

Abstract
Background
Gene expression profiling has shown its ability to identify with high accuracy low cytogenetic risk acute myeloid leukemia such as acute promyelocytic leukemia and leukemias with t(8;21) or inv(16). The aim of this gene expression profiling study was to evaluate to what extent suboptimal samples with low leukemic blast load (range, 2-59%) and/or poor quality control criteria could also be correctly identified.
Methods
Specific signatures were first defined so that all 71 acute promyelocytic leukemia, leukemia with t(8;21) or inv(16)-AML as well as cytogenetically normal acute myeloid leukemia samples with at least 60% blasts and good quality control criteria were correctly classified (training set). The classifiers were then evaluated for their ability to assign to the expected class 111 samples considered as suboptimal because of a low leukemic blast load (n = 101) and/or poor quality control criteria (n = 10) (test set).
Results
With 10-marker classifiers, all training set samples as well as 97 of the 101 test samples with a low blast load, and all 10 samples with poor quality control criteria were correctly classified. Regarding test set samples, the overall error rate of the class prediction was below 4 percent, even though the leukemic blast load was as low as 2%. Sensitivity, specificity, negative and positive predictive values of the class assignments ranged from 91% to 100%. Of note, for acute promyelocytic leukemia and leukemias with t(8;21) or inv(16), the confidence level of the class assignment was influenced by the leukemic blast load.
Conclusion
Gene expression profiling and a supervised method requiring 10-marker classifiers enable the identification of favorable cytogenetic risk acute myeloid leukemia even when samples contain low leukemic blast loads or display poor quality control criterion.
Background
Prognostic evaluation is a critical step in newly diagnosed patients with acute myeloid leukemia (AML) in order to identify those at high risk of relapse. For AML patients, cytogenetic abnormalities as well as gene mutations and/or hyper-expressions detected at diagnosis are the main prognostic factors guiding the initial treatment strategy in a risk-oriented manner [1-4]. Hypergranular acute promyelocytic leukemia (APL), as well as AMLs with either translocation t(8;21)(q22;q22) [t(8;21)-AMLs] or inversion inv(16)(p13q22)/t(16;16)(p13;q22) [inv(16)-AMLs], are well-defined entities associated with a favorable outcome [1,3]. They can be distinguished from all other AML subtypes based on specific chromosomal alterations and fusion genes: PML/RAR-alpha fusion gene with reciprocal translocation t(15;17)(q24;q21) for APLs, AML1/ETO (also called RUNX1/RUNX1T1) fusion gene for t(8;21)-AMLs, and CBFB/MYH11 fusion gene for AMLs with either inv(16)(q21;q22) or balanced reciprocal translocation t(16;16)(q21;q22). Of note, in up to 15 percent of APLs, no translocation t(15;17)(q24;q21) is detected by conventional cytogenetics at diagnosis, despite PML/RARA fusion gene is detected using molecular assays [5,6]. Similarly, cryptic t(8;21)(q22;q22) and inv(16)(q21;q22), undetected by conventional cytogenetics, have also been reported [7-9].
With samples containing a high leukemic blast load, microarray-based gene expression profiling (GEP) and class prediction analyses have demonstrated their ability to assign AML samples to one of these three well characterized favorable cytogenetic risk AML subtypes, with high accuracy and low error rates [10-18]. One of the largest class prediction analyses in AML achieved 100 percent classification accuracy with respect to APL, t(8;21)-AML, and inv(16)-AML subtypes, indeed [10]. However, in the majority of those studies, the minimum percentage of leukemic cells within each sample is most often above 60 percent [10-18], an arbitrary threshold that is significantly different from the one used by cytologists for the diagnosis of AML, i.e., excess of blasts greater or equal to 20 percent.
To our knowledge, the impact of the leukemic blast load and/or of the sample quality on microarray-derived class prediction results has not been specifically studied in AMLs. For centers wishing to integrate microarray-based GEP in a routine prognostic workflow for newly diagnosed AML patients, one critical issue is the ability to perform accurate class prediction analysis with suboptimal samples, i.e., containing as low as 20 percent blasts and even below, and/or not fulfilling all quality control criteria along their process.
In this study, GEP was first used to define a limited set of markers allowing to correctly classify 71 patients with either APL, t(8;21)-AML, inv(16)-AML or cytogenetically normal AML (NK-AML) based on samples containing at least 60 percent of leukemic blasts and characterized by good quality control criteria (training set including optimal samples). The classifiers derived from this first supervised analysis were then evaluated for their ability to assign to the correct class 111 suboptimal samples with low leukemic blast load (from 2 to 59 percent) and/or poor quality control criteria (test set including suboptimal samples), as well as duplicates of three AML cell lines.
Methods
Characteristics of the patients and samples
A total of 182 bone marrow or peripheral blood samples from 97 AML patients, diagnosed with either APL (n = 18), t(8;21)-AML (n = 19), inv(16)-AML (n = 29), or NK-AML (n = 31), followed at Angers University Hospital (n = 72) or Reims University Hospital (n = 25), were analyzed, as well as duplicated samples of NB4, Kasumi-1, and ME-1 cell lines (n = 6 samples), which are derived from patients with APL, t(8;21)-AML and inv(16)-AML, respectively. In addition, 18 samples of unique (n = 9) or pooled (n = 9) normal bone marrows obtained from 12 healthy volunteers were included in the study. The main characteristics of AML patients and samples are summarized in Tables 1 and 2. Conventional cytogenetic banding, fluorescence in situ hybridization, and RT-PCR analysis of fusion gene transcripts were used to identify patients with APL, t(8;21)-AML or inv(16)-AML, as previously reported [19]. Analysis of NPM1, FLT3 and CEBPA for mutations was also performed for all patients [20-23]. All participants gave their written informed consent, and the study was approved by the Ethical Committee of Angers University Hospital.
Table 1.
