

Abstract
Farnesylated prelamin A is a processing intermediate produced in the lamin A maturation pathway. Accumulation of a truncated farnesylated prelamin A form, called progerin, is a hallmark of the severe premature ageing syndrome, Hutchinson-Gilford progeria. Progerin elicits toxic effects in cells, leading to chromatin damage and cellular senescence and ultimately causes skin and endothelial defects, bone resorption, lipodystrophy and accelerated ageing. Knowledge of the mechanism underlying prelamin A turnover is critical for the development of clinically effective protein inhibitors that can avoid accumulation to toxic levels without impairing lamin A/C expression, which is essential for normal biological functions. Little is known about specific molecules that may target farnesylated prelamin A to elicit protein degradation. Here, we report the discovery of rapamycin as a novel inhibitor of progerin, which dramatically and selectively decreases protein levels through a mechanism involving autophagic degradation. Rapamycin treatment of progeria cells lowers progerin, as well as wild-type prelamin A levels, and rescues the chromatin phenotype of cultured fibroblasts, including histone methylation status and BAF and LAP2α distribution patterns. Importantly, rapamycin treatment does not affect lamin C protein levels, but increases the relative expression of the prelamin A endoprotease ZMPSTE24. Thus, rapamycin, an antibiotic belonging to the class of macrolides, previously found to increase longevity in mouse models, can serve as a therapeutic tool, to eliminate progerin, avoid farnesylated prelamin A accumulation, and restore chromatin dynamics in progeroid laminopathies.
Key words: prelamin A, autophagy, protein degradation, laminopathies, progeria, Rapamycin.
Introduction
Hutchinson-Gilford progeria syndrome (HGPS) is an autosomal dominant condition which develops in the first or second year of life, followed by severe and rapid premature senescence, which involves almost all tissues.1,2 Patients present delayed growth, short stature, alopecia, thinning of skin, loss of subcutaneous fat, midface hypoplasia, skeletal involvement with osteolysis and fractures, premature atherosclerosis, cardiac failure leading to death or stroke at about 13.5 years. A de novo missense mutation in exon 11 leads to creation of an abnormal splice donor site that results in expression of a truncated, permanently farnesylated prelamin A, termed progerin.3,4 This is the most common mutation in HGPS. Toxicity of progerin has been well documented in several papers.5–7 Lowering progerin levels or eliminating its expression has been shown to ameliorate the cellular phenotype both in mouse and human cell models.5,8 Moreover, accumulation of wild-type farnesylated prelamin A as a secondary effect of progerin expression, worsens the cellular phenotype.9–11 In the reported study, we explored the possibility that rapamycin, an antibiotic belonging to the class of macrolides, known to activate autophagy in cells through inactivation of the inhibitory mTOR-dependent pathway, could trigger progerin degradation.12 Our results show that rapamycin treatment of HGPS cells dramatically and selectively reduces progerin and prelamin A levels and elicits rescue of LAP2α, BAF and trimethylated H3K9 organisation, thus improving the chromatin epigenetic status.
Materials and Methods
Cell cultures and transfection
HGPS skin fibroblasts were obtained from a 6-year-old patient carrying the G608G LMNA mutation. Control skin fibroblast cultures were obtained from skin biopsies of healthy patients (mean age 12) undergoing orthopaedic surgery, following a written consent. The protocol had been approved by the local ethical committee and informed consent had been obtained from patients or families. Cell cultures had been established and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS) and antibiotics. The experiments were performed at passages 5–10. Transient transfections of HEK 293T cells were performed by calcium phosphate method. FLAG-tagged plasmids contained wild-type prelamin A (LA-WT) or prelamin A Δ50 (LA-Δ50), encoding a form devoid of the 50 aminoacids lacking in the progerin sequence.11 After transfection, cells were incubated for 72 h.
Drug treatments
Rapamycin (1 µM, Sigma, Milano, Italy) was applied to cultured fibroblasts for 6 days. This drug inhibits mTOR activity and releases the autophagic signaling pathway, which is inhibited by mTOR.13 In transfected HEK 293 cells, rapamycin was applied at 2 µM concentration for 72 h. To block lysosomal activity, chloroquine (Sigma, 25 µM for 10 h) was applied either in the presence or in the absence of rapamycin. To check proteasome-mediated degradation the ubiquitin proteasome inhbitor MG132 (1 µM for 10 h) was applied.
Antibodies
Antibodies employed for Western blot analysis or immunofluorescence labeling were: anti-lamin A/C, goat polyclonal (SC-6215, used at 1:1000 dilution for the Western blot analysis, Santa Cruz Biotechnology Inc., Segrate, MI, Italy); anti-prelamin A, goat polyclonal (SC-6214 used at 1:100 dilution for the immunofluorescence analysis, Santa Cruz); anti-prelamin A, rabbit polyclonal (antibody 1188-2, Diatheva, Fano, Italy), raised against the last 15 aminoacids of the prelamin A sequence including the farnesylated cysteine residue but not the SIM sequence;14 anti-LAP2α, rabbit polyclonal;15 anti-trimethyl-H3K9, rabbit polyclonal (Upstate, Lake Placid, NY, USA); anti-emerin, mouse monoclonal (Monosan, Uden, The Netherlands); anti-BAF, rabbit polyclonal (FL-89, Santa Cruz); anti-LC3 rabbit polyclonal antibody (ABR, Pierce); anti-FLAG, mouse monoclonal (M2, Sigma); anti-actin, goat polyclonal (Santa Cruz).
Western blot analysis
Western blot analysis was done as follows. Cells were lysed in lysis buffer containing 20 mM Tris-HCl, pH 7.0, 1% Nonidet P-40, 150 mM NaCl, 10% glycerol, 10 mM EDTA, 20 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM Na3VO4, 1 mM PMSF, 10 ?g/mL leupeptin and 10 µg/ml pepstatin at 4°C. Cell lysates were diluted in Laemmli buffer, subjected to SDS-PAGE (8%) and transferred to nitrocellulose membrane. Membranes were saturated with 4% BSA and incubated with primary antibodies for 1 h at room temperature. Secondary antibodies were used at 1:10000 dilution for 30 min. Immunoblotted bands were revealed by the Amersham ECL detection system. Intensity measurement was performed using a BioRad densitometer (GS 800) equipped with Quantity One Software.
Gene expression analysis
Total RNA was isolated using Rneasy Mini Kit (Qiagen GmbH, Hilden, Germany) from confluent fibroblast cultures following manufacturer instructions. RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Monza, Italy). LMNA or ZMPSTE24 expression was evaluated by Real Time PCR, performed using an Applied Biosystems StepOne termal cycler instrumentation (Applied Biosystems), by amplifying 1 µg of cDNA and the TaqMan Gene Expression Assays (Applied Biosystems). Probes and primers obtained by Applied Biosystems were: GAPDH, assay ID Hs99999905_m1, LMNA, assay ID Hs00153462_m1*, ZMPSTE24, assay ID Hs00195298_m1*. The amplification protocol was: 50°C for 2 min; 95°C for 10 min; 95°C for 15 s, 60°C for 1 min, for 40 cycles. The results were calculated by the 2 ΔΔ−CT method16 or as ratio between gene of interest and GAPDH reference gene.17 The experiments were performed in duplicate.
Immunofluorescence
Human fibroblasts grown on coverslips were fixed with 4% paraformaldehyde at 4°C for 10 min and permeabilized with 0.15% Triton X-100 for 5 min. Alternatively, cells were fixed with absolute methanol at −20°C. Samples were incubated with PBS containing 4% BSA to saturate non-specific binding and incubated with primary antibodies overnight at 4°C, and with secondary antibodies for 1 h at room temperature. Anti-prelamin A Sc-6214, anti-lamin A/C and anti-trimethyl H3K9 were used at 1:100 dilution. Anti-prelamin A 1188-2 and anti-BAF were applied at 1:10 dilution. Anti-LAP2α was applied at 1:300 dilution. Slides were mounted with an anti-fade reagent in glycerol and observed with a Nikon E 600 fluorescence microscope equipped with a digital camera.
Statistical analysis
Statistical analysis was performed using the non-parametric Mann-Whitney U test.18 Experiments were done in triplicate and differences were considered statistically significant for P<0.05.
Results
Rapamycin treatment of HGPS fibroblasts
In order to test rapamycin effect on prelamin A in laminopathic cells, we treated cultured fibroblasts with rapamycin for 6 days. HGPS nuclei accumulate the truncated prelamin A form, known as progerin, obtained by aberrant splicing of the LMNA gene due to the G608G mutation. In HGPS cells, progerin levels were strikingly reduced following administration of rapamycin (Figure 1A). Progerin was not degraded under basal conditions, as shown by absence of CQ or MG132-induced protein accumulation, showing that the protein minimally undergoes spontaneous degradation (Figure 1 A–D).
Figure 1.

Rapamycin reduces progerin level in HGPS cells. A) Western blotting evaluation of lamin A/C and progerin in control (CONTROL) and Hutchinson Gilford progeria cells (HGPS). Whole lysates from control and HGPS cells untreated (−) or treated (+) with rapamycin (Rapamycin), MG132 or chloroquine-diphosphate (Cq) were subjected to lamin A/C, LC3-B2 and actin antibodies detection; B) densitometric analysis of lamin A and C immunolabeled bands detected in control fibroblasts; C) densitometric analysis of lamin A and C immunolabeled bands detected in HGPS fibroblasts; P=0.0358 for lamin A (rapa), P=0.0298 for lamin A (Mg); D) densitometric analysis of progerin immunoblotted bands; P=0.0390 for progerin (rapa); P=0.0458 for progerin (Mg); E) prelamin A, FLAG and LC3-B2 protein levels evaluation in HEK-293 cells expressing FLAG-tagged wild type prelamin A (LA-WT) or progerin (LA-Δ50). Immunolabeled bands observed in untreated (−) or rapamycin (Rapamycin) and chloroquine-diphosphate (Cq) treated (+) cells are shown; F) densitometric of FLAG immunoblotted bands; G) RT-PCR analysis of ZMPSTE24 and LMNA mRNA expression in untreated (Nt) and rapamycintreated HGPS cells (Rapa) and control (control); 2 −ΔΔCT values are reported relative to untreated control samples. P=0.0236 for LA-Δ50 (rapa); H) ratio between ZMPSTE24 and LMNA mRNA expression. Values are means of duplicate experiments ± S.D. In B, C, D and F densitometric analysis of triplicate experiments was performed, and the mean values ± S.D. are reported; asterisk indicates statistically significant difference with respect to lamin A or progerin densitometry in untreated samples; statistical significance was calculated by the Mann-Whitney test vs untreated HGPS samples, or cells expressing LMNA constructs.
Transfection of HEK293 cells with an LMNA construct encoding progerin (LA-Δ50) was performed to support the above reported data (Figure 1E). Rapamycin treatment for 72 h strongly reduced LA-Δ50 level, while CQ administration did not cause protein accumulation (Figure 1E), supporting the finding that progerin does not undergo degradation in untreated cells. Quantitative analysis also supported the results showing progerin degradation by rapamycin (Figure 1 D,F). Importantly, accumulation of LC3-B2, which is a marker of autophagic activity, was observed in HGPS cells and, to a lower extent, in HEK293 cells expressing progerin, before rapamycin treament, but was increased by rapamycin treatment (Figure 1 A,E). The latter finding suggested that activation of the authophagic signaling is triggered by progerin itself.
RT-PCR analysis of LMNA or ZMPSTE24 mRNA was performed in HGPS before and after rapamycin treatment. LMNA and ZMPSTE24 expression were not decreased by rapamycin treatment, demonstrating that progerin elimination does not occur through downregulation of mRNA expression (Figure 1G). Figure 1H shows that the ratio between ZMPSTE24 and LMNA expression is increased following treatment, suggesting that not only degradation of progerin, but also processing of wild-type prelamin A is potentially increased by drug treatment.
Nuclear envelope in rapamycin treated cells
To test whether rapamycin could affect other nuclear envelope constituents, besides prelamin A, control and HGPS cells were examined before and after drug treatment. Figure 2 shows the western immunoblot of lamin B1, lamin B2, emerin, BAF and LAP2α (Figure 2A). Rapamycin treatment did not affect protein levels in control or laminopathic fibroblasts (Figure 2A). However, in HGPS fibroblasts, where LAP2α was downregulated, rapamycin administration restored protein levels comparable to controls (Figure 2 A,B). These results suggested that reduction of progerin and possibly wild-type prelamin A levels could restore nuclear integrity in HGPS cells.
Figure 2.
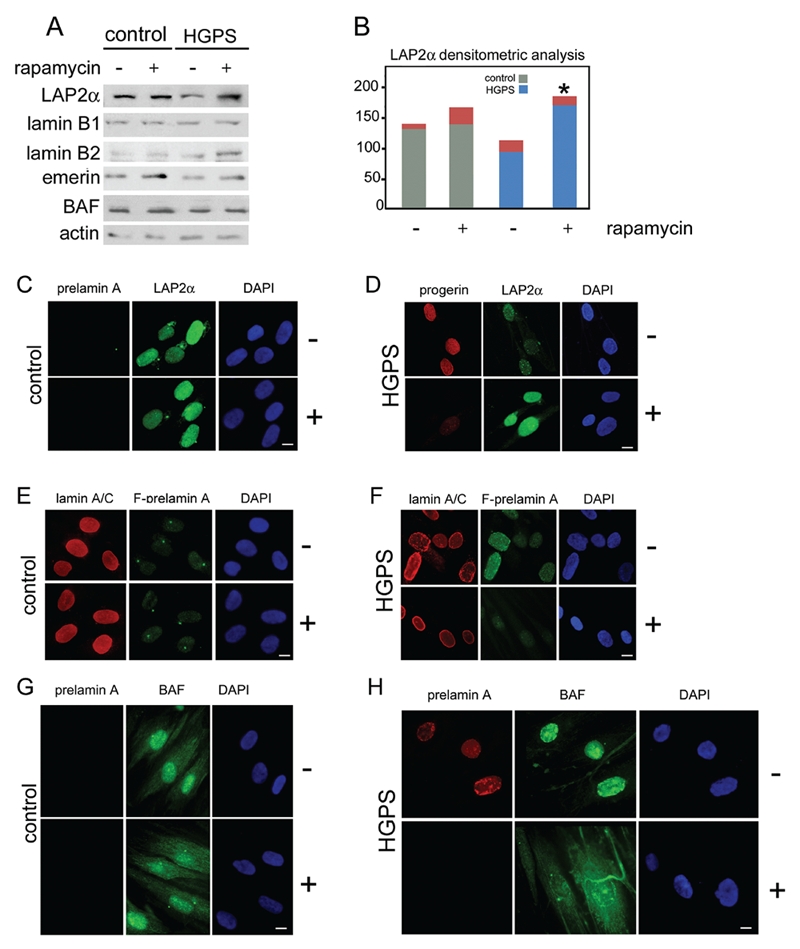
Nuclear envelope/lamina proteins in rapamycin treated cells. A) Western Blotting analysis of LAP2α (LAP2 ), lamin B1 (lamin B1), lamin B2 (lamin B2), emerin (emerin) and Barrier-to-autointegration factor (BAF) in control (control) and HGPS cells (HGPS) untreated (−) or treated (+) with rapamycin. Actin was detected as protein loading control. Immunolabeled bands are shown; B) densitometric analysis of LAP2α immunolabeled bands detected in Western blotting analysis performed in control and HGPS untreated (−) or rapamycin treated cells (+). Asterisk indicates statistically significant difference, P=0.0319 for HGPS + rapamycin (+) vs untreated HGPS (−); C) prelamin A and LAP2α immunofluorescence labeling performed in untreated (−) or rapamycin treated (+) control cells. Prelamin A was evaluated using a goat-polyclonal antibody visualized by TRITC-conjugated secondary antibody (red). LAP2α distribution was evaluated using a rabbitpolyclonal antibody visualized by FITC-conjugated secondary antibody (green); D) progerin and LAP2α immunolabeling detection performed in untreated (−) or rapamycin treated (+) HGPS cells. Progerin (progerin) detection was performed using a mouse-monoclonal antibody visualized by Cy3-conjugated secondary antibody (red). LAP2α distribution was evaluated using a rabbit-polyclonal antibody visualized by FITC-conjugated secondary antibody (green). In untreated HGPS cells progerin staining was observed, while LAP2α labeling was decreased. Rapamycin treatment dramatically reduced progerin labeling and restored LAP2α staining levels; E) Lamin A/C and farnesylated-prelamin A detection performed in untreated (−) or rapamycin treated (+) control cells. Lamin A/C (lamin A/C) was evaluated using a goat-polyclonal antibody visualized by TRITC-conjugated secondary antibody (red). Farnesylated-prelamin A (F-prelamin A) staining was performed using a rabbit-polyclonal antibody visualized by FITC-conjugated secondary antibody (green). Lamin A/C was detected in untreated or treated cells at the same levels. F-prelamin A was undetected in both samples; F) Lamin A/C and farnesylated-prelamin A detection performed in untreated (−) or rapamycin treated (+) HGPS cells. Lamin A/C (lamin A/C) was evaluated using a goat-polyclonal antibody visualized by TRITC-conjugated secondary antibody (red). Farnesylated-prelamin A (F-prelamin A) staining was performed using a rabbit-polyclonal antibody visualized by FITC-conjugated secondary antibody (green). Lamin A/C staining was observed at the nuclear lamina of both untreated and treated cells. F-prelamin A staining observed in untreated cells was strongly reduced by rapamycin treatment; G) prelamin A BAF detection performed in untreated (−) or rapamycin treated (+) control cells. Prelamin A was stained by a goat-polyclonal antibody visualized by TRITC-conjugated secondary antibody (red). BAF immunolabeling detection was performed by a rabbit-polyclonal antibody visualized by FITC-conjugated secondary antibody (green). Prelamin A was undetectable in untreated or rapamycin treated cells. BAF showed a normal cellular distribution in both samples; H) prelamin A and Barrier-to-autointegration (BAF) detection was performed in untreated (−) or rapamycin treated (+) HGPS cells. Prelamin A was stained by a goat-polyclonal antibody visualized by TRITC-conjugated secondary antibody (red). BAF was labeled by rabbit-polyclonal antibody and visualized by FITC-conjugated secondary antibody (green). Prelamin A staining was present at the nuclear lamina of untreated cells while in rapamycin treated cells lamin A precursor was undetectable. BAF nuclear localization was observed in HGPS untreated cells. The normal BAF nucleo-cytoplasmic cellular distribution was recovered by rapamycin treatment. In panel C, D, E, F,G and H nuclei were counterstained with DAPI. Scale bar, 10 µm.
Progerin and chromatin-binding proteins in rapamycin-treated cells
To support this hypothesis, progerin, farnesylated prelamin A and chromatin-binding prelamin A partners were examined by immunofluorescence before and after rapamycin treatment (Figure 2 C–H). Progerin and farnesylated prelamin A were detected at the nuclear rim of HGPS fibroblasts, but not in controls (Figure 2 C–F). Rapamycin dramatically reduced progerin and farnesylated prelamin A levels (Figure 2 D–F). Importantly, rapamycin treatment did not modify lamin A/C level and localization, thus confirming that lamin C is not substantially affected by rapamycin (Figure 2 E,F). Next, the expression pattern of the prelamin A-binding protein LAP2α was examined. LAP2α was evenly distributed in the nucleoplasm of control fibroblasts, but was mislocalized to the nuclear periphery in HGPS fibroblasts (Figure 2 C,D). Rapamycin restored both LAP2α level (Figure 2 A,B) and the proper distribution of LAP2α throughout the nucleoplasm (Figure 2 C,D). Next, the effect of rapamycin treatment on the localization of the prelamin A and chromatin-binding protein BAF, was assessed. BAF was recruited to the nuclear envelope in laminopathic cells,11 while in a major proportion of control cells it was located in the cytoplasm and, to a lower amount in the nucleus (Figure 2 G,H). Treatment with rapamycin, restored the proper distribution of BAF in HGPS cells (Figure 2 H).
Chromatin organization is recovered in rapamycin treated laminopathic cells
The above reported recovery of LAP2α and BAF expression patterns obtained by rapamycin treatment, suggested that loss of heterochromatin, which is known to occur in HGPS cells could have been similarly rescued. The methylation status of histone H3 lysine 9 (H3K9) was determined by specific labeling of the trimenthylated lysine. We had previously shown altered methylation pattern of H3K9 in HGPS cells.5 Here, we could observe recovery of trimethyl- H3K9 distribution pattern in rapamycin-treated HGPS nuclei (Figure 3). Fluorescence intensity measurement showed that trimethylated H3K9 levels were strongly reduced in HGPS nuclei; morevover, the clustered distribution of the histone observed in control cells (Figure 3 A,B) was completely lost (Figure 3 C,D). Trimethyl-H3K9 was not affected by rapamycin treatment in control cells (Figure 3 E,F). However, the proper organization and expression level of trimethyl-H3K9 was restored by rapamycin treatment of HGPS cells (Figure 3 G,H,I), which indicated recovery of heterochromatin organization. Thus, we conclude that rapamycin may be effectively used to restore the chromatin phenotype in laminopathies.
Figure 3.
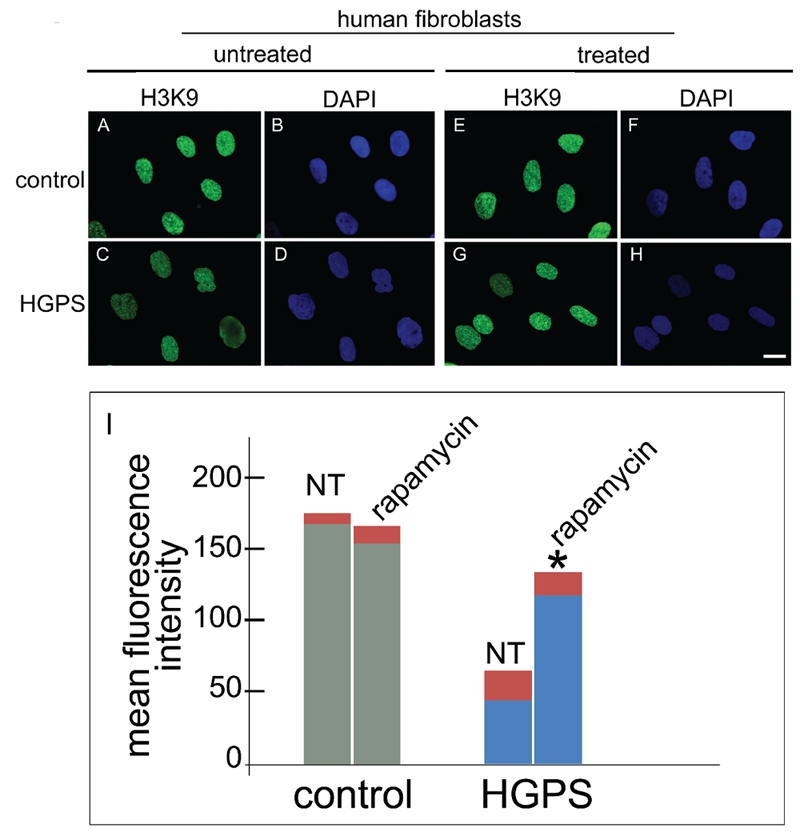
Rapamycin treatment rescues the heterochromatin marker trimethyl-H3K9 in HGPS cells. Immunofluorescence labeling of trimethyl-H3K9 (H3K9) in control and HGPS cells untreated (untreated) or treated (treated) with rapamycin using a rabbit-polyclonal antibody visualized by FITC-conjugated secondary antibody (green). In control untreated cells (A) a proper trimethyl-H3K9 staining was observed, while a clearly detectable decrease of immunolabeling was observed in untreated HGPS cells (C). Rapamycin treatment of control cells did not affect trimethyl-H3K9 staining (E), while rescued trimethyl-H3K9 distribution in HGPS cells (G). Nuclei in B, D, F and H were counterstained with DAPI. Scale bar = 10 µm; I) The mean fluorescence intensity of 300 nuclei per sample stained for trimethyl-H3K9 was measured by the NIS software and plotted for untreated (NT) or rapamycin-treated (treated) control and HGPS nuclei. Asterisk indicates statistically significant difference, P=0.0331 for HGPS + rapamycin vs untreated HGPS.
Discussion
The study here reported shows that activation of the mTOR-dependent authophagic pathway using rapamycin can counteract progerin and farnesylated prelamin A accumulation in HGPS cells, leading to rescue of the chromatin phenotype of senescent cells.
The toxicity of farnesylated prelamin A and progerin is well established.5,14,19–21 Several studies have shown that reduction of prelamin A or progerin levels either by molecular approaches6,8 or by drug treatment5,21 improves the cellular phenotype in progeroid cells. Here, we used a well-known drug, rapamycin, which interferes with m-TOR inhibition of autophagy, thus triggering the authophagic pathway.13 Authophagy is a physiological degradation mechanism, mostly aimed at scavenging damaged organelles, but also implied in the elimination of altered proteins.13 Our results show a major role of lysosomal degradation in the reduction of prelamin A levels triggered by rapamycin, as demonstrated by the effective accumulation of progerin, following cloroquine inhibition of rapamycin-treated cells. Moreover, our study shows that degradation of progerin does not occur at detectable levels in untreated cells. Thus, toxicity of progerin, which is likely associated with aberrant intermolecular interactions mediated by the farnesylated C-terminus of the molecule,11,22 is further increased by the resistance to degradation of that prelamin A form.
In this scenario, the availability of a drug capable of triggering farnesylated prelamin A degradation paves the way to promising therapeutic perspectives. Activation of authophagy by prelamin A accumulation has been reported23 and it is supported by our evidence of LC3-B2 accumulation24 in untreated HGPS cells, as well as by electron microscopy studies (not shown). However, since progerin is not or minimally degraded in untreated HGPS fibroblasts, while undergoes degradation upon rapamycin stimulus, we suggest that the function of rapamycin in laminopathic cells is to target progerin and farnesylated prelamin A to degrading enzymes. This event could be mediated by the prelamin A-specific C-terminus, since lamin C appears to be substantially unaffected by drug treatment. In this context, we speculate that a physiological role of prelamin A processing could be to provide a pool of farnesylated lamin A precursors to be either degraded or processed to mature protein, depending on the cell requirement. The unexpected finding that the relative amount of ZMPSTE mRNA is increased in rapamycin-treated cells, suggests that not only prelamin A degradation, but also prelamin A processing may be enhanced by drug treatment. In this context, the restoring ability of rapamycin we describe in HGPS may be extended to the treatment of other laminopathies featuring accumulation of farnesylated prelamin A forms different from progerin.
Acknowledgements:
the authors wish to thank Prof. Roland Foisner for kindly providing the anti-LAP2alpha antibody, A. Valmori, O. Fiorani, S. Grasso and D. Zini for the technical assistance and the Italian Network for Laminopathies for continous collaboration.
This work was supported by grants from the A.I.Pro.Sa.B., Italy, the Italian MIUR PRIN 2008 to G.L. and FIRB to N.M.M. and Fondazione Carisbo, Italy.
References
- 1.Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–24. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 2.Dominguez-Gerpe L, Araujo-Vilar D. Prematurely aged children: molecular alterations leading to Hutchinson-Gilford progeria and Werner syndromes. Curr Aging Sci. 2008;1:202–12. doi: 10.2174/1874609810801030202. [DOI] [PubMed] [Google Scholar]
- 3.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA. Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet. 2004;41:304–8. doi: 10.1136/jmg.2003.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Columbaro M, Capanni C, Mattioli E, Novelli G, Parnaik VK, Squarzoni S, et al. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell Mol Life Sci. 2005;62:2669–78. doi: 10.1007/s00018-005-5318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–5. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA. 2006;103:8703–8. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang SH, Qiao X, Farber E, Chang SY, Fong LG, Young SG. Eliminating the synthesis of mature lamin A reduces disease phenotypes in mice carrying a Hutchinson-Gilford progeria syndrome allele. J Biol Chem. 2008;283:7094–9. doi: 10.1074/jbc.M708138200. [DOI] [PubMed] [Google Scholar]
- 9.Haque F, Mazzeo D, Patel JT, Smallwood DT, Ellis JA, Shanahan CM, et al. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem. 2010;285:3487–98. doi: 10.1074/jbc.M109.071910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2004;101:8963–8. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capanni C, Cenni V, Haraguchi T, Squarzoni S, Schuchner S, Ogris E, et al. Lamin A precursor induces barrier-to-autointegration factor nuclear localization. Cell Cycle. 2010;9:2598–608. doi: 10.4161/cc.9.13.12080. [DOI] [PubMed] [Google Scholar]
- 12.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2011;16:1313–20. doi: 10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- 14.Dominici S, Fiori V, Magnani M, Schena E, Capanni C, Camozzi D, et al. Different prelamin A forms accumulate in human fibroblasts: a study in experimental models and progeria. Eur J Histochem. 2009;53:43–52. doi: 10.4081/ejh.2009.43. [DOI] [PubMed] [Google Scholar]
- 15.Dechat T, Gajewski A, Korbei B, Gerlich D, Daigle N, Haraguchi T, et al. LAP2alpha and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J Cell Sci. 2004;117:6117–28. doi: 10.1242/jcs.01529. [DOI] [PubMed] [Google Scholar]
- 16.Mattioli E, Columbaro M, Capanni C, Maraldi NM, Cenni V, Scotlandi K, et al. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011;18:1305–15. doi: 10.1038/cdd.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avnet S, Pallotta R, Perut F, Baldini N, Pittis MG, Saponari A, et al. Osteoblasts from a mandibuloacral dysplasia patient induce human blood precursors to differentiate into active osteoclasts. Biochim Biophys Acta. 2011;1812:711–8. doi: 10.1016/j.bbadis.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Araujo-Vilar D, Lattanzi G, Gonzalez-Mendez B, Costa-Freitas AT, Prieto D, Columbaro M, et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipody-strophy. J Med Genet. 2009;46:40–8. doi: 10.1136/jmg.2008.059485. [DOI] [PubMed] [Google Scholar]
- 19.Lattanzi G, Columbaro M, Mattioli E, Cenni V, Camozzi D, Wehnert M, et al. Pre-Lamin A processing is linked to heterochromatin organization. J Cell Biochem. 2007;102:1149–59. doi: 10.1002/jcb.21467. [DOI] [PubMed] [Google Scholar]
- 20.Maraldi NM, Lattanzi G, Capanni C, Columbaro M, Merlini L, Mattioli E, et al. Nuclear envelope proteins and chromatin arrangement: a pathogenic mechanism for laminopathies. Eur J Histochem. 2006;50:1–8. [PubMed] [Google Scholar]
- 21.Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–72. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- 22.Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, et al. Identification of differential protein interactors of lamin A and progerin. Nucleus. 2011;1:513–25. doi: 10.4161/nucl.1.6.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marino G, Ugalde AP, Salvador-Montoliu N, Varela I, Quiros PM, Cadinanos J, et al. Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum Mol Genet. 2008;17:2196–211. doi: 10.1093/hmg/ddn120. [DOI] [PubMed] [Google Scholar]
- 24.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]