Comparison of the 1H NMR spectral data (D2O) documenting the formation of the two pentuloses in the reaction of DHF and dl-glyceraldehyde. top (blue), authentic ribulose; middle (green), crude reaction mixture; bottom (red), authentic xylulose.17b
We then set out to see whether there were conditions that would affect the ratio of xylulose to ribulose. We set up the following experiments: Li2DHF was treated with glyceraldehyde at 4 °C. After 5 min, 13C NMR showed the formation of a six-membered ring intermediate (16-p) along with signals for five-membered ring intermediates (15, 16-f), with no signals corresponding to the starting materials. At this stage the reaction was divided into five parts:
-
(a)
Portion 1 was immediately treated with 2.0 equiv of ZnCl2 and kept at rt for 24 h. The reaction was worked up and analyzed by 1H NMR.
-
(b)
Portion 2 was maintained 4 °C for 48 h and then treated with 2.0 equiv of ZnCl2 and kept at rt for 24 h. The reaction was worked up and analyzed by 1H NMR.
-
(c)
Portions 3, 4, and 5 were brought to rt and kept for 3, 23, and 48 h and then treated with 2.0 equiv of ZnCl2, monitored by 13C NMR, worked up, and analyzed by 1H NMR.
13C NMR spectral monitoring of the five different reactions at regular intervals, combined with the 1H NMR spectral analysis of the (crude) pentuloses produced in the above five reactions revealed the following: (a) the cyclic intermediates were present in two furanose and one pyranose forms; the threo configured adduct existed predominantly in the pyranose form (16) with a minor furanose form, while the erythro configured adduct existed only in the furanose form (15); (b) the pyranose form (16-p) of the threo configured adduct does equilibrate to the furanose form (16-f), while the eythro configured adduct (15) seems not to equilibrate to its pyranose form; (c) the ribulose/xylulose ratio is set at the stage of the formation of adducts and is not influenced after the fact, by time, the nature of the divalent metal ion, or temperature.
This indicated that in order to produce more ribulose, we must influence the reaction before (or as) DHF and glyceraldehyde come together. To that end, we found that mixing the starting materials at a higher temperature affected the xylulose/ribulose ratio. For example, mixing the reaction partners at rt (instead of 4 °C) increased the amount of ribulose that is formed (ribulose/xylulose = 33:67). However, changing the concentration seemed to have no effect; for example, conducting the reaction at a lower concentration (0.055 M at 4 °C) did not affect the ratio of xylulose and ribulose. The reaction at 0.055 M was found to go to completion with 1H and 13C NMR of the reaction mixture qualitatively similar to the ones from the reactions at higher concentrations, demonstrating the efficiency of the reaction even at this lower concentration.
Reaction of DHF with Dihydroxyacetone
The formation of dihydroxyacetone as one of the products in the reaction between Li2DHF and sodium glyoxylate, opened up the possibility that DHF could react further with dihydroxyacetone. To explore this avenue, 1.0 equiv (0.3–0.5 M) of Li2DHF was mixed with 1–2 equiv of dihydroxyacetone at 4 °C; pH of the reaction mixtures ranged from 8 to 9.
The reaction between DHF with dihydroxyacetone was found to proceed slowly (monitored by 13C NMR spectroscopy), compared with the reaction of DHF with aldehydes at comparable time intervals. Pentulosonic acid (3) was the major product (deriving from reaction of DHF with itself)8 along with a branched pentulose 20 (Scheme 7). 13C NMR spectral data of the reaction was consistent with the formation of a cyclic adduct (19), deriving from the first condensation step of DHF with dihydroxyacetone; subsequent decarboxylations of this cyclic intermediate led to the formation of the known branched pentulose 20(18) as a minor product. Unreacted dihydroxyacetone constituted a major portion of the reaction mixture.
Scheme 7. Reaction of DHA with DHF (as Followed by 13C NMR Spectroscopy).

13C NMR chemical shifts (δ, ppm) are indicated.
The sluggishness of the reaction and the preponderance of the self-reaction of DHF to form pentulosonic acid seem to be the direct consequence of a combination of an intrinsically less reactive keto carbonyl and steric hindrance around the keto carbonyl group in dihydroxyacetone. In line with this reasoning, no reaction of DHF with (less reactive) acetone was observed.
Furthermore, when a 1:1 mixture of dihydroxyacetone and glyceraldehyde was reacted with DHF, 13C NMR spectrum showed predominant signals attributable to cyclic intermediates 15 and 16 formed by reaction between DHF and glyceraldehyde; the other dominant signals belonged to the unreacted dihydroxyacetone. Signals corresponding to 19, the intermediate derived from DHF and DHA, were not observed. When this reaction was allowed to go to completion (with addition of ZnCl2), pentuloses were formed, with xylulose as the predominant product indicating that the presence of dihydroxyacetone has not interfered by reacting with DHF in the reaction pathway.19
Discussion
The proposal1b that DHF chemistry could act as an abiotic source of biologically relevant molecules exploits the chemical reactivity of the C(2)–C(3)-ene-diol moiety present in DHF, which could act as a carbon nucleophile at either end and react with carbon electrophiles. The resulting 2-keto-1,3-dicarboxylate adduct, as proposed, would be expected to undergo stepwise decarboxylations, followed by carbonyl migrations, that would drive the overall chemistry. Such a proposition has been experimentally realized, herein, demonstrating that the carbon–carbon bond forming reaction of DHF with α-hydroxy-aldehydes can generate biologically relevant ketoses in a highly efficient, clean, and selective manner (Scheme 8). In all of the reactions explored here, DHF exhibits a proclivity for acting as the nucleophile. Only in the case of the self-condensation reaction has it shown the ability to be, also, an electrophile.8
Scheme 8. The “Expanded” Reaction Spectrum of Aqueous Chemistry of DHF.

M = H, Li, Cs, or Mg. The formation of dioxosuccinate may not be relevant in the context of potential prebiotic chemistry.
Interestingly, DHF is highly reactive toward aldehydes while being sluggish in its reactivity with ketoses. This result indicates that DHF would continue to react preferentially with aldoses, even if compounds containing keto groups would be formed (or be present) in the reaction mixture.
Therefore, no “interference” from the product ketoses, and no formation of “branched sugars” would be expected. Such a reactivity preference seems to lie at the heart of the sparseness20 of the products and cleanliness of these reactions. Also, under the reaction conditions, the products formed are stable and do not seem to be broken down or react further. In these two aspects, the reactions described in this study stands in stark contrast to the classical formose reaction,7 wherein (a) aldoses and ketoses, both linear and branched, are formed to a significant extent and (b) the final products are degraded (under the reaction conditions of the classical formose reaction).
The generation of carbohydrates (especially ribose) in the context of origin of life research is yet an unresolved issue, with the formose reaction still continuing to attract much attention, highlighting the desire to come-up with solutions to the complex mixtures generated in this reaction. Intensive efforts have been expended in simplifying and “cleaning-up” the classical formose reaction and to channel it toward ribose production.
One approach was to replace glycolaldehyde in the formose reaction with glycolaldehyde phosphate.21 This substitution resulted in vastly limiting the reaction products to linear aldose-phosphates, due to the presence of 2-O-phosphate in the intermediates and products, which prevented further keto–enol tautomerization. It is interesting to note that the DHF reactions in this paper bear some resemblance to the glycolaldehyde phosphate variant in two aspects: (a) the simplification and the “cleanliness” of the overall reaction and (b) the presence of carboxyl groups, which emulate the phosphate group in not only imparting charge to the adducts but also controlling the direction of carbonyl migration in the products and the site of carbon–carbon bond formation (toward linear and not branched ketoses).
Other solutions for modulating the formose reaction range from channelizing the reaction pathways via interaction with plumbous ions22 to interaction with borate23 or silicate24 minerals. Of these, the borate-moderated formose reaction has been extensively investigated, by Benner and co-workers; they have proposed that borate minerals, in conjunction with molybdate minerals, could guide the synthesis of carbohydrates starting from formaldehyde and glycolaldehyde.23b
All of the above, and many variations thereof, still deal with formaldehyde as the main starting molecule.25 Herein, we have taken up an unconventional “glyoxylate scenario”1b and experimentally shown that there could be another pathway to carbohydrates starting from an “alternate” source molecule: dihydroxyfumaric acid (formally a dimer of glyoxylic acid, a “carboxylated formaldehyde”, and formally an aquo-dimer of CO), interacting with other small aldehydes, leading to clean formation of ketoses, as the primary sugar products. It should be noted in the early biochemical literature, DHF had been considered4 as a source of sugars, but by a very different pathway: decarboxylative conversion of DHF to glycolaldehyde (“glycolic aldehyde”), which can react further by aldolization, has been proposed as a possible pathway in the biosynthesis of sugars.
In view of the efficient and selective generation of ketoses from DHF (and formally, glyoxylate), the proposal,1 whether the glyoxylate scenario could be considered (in a prebiotic context) as an alternative to the formose reaction, becomes enticing. This immediately presents two obvious caveats: (1) the prebiotic availability of glyoxylate and (2) the prebiotic availability of dihydroxyfumarate. The first point seems to be more easily addressed by the fact there exists experimentally demonstrated reductive conversion of CO2 or CO to glyoxylate,26 which may be reasonably “stretched” to primeval earth scenarios. However, the second point, the prebiotic availability of DHF, seems to be more challenging; the two plausible sources for DHF proposed are (a) dimerization of glyoxylate1b and (b) the controlled hydrolysis of the diamino maleonitrile1a (DAMN, “HCN tetramer”). While, these two pathways have not yet been experimentally demonstrated, the results described above are sure to stimulate and initiate a search for the formation of DHF in a prebiotic context. Additionally, prebiotically acceptable replacements for the lithium and cesium cations need to be found; such endeavors are currently underway.27
In summary, experimental investigations carried out in the context of the “glyoxylate scenario” have uncovered reaction conditions where DHF reacts with aldehydes (glyoxylate, formaldehyde, glycolaldehyde, and glyceraldehyde) to afford selectively the keto-sugars as the main products. Such a reaction pathway could be considered as supplant for the formose pathway for the generation of carbohydrates.1 While this may be pertinent from a synthetic chemistry standpoint,28 whether such a statement could be also made from a “prebiotic chemistry” viewpoint needs to be substantiated and can be a motivation and inducement for further experimental endeavors.
Experimental Section
General Procedures
Degassed water was used throughout the experiments involving DHF. Anhydrous lithium hydroxide (reagent grade) (no. 44241-0; batch no. 10213CJ; CAS-1310-65-2); dihydroxyfumaric acid hydrate, 98%, solid; formalin, ACS grade, 37 wt % in H2O, contains 10–15% MeOH as stabilizer; glycolaldehyde dimer, crystalline solid; dl-glyceraldehyde dimer, ≥ 97%, solid; 1,3-dihydroxyacetone dimer, 97%, solid; (S)-(+)-erythrulose hydrate (syrup); d-xylulose, 95%, syrup; d-ribulose, 95%, syrup; cesium carbonate, 99.995%, solid; and sodium hydroxide, 97%, solid, were obtained from Sigma-Aldrich. Glyoxylic acid monohydrate, 98%, solid was obtained from Acros organics. Doubly 13C labeled glyoxylic acid, 99%, solid [contains glycolic acid (3%) and oxalic acid (1.5%)] was obtained from Cambridge Isotope Laboratories Inc. 1H NMR and 13C NMR spectra were recorded with a Bruker WM 600 MHz spectrometer and on a Bruker DRX-600 equipped with a 5 mm DCH cryoprobe. Samples for NMR studies were prepared by adding either 10% D2O or a drop of DMSO-d6. Chemical shifts δ values are expressed in ppm and coupling constants J in Hz. FAB-MS (ESI) was performed on a VG ZAB2 SEQ double focusing high-resolution mass spectrometer equipped with a cesium ion gun using a 3-nitrobenzyl alcohol/CsI matrix.
Preparation of Dihydroxyfumarate Dilithium Salt [Lithium (E)-2,3-Dihydroxybut-2-enedioate]
Dihydroxyfumaric acid (500 mg, 3.38 mmol, 1.0 equiv) was placed in a round-bottom flask and kept under vacuum for 10 min. To this flask, 7.5 mL of 0.9 M aq. LiOH solution was added through syringe at rt under nitrogen atmosphere and stirred at room temperature until it became a clear dark brown/red solution (in approximately 3–5 min.); pH of the resulting solution was 8.9.9
Preparation of Sodium Glyoxylate [Sodium 2,2-Dihydroxyethanoate]
Glyoxylic acid monohydrate (331 mg, 3.38 mmol, 1.0 equiv) was taken into round-bottom flask under nitrogen atmosphere and to this was added freshly prepared 7.2 M aqueous NaOH solution (0.47 mL, 1.0 equiv). Immediately heat was generated and the solution was cooled to room temperature (care has to be taken to avoid formation of precipitate). An alternative preparation used 331 mg (3.38 mmol) of glyoxylic acid and 3.6 M NaOH (0.94 mL).
Reaction of Dihydroxyfumarate with Glyoxylate
After the preparation of both salt solutions, aqueous “dihydroxyfumarate dilithium salt” solution was added dropwise to aqueous sodium glyoxylate solution at room temperature (or at 4 °C) under nitrogen atmosphere and stirring was continued at the same temperature. After the addition, the reaction became a very clear solution, and pH of the resulting reaction mixture was 8.3 (by pH meter).9 After 30 min, 0.5 mL of reaction mixture was transferred to an NMR tube and monitored by 13C NMR (H2O/D2O).
Reaction of Dihydroxyfumarate with Formaldehyde
To a cold (4 °C) aqueous solution of 1.0 equiv of DHF dilithium salt (0.8 M), 1.0 equiv of 37% formalin in water (∼0.30 mL, contains 10–15% MeOH as stabilizer) was added in one portion under nitrogen atmosphere at 4 °C. The reaction mixture became a clear solution, and the pH of the resulting solution was 8–9 (by pH paper). After ∼2 min, the “reaction flask” was placed at rt and monitored by 13C NMR.
Reaction of Dihydroxyfumarate with Glycolaldehyde
To a cooled (4 °C) aqueous solution of DHF dilithium salt was added aqueous glycolaldehyde solution (1 equiv, 0.5 M solution in water, stored overnight at rt for hydrolysis to monomer) at 4 °C under nitrogen atmosphere. After the addition, the reaction became a clear solution, and pH of the resulting reaction mixture was 8–9 (pH paper). The reaction mixture was monitored by 13C NMR.
Reaction of Dihydroxyfumarate with Glyceraldehyde
Glyceraldehyde (303 mg, 1.0 equiv, 0.33 M) was suspended in degassed water (at 4 °C). To this yellowish suspension, at 4 °C, a solution of Li2DHF [0.33 M, prepared by addition of aq. LiOH (161 mg, 2.0 equiv, 0.84 M solution) to solid DHF (500 mg, 1.0 equiv) kept at 4 °C; the Li2DHF solution was dark brown in color] was added. The resulting reaction mixture turned into a pale yellow solution over a period of 5 min; pH of the reaction mixture was 10.7 (at 4 °C). Reaction was kept at 4 °C for 2 h; 13C NMR (measured at 4 °C) at 2 h showed only pyranosyl adduct formation (16-p) with no 13C signals corresponding to starting materials visible. The reaction mixture was added, at 4 °C, to ZnCl2 (920 mg, 2.0 equiv). The reaction mixture acquired a dark violet color. After complete addition, the reaction mixture was left at rt; the pH of the mixture (after the immediate addition and bringing to rt) was 4.39. The reaction mixture was left at rt overnight (pH ≈ 5.6). Formation of a suspension was observed after 30 min of mixing, and after overnight reaction, more precipitation was seen (the suspension was violet in color, while the supernatant was pale yellow in color). The reaction mixture was centrifuged, and the pale yellow filtrate was separated, checked by 13C NMR, and concentrated to dryness to give a syrup (yellow in color). A portion (17 mg) of this syrup was dissolved in 0.5 mL of D2O; a small amount of undissolved white particles were separated by centrifugation, and the clear solution was analyzed by 1H NMR.
Acknowledgments
The work was initiated jointly by Eschenmoser and Krishnamurthy at TSRI and is published with the consent of Professor Eschenmoser. We are grateful to Professor A. Eschenmoser for the encouragement, support, and productive discussions. This work was supported by the Skaggs Research Foundation, NASA Astrobiology: Exobiology and Evolutionary Biology Program (Grant NNX09AM96G), and was jointly supported by NSF and the NASA Astrobiology Program under the NSF Center for Chemical Evolution, Grant CHE-1004570. V.N.S., F.H., and V. P. are Skaggs Postdoctoral Fellows. We thank the referees for their constructive comments.
Supporting Information Available
Full experimental details and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Eschenmoser A. Chem. Biodiversity 2007, 4, 554–573. [DOI] [PubMed] [Google Scholar]; b Eschenmoser A. Tetrahedron 2007, 63, 12821–12844. [Google Scholar]
- a Butlerow A. Justus Liebigs Ann. Chem. 1861, 120, 295–298. [Google Scholar]; b Butlerow A. C. R. Acad. Sci. 1861, 53, 145–147. [Google Scholar]; c Fischer E. Ber. Deutsch. Chem. Ges. 1888, 21, 988–992. [Google Scholar]; d Partridge R. D.; Weiss A. H.; Todd D. Carbohydr. Res. 1972, 24, 29–44. [Google Scholar]; e Mizuno T.; Weiss A. H. Adv. Carbohydr. Chem. Biochem. 1974, 29, 173–227. [Google Scholar]; f Decker P.; Schweer H.; Pohlmann R. J. Chromatogr. 1982, 244, 281–291. [Google Scholar]; g Decker P.; Schweer H. Orig. Life Evol. Biosphere 1984, 14, 335–342. [Google Scholar]; h Shapiro R. Orig. Life Evol. Biosphere 1988, 18, 71–85. [DOI] [PubMed] [Google Scholar]; i Schwartz A. W.; de Graaf R. M. J. Mol. Evol. 1993, 36, 101–106. [Google Scholar]
- a Loh T.-P.; Wei L.-L.; Feng L.-C. Synlett 1999, 1059–1060. [Google Scholar]; b Loh T.-P.; Feng L.-C.; Wei L.-L. Tetrahedron 2001, 57, 4231–4236. [Google Scholar]
- DHF reactions have been investigated for well over a century, starting with the discovery and extensive study of DHF in the late 1800s:; a Bourgoin E. Compt. Rend. 1874, 79, 1053–1054. [Google Scholar]; b Fenton H. J. H. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar]; c Fenton H. J. H. J. Chem. Soc. Trans. 1896, 69, 546–562. [Google Scholar]; d Fenton H. J. H. J. Chem. Soc. Trans. 1905, 87, 804–818. [Google Scholar]; e Fenton H. J. H.; Wilks W. A. R. J. Chem. Soc. Trans. 1912, 101, 1570–1582. [Google Scholar]; For X-ray analysis of DHF, see:; f Gupta M. P. J. Am. Chem. Soc. 1953, 75, 6312–6313. [Google Scholar]; g Gupta M. P.; Gupta N. P. Acta Crystallogr. 1968, B24, 631–636. [Google Scholar]; h Hartree E. F. J. Am. Chem. Soc. 1953, 75, 6244–6249. [Google Scholar]; i Goodwin S.; Witkopf B. J. Am. Chem. Soc. 1954, 76, 5012–5014. [Google Scholar]; j Yalpani M.; Wilke G. Chem. Ber. 1985, 118, 661–669. [Google Scholar]; k Kemp D. S.; Bowen B. J. Tetrahedron Lett. 1988, 29, 5077–5080. [Google Scholar]; l Jung M. E.; Blum R. E.; Gaede B. J.; Gisler M. R. Heterocycles 1989, 28, 93–97. [Google Scholar]; m Reithel F. J.; West E. S. J. Am. Chem. Soc. 1948, 70, 898–900. [DOI] [PubMed] [Google Scholar]
- a Fenton H. J. H. J. Chem. Soc. Trans. 1895, 67, 774–780. [Google Scholar]; b Fanke W.; Brathun G. Justus Liebigs Ann. Chem. 1931, 487, 1–52. [Google Scholar]; c Hay R. W.; Harvie S. Aust. J. Chem. 1965, 18, 1197–1209. [Google Scholar]; d Fleury M. Compt. Rend. 1965, 260, 5563–5566. [Google Scholar]; e Wong C. H.; Whitesides G. M. J. Am. Chem. Soc. 1983, 105, 5599–5603. [Google Scholar]; f Dazou D.; Karabinas P.; Jannakoudkis D. J. Electroanal. Chem. 1984, 176, 225–234. [Google Scholar]; g Garcia B.; Ruiz R.; Leal J. M. J. Phys. Chem. A 2008, 112, 4921–4928. [DOI] [PubMed] [Google Scholar]
- a Locke A. J. Am. Chem. Soc. 1924, 46, 1246–1252. [Google Scholar]; b Calvin M. J. Chem. Educ. 1949, 26, 639–657. [Google Scholar]; c Houg L.; Jones J. K. N. Nature 1951, 167, 180–183. [DOI] [PubMed] [Google Scholar]; Many enzymatic reactions of DHF have been carried in order to produce sugars; for early work, see:; d Neuberg C.; Schwenk E. Biochem. Z. 1915, 71, 104–113. [Google Scholar]; e Akabori S.; Uhera K.; Murumatsu I. Proc. Jpn. Acad. 1952, 28, 39–43. [Google Scholar]; f Fukunga K. J. Biochem. 1960, 47, 741–754. [Google Scholar]
- a Wieland H.; Franke W. Justus Liebigs Ann. Chem. 1928, 464, 101–126. [Google Scholar]; b Chow C. T.; Vennesland B. J. Biol. Chem. 1958, 233, 997–1002. [PubMed] [Google Scholar]; c Abrahams B. F.; Hudson T. A.; Robson R. J. Am. Chem. Soc. 2004, 126, 8624–8625. [DOI] [PubMed] [Google Scholar]
- Sagi V. N.; Karri P.; Hu F.; Krishnamurthy R. Angew. Chem., Int. Ed. 2011, 50, 8127–8130. [DOI] [PubMed] [Google Scholar]
- In some instances, the pH of the reaction mixture fluctuated as high as 10; such a behavior could be traced to the slight excess of base (NaOH or LiOH) added during the preparation. In cases where the pH was higher than 10, the 13C-NMR spectra showed minor signals ascribed to the formation of oxalate and glycolic acid, resulting from the Canizzaro reaction of glyoxylate, for example:; Honma H.; Kobayashi T. J. Electrochem. Soc. 1994, 141, 730–733. [Google Scholar]; Alternatively, this could be ascribed to decomposition of DHF under strong basic conditions, ref (8).
- However, there could be unreacted DHF or glyoxylate (formed reversibly from 5) present in amounts below the detection limits of the NMR instrument.
- We have separately reacted intermediates, dicarboxylated DHA 6, monocarboxylated DHA 7, and DHA 4, with glyoxylate and find that these reactions are comparatively slow and more complex. Monitoring by 13C NMR spectroscopy indicates that 6 reacts within hours (not minutes) with glyoxylate and gives pentulosonic acid and DHA 4 as the main products; 7 reacts more slowly (overnight) with glyoxylate and leads to more complicated reaction products, while 4 reacts even more slowly with glyoxylate, such that glyoxylate is not consumed even after 10 days at rt (see Supporting Information).
- When excess (5 equiv) glyoxylate is reacted with DHF, no dicarboxyl-DHA or monocarboxyl-DHA or dihydroxyacetone formation was observed.
- Wu J.; Serianni A. S. Carbohydr. Res. 1991, 210, 51–70. [DOI] [PubMed] [Google Scholar]
- A lower pH or the presence of divalent metals from the very beginning of the reaction was found to interfere with the desired pathway by accelerating the conversion of DHF to glycolaldehyde or by precipitating the DHF. These observations pointed to the need for a different strategy wherein we have to postpone such active intervention till after the desired intermediates (5, 6, or 7) are formed.
- Attempts were made to acidify at 4 °C after 6 h (when the dicarboxyl (6) and monocarboxyl (7) intermediates are dominant) or at 4 °C after 24 h (the monocarboxyl (7) is the dominant species). In both these cases, relatively more pentulosonic acid formation was observed.
- That this diketone 12 was not formed from tetrulose 11 was confirmed by separately acidifying tetrulose, which under identical conditions was found to be stable.
- a Vuorinen T.; Serianni A. S. Carbohydr. Res. 1991, 209, 13–31. [DOI] [PubMed] [Google Scholar]; b http://www.bmrb.wisc.edu/metabolomics/mol_summary/show_data.php?molName=D_xylulose&id=bmse000027&whichTab=1
- Simonov A. N.; Matvenko L. G.; Pestunova O. P.; Parmon V. N.; Komandrova N.; Denisenko V. A.; Vas’kovskii V. E. Kinet. Catal. 2007, 48, 550–555. [Google Scholar]
- The other side products that were seen in the 13C NMR spectrum seem to be best explained by compounds that would arise from the reaction of dihydroxyacetone with glyceraldehyde, under the mixing and reaction conditions. In this context, see the Zn–proline complex catalyzed reaction of glyceraldehyde/dihydroxyacetone:; Kofoed J.; Reymond J.-L.; Darbre T. Org. Biomol. Chem. 2005, 3, 1850–1855. [DOI] [PubMed] [Google Scholar]
- Copley S. D.; Smith E.; Morowitz H. J. J. Cosmol. 2010, 10, 3345–3361. [Google Scholar]
- Mueller D.; Pitsch S.; Kittaka A.; Wagner E.; Wintner C. E.; Eschenmoser A. Helv. Chim. Acta 1990, 73, 1410–1468. [Google Scholar]
- Zubay G. Orig. Life Evol. Biosphere 1998, 28, 13–26. [DOI] [PubMed] [Google Scholar]
- a Ricardo A.; Carrigan M. A.; Olcott A. N.; Benner S. A. Science 2004, 303, 196. [DOI] [PubMed] [Google Scholar]; b Kim H.-J.; Ricardo A.; Illangkoon H. I.; Kim M. J.; Carrigan M. A.; Fabianne F.; Benner S. A. J. Am. Chem. Soc. 2011, 133, 9457–9468. [DOI] [PubMed] [Google Scholar]
- a Degens E. T. Chem. Geol. 1974, 13, 1–10. [Google Scholar]; b Lambert J. B.; Gurusamy-Thangavelu S. A.; Ma K. Science 2010, 327, 984–986. [DOI] [PubMed] [Google Scholar]
- With the intention of producing pentoses, experimental investigations have been carried out, by starting from glycolaldehyde and glyceraldehyde. For example, see:; Pizzarello S.; Weber A. Orig. Life Evol. Biospheres 2010, 40, 3–10. [DOI] [PubMed] [Google Scholar]; See also ref (17a).
- a Park H. R.; Getoff N. Z. Naturforsch. 1988, 43a, 430–434. [Google Scholar]; b Eggins B. R.; Irvine J. T.S.; Murphy E. P.; Grimshaw J. J. Chem. Soc., Chem. Commun. 1988, 1123–1124. [Google Scholar]; c Eggins B. R.; Brown E. M. B.; McNeil E. A.; Grinshaw J. Tetrahedron Lett. 1988, 29, 945–948. [Google Scholar]; d Irvine J. T. S.; Eggins B. R.; Grimshaw J. Sol. Energy 1990, 45, 27–33. [Google Scholar]; e Eggins B. R.; Robertson P.K. J.; Stewart J. H.; Woods E. J. Chem. Soc., Chem. Commun. 1993, 349–350. [Google Scholar]; f Eggins B. R.; Ennis C.; McConnell R.; Spence M. J. Appl. Electrochem. 1997, 27, 706–712. [Google Scholar]
- For example, we have found that MgO (found as mineral periclase) serves as good candidate to replace Li+ and Cs+ and have preliminary results that are encouraging.
- The DHF chemistry demonstrated here could have applications in organic synthesis and, in combination with chiral catalysts, be further enhanced.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.