Characteristics of the patients.
| Covariates | Overall | APLs | t(8;21)-AMLs | inv(16)-AMLs | NK-AMLs |
|---|---|---|---|---|---|
| Patients | N = 97 | N = 18 | N = 19 | N = 29 | N = 31 |
| Gender | |||||
| Males | 44 | 7 | 13 | 15 | 9 |
| Females | 53 | 11 | 6 | 14 | 22 |
| Age at diagnosis | |||||
| Median (years) | 54 | 56 | 53 | 38 | 60 |
| Range | 18-87 | 19-87 | 18-84 | 18-70 | 25-78 |
| Leukocytosis at diagnosis | |||||
| WBC ≥ 30 G/L | 39 | 2 | 6 | 12 | 19 |
| FAB classification | |||||
| M1 or M2 | 45 | 0 | 18 | 6 | 21 |
| M3 | 18 | 18 | 0 | 0 | 0 |
| M4 or M5 | 34 | 0 | 0 | 24 | 10 |
| Cytogenetics | |||||
| Expected anomaly* | 62 | 16 | 19 | 27 | 0 |
| Normal karyotype | 34 | 2 | 0 | 1 | 31 |
| Karyotype failure | 1 | 0 | 0 | 1 | 0 |
| Gene mutations | |||||
| FLT3-ITD$ | 19 | 2 | 1 | 2 | 14 |
| FLT3-D835& | 7 | 2 | 2 | 2 | 1 |
| NPM1 | 18 | 0 | 0 | 1 | 17 |
| CEBPA (mono-allelic) | 4 | 0 | 2 | 1 | 1 |
WBC, white blood cells. * t(15;17), t(8;21), inv(16) or t(16;16) for APL, t(8;21)-AML and inv(16)-AML, respectively detected. $ Internal tandem duplication. &Mutation Asp835 of the tyrosine kinase domain.
Table 2.
Characteristics of the samples.
| Covariates | Overall | APLs | t(8;21)- AMLs | inv(16)-AMLs | NK-AMLs |
|---|---|---|---|---|---|
| Samples* | N = 206 | N = 40 | N = 39 | N = 52 | N = 57 |
| Groups | |||||
| Training Set AML samples | 71 | 14 | 14 | 15 | 28 |
| Training Set NBM&samples | 18 | -- | -- | -- | -- |
| Test Set AML samples* | 111 | 24 | 23 | 35 | 29 |
| Test Set AML cell lines | 6 | 2 | 2 | 2 | -- |
| Samples with optimal QCC# | |||||
| Blast % < 60% (undiluted) | 22 | 3 | 5 | 11 | 3 |
| Diluted at 50% | 28 | 5 | 5 | 5 | 13 |
| Diluted at 75% | 27 | 5 | 4 | 5 | 13 |
| Diluted at 90% | 12 | 4 | 4 | 4 | 0 |
| Diluted at 95% | 12 | 4 | 4 | 4 | 0 |
| Overall, blasts < 60% | 101 | 21 | 22 | 29 | 29 |
| Overall, blasts ≥ 40% < 60% | 23 | 2 | 2 | 6 | 13 |
| Overall, blasts ≥ 20% < 40% | 40 | 6 | 9 | 10 | 15 |
| Overall, blasts ≥ 10% < 20% | 15 | 4 | 4 | 6 | 1 |
| Overall, blasts ≥ 5% < 10% | 14 | 5 | 4 | 5 | 0 |
| Overall, blasts < 5% | 9 | 4 | 3 | 2 | 0 |
| Samples with poor QCC# | 10 | 3 | 1 | 6 | 0 |
| cRNA $ < 750 ng | 7 | 1 | 1 | 5 | 0 |
| Low RIN% | 2 | 2 | 0 | 0 | 0 |
| Low RIN + low cRNA | 1 | 0 | 0 | 1 | 0 |
* including the three AML cell lines. # quality control criteria. $ labeled cRNA to be hybridized on Illumina Beadchips.
% RNA integrity number lower than 7.00. &NBM, normal bone marrow.
The gene expression dataset, generated from 206 samples (182 AML samples, duplicated samples of NB4, Kasumi-1, and ME-1 cell lines, 18 samples of unique or combined normal bone marrows), was divided into a Training Set (n = 89 samples) and a Test Set (n = 117 samples). The Training Set, used to identify specific class markers (classifiers), included 18 samples of unique or pooled normal bone marrows (see details about the pools below) as well as 71 AML samples obtained from patients (bone marrow samples, n = 48; peripheral blood samples, n = 23) newly diagnosed with APL (n = 14 patients), t(8;21)-AML (n = 14 patients), inv(16)-AML (n = 15 patients) or NK-AML (n = 28 patients), which contained at least 60 percent blasts. The class prediction analysis was performed on a Test Set including 111 suboptimal AML samples (bone marrow samples, n = 78; peripheral blood samples, n = 33 obtained from 53 patients), i.e., with a low leukemic blast load (n = 101 samples containing from 2 to 56 percent blasts) and/or poor quality control criteria (n = 10 samples) as well as duplicated samples of NB4, Kasumi-1, and ME-1 cell lines (n = 6 samples). Among test samples with optimal quality control criteria, 22 originally contained less than 60% blasts (range, 5-56 percent blasts), whereas 79 were high blast load ones artificially diluted within one of the five pools of normal bone marrows (see details below and Tables 2 and 3). The 10 samples with suboptimal quality control criteria were characterized by either a low RNA integrity number suggestive of RNA degradation (n = 2), a low cRNA amount obtained after total RNA amplification and labeling (cRNA hybridized on BeadChips < 750 micrograms) (n = 7) or both (n = 1) (Tables 2 and 3).
Table 3.
Characteristics and class assignment of the 117 Test Set samples.
| UPN | Cell Source | Time | Blasts % | Dilution | QCC | Real Class | Assigned Class | Confidence |
|---|---|---|---|---|---|---|---|---|
| NB4 | BM | Diagnosis | 100 | No Dilution | OK | APLs Test Set | APLs | 10.802 |
| NB4 | BM | Diagnosis | 100 | No Dilution | OK | APLs Test Set | APLs | 17.507 |
| UPN53 | BM | Diagnosis | 43 | Dilution 50% | OK | APLs Test Set | APLs | 26.028 |
| UPN22 | BM | Diagnosis | 40 | Dilution 50% | OK | APLs Test Set | APLs | 29.91 |
| UPN65 | BM | Diagnosis | 39 | Dilution 50% | OK | APLs Test Set | APLs | 26.403 |
| UPN40 | BM | Diagnosis | 33 | Dilution 50% | OK | APLs Test Set | APLs | 29.91 |
| UPN50 | PB | Diagnosis | 27 | Dilution 50% | OK | APLs Test Set | APLs | 19.2 |
| UPN53 | BM | Diagnosis | 22 | Dilution 75% | OK | APLs Test Set | APLs | 20.904 |
| UPN22 | BM | Diagnosis | 20 | Dilution 75% | OK | APLs Test Set | APLs | 29.91 |
| UPN65 | BM | Diagnosis | 20 | Dilution 75% | OK | APLs Test Set | APLs | 15.763 |
| UPN40 | BM | Diagnosis | 16 | Dilution 75% | OK | APLs Test Set | APLs | 26.425 |
| UPN5 | PB | Diagnosis | 15 | No Dilution | OK | APLs Test Set | APLs | 17.193 |
| UPN50 | PB | Diagnosis | 14 | Dilution 75% | OK | APLs Test Set | APLs | 10.46 |
| UPN48 | PB | Diagnosis | 11 | No Dilution | OK | APLs Test Set | APLs | 23.005 |
| UPN53 | BM | Diagnosis | 9 | Dilution 90% | OK | APLs Test Set | APLs | 16.749 |
| UPN22 | BM | Diagnosis | 8 | Dilution 90% | OK | APLs Test Set | APLs | 15.781 |
| UPN40 | BM | Diagnosis | 7 | Dilution 90% | OK | APLs Test Set | APLs | 10.143 |
| UPN86 | PB | Diagnosis | 7 | No Dilution | OK | APLs Test Set | APLs | 11.641 |
| UPN50 | PB | Diagnosis | 6 | Dilution 90% | OK | APLs Test Set | APLs | 2.661 |
| UPN22 | BM | Diagnosis | 4 | Dilution 95% | OK | APLs Test Set | APLs | 7.665 |
| UPN53 | BM | Diagnosis | 4 | Dilution 95% | OK | APLs Test Set | APLs | 1.201 |
| UPN40 | BM | Diagnosis | 3 | Dilution 95% | OK | APLs Test Set | APLs | 10.667 |
| UPN50 | PB | Diagnosis | 3 | Dilution 95% | OK | APLs Test Set | APLs | 4.337 |
| UPN59 | PB | Diagnosis | 70 | No Dilution | Low RIN | APLs Test Set | APLs | 26.497 |
| UPN82 | BM | Diagnosis | 64 | No Dilution | Low RIN | APLs Test Set | APLs | 22.822 |
| UPN13 | BM | Diagnosis | 30 | No Dilution | Low cRNA | APLs Test Set | APLs | 18.651 |
| Kasumi-1 | BM | Diagnosis | 100 | No Dilution | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 5.184 |
| Kasumi-1 | BM | Diagnosis | 100 | No Dilution | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 1.525 |
| UPN30 | PB | Diagnosis | 49 | Dilution 50% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 19.199 |
| UPN69 | PB | Diagnosis | 41 | Dilution 50% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 30.674 |
| UPN51 | BM | Relapse | 38 | Dilution 50% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 20.609 |
| UPN76 | PB | Diagnosis | 31 | No Dilution | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 32.067 |
| UPN69 | BM | Diagnosis | 30 | Dilution 50% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 25.183 |
| UPN26 | BM | Diagnosis | 27 | No Dilution | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 14.89 |
| UPN30 | PB | Diagnosis | 25 | Dilution 75% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 17.719 |
| UPN61 | BM | Diagnosis | 24 | Dilution 50% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 30.674 |
| UPN69 | PB | Diagnosis | 21 | Dilution 75% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 27.738 |
| UPN21 | BM | Diagnosis | 21 | No Dilution | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 24.116 |
| UPN95 | BM | Diagnosis | 20 | No Dilution | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 30.674 |
| UPN51 | BM | Relapse | 19 | Dilution 75% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 10.519 |
| UPN94 | PB | Diagnosis | 16 | No Dilution | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 28.631 |
| UPN69 | BM | Diagnosis | 15 | Dilution 75% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 22.744 |
| UPN30 | PB | Diagnosis | 10 | Dilution 90% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 16.958 |
| UPN69 | PB | Diagnosis | 6 | Dilution 90% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 13.468 |
| UPN30 | PB | Diagnosis | 5 | Dilution 95% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 13.581 |
| UPN61 | BM | Diagnosis | 5 | Dilution 90% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 28.443 |
| UPN51 | BM | Diagnosis | 4 | Dilution 90% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 3.096 |
| UPN69 | PB | Diagnosis | 3 | Dilution 95% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 12.875 |
| UPN51 | BM | Diagnosis | 2 | Dilution 95% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 4.6 |
| UPN61 | BM | Diagnosis | 2 | Dilution 95% | OK | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 19.237 |
| UPN30 | PB | Diagnosis | 25 | No Dilution | Low cRNA | t(8;21)-AMLs Test Set | t(8;21)-AMLs | 15.256 |
| ME-1 | BM | Diagnosis | 100 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 15.836 |
| ME-1 | BM | Diagnosis | 100 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 18.901 |
| UPN52 | BM | Diagnosis | 56 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 26.467 |
| UPN45 | BM | Diagnosis | 50 | Dilution 50% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 30.188 |
| UPN31 | BM | Diagnosis | 50 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 29.511 |
| UPN88 | BM | Diagnosis | 46 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 31.576 |
| UPN47 | BM | Diagnosis | 41 | Dilution 50% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 30.517 |
| UPN37 | BM | Diagnosis | 40 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 28.551 |
| UPN9 | BM | Diagnosis | 35 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 31.083 |
| UPN87 | PB | Diagnosis | 35 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 29.511 |
| UPN10 | BM | Diagnosis | 33 | Dilution 50% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 17.561 |
| UPN43 | PB | Diagnosis | 31 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 24.289 |
| UPN52 | BM | Diagnosis | 28 | Dilution 50% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 24.608 |
| UPN45 | BM | Diagnosis | 25 | Dilution 75% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 23.687 |
| UPN67 | PB | Diagnosis | 24 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 31.576 |
| UPN36 | PB | Diagnosis | 23 | No Dilution | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 27.432 |
| UPN47 | BM | Diagnosis | 20 | Dilution 75% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 16.733 |
| UPN37 | BM | Diagnosis | 20 | Dilution 50% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 26.644 |
| UPN10 | BM | Diagnosis | 16 | Dilution 75% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 10.252 |
| UPN52 | BM | Diagnosis | 14 | Dilution 75% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 6.543 |
| UPN37 | PB | Relapse | 14 | No Dilution | OK | inv(16)-AMLs Test set | t(8;21)-AMLs | 4.33 |
| UPN45 | BM | Diagnosis | 10 | Dilution 90% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 14.568 |
| UPN47 | BM | Diagnosis | 10 | Dilution 90% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 15.298 |
| UPN37 | BM | Diagnosis | 10 | Dilution 75% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 20.423 |
| UPN10 | BM | Diagnosis | 7 | Dilution 90% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 8.479 |
| UPN52 | BM | Diagnosis | 6 | Dilution 90% | OK | inv(16)-AMLs Test set | APLs | 0.6 |
| UPN45 | BM | Diagnosis | 5 | Dilution 95% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 15.056 |
| UPN47 | BM | Diagnosis | 5 | Dilution 95% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 2.293 |
| UPN7 | BM | Relapse | 5 | No Dilution | OK | inv(16)-AMLs Test set | t(8;21)-AMLs | 0.473 |
| UPN10 | BM | Diagnosis | 4 | Dilution 95% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 3.715 |
| UPN52 | BM | Diagnosis | 3 | Dilution 95% | OK | inv(16)-AMLs Test set | inv(16)-AMLs | 3.79 |
| UPN6 | BM | Diagnosis | 74 | No Dilution | Low cRNA | inv(16)-AMLs Test set | inv(16)-AMLs | 27.753 |
| UPN45 | BM | Diagnosis | 25 | No Dilution | Low cRNA | inv(16)-AMLs Test set | inv(16)-AMLs | 31.005 |
| UPN47 | BM | Diagnosis | 20 | No Dilution | Low cRNA | inv(16)-AMLs Test set | inv(16)-AMLs | 26.009 |
| UPN79 | BM | Diagnosis | 47 | No Dilution | Low cRNA | inv(16)-AMLs Test set | inv(16)-AMLs | 29.75 |
| UPN90 | PB | Diagnosis | 9 | No Dilution | Low cRNA | inv(16)-AMLs Test set | inv(16)-AMLs | 2.693 |
| UPN23 | BM | Diagnosis | 27 | No Dilution | Low RIN + Low cRNA | inv(16)-AMLs Test set | inv(16)-AMLs | 24.111 |
| UPN60 | PB | Diagnosis | 50 | No Dilution | OK | NK-AMLs Test Set | inv(16)-AMLs | 0.948 |
| UPN72 | BM | Diagnosis | 49 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 26.707 |
| UPN8 | BM | Diagnosis | 48 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 27.354 |
| UPN44 | PB | Diagnosis | 47 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 28.541 |
| UPN73 | PB | Diagnosis | 47 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 19.624 |
| UPN97 | BM | Diagnosis | 47 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 22.934 |
| UPN1 | PB | Diagnosis | 46 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 26.707 |
| UPN66 | BM | Diagnosis | 46 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 23.454 |
| UPN78 | PB | Diagnosis | 46 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 28.541 |
| UPN25 | BM | Diagnosis | 43 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 21.699 |
| UPN38 | BM | Diagnosis | 40 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 26.707 |
| UPN74 | BM | Diagnosis | 40 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 19.777 |
| UPN89 | BM | Diagnosis | 40 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 29.115 |
| UPN19 | BM | Diagnosis | 39 | No Dilution | OK | NK-AMLs Test Set | NK-AMLs | 25.313 |
| UPN11 | BM | Diagnosis | 37 | Dilution 50% | OK | NK-AMLs Test Set | NK-AMLs | 29.776 |
| UPN96 | PB | Diagnosis | 35 | No Dilution | OK | NK-AMLs Test Set | NK-AMLs | 27.303 |
| UPN72 | BM | Diagnosis | 25 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 26.707 |
| UPN8 | BM | Diagnosis | 24 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 27.357 |
| UPN44 | PB | Diagnosis | 24 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 29.235 |
| UPN97 | BM | Diagnosis | 24 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 24.035 |
| UPN1 | PB | Diagnosis | 23 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 26.707 |
| UPN66 | BM | Diagnosis | 23 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 19 |
| UPN73 | PB | Diagnosis | 23 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 24.104 |
| UPN78 | PB | Diagnosis | 23 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 28.541 |
| UPN25 | BM | Diagnosis | 21 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 18.99 |
| UPN38 | BM | Diagnosis | 20 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 25.107 |
| UPN74 | BM | Diagnosis | 20 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 19.705 |
| UPN89 | BM | Diagnosis | 20 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 26.555 |
| UPN11 | BM | Diagnosis | 19 | Dilution 75% | OK | NK-AMLs Test Set | NK-AMLs | 24.587 |
UPN, unique patient number. BM, bone marrow. PB, peripheral blood. QCC, quality control criteria.
Low cRNA, amount of labeled cRNA hybridized on the BeadChip below 750 ng. Low RIN, RNA integrity number below 7.00.
Sample preparation for gene expression profiling
Blasts and mononuclear cells were purified by Ficoll-Hypaque density gradient centrifugation from bone marrow or peripheral blood samples (Nygaard, Oslo, Norway). Isolated cell samples were then immediately cryopreserved. Total RNAs were extracted from 107 thawed cells using RNEasy® Mini Kits (Qiagen Incorporation, Valencia, USA). Total RNA quantification was performed using the Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific Incorporation, Waltham, USA) according to manufacturer recommendations. Integrity of the extracted RNAs was assessed with the Bioanalyzer 2100 and the RNA6000 Nano kit (Agilent Technologies Incorporation, Santa Clara, USA). A RNA integrity number (RIN) greater or equal to 7.00 was achieved for 203 samples (RNA degradation observed for 3 AML samples). No sign of DNA contamination was detected in any of the 206 samples analyzed. The starting amount of total RNA used for the reactions was 200 nanograms per sample, for all samples. The Illumina Total Prep RNA Amplification Kit (Applied Biosystems/Ambion, Austin, USA) was used to generate biotinylated, amplified cRNA according to the manufacturer recommendations (see additional file 1 for details). To study the leukemic blast load effect on the class prediction accuracy, besides including 22 samples with good quality control criteria that originally contained less than 60 percent of leukemic blasts, 79 artificially generated low leukemic blast load samples, via a "dilution/mixture" approach, were also assessed (see additional file 1 for details for the generation of low leukemic blast load AML samples by a "dilution/mixture" approach). Hybridization on Illumina HumanHT-12 v3 Expression BeadChips, staining and detection of cRNAs on microarrays using an I-Scan system were performed according to Illumina's protocol (see additional file 1 for details).
Data analyses
GenomeStudio 2010.3 software (Illumina Inc., San Diego, USA) and its Gene Expression Analysis Module (version 1.8.0) were used for signal extraction and normalization (for the Training Set only). Briefly, the Invariant Rank normalization method was applied to the primary probe data obtained from the Training Set samples. Processed probe data were then filtered according to the following criteria: minimal signal intensity fold change of 1.50 across all samples; minimal probe signal intensity absolute change of 150 across all samples (choice based on the maximum expression levels of XIST transcript in males, and CYorf15B transcript in females); and maximal value for probe signal intensity of 50 000 across all samples. Among the 48 803 probes assessed on HumanHT-12 v3 Expression BeadChips, 11 779 satisfied these low stringency filtering criteria. Filtered data were then log-transformed and exported to appropriate softwares for the analyses. Regarding the Test Set samples processing, only data concerning the markers identified during the first analysis, restricted to the Training Set, were used. These raw data were log-transformed and merged to the one of the Training Set for class prediction analyses. The Class Prediction Module of ArrayMiner 5.3.3 (Optimal Design, Brussels, Belgium - http://www.optimaldesign.org), which uses a proprietary method based on grouping genetic algorithms, was used for all class prediction analyses (see additional file 1 for details) [24]. This module allowed calculating the confidence level of the class prediction for each sample, or for a given group of samples, and the fitness of the overall model. This estimator enabled optimizing the number of probes/markers to be used per class. Briefly, the confidence level of the class prediction analysis reflected the strength with which the probe/marker landed each sample into its predicted class. More precisely, for each sample, the following happened: (1) each probe/marker casted its vote for the sample being in each of the train classes, (2) the votes were summed up over all probes/markers, yielding a vote for each of the classes, (3) the score of the winner class (call it S1) was compared to the second best (call it S2), and the confidence level of the classification of the sample was computed as (S1-S2)/(S1+S2). In the Cross-validation analysis, the same was performed, except that the probe/marker set was recomputed each time, leaving out the sample being tested in order to yield an honest estimate of the worth of the method. The fitness of a class prediction model was computed on the test samples only, in order to estimate how good the class prediction (of the "unclassified" samples) was, and reflected the success with which the model was able to correctly (re)classify the already "classified" samples. The fitness of the model was in fact the result of a cross-validation (as described above) on the Training samples, with the integral part being equal to the number of correct (re)classifications, and the fractional part being computed as follows: (1) if all samples were correctly classified, the average confidence level (as defined above) of all samples, (2) if some samples were misclassified, the average confidence level of the correctly classified samples minus the average confidence level of the misclassified ones. If all samples were misclassified, the fitness of the model was zero. Omics Explorer 2.2 software (Qlucore, Lund, Sweden - http://www.qlucore.com) was used for principal-component analyses. S-Plus® 8.0 Enterprise Developer software (Insightful Corporation, Seattle, USA) was used for all other statistical analyses. The two datasets (Training Set and Test Set) discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE34823 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE34823. The dataset associated with the Training Set is also linked to the SubSeries accession number GSE34577. The dataset associated with the Test Set is also linked to the SubSeries accession number GSE34714.
Results
Identification of classifiers from optimal APL, t(8;21)-AML, inv(16)-AML and NK-AML samples
First, using the Class Prediction Module of ArrayMiner 5.3.3 software, classifiers associated with the APL, t(8;21)-AML, inv(16)-AML, NK-AML classes were defined based on 71 samples containing at least 60% of leukemic blasts and characterized by good quality control criteria (Optimal samples - Training Set). At this stage, unique and pooled normal bone marrow samples were also included in the model as the objective was to enhance the predictive capacity of the classifiers, especially for Test Set samples containing a majority of residual normal cells and a low leukemic blast load. Evaluating classifiers which included from 1 to 100 markers per class, all Training Set samples were assigned to the correct class when selecting from 3 to 46 markers per class (class prediction accuracy, sensitivity, specificity, negative and positive predictive values, 100% for each class - error rate, 0%), whether the source of leukemic cells was bone marrow or peripheral blood. The best model fitness was obtained with 8, 9 or 10 markers per classifier. To select the optimal model among these three, a principal-component analysis was performed. The highest cumulative percent of variance accounted for by the first three components was obtained with the 10-marker classifiers (cumulative percent of variance with classifiers including 10 markers, 79 percent - additional file 1, Figure S1), which led to select these for subsequent analyses (Figure 1). The best median confidence levels for the correct assignment of the Training Set samples were achieved with 10, 2, 1, and 9 markers for APLs, t(8;21)-AMLs, inv(16)-AMLs, and NK-AMLs, respectively (additional file 1, Figure S2). Regarding NK-AMLs and in agreement with previous reports, the highest median confidence levels were observed for NK-AMLs with mutated NPM1 and the lowest ones for NK-AMLs with neither NPM1 nor FLT3 mutations (additional file 1, Figure S3) [22-26].
Figure 1.

Heatmap of the 40 markers used to define the four AML classifiers allowing the assignment of all Training Set AML samples to the correct class. From the top to the bottom: APL, t(8;21)-AML, inv(16)-AML, and NK-AML classes - 10 markers per class (normal bone marrow class is not represented). Each column represents a sample; each row represents a marker (gene transcript). The log2 relative gene expression scale is depicted on the bottom left.
Class prediction analysis for suboptimal AML samples - Impact of the leukemic blast load
Using the 10-marker classifiers, the GGA-based supervised method was subsequently applied to a series of 101 Test Set samples, with optimal quality control criteria, for which the leukemic blast load was originally below 60 percent or had been artificially lowered to less than 60 percent blasts by dilution series. Only the classifiers associated with one of the four AML classes were considered for this analysis. All APL and t(8;21)-AML Test Set samples were correctly classified, even when containing as low as 2 percent blasts (Table 3 - Figure 2). Twenty six of the 29 inv(16)-AML samples were assigned to the expected class, including the 95 percent-diluted ones which contained as low as 3 percent blasts (error rate, 10 percent). In all three inv(16)-AML Test Set samples that were incorrectly classified, CBFB-MYH11 fusion gene was detected by FISH assay. For APL, t(8;21)-AML and inv(16)-AML classes, the blast load significantly influenced the confidence level of the class assignment (Figures 3 - additional file 1, Figure S4). For NK-AMLs, all but 1 of the 29 Test Set samples were correctly classified (error rate, 3 percent) (Table 3). This misclassified sample, obtained at diagnosis from UPN60, had no NPM1, FLT3 or CEBPA mutations. Its class assignment, characterized by a low confidence level (given its blast load - Table 1, additional file 1, Figure S4), was not confirmed as no PML/RARA fusion gene was detected by FISH analysis and RT-PCR assay. Across the range of leukemic blast loads studied in the Test Set, the confidence level associated with the class assignment of diluted AML samples was not different from the one achieved for undiluted low leukemic blast load samples (Table 3 - additional file 1, Figure S5). For those 101 AML Test set samples with low leukemic blast load and optimal quality control criteria, the overall error rate was 3.9 percent.
Figure 2.

Results of the class assignment for the 107 AML Test Set samples fulfilling all quality control criteria based on the 10-marker classifiers characterizing the APL, t(8;21)-AML, inv(16)-AML and NK-AML classes (all three AML cell lines run in duplicates included). Each column represents a sample; each row represents a marker (gene transcript). The log2 relative gene expression scale is depicted on the bottom left.
Figure 3.
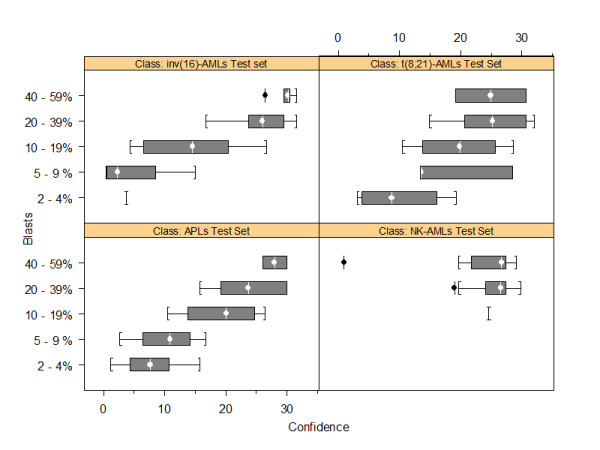
Box plots of the confidence levels for the class assignment of the APL, t(8;21)-AML, inv(16)-AML and NK-AML Test Set samples according to their leukemic blast load. The white vertical line and circle in the interior of the dark gray box is located at the median of the data. The width of the box is equal to the interquartile distance, which is the difference between the third and first quartiles of the data. The interquartile distance indicates the spread of the distribution for the data. The whiskers (the lines extending from the left and right parts of the box) go to the nearest value not beyond the span from the quartiles, i.e., 1.5 times the interquartile distance from the center of the data. Points beyond the whiskers are considered outliers and are drawn individually, indicated in black (+).
Class prediction analysis for suboptimal AML samples - Impact of quality control criteria
When the performance of the 10-marker classifiers was assessed on 10 samples that did not fulfill all quality control criteria, all suboptimal samples were assigned to the expected class, even though their leukemic blast content was as low as 9 percent (median, 55 percent; range 9 to 100 percent) (Table 3 - additional file 1, Figure S6). The confidence level for the class assignment of these samples was not different from the one achieved for the other Test Set samples (Data not shown).
Overall, the error rate was 3.6 percent for the entire Test Set (excluding AML cell lines that were considered as controls), and for each class, sensitivity and specificity, negative and positive predictive values of the class assignments ranged from 91 to 100 percent (additional file 1, Figure S7).
Discussion
The integration of microarray-derived data to the current workflow dealing with the prognostic evaluation of AML patients requires the technology to deliver informative data for the majority of samples, including those with suboptimal characteristics. The current study focused on this kind of samples - either with a low leukemic blast load, poor quality control criteria or both - and the ability to identify favorable-risk karyotype AMLs in such situations, using microarray-based GEP and a class prediction method based on GGA. Apart from the study of de Ridder et al, which addressed the impact of random, fixed and group-specific impurities on the results of differential gene expression analyses, using a limited set of samples and computer simulations, the influence of the leukemic blast load on class prediction accuracy based on GEP data has never been specifically studied so far [25]. To evaluate the capacity of microarray-derived predictors to correctly assigned samples with a low leukemic blast load, besides using "real" AML samples with low blast percentage, i.e., less than 60 percent, a "dilution/mixture" approach was considered [26,27]. This strategy has already been successfully applied to assess the efficiency of various normalization methods applied to microarray datasets. It consisted in diluting labeled cRNAs of originally high blast load AML samples within pools of normal bone marrow labeled cRNAs. This approach was also considered as in some AML subtypes, such as t(8;21)-AMLs, more differentiated cells (not counted as blasts) are frequently of leukemic origin, leading to under-estimate the leukemic load of some "low blast content" samples. Consequently, it became possible to accurately control the percentage of leukemic blasts within the Test Set sample down to 2 percent.
Overall, considering the 111 Test Set samples with low leukemic blast load and/or suboptimal quality control criteria, whether being peripheral blood or bone marrow samples, the predictive capacity of the GGA-derived model with 10-marker classifiers was encouraging, with specificities ranging from 0.98 to 1.00, and sensitivities ranging from 0.91 to 1.00. Moreover, considering AML samples containing 20 to 56 percent blasts, the overall error rate was lower than 1.4 percent (1 misclassified NK-AML sample out of 72). Finally, APL, t(8;21)-AML, and inv(16)-AML samples containing as low as 2 percent blasts could be correctly classified, whether the classifiers mainly relied on a single marker, such as for inv(16)-AMLs and t(8;21)-AMLs, or on the overall set of 10 markers defining the classifier, as for APL samples. However, regarding APL, t(8;21)-AML and inv(16)-AML samples, the confidence level of the class assignment was correlated to the percentage of leukemic blasts within the studied samples. For NK-AML samples, such a finding was not observed, probably because NK-AMLs represent a more heterogeneous group of leukemias as compared to t(8;21)-AMLs, inv(16)-AMLs or APLs in term of oncogenic processes. This heterogeneity likely led to a higher variance of gene expression profiles within the NK-AML class, to the detriment of the interclass variance, hence influencing the class prediction confidence level more than the leukemic blast load. This hypothesis was supported by the fact that, within the NK-AML class, the best predictive capacity of the GGA was achieved for NPM1-mutated samples, in accordance with previous studies, and the worst one for samples with no NPM1 and FLT3 mutations [28-31].
Unexpectedly, the worst results of the class prediction analysis were achieved for inv(16)-AML Test set samples (Sensitivity, 0.91; negative predictive value, 0.96). Among the three misclassified inv(16)-AML samples and for which the presence of CBFB/MYH11 fusion gene was confirmed by FISH analysis, all contained less than 20 percent blasts and two were obtained at the time of relapse - from patients UPN7 and UPN37 -. Regarding these two samples, which respectively contained 5 and 14 percent blasts, they were characterized by a low expression level of most markers defining the inv(16)-AML specific signature; especially MYH11, which expression level was within the estimated background signal range. It is noteworthy that for one of those two patients, UPN37, the bone marrow sample obtained at diagnosis was correctly classified, even when diluted at 50 and 75 percent (blast content, 20 and 10 percent, respectively). For this patient, a clonal evolution of the disease was suspected as CD2, CD13, CD34 and CD117 expression values, assessed by flow cytometry on gated leukemic cells, and MYH11 expression level (microarray data) were significantly lower in the relapse sample as compare to the ones observed in the diagnostic sample. Verhaak et al in their study using Affymetrix GeneChips reported that MYH11 expression level was sufficient to identify inv(16)-AMLs [10]. The present results suggest that the use of MYH11 as the sole marker for the inv(16)-AML class could lead to unstable results in a class prediction model, especially in low leukemic blast load samples or in case of a clonal evolution at relapse. Of note, the inv(16)-AML signature developed on the AMLProfiler™ kit http://www.skyline-diagnostics.com, which uses the Affymetrix™ platform, also includes a set of 19 markers [32].
As reported by Kohlmann et al., sub-optimal AML samples with poor quality control criteria, mainly because of low amounts of hybridized cRNAs, were all correctly classified, even though half of these contained less than 60 percent blasts (down to 9 percent for UPN90) [33]. Studying the impact of RNA degradation on GEP analyses, similar results have been recently reported by Opitz et al on a different microarray platform [34]. As in the present study, they showed that useful information could still be obtained from thermically degraded RNA; at least to a certain extent, and depending on the length of the mRNA molecules. Interestingly, even AML samples for which a third of the required cRNA amount was hybridized on BeadChips could be assigned to the correct class in the present study, suggesting that the biological variance between the AML classes was higher than the one related to technical variability or artifacts. This result could also be related to the fact that, during the sample processing, the TotalPrep RNA Amplification kit that was used, did not required any fragmentation step, which could further alter poor quality RNA or lower the already reduced cRNA amounts. These encouraging results, on a limited set of poor quality samples, could also be related to the classification method used, as the GGA-based class prediction strategy aimed at finding the most influential markers for each AML class (top markers with the highest inter-class variance and the lowest intra-class variance). This GGA-based class prediction method was indeed associated with the lowest error rate and the highest predictive accuracy when compared to other supervised methods, such as support vector machines, random forests, artificial neural networks, k-Nearest Neighbor or nearest shrunken centroids (also known as PAM) (data not shown).
Although, in most cases, these favorable prognostic chromosomal abnormalities and/or their related gene products can be detected by karyotype, FISH or PCR assay, they represent main targets for a microarray-based class prediction analysis to be identified before considering GEP as a useful tool in a routine workflow for prognostic assessment of AML patients. The fact that the AMLProfiler, a microarray-derived kit based on the Affymetrix™ platform, has been recently commercialized with the aim of achieving a molecular diagnosis for APLs, t(8;21)-AMLs and inv(16)-AMLs, respectively using 27, 31 and 19 markers, emphasizes this assumption [32].
Finally, regarding the classifiers associated with APLs, t(8;21)-AMLs or inv(16)-AMLs, 18 of the 28 markers identified in this study (64 percent) had already been reported in previous studies using various microarray technologies (additional file 1, Table S1). These findings (1) confirm that the Illumina bead-based technology is as reliable, robust and sensitive as other microarray technologies developed by commercial manufacturers or academic facilities, and (2) suggest that the GGA-derived class prediction approach is a highly efficient one as it required a limited set of ten markers per class to achieve accurate class assignments. Furthermore, this study has identified new AML markers that will need further studies to delineate their role in the leukemogenic events involved in APLs (CERCAM, COL23A1, LOC643201, LOXL4, SLC39A11), t(8;21)-AMLs (LOC440030, TNFRSF21), and inv(16)-AMLs (EFHC2, GPR12, MEGF10).
Conclusions
In more than 96 percent of the suboptimal cases, using a microarray-derived GEP approach and a GGA-based class prediction method, favorable cytogenetic risk AML samples with low leukemic blast load and/or poor quality control criteria could be correctly assign to the appropriate class with a limited set of markers, allowing to consider GEP as a useful tool in a routine workflow for prognostic assessment of AML patients.
List of abbreviations used
APL: acute promyelocytic leukemia; AML: acute myeloid leukemia; GEP: gene expression profiling; GGA: grouping genetic algorithms; NK-AML: cytogenetically normal acute myeloid leukemia; RIN: RNA integrity number.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PhG was the principal investigator and takes primary responsibility for the paper. DRB, OB and PhG contribute to the conception of design. DRB, PCL, AC, LB, FG, IL, MHB, AB, AST, MZ, YD, NI, OB, PhG contribute to the collection and assembly of data. DRB, OB, YD, MZ, NI, PhG contribute to the data analysis and interpretation. DRB, PhG contribute to manuscript writing. DRB, OB, PCL, AC, LB, FG, IL, MHB, AB, AST, MZ, YD, NI, PhG contribute to the drafting of the article, revise it and approved the final version. PhG coordinated the research.
Authors' information
PhG is the Head of Plateforme SNP, Transcriptome & Epigénomique, i.e., the genomic platform of Angers University Hospital, and is also an Associate Editor at BMC Genomics and a member of the American Society of Human Genetics.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Sample preparation for gene expression profiling. - Generation of biotinylated, amplified cRNA using The Illumina Total Prep RNA Amplification Kit (Applied Biosystems/Ambion, Austin, USA). - Generation of low leukemic blast load AML samples by a "dilution/mixture" approach. - Hybridization on Illumina HumanHT-12 v3 Expression BeadChips, staining and detection of cRNAs on microarrays using an I-Scan system. Additional table S1 - Citations in previous studies of the identified markers. Additional figures S1 to S7. Figure S1. Three-dimensional projection of the 3 principal components in a principal-components analysis of APL, t(8;21)-AML, inv(16)-AML, NK-AML, and Normal Bone Marrow samples belonging to the Training Set, with the use of the 10-marker classifiers. Figure S2. Per class median confidence level of the assignment for APL, t(8;21)-AML, inv(16)-AML, NK-AML and normal bone marrow samples belonging to the Training Set according to the number of markers per class (from 1 to 100 markers per class). Figure S3. Median confidence level of the class assignments for the NK-AML samples belonging to the Training Set according to FLT3 and NPM1 mutational status and the number of markers per class (from 1 to 100 markers per class). Figure S4. Relationship between the percentage of leukemic blasts within the 101 Test Set samples (X axis) and the confidence level of their class assignments (Y axis) (samples with poor quality control criteria and AML cell lines were excluded). Figure S5. Confidence level of the class assignments according to the percentage of leukemic blasts including comparisons between diluted and not diluted samples. Figure S6. Results of the class assignment for the 10 AML Test Set samples with suboptimal quality control criteria based on the 10-marker classifiers characterizing the APL, t(8;21)-AML, inv(16)-AML and NK-AML classes. Figure S7. Sensitivity, specificity, negative and positive predictive values of the prediction model for the class assignment of the 111 Test Set samples (all AML samples with or without optimal quality control criteria - AML cell line samples excluded) to the APL, t(8;21)-AML, inv(16)-AML and NK-AML classes with 10-marker classifiers.
Contributor Information
Diane Raingeard de la Blétière, Email: DIANE.LAMBERT@free.fr.
Odile Blanchet, Email: odblanchet@chu-angers.fr.
Pascale Cornillet-Lefèbvre, Email: plefebvre@chu-reims.fr.
Anne Coutolleau, Email: ANNE.COUTOLLEAU@orange.fr.
Laurence Baranger, Email: labaranger@chu-angers.fr.
Franck Geneviève, Email: frgenevieve@chu-angers.fr.
Isabelle Luquet, Email: iluquet@chu-reims.fr.
Mathilde Hunault-Berger, Email: mahunault@chu-angers.fr.
Annaelle Beucher, Email: ANNAELLEBEUCHER@hotmail.fr.
Aline Schmidt-Tanguy, Email: alschmidt@chu-angers.fr.
Marc Zandecki, Email: mazandecki@chu-angers.fr.
Yves Delneste, Email: yves.delneste@univ-angers.fr.
Norbert Ifrah, Email: noifrah@chu-angers.fr.
Philippe Guardiola, Email: phguardiol@aol.com.
Acknowledgements and funding
PhG from Plateforme SNP, Transcriptome & Epigénomique received a grant from La Ligue Contre le Cancer/Comité Départemental 49 to support this work.
References
- Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD. European LeukemiaNet. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- Baldus CD, Mrózek K, Marcucci G, Bloomfield CD. Clinical outcome of de novo acute myeloid leukaemia patients with normal cytogenetics is affected by molecular genetic alterations: a concise review. Br J Haematol. 2007;137:387–400. doi: 10.1111/j.1365-2141.2007.06566.x. [DOI] [PubMed] [Google Scholar]
- Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, Goldstone A. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92:2322–33. [PubMed] [Google Scholar]
- Breems DA, Van Putten WL, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KB, Mellink CH, Nieuwint A, Jotterand M, Hagemeijer A, Beverloo HB, Löwenberg B. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26:4791–7. doi: 10.1200/JCO.2008.16.0259. [DOI] [PubMed] [Google Scholar]
- Iqbal S, Grimwade D, Chase A, Goldstone A, Burnett A, Goldman JM, Swirsky D. Identification of PML/RARalpha rearrangements in suspected acute promyelocytic leukemia using fluorescence in situ hybridization of bone marrow smears: a comparison with cytogenetics and RT-PCR in MRC ATRA trial patients. MRC Adult Leukaemia Working Party. Leukemia. 2000;14:950–3. doi: 10.1038/sj.leu.2401688. [DOI] [PubMed] [Google Scholar]
- Grimwade D, Biondi A, Mozziconacci MJ, Hagemeijer A, Berger R, Neat M, Howe K, Dastugue N, Jansen J, Radford-Weiss I, Lo Coco F, Lessard M, Hernandez JM, Delabesse E, Head D, Liso V, Sainty D, Flandrin G, Solomon E, Birg F, Lafage-Pochitaloff M. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood. 2000;96:1297–308. [PubMed] [Google Scholar]
- Mitterbauer M, Kusec R, Schwarzinger I, Haas OA, Lechner K, Jaeger U. Comparison of karyotype analysis and RT-PCR for AML1/ETO in 204 unselected patients with AML. Ann Hematol. 1998;76:139–43. doi: 10.1007/s002770050378. [DOI] [PubMed] [Google Scholar]
- Rowe D, Cotterill SJ, Ross FM, Bunyan DJ, