

Abstract
Replication-deficient adenovirus and modified vaccinia virus Ankara (MVA) vectors expressing single pre-erythrocytic or blood-stage Plasmodium falciparum antigens have entered clinical testing using a heterologous prime-boost immunization approach. Here we investigated the utility of the same immunization regime when combining viral vectored vaccines expressing the 42kDa C-terminus of the blood-stage antigen merozoite surface protein 1 (MSP142) and the pre-erythrocytic antigen circumsporozoite protein (CSP) in the P. yoelii mouse model. We find that vaccine co-administration leads to maintained antibody responses and efficacy against blood-stage infection, but reduced secondary CD8+ T cell responses against both antigens and efficacy against liver-stage infection. CD8+ T cell interference can be minimized by co-administering the MVA vaccines at separate sites, resulting in enhanced liver-stage efficacy in mice immunized against both antigens compared to just one. CD8+ T cell interference (following MVA co-administration as a mixture) may partly be caused by a lack of physiological space for high magnitude responses against multiple antigens, but is not caused by competition for presentation of antigen on MHC class I molecules, nor is it due to restricted T cell access to APCs presenting both antigens. Instead, enhanced killing of peptide-pulsed cells is observed in mice possessing pre-existing T cells against two antigens, in comparison to just one, suggesting priming against multiple antigens may in part reduce the potency of multi-antigen MVA vectors to stimulate secondary CD8+ T cell responses. These data have important implications for the development of a multi-stage or multi-component viral vectored malaria vaccine for use in humans.
Introduction
Malaria remains a significant global health problem. Plasmodium spps infected approximately 240 million people and caused approximately 860,000 deaths worldwide in 2008 (1). It is widely recognized that a highly effective vaccine against malaria remains urgently needed. One promising approach is the use of replication-deficient recombinant viral vectored vaccines (2, 3), whereby an adenovirus (Ad)-MVA prime-boost approach has been shown to induce strong T cell responses as well as high-titer antibodies against malaria antigens in pre-clinical studies in mice, rabbits and rhesus macaques (4-10). Phase I/IIa clinical trials using this strategy are currently underway in Oxford, UK (2, 11). The clinical vaccine candidates comprise chimpanzee adenovirus 63 (ChAd63) and the orthopoxvirus modified vaccinia virus Ankara (MVA), administered eight weeks apart, expressing the P. falciparum antigens ME-TRAP (a string of multiple epitopes from liver-stage malaria antigens fused to the thrombospondin-related adhesion protein) (12), or the blood-stage malaria antigens merozoite surface protein 1 (MSP1) (6) or apical membrane antigen 1 (AMA1) (9, 10).
However, it is widely acknowledged that a multi-stage and/or multi-antigen formulation will likely be necessary to provide high-level efficacy. Any such vaccine will only warrant deployment if it provides significantly greater efficacy than pre-existing pre-erythrocytic control measures as well as RTS,S (a pre-erythrocytic-stage vaccine targeting the circumsporozoite protein (CSP) currently in Phase III trials across Africa) (13). It is suggested that a second generation vaccine will need to provide greater than 80% efficacy for at least four years in order to justify deployment (14). One possible strategy that has been investigated pre-clinically in rhesus macaques is the combination of RTS,S/AS02A with protein-in-adjuvant vaccine candidates for the blood-stage antigens MSP1 and AMA1 (15). This study found that AMA1-specific antibody responses are reduced when AMA1 is co-administered with either RTS,S or the 42kDa C-terminus of MSP1 (MSP142). A substantial body of work has also been carried out investigating mixtures of DNA and poxvirus vaccines encoding four to nine malaria antigens in mice and macaques (16-21). This work highlighted the issue of antigenic competition in multi-component malaria vaccine formulations, and demonstrated that immune interference may be complex and antigen dependent.
Despite these findings the majority of Phase I/IIa clinical trials investigating multi-antigen and/or multi-stage malaria vaccines have not investigated the immunogenicity of each antigenic component alone nor assessed individual contributions to efficacy. These include protein vaccines such as GMZ2 (22), PfCP2.9 (23) and combination B (24), as well as multi-antigen strings expressed in DNA plasmids or viral vectors such as ME-TRAP (12, 25), NYVAC-Pf7 (26), and L3SEPTL polyprotein (27). Similarly, studies in the field of HIV-1 vaccines have used mixtures of 3-4 DNA and/or poxvirus or human adenovirus serotype 5 (AdHu5) vaccines, but any potential effect(s) of immune interference between the individual components and their encoded antigens have not been reported (28, 29). Two studies have been carried out in which individual components in a multi-antigen virosome/viral vectored malaria vaccine have been directly assessed in humans when administered alone and in combination (30, 31). In both cases total IgG responses were not affected by co-administration of antigens, however T cell responses were not directly assessed. To the best of our knowledge, no study has directly assessed the impact of co-administration of malaria antigens on T cell responses in humans. These data clearly highlight the importance of investigating the individual antigenic components within a multi-antigen vaccine and their impact on vaccine immunogenicity and efficacy in both pre-clinical and clinical studies.
Crucially, a combination vaccine strategy has not previously been assessed with the clinically relevant Ad-MVA prime-boost vectored vaccine platform. Here, we have evaluated in mice the co-administration of vectors expressing the P. yoelii antigens CSP (PyCSP) and MSP142 in an AdHu5-MVA prime-boost regime. CSP is expressed on the surface of sporozoites and antibodies against this antigen are thought to prevent sporozoite invasion, whilst CSP-specific CD8+ T cell responses, induced by recombinant human adenoviral vectors in mice (32-34), as well as CD4+ T cell responses in humans (35), have been associated with protective outcome. MSP142 is the 42kDa C-terminus of MSP1 and is expressed during both the late liver-stage as well as on the surface of merozoites during blood-stage infection. During merozoite invasion of red blood cells, MSP142 is cleaved into a 33kDa (MSP133) and a 19kDa (MSP119) fragment (36). MSP119 remains associated with the merozoite surface and antibodies against this antigen are capable of providing blood-stage protection. We have shown that vectors expressing P. yoelii MSP142 can provide partial liver-stage efficacy due to CD8+ T cells targeting PyMSP133 (4), and protective blood-stage efficacy due to antibody responses against PyMSP119 (4, 5). In these studies, efficacy was further enhanced if the PyMSP142 vaccine construct included the core domain from the murine α-chain of complement C4 binding protein (mC4bp / IMX108) fused to the C-terminus (5, 37).
In this study using an AdHu5-MVA vectored vaccine platform for PyCSP and PyMSP142 we demonstrate an absence of interference for antigen-specific antibodies following administration of the vaccine mixture, however significant CD8+ T cell interference does occur after the MVA boost resulting in reduced efficacy against sporozoite challenge. This effect is associated with enhanced killing of infected cells in vivo at the time of the boosting immunization in mice that are previously primed against two antigens rather than one. CD8+ T cell interference can however be minimized by separate site immunization, which leads to enhanced efficacy of the dual-antigen vaccine regime. These data have important implications for development of multi-component viral vectored vaccines against numerous difficult infectious diseases.
Materials and Methods
Recombinant AdHu5 and MVA vaccines
Construction of AdHu5 and MVA vectors expressing PyCSP and PyMSP142-mC4bp (IMX108), referred to throughout as PyMSP142, has been previously described (4, 5, 37). Control vectors were made using similar methods. AdHu5-Control included the transgene promoter and polyA tail, but no antigenic insert. MVA-Control contained GFP.
Animals and immunizations
Procedures were carried out according to the UK Animals (Scientific Procedures) Act 1986 and were approved by the University of Oxford Animal Care and Ethical Review Committee. 6-8 week old female BALB/c (H-2d) and C57BL/6 (H-2b) mice were obtained from Harlan, UK, and housed in specific pathogen-free conditions. Immunizations were carried out as described in Table I, unless stated otherwise. All vaccines were formulated in endotoxin-free PBS and given intradermally (i.d.) in a total volume of 50μL (25μL/ear). There was an eight-week interval between priming and boosting immunizations in all cases. When only a single malaria antigen vaccine was given, control vaccines were included to equalize viral load.
Table I. Standard immunization regimes.
In all experiments mice were vaccinated with the indicated regimes, unless stated otherwise. The viruses used were mixed, and then mice were vaccinated i.d. in the ear in a total volume of 50μL, with 25μL given into each ear. There was an eight week interval between prime and boost. Where only a single malaria antigen was given control vaccines were included to control for viral load.
| Antigens | AdHu5 Prime (Mixed) | MVA Boost (Mixed) |
|---|---|---|
|
| ||
| MSP1 + CSP | 1×1010 vp AdHu5-PyMSP142 | 1×107 pfu MVA-PyMSP142 |
| 1×1010 vp AdHu5-PyCSP | 1×107 pfu MVA-PyCSP | |
|
| ||
| MSP1 + Control | 1×1010 vp AdHu5-PyMSP142 | 1×107 pfu MVA-PyMSP142 |
| 1×1010 vp AdHu5-Control | 1×107 pfu MVA-Control | |
|
| ||
| CSP + Control | 1×1010 vp AdHu5-Control | 1×107 pfu MVA-Control |
| 1×1010 vp AdHu5-PyCSP | 1×107 pfu MVA-PyCSP | |
Isolation of splenocytes
Spleens were dissected into PBS, mechanically homogenized and passed through a 70μm cell strainer. Erythrocytes were removed by re-suspension of cells in ACK lysis buffer (0.15M NH4Cl + 1mM KHCO3 + Na2EDTA dissolved in 1L dH2O and adjusted to pH 7.2-7.4 with 1M HCl (all from Sigma)) for 5 min, before the addition of excess PBS. Splenocytes were pelleted by centrifugation and re-suspended in complete MEM (MEM α-modification, 10% FCS, 100U/mL penicillin, 100μg/mL streptomycin, L-glutamine [4mM] and 2-mercaptoethanol [50μM]).
Isolation of PBMC from blood
7-10 drops of blood were collected from the tail vein into 200μL 10mM EDTA in PBS. Erythrocytes were removed by incubation with ACK lysis buffer and cells were re-suspended in complete MEM.
Isolation of liver mononuclear cells
Livers were perfused with PBS and mechanically homogenized. Cells were incubated for 1 h in complete MEM containing 0.7 mg/mL collagenase (Sigma) and 0.03 mg/mL DNase (Sigma) then passed through a 70μm cell strainer. Cells were washed in PBS and under laid with Ficoll-Paque™ PLUS (GE Healthcare) before spinning at 2000 rpm without the brake for 45 min at 21°C to separate cells. Cells at the interphase were carefully removed, washed twice with PBS and re-suspended in complete MEM.
Peptides
PyCSP-specific responses were measured using peptide against the dominant CD8+ epitope, PyCSP280-288 (38), the sub-dominant CD8+ epitope, PyCSP59-67 (39), or the CD4+ epitope, PyCSP59-79 (40). PyMSP1-specific T cell responses were measured following stimulation with a pool of peptides spanning the entire length of PyMSP133. This consisted of 52 15mers overlapping by 10 amino acids (αα) (5). For all ICS assays, cells were stimulated with 1μg/mL of each peptide unless stated otherwise. For peptide-pulsing CD11c+ cells only peptides encoding the seven PyMSP133 epitopes (41) were used. These are all predicted to be H-2 Kd-restricted using the epitope prediction section of the SYFPEITHI website (http://www.syfpeithi.de/Scripts/MHCServer.dll/EpitopePrediction.htm) developed at the University of Tübingen. MVA-specific responses were measured using peptides against E3 and F2(G) (42). Individual peptides are described in Table II.
Table II.
Immunogenic peptides and murine MHC class I restriction.
| Species | Antigen | Residues (aa) | Sequence | MHC class I restriction |
|---|---|---|---|---|
| P. yoelii | CSP | 58 - 67 | IYNRNIVNRL | Kd |
| CSP | 280 - 288 | SYVPSAEQI | Kd | |
| CSP | 59 - 79 | YNRNIVNRLLGDALNGKPEEK | MHC class II |
|
| MSP133 | 1421 - 1429 | TYKSIKKHML | Kd * | |
| MSP133 | 1453 - 1462 | DFLEVLSHEL | Kd * | |
| MSP133 | 1473 - 1481 | YVIRNPYQL | Kd * | |
| MSP133 | 1513 - 1521 | KFFNKMVEL | Kd * | |
| MSP133 | 1550 - 1558 | KYIPILEDL | Kd * | |
| MSP133 | 1571 - 1580 | EYSEELQNRL | Kd * | |
| MSP133 | 1599 - 1607 | KYIQIDEKL | Kd * | |
| MVA | E3 | 140 - 148 | VGPSNSPTF | Dd |
| F2(G) | 26 - 34 | SPGAAGYDL | Ld |
Predicted MHC class I restriction.
Intracellular cytokine staining (ICS)
Cellular immune responses in blood, spleen and liver were measured by ICS as previously described (4). Briefly, cells were re-stimulated for 5 h at 37°C in the presence of GolgiPlug (BD Biosciences) and peptides specific for PyMSP1, PyCSP or MVA vector at a final concentration of 1μg/mL each peptide, before storage at 4°C overnight.
Cells were then incubated for 15 min in the presence of anti-mouse CD16/CD32 (Fc-block), before surface staining for 30 min with anti-mouse CD8α-PerCP-Cy5.5 (clone 53-6.7) and CD4-Pacific Blue (clone GK1.5). Permeabilization was performed using Cytofix/Cytoperm solution (BD Biosciences) according to the manufacturer’s instructions. Cells were stained intracellularly for 30 min with anti-mouse IFN-γ-APC (clone XMG1.2), IL-2-PE (clone JES6-5H4) and TNFα-FITC (clone MP6-XP22). All incubations were carried out at 4°C with antibodies from eBioscience. Cells were re-suspended in 1% formalin solution and responses measured using a CyAn ADP Flow Cytometer (Dako, Ely, UK) or LSRII (BD Biosciences). Data were analyzed using Flowjo version 8.7 (Tree Star, Inc.; USA). For some experiments, analysis of distributions was performed using SPICE version 5.1, downloaded from http://exon.niaid.nih.gov/spice (43). Results show the stimulated response with background responses from the un-stimulated control subtracted (typically < 0.05%).
ELISA
Blood was collected by tail bleed into microvette tubes, stored at 4°C overnight, and spun at 13,000 rpm for 4 min. Serum was removed and stored at −20°C until use. 2μg/mL GST-PyMSP-119 protein (5) or a branched PyCSP peptide (MAP4-Py1T-Py3) (44) in PBS was adsorbed to 96-well Nunc-Immuno Maxisorp plates (Fisher Scientific) by overnight incubation at room temperature (RT). Plates were washed 6 times in PBS 0.05% Tween 20 (PBS/T) and blocked for 1 h in PBS/T containing 10% skimmed milk powder. Pre-boost samples sera were diluted 1:100 and post-boost 1:1000 before serial dilution down the plate and incubation for 2 h at RT. Plates were then washed in PBS/T and bound antibodies detected by incubation for 1 h with alkaline phosphatase conjugated goat anti-mouse total IgG (Sigma). Alternatively bound antibody isotypes were detected using biotin-conjugated rat anti-mouse IgG1 or IgG2a (BD Biosciences) at a concentration of 1 μg/mL in PBS/T for 1 h, before washing and adding ExtrAvidin (Sigma) conjugated to alkaline phosphatase and incubation for 30 min. Plates were then washed and developed using 1mg/mL p-nitrophenylphosphate substrate pNPP (Sigma) in diethanolamine buffer (Fisher Scientific UK Ltd). Optical density was read at 405nm. Endpoint titers were taken as the x-axis intercept of the dilution curve at an absorbance value 3 S.D. greater than the OD405 for naïve mouse serum. All samples were tested in duplicate and a high titer reference control sample was included in all experiments.
P. yoelii sporozoite and parasitized red blood cell (pRBC) challenge
P. yoelii YM sporozoite and pRBC challenges were carried out as previously described (4). Briefly, sporozoites were obtained by homogenization of salivary glands from infected Anopheles stephensi mosquitoes in RPMI 1640 medium (Sigma). Infected erythrocytes (pRBC) were stored by cryopreservation and passaged in a donor mouse prior to challenge. Mice were infected by intravenous (i.v.) injection with 50, 250 or 1000 sporozoites or 104 pRBC and blood-stage parasitemia was monitored post-challenge by Giemsa-stained thin blood smear from day 2 for pRBC challenge and day 4 or 5 for sporozoite challenge. Parasitemia was calculated as percentage of infected RBCs. Mice were considered uninfected if no parasites were observed in 50 fields of view containing approximately 500 RBC per field. The lower limit of detection was therefore 0.004%. Mice were culled if they reached ≥ 80% infected RBC.
P. yoelii sporozoite immunofluorescence antibody test (IFAT)
P. yoelii YM sporozoites were isolated from the salivary glands of infected mosquitoes and were dissected in PBS containing azide. Samples were diluted to 3 ×105 sporozoites/mL and 10μL placed in each well on an 8-well microscope slide, such that each well contained approximately 3000 sporozoites. Slides were air-dried, wrapped in foil and stored with desiccant at −20°C until use. For the IFAT, all incubation steps were carried out at RT in a dark humidified chamber and slides were washed three times in PBS between each step. Wells were blocked for 2 h using 1% BSA in PBS. Following washing, serum samples were added at a dilution of 1:100 in PBS for 1 h. Slides were washed again and Alexa Fluor® 488 conjugated goat anti-mouse IgG secondary antibody (Invitrogen) added at a dilution of 1:200 (in 1% BSA, PBS. Slides were incubated for 30 min, washed, and mounted with Mowiol® and a coverslip. Slides were dried at RT overnight in the dark. Slides were viewed under a Leica DMI3000 microscope.
Isolation and peptide-pulsing of CD11c+ cells
Spleens from naïve BALB/c mice were chopped, re-suspended in complete MEM and rotated at RT for 30 min in the presence of 0.3 mg/mL collagenase-dispase (Gibco) and 0.02 mg/mL DNase I (Sigma), before addition of 0.1M EDTA for a further 5 min to inactivate collagenase and disrupt dendritic cell (DC)-T cell interactions. Spleens were then homogenized, passed through a 70μm cell strainer and erythrocytes were lysed by re-suspension in ACK lysis buffer for 5 min, before washing in PBS, centrifugation and re-suspension in cold MACS buffer (PBS pH7.2, 0.5% BSA, 2mM EDTA) at a concentration of 2.5 × 108 cells/mL. Cells were incubated for 10 min at 4°C with 1:100 naïve BALB/c serum and 1:100 Fc-block (eBioscience) before addition of 100μL CD11c MicroBeads per 108 cells and incubation for a further 15 min at 4°C. Cells were then washed and re-suspended at 2 × 108 cells/mL in MACS buffer. CD11c+ cells were isolated by passing splenocytes through an LS MACS column according to the manufacturer’s instructions (Miltenyi Biotech Ltd). After positive selection, CD11c+ cells were washed thoroughly, re-suspended in complete MEM and pulsed with 10μg/mL peptide for 16 h. Cells were either pulsed with the seven PyMSP133 peptides, the two PyCSP peptides, or a mixture of all nine peptides. The concentration of DMSO was controlled across all wells at 0.24%. After pulsing, cells were washed thoroughly in PBS. Mice were subsequently injected i.d. in the ear with 2 × 105 CD11c+ cells in a volume of 100μL (50μL/ear). In mice that received cells that had been pulsed separately, cells pulsed with each antigen were combined in a 1:1 ratio immediately before injection (2 × 105 total cells).
In vivo CTL assay
Naïve BALB/c splenocytes were prepared by mechanical disruption (as described) as targets for in vivo killing. Cells were washed and re-suspend at 5 × 107 cells/mL in warm MEM (without additions, 37°C), then split into two populations. One population of cells was labeled with a high concentration of CFSE (5μM) and the other with a low concentration of CFSE (0.5μM) (Invitrogen). After addition of CFSE, cells were immediately incubated for 10 min at 37°C, with regular mixing by inversion. The reaction was stopped by addition of cold complete MEM. Cells were then washed twice in complete MEM and re-suspended at 1 × 107 cells/mL. Cells labeled with a high concentration of CFSE were split into sub-populations and incubated at 37°C for 1 h with PyMSP133 peptide pool, PyCSP280-288 or with both PyMSP133 peptide pool and PyCSP280-288. All peptides were used at 200 ng/mL. Cells were then washed twice in PBS. For i.v. injection, a 1:1 ratio of high and low CFSE labeled cells were then combined such that each mouse received 1 × 107 cells labeled with a low concentration of CFSE that were not peptide pulsed, and 1 × 107 cells labeled with a high concentration of CFSE that were pulsed with the indicated peptides. These were then injected in a volume of 200μL in PBS into BALB/c mice that had been previously primed with AdHu5 vaccines as indicated. Immunized mice were culled 5 h later and a single-cell suspension of splenocytes prepared. The CFSE intensity of each population was analyzed by flow cytometry. Specific killing was calculated as follows: ratio = (percentage CFSE low / percentage CFSE high) and percentage specific killing = (1 - (mean ratio control primed / ratio primed)) × 100%. In one experiment splenocytes were labeled with CD11c-APC prior to flow cytometry to allow assessment of killing of CD11c+ cells.
Statistics
Data were analyzed using GraphPad Prism v5.01. The normality of the data was determined using the Kolmogorov-Smirnov one-sample test. For non-parametric data, two groups were compared using a Mann-Whitney U test and multiple groups were analyzed by Kruskall-Wallis test. Data are presented as median ± range or as individual data points. ELISA titers were logarithmically (log 10) transformed in order to normalize the data and allow parametric analysis. Parametric data were analyzed using an unpaired t-test for two groups and are presented as mean ± standard deviation (SD). In all cases P ≤ 0.05 was considered significant (*P ≤ 0.05, ** P ≤ 0.01 and ***P ≤ 0.001).
Results
Antibody responses to PyMSP142 and PyCSP vaccines
BALB/c mice were immunized against the PyMSP142 and/or PyCSP antigens either individually or as a mixture, utilizing a previously described AdHu5-MVA (Ad-MVA) prime-boost immunization regime (4, 5). Details of the specific immunizations regimes are shown in Table I. The kinetics of the total IgG antibody responses against the PyMSP119 and PyCSP antigens were measured by ELISA over time (Fig. 1, A and B). Antibody responses to each antigen were specific, with no cross-reactivity observed. Responses to both antigens were also detectable two weeks after the Ad prime, and were not significantly affected following co-administration of the two AdHu5 vaccines as a mixture. Following the MVA immunization at day 56, responses to both antigens were significantly boosted (P < 0.0001 by paired t-test for both antigens) and no significant differences in PyMSP119- or PyCSP-specific IgG titers were observed, either at week 10 or 18, between mice that had received vaccines targeting a single antigen and those that had received the vaccine mixture targeting both antigens. Furthermore, administration of the vaccine mixture did not affect the titer of PyMSP119-specific or PyCSP-specific IgG1 or IgG2a two weeks after the MVA boost (Fig. 1, C and D). Total IgG titers against each antigen were also measured two weeks after MVA in C57BL/6 mice that had been similarly immunized (Table I). PyCSP-specific IgG was unaffected following vaccine mixture co-administration, but the titer of PyMSP119-specific IgG was marginally and significantly higher in mice receiving the two-antigen regime (Fig. 1E). The ability of Ad-MVA vaccination to induce functional PyMSP119-specific IgG antibody responses that inhibit blood-stage parasite growth is well established (4, 5). The ability of antibodies induced by Ad-MVA PyCSP to bind P. yoelii sporozoites was also confirmed by IFAT (Supplementary Figure 1).
Figure 1.

Antigen-specific IgG is maintained in a multi-stage vaccine regime. BALB/c mice were immunized with the AdHu5-MVA vaccine regimes described in Table I. Serum was collected two and eight weeks post-prime and two and nine weeks post-boost (week 10 and 17). Total IgG responses at all time points were measured against PyMSP119-GST protein (A) and PyCSP-specific MAP4-Py1T-Py3 branched peptide (B) by ELISA. Data are means ± SD for six mice per group. These data are representative of two independent experiments. IgG1 and IgG2a responses were measured two weeks post-boost against PyMSP119 (C) and PyCSP (D). C57BL/6 mice were also immunized with the vaccines described in Table I. Serum was collected two weeks post-boost and total IgG responses measured against both antigens (E). Bars indicate median responses. The dashed line indicates the limit of detection. * P = 0.018 by Student’s t test.
T cell responses to PyMSP142 and PyCSP vaccines
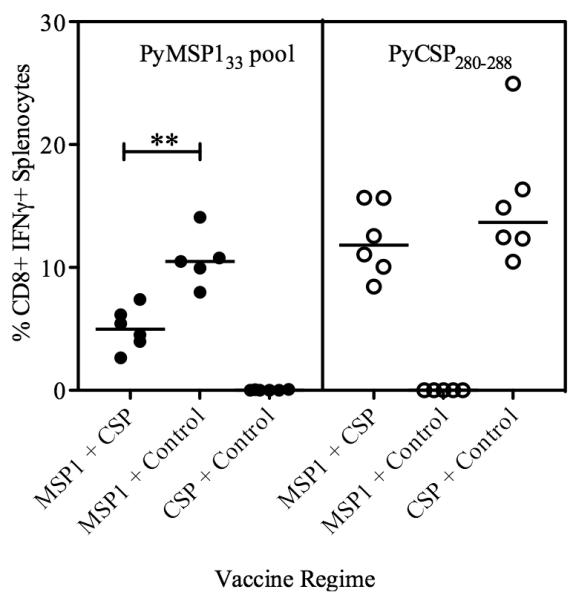
BALB/c mice were immunized as before (Table I) and PBMC were isolated from the blood two and eight weeks after the Ad prime and two and nine weeks after the MVA boost, in order to assess the affect of mixing the Ad and MVA vaccines on transgene-specific T cell responses. Production of IFN-γ from CD8+ cells was assessed by intracellular cytokine staining (ICS) following re-stimulation with a pool of overlapping peptides against PyMSP133 (Fig. 2A) or PyCSP280-288 peptide (Fig. 2B). T cell responses could not be measured against full-length PyCSP due to lack of availability of peptides and were not measured against PyMSP119, as they were previously found to be undetectable (4). Following Ad priming there was comparable production of antigen (Ag)-specific IFN-γ from CD8+ T cells in mice immunized with either a single antigen or both antigens. However two weeks after the MVA boost, the two-antigen vaccine regime resulted in significantly lower Ag-specific CD8+ IFN-γ+ responses to both PyMSP1 and PyCSP in comparison to the single antigen regimes. CD8+ IFN-γ+ responses were also measured in the spleen two weeks post-MVA (Fig. 2C). These data confirmed the responses observed in the blood, with significantly lower responses to the PyMSP133 peptide pool and PyCSP280-288 in the two antigen regime than if either antigen had been given alone. Additionally, due to the availability of cells, responses were also measured against the sub-dominant PyCSP epitope, PyCSP58-67 (39). These data confirmed that responses to both reported class I H-2d epitopes within PyCSP are lower in mice receiving the vaccine mixture in comparison to when Ad-MVA PyCSP is given alone. These data also indicated that the total magnitude of the CD8+ IFN-γ+ T cell response in the two-antigen regime is no greater than when the PyCSP vaccines are given alone. In addition to CD8+ T cell responses, CD4+ T cell responses were assessed in the spleen two weeks post-boost (Fig. 2D). Cells were stimulated with PyMSP133, as well as the published PyCSP CD4+ T cell epitiope, PyCSP59-79, and the overlapping eptiope, PyCSP58-67. Responses to each peptide are shown, but overall were extremely low at < 0.1% CD4+ T cells and although they trend towards being lower in the dual-antigen regime, were not significantly lower. Given the low level of these responses, they were not investigated further in this study. CD8+ T cell responses were also assessed in C57BL/6 mice. Two weeks after the MVA boost, responses were measured in the blood by ICS against the pool of PyMSP133 peptides (Fig. 2E). Responses tended towards being lower in the PyMSP142 + PyCSP immunized mice, although this did not reach statistical significance. No responses were detected following stimulation with PyCSP280-288 (data not shown) and peptides were not available against full length PyCSP.
Figure 2.

Antigen-specific CD8+ T cell responses are lower post-boost following a two-antigen vaccine regime than a single antigen regime. BALB/c mice were immunized with the regimes described in Table I. The percentages of CD8+ PBMCs producing IFN-γ were measured in the blood by ICS at the indicated time-points. PBMCs were re-stimulated with the PyMSP133 peptide pool (A) or PyCSP280-288 (B). Data are median ± interquartile range for 6-12 mice per group. Two weeks post boost CD8+ IFN-γ+ (C) and CD4+ IFN-γ+ (D) responses were measured in the spleen against the indicated peptides. Bars represent median responses ± range for six mice per group. * P < 0.05, ** P < 0.01 by Mann Whitney test, excluding negative controls. C57BL/6 mice were also immunized and the percentage of CD8+ PBMCs producing IFN-γ was measured two weeks after the MVA boost following re-stimulation with the PyMSP133 peptide pool (E).
It has previously been shown that CD4+ T cells simultaneously producing IFN-γ, TNFα and IL-2 are associated with protection against Leishmania major in a murine challenge model (45), and also that IFN-γ and TNFα are important components of the non-cytolytic pathways underlying CD8+ T cell-mediated efficacy against intracellular liver-stage malaria infection in mice (7, 46). Therefore, in addition to IFN-γ, production of TNFα and IL-2 was also measured and the multi-functionality of the CD8+ T cells was assessed. The so-called “quality” of the antigen-specific cells was analyzed in terms of production of all three cytokines in blood, spleen and liver two weeks after the MVA boost immunization (Fig. 3). Responses in the blood and spleen were measured against both the PyMSP133 peptide pool (Fig. 3A) and PyCSP280-288 peptide (Fig. 3B). As seen previously for IFN-γ, total production of antigen-specific TNFα and IL-2 was significantly reduced in mice immunized against both antigens in comparison to a single antigen. However, within each compartment the proportion of cells (Fig. 3, A and B pie charts) that were producing all three cytokines (IFN-γ+TNFα+IL2), two cytokines (predominantly (IFN-γ+TNFα) or a single cytokine (predominantly IFN-γ), was largely unaffected by immunizing against both antigens. Due to the relatively small number of cells isolated, mononuclear cells could not be stimulated with each antigen separately. Instead, they were stimulated with a single peptide pool containing PyMSP133 peptide pool, PyCSP280-288 and PyCSP58-67 (Fig. 3C). Whilst conclusions about the contribution of each individual antigen cannot be made from these data, it was clear that the total cytokine production from liver mononuclear cells of mice that had received the MSP1 + CSP vaccine regime was no higher than in mice that had received the PyCSP vaccines alone – in agreement with data from the spleen and blood. Interestingly, however, CD8+ T cells expressing IL-2 following re-stimulation were undetectable in the blood, but observed in both the spleen and liver.
Figure 3.

The quantity, but not the quality, of CD8+ T cells is affected following immunization against two antigens. In addition to IFN-γ, TNFα and IL-2 were measured by ICS in both the blood and the spleen following re-stimulation with the PyMSP133 peptide pool (A) or PyCSP280-288 (B), or in the liver following re-stimulation with a mixed peptide pool containing the PyMSP133 peptide pool, PyCSP280-288 and PyCSP58-67 (C). Bars indicate the median percentage of cells producing each of the three cytokines. The pies indicate the proportion of cells producing all three cytokines (black), two cytokines (dark grey) or a single cytokine (light grey) and the numbers above the pies indicate the total percentage of responding cells (± range) producing any cytokine in response to stimulation with the indicated peptides.
Protection against P. yoelii sporozoite challenge
CD8+ T cell responses, induced by viral vectored vaccines encoding either PyMSP142 or PyCSP, have been previously reported to provide partial or sterilizing liver-stage protection against P. yoelii sporozoite challenge (4, 32-34). In order to assess whether the reduced CD8+ T cell responses post-boost in the MSP1 + CSP vaccine regime had an impact on efficacy against sporozoite challenge, mice were challenged intravenously 2 or 10 weeks after the MVA boost with either 1000 or 50 sporozoites respectively (Table III). 50 sporozoites are routinely used in the P. yoelii challenge model (4, 5, 47), however, we had already reported that the Ad-MVA PyMSP142 vaccine regime could protect against a high dose challenge of 250 sporozoites (48) two weeks after the MVA boost immunization (4). Therefore in order to maximize any protective differences between immunization groups, an even higher challenge dose of 1000 sporozoites was used at the two weeks post boost challenge time-point (Table III and Fig. 4). In both experiments fewer mice were sterilely protected following immunization with the MSP1 + CSP regime in comparison to the CSP alone regime. Following challenge two weeks post boost, only 1/6 mice was sterilely protected in the MSP1 + CSP group, compared to 4/6 in the CSP alone group, and a similar trend was observed at the late 10 week challenge time-point. Sterilizing liver-stage efficacy had therefore been reduced following co-administration of the MSP1 + CSP vaccines, indicating that a mixed population of CD8+ T cells against PyMSP133 and PyCSP is less protective in this model than a similar quantity of cells targeting PyCSP alone. However, despite this reduced efficacy at the liver-stage and in agreement with the PyMSP119 antibody data (Fig. 1A), overall survival in the MSP1 + CSP vaccinated mice remained high following the onset of blood-stage parasitemia, with 5/6 (83%) surviving in each challenge experiment in comparison to 100% survival following immunization with the MSP1 vaccine alone. For mice challenged ten weeks post-boost, sera were collected and peak parasitemia correlated significantly with pre-challenge total PyMSP119-specific IgG ELISA titer (Fig. 4). These data suggest that, following the onset of blood-stage parasitemia, survival in this model remains dependent on PyMSP119 antibody titers. These data also confirmed that the Ad-MVA PyMSP142 vaccine regime by itself maintains 100% survival following both a high dose challenge with 1000 sporozoites at the day 70 time-point, and also following a challenge ten weeks post-boost with 50 sporozoites.
Table III. Efficacy of the vaccine mixture against P. yoelii sporozoite challenge.
BALB/c mice were immunized with the regimes described in Table I and challenged i.v. with sporozoites at the indicated time-points. Parasitemia was monitored as described.
| Immunization Regime | No. survived / No. challenged (% survival) |
No. sterilely protected / No. challenged (% survival) |
Geometric mean (range) peak parasitemia of surviving mice |
|---|---|---|---|
| A) 1000 sporozoites, 2 weeks post boost | |||
| MSP1 + CSP | 5/6 (83%) | 1/6 (16%) | 4.62% (0 – 28%) |
| MSP1 + Control | 6/6 (100%) | 1/6 (16%) | 1.80% (0 – 7.9%) |
| CSP + Control | 4/6 (66%) | 4/6 (66%) | N/A |
| Naïve unimmunized | 0/6 (0%) | 0/6 (0%) | N/A |
|
| |||
| B) 50 sporozoites, 10 weeks post boost | |||
| MSP1 + CSP | 5/6 (83%) | 2/6 (33%) | 4.53% (0 – 11.8%) |
| MSP1 + Control | 6/6 (100%) | 1/6 (16%) | 0.20% (0 – 17.6%) |
| CSP + Control | 3/6 (50%) | 3/6 (50%) | N/A |
| Naïve unimmunized | 0/6 (0%) | 0/6 (0%) | N/A |
Figure 4.

Survival and peak parasitemia following sporozoite challenge. Mice were challenged with sporozoites as described in Table III. Kaplan-Meier curves show survival following sporozoite challenge two weeks post-boost (A) and ten weeks post-boost (B). Following challenge ten weeks post-boost PyMSP119-specific total IgG titers correlate with peak parasitemia (C). rs = −0.7541 by Spearman’s Rank correlation analysis.
CD8+ T cell interference is not completely overcome by lowering vaccine dose
One possible explanation for the observed CD8+ T cell interference following the co-administration of the MVA PyMSP142 and PyCSP booster vaccines (but not AdHu5 priming vaccines), was that it was a systemic or spatial effect. Given the relatively high overall magnitude of the responses induced against each antigen individually, it may not be physiologically possible to expand both antigen-specific populations to the same extent when both vaccines were co-administered. T cell responses induced by vectored vaccines follow a dose response curve (49), and it was therefore hypothesized that lowering the dose of the vaccines might reduce the overall magnitude of the response induced and thus prevent the observed CD8+ T cell interference. BALB/c mice were thus immunized as described in Table I, but primed with 108 vp of each AdHu5 vaccine (rather than 1010 vp) and boosted with 106 pfu of each MVA vaccine (rather than 107 pfu). Two weeks after the MVA boost, mice were culled and splenocytes were re-stimulated with the PyMSP133 peptide pool or PyCSP280-288 before assaying responses by ICS (Fig. 5). In this case, PyCSP-specific responses were significantly lower (P = 0.0087 by Mann Whitney test) than those observed previously with a median response of 13.7% of CD8+ T cells producing IFN-γ after CSP only vaccination, in comparison to 25.4% observed in the previous experiment (Fig. 2C), and now significant CD8+ T cell inference was not observed for this antigen. However, despite the reduced magnitude of the PyCSP CD8+ T cell population, significant interference was still observed following vaccine co-administration for the relatively weaker responses against PyMSP133. Given overall responses of greater total magnitude were previously observed in the spleen (Fig. 2C), these data indicate that physiological space is not the only contributing factor to CD8+ T cell interference in this model.
Figure 5.
CD8+ T cell competition is not completely overcome by lowering doses of vaccines. BALB/c mice were immunized with the same regimes as described in Table I, but using lower doses of vaccines (108 vp of each AdHu5 and 106 pfu of each MVA). Antigen-specific CD8+ IFN-γ+ responses were measured as before in the spleen two weeks post-boost by ICS. ** P < 0.01 by Mann Whitney test, excluding negative controls.
CD8+ T cell interference can be minimized by immunization at separate sites
A second possibility was that CD8+ T cell interference was a local effect, occurring at the draining lymph node sites of T cell boosting, that could be overcome by immunizing with the vectored vaccines at physically separated sites. To investigate this, BALB/c mice were primed as usual with a mixture of 1010 vp of each AdHu5 vaccine and then boosted eight weeks later with 107 pfu of each MVA vaccine, except that the two different MVA vaccines were given into separate ears. Two weeks after the MVA boost, PBMC were isolated and re-stimulated as before with the indicated peptides before analysis by ICS (Fig. 6A). Comparable PyCSP-specific CD8+ IFN-γ+ T cell responses were now observed in mice either immunized against both PyMSP142 + PyCSP or against PyCSP alone. Importantly, these PyCSP-specific responses were maintained at a similar magnitude to those seen previously when the vaccines had been given as a mixture, rather than at the lower magnitude observed following reduced dose immunization. PyMSP133-specific CD8+ IFN-γ+ T cell responses were, however, still lower in mice immunized against PyMSP142 + PyCSP compared to PyMSP142 alone. Similar results were observed for TNFα and IL-2, and the multi-functionality of the cells was unaffected by immunizing at separate sites, in agreement with that observed previously (data not shown). Furthermore, total IgG responses against both antigens remained unaffected by immunizing against both antigens at separate sites, and were maintained at the same level as when the two MVAs had been given as a mixture (Fig. 6B and C).
Figure 6.

BALB/c mice were primed with a mixture of AdHu5 vaccines as described in Table I but eight weeks later the two MVA vaccines were co-administered at separate sites – one given in each ear. Twelve days after immunization with MVA, the production of antigen-specific IFN-γ from CD8+ PBMCs was measured by ICS (A). Bars indicate median responses. * P < 0.05, by Mann Whitney test, excluding negative controls. These data are representative of two independent experiments. Total IgG against both PyMSP119 (B) and PyCSP (C) were also measured. The dashed line indicates the limit of detection and bars indicate mean response.
Immunization at separate sites improves efficacy against P. yoelii sporozoite challenge
Two weeks following immunization with the MVA vaccines at separate sites, mice were challenged by i.v. injection of 1000 sporozoites. In this instance, on day five post-challenge, parasitemia was undetectable in mice immunized against PyMSP142 + PyCSP, whereas mice immunized with either antigen alone had already begun to develop patent blood-stage infection (Fig. 7A). Given PyMSP119 antibody responses remained comparable in the MSP1 + CSP and MSP1-only immunized groups, and given the previously observed reduced interference in CD8+ T cell responses against both antigens, this reduction in patent parasitemia at day 5 post-challenge indicates that when the two recombinant MVA vaccines are given at separate sites, mice immunized against both antigens had enhanced liver-stage protection compared to when they were immunized with either antigen alone. However, all mice in the co-administration and MSP1-only immunized groups went on to eventually develop a patent blood-stage parasitemia but, like before, high-level survival was maintained in both groups of mice possessing antibodies against PyMSP119 (Fig. 7B). Ultimately, delayed onset blood-stage parasitemia combined with subsequent survival in all mice, indicated a more beneficial effect of PyMSP142 and PyCSP co-immunization following separate site administration of the MVA boosting vaccines in comparison to that previously seen when the vaccines were administered as a mixture.
Figure 7.

Efficacy against sporozoite challenge in mice immunized with MVA at separate sites. BALB/c mice that had been immunized with MVA at separate sites were challenged i.v. with 1000 sporozoites fourteen days after MVA. (A) Parasitemia on day 5 post-challenge. Bars represent geometric mean parasitemia. (B) Parasitemia over time for the different vaccine groups. Lines indicate percentage of infected RBC for individual mice of six per group. Crosses indicate mice that were culled. *** P ≤ 0.001
CD8+ T cell interference is not due to competition for presentation on MHC class I
Having identified that CD8+ T cell interference could be overcome for PyCSP-specific responses and minimized for PyMSP133-specific responses by immunization with the two recombinant MVAs at separate sites, we sought to identify the mechanism of interference. One possibility was that it was due to peptides competing for presentation on MHC class I molecules on the surface of APCs. In BALB/c mice all seven dominant epitopes within the PyMSP133 peptide pool are predicted to be H-2Kd-restricted, as well as PyCSP280-288 (38) and PyCSP58-67 (39). To investigate whether this was the reason for interference between these multiple CD8+ T cell responses, we measured responses against epitopes with a different MHC class I restriction within the MVA vector backbone. Splenocytes, harvested two weeks after the MVA boosting immunization, were re-stimulated with F2(G) (H-2Ld-restricted) and E3 (H-2Dd-restricted) peptides (42) (Fig. 8). For both epitopes encoded within the MVA vector, IFN-γ production from de novo primed CD8+ T cells was also significantly lower in mice that had been immunized with the two malaria antigen regime compared to when mice were immunized with either antigen alone, despite all mice receiving the same viral load. CD8+ T cell interference was therefore occurring with respect to MVA-specific responses as well as antigen-specific responses regardless of epitope MHC class I restriction.
Figure 8.

CD8+ T cell interference is not due to competition for presentation on MHC class I molecules. BALB/c mice were immunized with the regime described in Table I. Two weeks after the boost MVA-specific responses were measured in the spleen by ICS. CD8+ IFN-γ+ responses were measured following re-stimulation with the H-2Ld-restricted peptide F2(G) (A), and H-2Dd-restricted peptide E3 (B). * P < 0.05, ** P < 0.001 by Dunn’s multiple comparison test.
CD8+ T cell interference is not due to presentation of both antigens on the same APC
We hypothesized that interference could also be due to some other aspect of presentation of both antigens on the same APC. It has previously been demonstrated that CD8+ T cell interference can occur when CD8+ T cells compete for access to APCs when both antigens are presented on the same APC surface (50). Whilst the exact cause of the competition is not known, it has been suggested it may be due to limited space around the APC, perhaps limiting access to antigen, co-stimulatory molecules and/or survival factors. In order to investigate this phenomenon here, BALB/c mice were immunized with a mixture containing 1 × 1010 vp each of AdHu5-PyMSP142 and AdHu5-PyCSP. Eight weeks later, rather than boosting with MVA, the responses were boosted with 2 × 105 peptide-pulsed CD11c+ dendritic cells (DCs), as previously reported (51). The DCs were pulsed with PyMSP133 peptides and PyCSP peptides, either mixed together or pulsed separately and with cells mixed immediately prior to i.v. injection. Two weeks later CD8+ IFN-γ+ responses were measured against the two antigens in the blood by ICS (Fig. 9). In the majority of mice both PyMSP133 and PyCSP-specific responses were boosted by injection of peptide-pulsed CD11c+ DC compared to non-pulsed DC, but the level of boosting did not reach statistical significant due to at least one non-responder in each group. Nevertheless it demonstrated that responses were not different, irrespective of whether the DCs had been pulsed with peptides specific for both antigens either separately or as a mixture. These data thus suggest that T cell interference is not occurring due to CD8+ T cells competing when both antigens are presented on the same APC.
Figure 9.

Enhanced immunogenicity when vaccines are given at separate sites is not due to presentation of antigens on separate APCs. BALB/c mice were primed with a mixture of AdHu5 vaccines as described in Table I. Eight weeks later they were boosted i.d. with 2 × 105 CD11c+ cells pulsed with either a mixture of the seven known PyMSP133 peptide epitopes and the two known PyCSP peptide epitopes, or pulsed separately with the PyMSP133 peptides and PyCSP peptides which were then mixed in a 1:1 ratio immediately prior to injection to give a total of 2 × 105 cells. Two weeks after immunization with peptide pulsed DCs, PBMCs were isolated and re-stimulated with peptides specific for PyMSP133 (A) and PyCSP (B) before ICS staining for IFN-γ.
Cells presenting two antigens are better targets for killing at the time of MVA boost immunization
We hypothesized that secondary CD8+ T cell responses may be lower when the two recombinant MVA vaccines expressing both antigens are given together at the same site due to dual infection of cells at the vaccination site. Infection of a single cell with more than one MVA has been demonstrated in vitro (52), and is likely to occur in vivo given the dose of MVA used. These cells may be better targets for killing by pre-existing transgene-specific cytotoxic CD8+ T cells specific for both PyMSP133 and PyCSP, allowing less time for antigen expression, processing and/or cross-presentation to APCs, in comparison to when the two vaccines are given separately. In order to investigate this an in vivo CTL killing assay was carried out. In the first experiment, BALB/c mice were primed by i.d. immunization with 2 × 1010 vp AdPyMSP142 + AdPyCSP, AdPyMSP142 + AdControl or AdControl only and then left to rest for eight weeks. At the time the MVA-boost immunization would normally be given, the mice were injected with CFSE-labeled, peptide-pulsed splenocytes as described. Five hours after the cells were injected, the mice were culled and antigen-specific killing was calculated by analyzing CFSE-fluorescence intensity (Fig. 10A). These data showed that if cells were pulsed with PyMSP133 and PyCSP peptides mixed together (and therefore presenting both antigens) then in mice primed against both antigens approximately 40% of the cells are killed within 5 hours. This was in comparison to approximately 20% killing seen when mice had been primed only against MSP142, or when cells pulsed with only MSP133 peptides were transferred into mice primed against either MSP142 alone or both antigens (Fig. 10B). These data imply that priming mice against two malaria antigens could lead to a greater level of cellular killing following co-administration of two recombinant MVA booster vaccines as mixture, and this is associated with reduced boosting of CD8+ T cell responses against both antigens. However, in a second experiment, similar observations were made when mice primed against both antigens were injected with splenocytes that had been pulsed with the PyMSP133 and PyCSP peptides separately (Fig. 10C). This was the same outcome, irrespective of whether the total splenocyte population or only CD11c+ cells were analyzed (data not shown). Although enhanced killing is observed when mice are previously primed against both antigens, the observations using this model assay failed to account for the improved CD8+ T cell immunogenicity seen against the PyCSP antigen following co-administration of the two MVA vaccines at separate sites.
Figure 10.

Antigen-specific killing occurs at the time of the MVA boost. To analyze antigen-specific killing, an in vivo cytotoxic T cell killing assay was used. Naïve splenocytes labelled with a high concentration of CFSE (CFSE high) were pulsed with the PyMSP133 peptide pool and PyCSP280-288 peptides as indicated. To control for non-specific killing un-pulsed cells were labelled with a low concentration of CFSE (CFSE low). 2 × 107 cells containing an equal number of CFSE high and CFSE low cells were injected i.v. into BALB/c mice that had been immunized eight weeks earlier with a total of 2 × 1010 vp AdHu5. In the first experiment mice were immunized with the indicated vaccines (A and B), in the second experiment all mice were immunized with AdHu5-PyMSP142 + AdHu5-PyCSP (C). In both experiments, cells were also transferred into mice that had been primed with AdHu5-Control vaccine. After 5 h mice were culled and splenocytes analyzed by flow cytometry and antigen specific killing was calculated as described. Representative histograms of the intensity of CFSE staining showing the proportion of CFSE high to CFSE low cells recovered in the first experiment are shown (A) and antigen specific killing was calculated (B). ** P = 0.0032 by Mann Whitney test. In the second experiment all mice were immunized with AdHu5-PyMSP142 + AdHu5-PyCSP and the indicated cells were transferred (C). * P < 0.05 by Dunn’s multiple comparison test. Bars indicate median response.
Discussion
Here we demonstrate the feasibility of a multi-stage and multi-antigen vaccine regime using a clinically-relevant viral vectored vaccine delivery platform. In the P. yoelii mouse model we show that co-administration of a mixture of viral vectored vaccines expressing two different malaria antigens in an AdHu5-MVA prime-boost regime results in strong antibody responses against both antigens. However, CD8+ T cell responses were significantly reduced after the MVA boost immunization, if the two vaccines were given as a mixture, but this effect was minimized if the two MVA boost vaccines were co-administered at separate sites. Importantly, maintaining high-level CD8+ T cell and antibody responses against both the PyCSP and PyMSP142 antigens was associated with the highest levels of efficacy against a stringent high dose challenge with P. yoelii sporozoites.
An important finding from these experiments was that antibody responses were maintained against both antigens following Ad-MVA vaccination in two strains of mice. This was confirmed by sporozoite challenge in which no significant differences were observed in survival between mice immunized against PyMSP142 alone, as compared to mice immunized against both PyMSP142 and PyCSP. These results are in agreement with a study combining the RTS,S (CSP vaccine) with MSP142 and AMA1 protein vaccines in rhesus macaques (15), in which there was no interference between antibody responses against RTS,S and MSP142. However the authors did report that MSP142 immunization interfered with both AMA1-specific antibodies and IFN-γ production, and it will be important in future studies to ascertain whether antigenic interference is an antigen-specific phenomenon, or related to specific vaccine delivery platforms.
Here we found that following an Ad-MVA vaccination regime the magnitude of the total CD8+ IFN-γ+ T cell response was the same whether mice were immunized with the PyCSP vaccines alone, or with both the PyMSP142 and PyCSP vaccines. Despite this, following sporozoite challenge, liver-stage protection was reduced in mice receiving the two-antigen regime, showing that in this case it was more beneficial to have a high number of T cells against PyCSP than the same number of T cells targeting PyCSP and PyMSP133. Given the potential for antigenic interference, this highlights the importance of careful consideration of the antigens used in a multi-antigen vaccine. It also suggests that in the P. yoelii mouse model CD8+ T cells against PyCSP are more efficient at killing the pre-erythrocytic parasites than those against PyMSP133. This may be because CSP is expressed throughout the sporozoite and early pre-erythrocytic stages of the parasite life-cycle (53), whilst PyMSP1 is expressed only during the late-liver stage (54).
CD8+ T cell interference may occur for a number of reasons. Firstly, we hypothesized that it could be a systemic or spatial effect, due to the large magnitude of the CD8+ T cell responses induced in BALB/c mice against these two antigens. In order to address this question we lowered the doses of vaccines, giving 100-fold less AdHu5 and 10-fold less MVA. This reduced the overall magnitude of the antigen-specific immune responses, in agreement with other dosing studies in mice for similar viral vectored vaccines (49), and PyCSP-specific CD8+ T cell interference was no longer observed, although the PyMSP133-specific response was still reduced by more than 50% in the two-antigen regime, indicating that whilst the magnitude may have been contributing to competition, it is not the sole cause. Additionally, altering the ratio of the vaccines, such that mice were boosted with 10-fold more MVA-PyMSP142 than MVA-PyCSP in the two-antigen regime, could not overcome the PyMSP133-specific CD8+ T cell competition (data not shown).
It has been found that co-administering vaccines at separate sites can overcome CD8+ T cell interference (16). In agreement with that, we found that administration of the two recombinant MVA vectors at separate sites eliminated PyCSP-specific CD8+ T cell interference. PyMSP133-specific competition did still occur, but in this case it was minimized, with 70% of the response maintained in the mice immunized against both antigens, whilst in previous experiments the response had been reduced by more than 50%. Furthermore, when these mice were challenged with P. yoelii sporozoites those immunized at separate sites demonstrated enhanced liver-stage efficacy, as determined by day 5 blood-stage parasitemias.
A number of studies have found that CD8+ T cell interference occurs when CD8+ T cells compete for antigenic epitopes presented on the same APC surface (50). The cause is not known, but it may be due to limited space around the APC, perhaps limiting access to co-stimulatory molecules or survival factors. It has previously been shown that peptide-pulsed DCs can boost MVA primed immune responses (51), and so to investigate whether this was the case here, we boosted mice primed with the mixture of Ad-PyCSP and Ad-PyMSP142 with peptide-pulsed CD11c+ DCs that had either been pulsed with peptides from both antigens together or separately. We found that a modest increase in the production of IFN-γ from CD8+ T cells was observed when Ad-primed responses were boosted with peptide-pulsed DCs, but there was no significant difference in the magnitude of the CD8+ IFN-γ+ response between those that had been pulsed with peptide separately or together. This is in contrast to the findings of Kedl et al. (50), who show that when mice are immunized with DC pulsed with two different peptides, in the presence of high-affinity T cells specific for one of the peptides, then responses to both peptides are impaired if they are presented on the same APC, but only to one peptide if they are presented on separate APC.
We subsequently hypothesized that the killing of MVA infected cells at the time of the boost by cytotoxic CD8+ T cells, previously primed by the AdHu5 vaccines, may also be a crucial factor in determining the magnitude of the secondary CD8+ T cell response. A study of secondary influenza infection showed that CD8+ T cells secreting perforin rapidly terminate antigen presentation in this model (55). Furthermore, studies with MVA and vaccinia virus have shown that during secondary responses the timing of antigen presentation is crucial, with responses only detected to antigens expressed early during the viral life cycle. This was dependent on the presence of primed T cells, although a mechanism was not identified (56). Our own results using an in vivo killing assay showed that following injection of peptide-pulsed splenocytes, cells pulsed with both PyMSP133 and PyCSP peptides together were almost twice as likely to be killed within 5 hours as those that had been pulsed with peptides corresponding to a single antigen. Taken together these data suggest that the more rapid clearance of MVA co-infected cells that express both the PyMSP142 and PyCSP antigens, results in a reduced period of antigen expression and presentation, and consequently a reduced secondary CD8+ T cell response. We also observed that enhanced killing occurred when cells were pulsed with peptides separately and the cells mixed immediately before injection. This does not appear to be in agreement with the immunogenicity data and may reflect differences in systemic compared to local CTL activity, or otherwise, alternative mechanisms may yet explain the reasons for improved immunogenicity following separate site immunization with two recombinant MVAs.
The outcomes from this study imply that immunizing at separate sites may be necessary to prevent antigenic interference. However, this may be neither practical, nor financially viable in the field. A more feasible approach is the development of multivalent MVA vectors, which express a number of malaria antigens from a single virus. Establishing whether CD8+ T cell interference occurs in humans, and the mechanism of interference, will be crucial in deciding which is the more effective approach to developing a multi-antigen or multi-stage malaria vaccine. Phase I/IIa clinical trials are currently underway at Oxford University to assess the efficacy of AdCh63-MVA vectored vaccines expressing P. falciparum MSP1, AMA1 and ME-TRAP alone and in combination (2). Based on the pre-clinical work presented here, vaccines are being co-administered at separate sites in the first instance.
In summary, we show that Ad-MVA viral vectored vaccines expressing the antigens PyCSP and PyMSP142, can induce enhanced protection against P. yoelii sporozoite challenge compared to either antigen given alone, provided MVA vaccinations are given at separate sites. When MVA vaccines are given as a mixture reduced CD8+ T cell responses are observed. This CD8+ T cell interference is not due to peptide epitope competition for presentation on same MHC class I molecules, nor is it at the level of the APC. Enhanced killing of cells presenting both antigens at the time of the boost occurs regardless of whether the vaccines are given at separate sites or as a mixture. The exact mechanism of CD8+ T cell interference in a vaccine mixture thus remains to be fully elucidated. Classical protein-in-adjuvant vaccines do not induce strong cellular immunity and therefore T cell interference in multi-antigen vaccination regimes is not routinely assessed. However, multi-antigen viral vectored and DNA vaccines which induce strong T cell responses are now being tested for a wide variety of diseases including HIV-1, tuberculosis, malaria and influenza (3). Understanding T cell interference will be critical in developing effective vaccines against these difficult infectious diseases.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Spencer, J. Furze, M. Tunnicliff, M. Dicks, S. Douglas, S. de Cassan, A. Goodman, D. Worth, B. Bolinger as well as the Jenner Institute Viral Vector and Flow Cytometry Core Facilities for assistance.
ABBREVIATIONS
- AdHu5
human adenovirus serotype 5
- AMA1
apical membrane antigen 1
- CSP
circumsporozoite protein
- EPI
expanded program on immunization
- ICS
intracellular cytokine staining
- i.d.
intradermal
- MSP1
merozoite surface protein 1
- MVA
modified vaccinia virus Ankara
- pRBC
parasitized red blood cell
Footnotes
This work was funded by the Wellcome Trust and the EMVDA (European Malaria Vaccine Development Association, a European Commission FP6-funded consortium). SCG, AVSH and SJD are Jenner Investigators, and AVSH is also a Wellcome Trust Principal Research Fellow. SJD is a MRC Career Development Fellow.
References
- 1.World Health Organisation . World Malaria Report. 2009. [Google Scholar]
- 2.Hill AV, Reyes-Sandoval A, O’Hara G, Ewer K, Lawrie A, Goodman A, Nicosia A, Folgori A, Colloca S, Cortese R, Gilbert SC, Draper SJ. Prime-boost vectored malaria vaccines: progress and prospects. Hum Vaccin. 2010;6:78–83. doi: 10.4161/hv.6.1.10116. [DOI] [PubMed] [Google Scholar]
- 3.Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol. 2010;8:62–73. doi: 10.1038/nrmicro2240. [DOI] [PubMed] [Google Scholar]
- 4.Draper SJ, Goodman AL, Biswas S, Forbes EK, Moore AC, Gilbert SC, Hill AV. Recombinant viral vaccines expressing merozoite surface protein-1 induce antibody- and T cell-mediated multistage protection against malaria. Cell Host Microbe. 2009;5:95–105. doi: 10.1016/j.chom.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Draper SJ, Moore AC, Goodman AL, Long CA, Holder AA, Gilbert SC, Hill F, Hill AV. Effective induction of high-titer antibodies by viral vector vaccines. Nat Med. 2008;14:819–821. doi: 10.1038/nm.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodman AL, Epp C, Moss D, Holder AA, Wilson JM, Gao GP, Long CA, Remarque EJ, Thomas AW, Ammendola V, Colloca S, Dicks MD, Biswas S, Seibel D, van Duivenvoorde LM, Gilbert SC, Hill AV, Draper SJ. New candidate vaccines against blood-stage P. falciparum malaria: Prime-boost immunization regimes incorporating human and simian adenoviral vectors and poxviral vectors expressing an optimized antigen based on MSP-1. Infect Immun. 2010;78:4601–12. doi: 10.1128/IAI.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reyes-Sandoval A, Berthoud T, Alder N, Siani L, Gilbert SC, Nicosia A, Colloca S, Cortese R, Hill AV. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect Immun. 2010;78:145–153. doi: 10.1128/IAI.00740-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Douglas AD, de Cassan SC, Dicks MD, Gilbert SC, Hill AV, Draper SJ. Tailoring subunit vaccine immunogenicity: Maximizing antibody and T cell responses by using combinations of adenovirus, poxvirus and protein-adjuvant vaccines against Plasmodium falciparum MSP1. Vaccine. 2010;28:7167–7178. doi: 10.1016/j.vaccine.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draper SJ, Biswas S, Spencer AJ, Remarque EJ, Capone S, Naddeo M, Dicks MDJ, Faber BW, de Cassan SC, Folgori A, Nicosia A, Gilbert SC, Hill AVS. Enhancing blood-stage malaria subunit vaccine immunogenicity in rhesus macaques by combining adenovirus, poxvirus, and protein-in-adjuvant vaccines. J Immunol. 2010;185:7583–7595. doi: 10.4049/jimmunol.1001760. [DOI] [PubMed] [Google Scholar]
- 10.Biswas S, Dicks MDJ, Long CA, Remarque EJ, Siani L, Colloca S, Cottingham MG, Holder AA, Gilbert SC, Hill AVS, Draper SJ. Transgene Optimization, Immunogenicity and In Vitro Efficacy of Viral Vectored Vaccines Expressing Two Alleles of Plasmodium falciparum AMA1. PLoS ONE. 2011;6:e20977. doi: 10.1371/journal.pone.0020977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman AL, Draper SJ. Blood-stage malaria vaccines - recent progress and future challenges. Ann Trop Med Parasitol. 2010;104:189–211. doi: 10.1179/136485910X12647085215534. [DOI] [PubMed] [Google Scholar]
- 12.McConkey SJ, Reece WH, Moorthy VS, Webster D, Dunachie S, Butcher G, Vuola JM, Blanchard TJ, Gothard P, Watkins K, Hannan CM, Everaere S, Brown K, Kester KE, Cummings J, Williams J, Heppner DG, Pathan A, Flanagan K, Arulanantham N, Roberts MT, Roy M, Smith GL, Schneider J, Peto T, Sinden RE, Gilbert SC, Hill AV. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003;9:729–735. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 13.Greenwood B, Targett G. Do we still need a malaria vaccine? Parasite Immunol. 2009;31:582–586. doi: 10.1111/j.1365-3024.2009.01140.x. [DOI] [PubMed] [Google Scholar]
- 14.Malaria Vaccine Technology Roadmap. 2006 http://www.malariavaccine.org/files/Malaria_Vaccine_TRM_Final.pdf.
- 15.Pichyangkul S, Tongtawe P, Kum-Arb U, Yongvanitchit K, Gettayacamin M, Hollingdale MR, Limsalakpetch A, Stewart VA, Lanar DE, Dutta S, Angov E, Ware LA, Bergmann-Leitner ES, House B, Voss G, Dubois MC, Cohen JD, Fukuda MM, Heppner DG, Miller RS. Evaluation of the safety and immunogenicity of Plasmodium falciparum apical membrane antigen 1, merozoite surface protein 1 or RTS,S vaccines with adjuvant system AS02A administered alone or concurrently in rhesus monkeys. Vaccine. 2009;28:452–462. doi: 10.1016/j.vaccine.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 16.Sedegah M, Charoenvit Y, Minh L, Belmonte M, Majam VF, Abot S, Ganeshan H, Kumar S, Bacon DJ, Stowers A, Narum DL, Carucci DJ, Rogers WO. Reduced immunogenicity of DNA vaccine plasmids in mixtures. Gene Ther. 2004;11:448–456. doi: 10.1038/sj.gt.3302139. [DOI] [PubMed] [Google Scholar]
- 17.Rogers WO, Weiss WR, Kumar A, Aguiar JC, Tine JA, Gwadz R, Harre JG, Gowda K, Rathore D, Kumar S, Hoffman SL. Protection of rhesus macaques against lethal Plasmodium knowlesi malaria by a heterologous DNA priming and poxvirus boosting immunization regimen. Infect Immun. 2002;70:4329–4335. doi: 10.1128/IAI.70.8.4329-4335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang G, Charoenvit Y, Moreno A, Baraceros MF, Banania G, Richie N, Abot S, Ganeshan H, Fallarme V, Patterson NB, Geall A, Weiss WR, Strobert E, Caro-Aquilar I, Lanar DE, Saul A, Martin LB, Gowda K, Morrissette CR, Kaslow DC, Carucci DJ, Galinski MR, Doolan DL. Induction of multi-antigen multi-stage immune responses against Plasmodium falciparum in rhesus monkeys, in the absence of antigen interference, with heterologous DNA prime/poxvirus boost immunization. Malar J. 2007;6:135. doi: 10.1186/1475-2875-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang G, Shi M, Conteh S, Richie N, Banania G, Geneshan H, Valencia A, Singh P, Aguiar J, Limbach K, Kamrud KI, Rayner J, Smith J, Bruder JT, King CR, Tsuboi T, Takeo S, Endo Y, Doolan DL, Richie TL, Weiss WR. Sterile protection against Plasmodium knowlesi in rhesus monkeys from a malaria vaccine: comparison of heterologous prime boost strategies. PLoS One. 2009;4:e6559. doi: 10.1371/journal.pone.0006559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R, Richie TL, Baraceros MF, Rahardjo N, Gay T, Banania JG, Charoenvit Y, Epstein JE, Luke T, Freilich DA, Norman J, Hoffman SL. Boosting of DNA vaccine-elicited gamma interferon responses in humans by exposure to malaria parasites. Infect Immun. 2005;73:2863–2872. doi: 10.1128/IAI.73.5.2863-2872.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sedegah M, Charoenvit Y, Aguiar J, Sacci J, Hedstrom R, Kumar S, Belmonte A, Lanar DE, Jones TR, Abot E, Druilhe P, Corradin G, Epstein JE, Richie TL, Carucci DJ, Hoffman SL. Effect on antibody and T-cell responses of mixing five GMP-produced DNA plasmids and administration with plasmid expressing GM-CSF. Genes Immun. 2004;5:553–561. doi: 10.1038/sj.gene.6364125. [DOI] [PubMed] [Google Scholar]
- 22.Esen M, Kremsner PG, Schleucher R, Gassler M, Imoukhuede EB, Imbault N, Leroy O, Jepsen S, Knudsen BW, Schumm M, Knobloch J, Theisen M, Mordmuller B. Safety and immunogenicity of GMZ2 - a MSP3-GLURP fusion protein malaria vaccine candidate. Vaccine. 2009;27:6862–6868. doi: 10.1016/j.vaccine.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 23.Malkin E, Hu J, Li Z, Chen Z, Bi X, Reed Z, Dubovsky F, Liu J, Wang Q, Pan X, Chen T, Giersing B, Xu Y, Kang X, Gu J, Shen Q, Tucker K, Tierney E, Pan W, Long C, Cao Z. A phase 1 trial of PfCP2.9: an AMA1/MSP1 chimeric recombinant protein vaccine for Plasmodium falciparum malaria. Vaccine. 2008;26:6864–6873. doi: 10.1016/j.vaccine.2008.09.081. [DOI] [PubMed] [Google Scholar]
- 24.Saul A, Lawrence G, Smillie A, Rzepczyk CM, Reed C, Taylor D, Anderson K, Stowers A, Kemp R, Allworth A, Anders RF, Brown GV, Pye D, Schoofs P, Irving DO, Dyer SL, Woodrow GC, Briggs WR, Reber R, Sturchler D. Human phase I vaccine trials of 3 recombinant asexual stage malaria antigens with Montanide ISA720 adjuvant. Vaccine. 1999;17:3145–3159. doi: 10.1016/s0264-410x(99)00175-9. [DOI] [PubMed] [Google Scholar]
- 25.Webster DP, Dunachie S, Vuola JM, Berthoud T, Keating S, Laidlaw SM, McConkey SJ, Poulton I, Andrews L, Andersen RF, Bejon P, Butcher G, Sinden R, Skinner MA, Gilbert SC, Hill AV. Enhanced T cell-mediated protection against malaria in human challenges by using the recombinant poxviruses FP9 and modified vaccinia virus Ankara. Proc Natl Acad Sci U S A. 2005;102:4836–4841. doi: 10.1073/pnas.0406381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ockenhouse CF, Sun PF, Lanar DE, Wellde BT, Hall BT, Kester K, Stoute JA, Magill A, Krzych U, Farley L, Wirtz RA, Sadoff JC, Kaslow DC, Kumar S, Church LW, Crutcher JM, Wizel B, Hoffman S, Lalvani A, Hill AV, Tine JA, Guito KP, de Taisne C, Anders R, Ballou WR, et al. Phase I/IIa safety, immunogenicity, and efficacy trial of NYVAC-Pf7, a pox-vectored, multiantigen, multistage vaccine candidate for Plasmodium falciparum malaria. J Infect Dis. 1998;177:1664–1673. doi: 10.1086/515331. [DOI] [PubMed] [Google Scholar]
- 27.Porter DW, Thompson FM, Berthoud TK, Hutchings CL, Andrews L, Biswas S, Poulton I, Prieur E, Correa S, Rowland R, Lang T, Williams J, Gilbert SC, Sinden RE, Todryk S, Hill AV. A human Phase I/IIa malaria challenge trial of a polyprotein malaria vaccine. Vaccine. 2011;29:7514–7522. doi: 10.1016/j.vaccine.2011.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catanzaro AT, Roederer M, Koup RA, Bailer RT, Enama ME, Nason MC, Martin JE, Rucker S, Andrews CA, Gomez PL, Mascola JR, Nabel GJ, Graham BS. Phase I clinical evaluation of a six-plasmid multiclade HIV-1 DNA candidate vaccine. Vaccine. 2007;25:4085–4092. doi: 10.1016/j.vaccine.2007.02.050. [DOI] [PubMed] [Google Scholar]
- 29.Catanzaro AT, Koup RA, Roederer M, Bailer RT, Enama ME, Moodie Z, Gu L, Martin JE, Novik L, Chakrabarti BK, Butman BT, Gall JG, King CR, Andrews CA, Sheets R, Gomez PL, Mascola JR, Nabel GJ, Graham BS. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J Infect Dis. 2006;194:1638–1649. doi: 10.1086/509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genton B, Pluschke G, Degen L, Kammer AR, Westerfeld N, Okitsu SL, Schroller S, Vounatsou P, Mueller MM, Tanner M, Zurbriggen R. A randomized placebo-controlled phase Ia malaria vaccine trial of two virosome-formulated synthetic peptides in healthy adult volunteers. PLoS One. 2007;2:e1018. doi: 10.1371/journal.pone.0001018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson FM, Porter DW, Okitsu SL, Westerfeld N, Vogel D, Todryk S, Poulton I, Correa S, Hutchings C, Berthoud T, Dunachie S, Andrews L, Williams JL, Sinden R, Gilbert SC, Pluschke G, Zurbriggen R, Hill AV. Evidence of Blood Stage Efficacy with a Virosomal Malaria Vaccine in a Phase IIa Clinical Trial. PLoS ONE. 2008;3:e1493. doi: 10.1371/journal.pone.0001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodrigues EG, Zavala F, Eichinger D, Wilson JM, Tsuji M. Single immunizing dose of recombinant adenovirus efficiently induces CD8+ T cell-mediated protective immunity against malaria. J Immunol. 1997;158:1268–1274. [PubMed] [Google Scholar]
- 33.Bruna-Romero O, Gonzalez-Aseguinolaza G, Hafalla JC, Tsuji M, Nussenzweig RS. Complete, long-lasting protection against malaria of mice primed and boosted with two distinct viral vectors expressing the same plasmodial antigen. Proc Natl Acad Sci U S A. 2001;98:11491–11496. doi: 10.1073/pnas.191380898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ophorst OJ, Radosevic K, Havenga MJ, Pau MG, Holterman L, Berkhout B, Goudsmit J, Tsuji M. Immunogenicity and protection of a recombinant human adenovirus serotype 35-based malaria vaccine against Plasmodium yoelii in mice. Infect Immun. 2006;74:313–320. doi: 10.1128/IAI.74.1.313-320.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reece WH, Pinder M, Gothard PK, Milligan P, Bojang K, Doherty T, Plebanski M, Akinwunmi P, Everaere S, Watkins KR, Voss G, Tornieporth N, Alloueche A, Greenwood BM, Kester KE, McAdam KP, Cohen J, Hill AV. A CD4(+) T-cell immune response to a conserved epitope in the circumsporozoite protein correlates with protection from natural Plasmodium falciparum infection and disease. Nat Med. 2004;10:406–410. doi: 10.1038/nm1009. [DOI] [PubMed] [Google Scholar]
- 36.Holder AA, Sandhu JS, Hillman Y, Davey LS, Nicholls SC, Cooper H, Lockyer MJ. Processing of the precursor to the major merozoite surface antigens of Plasmodium falciparum. Parasitology. 1987;94(Pt 2):199–208. doi: 10.1017/s0031182000053889. [DOI] [PubMed] [Google Scholar]
- 37.Ogun SA, Dumon-Seignovert L, Marchand JB, Holder AA, Hill F. The oligomerization domain of C4-binding protein (C4bp) acts as an adjuvant, and the fusion protein comprised of the 19-kilodalton merozoite surface protein 1 fused with the murine C4bp domain protects mice against malaria. Infect Immun. 2008;76:3817–3823. doi: 10.1128/IAI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodrigues MM, Cordey AS, Arreaza G, Corradin G, Romero P, Maryanski JL, Nussenzweig RS, Zavala F. CD8+ cytolytic T cell clones derived against the Plasmodium yoelii circumsporozoite protein protect against malaria. Int Immunol. 1991;3:579–585. doi: 10.1093/intimm/3.6.579. [DOI] [PubMed] [Google Scholar]
- 39.Franke ED, Sette A, Sacci J, Jr., Southwood S, Corradin G, Hoffman SL. A subdominant CD8(+) cytotoxic T lymphocyte (CTL) epitope from the Plasmodium yoelii circumsporozoite protein induces CTLs that eliminate infected hepatocytes from culture. Infect Immun. 2000;68:3403–3411. doi: 10.1128/iai.68.6.3403-3411.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grillot D, Michel M, Muller I, Tougne C, Renia L, Mazier D, Corradin G, Lambert PH, Louis JA, Del Guidice G. Immune responses to defined epitopes of the circumsporozoite protein of the murine malaria parasite, Plasmodium yoelii. Eur J Immunol. 1990;20:1215–1222. doi: 10.1002/eji.1830200604. [DOI] [PubMed] [Google Scholar]
- 41.Sridhar S, Reyes-Sandoval A, Draper SJ, Moore AC, Gilbert SC, Gao GP, Wilson JM, Hill AV. Single-dose protection against Plasmodium berghei by a simian adenovirus vector using a human cytomegalovirus promoter containing intron A. J Virol. 2008;82:3822–3833. doi: 10.1128/JVI.02568-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tscharke DC, Woo WP, Sakala IG, Sidney J, Sette A, Moss DJ, Bennink JR, Karupiah G, Yewdell JW. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J Virol. 2006;80:6318–6323. doi: 10.1128/JVI.00427-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roederer M, Nozzi JL, Nason MX. SPICE: Exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A. 2011;79:167–74. doi: 10.1002/cyto.a.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marussig M, Renia L, Motard A, Miltgen F, Petour P, Chauhan V, Corradin G, Mazier D. Linear and multiple antigen peptides containing defined T and B epitopes of the Plasmodium yoelii circumsporozoite protein: antibody-mediated protection and boosting by sporozoite infection. Int Immunol. 1997;9:1817–1824. doi: 10.1093/intimm/9.12.1817. [DOI] [PubMed] [Google Scholar]
- 45.Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, Roederer M, Seder RA. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 46.Butler NS, Schmidt NW, Harty JT. Differential effector pathways regulate memory CD8 T cell immunity against Plasmodium berghei versus P. yoelii sporozoites. J Immunol. 2010;184:2528–2538. doi: 10.4049/jimmunol.0903529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belmonte M, Jones TR, Lu M, Arcilla R, Smalls T, Belmonte A, Rosenbloom J, Carucci DJ, Sedegah M. The infectivity of Plasmodium yoelii in different strains of mice. J Parasitol. 2003;89:602–603. doi: 10.1645/0022-3395(2003)089[0602:TIOPYI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 48.Sedegah M, Brice GT, Rogers WO, Doolan DL, Charoenvit Y, Jones TR, Majam VF, Belmonte A, Lu M, Belmonte M, Carucci DJ, Hoffman SL. Persistence of protective immunity to malaria induced by DNA priming and poxvirus boosting: characterization of effector and memory CD8(+)-T-cell populations. Infect Immun. 2002;70:3493–3499. doi: 10.1128/IAI.70.7.3493-3499.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ophorst OJ, Radosevic K, Klap JM, Sijtsma J, Gillissen G, Mintardjo R, van Ooij MJ, Holterman L, Companjen A, Goudsmit J, Havenga MJ. Increased immunogenicity of recombinant Ad35-based malaria vaccine through formulation with aluminium phosphate adjuvant. Vaccine. 2007;25:6501–6510. doi: 10.1016/j.vaccine.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 50.Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, Marrack P. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. 2000;192:1105–1113. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Behboudi S, Moore A, Hill AV. Splenic dendritic cell subsets prime and boost CD8 T cells and are involved in the generation of effector CD8 T cells. Cell Immunol. 2004;228:15–19. doi: 10.1016/j.cellimm.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 52.Vasilakis N, Falvey D, Gangolli SS, Coleman J, Kowalski J, Udem SA, Zamb TJ, Kovacs GR. Transfection-independent production of alphavirus replicon particles based on poxvirus expression vectors. Nat Biotechnol. 2003;21:932–935. doi: 10.1038/nbt845. [DOI] [PubMed] [Google Scholar]
- 53.Bodescot M, Silvie O, Siau A, Refour P, Pino P, Franetich JF, Hannoun L, Sauerwein R, Mazier D. Transcription status of vaccine candidate genes of Plasmodium falciparum during the hepatic phase of its life cycle. Parasitol Res. 2004;92:449–452. doi: 10.1007/s00436-003-1061-9. [DOI] [PubMed] [Google Scholar]
- 54.Kawabata Y, Udono H, Honma K, Ueda M, Mukae H, Kadota J, Kohno S, Yui K. Merozoite surface protein 1-specific immune response is protective against exoerythrocytic forms of Plasmodium yoelii. Infect Immun. 2002;70:6075–6082. doi: 10.1128/IAI.70.11.6075-6082.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belz GT, Zhang L, Lay MD, Kupresanin F, Davenport MP. Killer T cells regulate antigen presentation for early expansion of memory, but not naive, CD8+ T cell. Proc Natl Acad Sci U S A. 2007;104:6341–6346. doi: 10.1073/pnas.0609990104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kastenmuller W, Gasteiger G, Gronau JH, Baier R, Ljapoci R, Busch DH, Drexler I. Cross-competition of CD8+ T cells shapes the immunodominance hierarchy during boost vaccination. J Exp Med. 2007;204:2187–2198. doi: 10.1084/jem.20070489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.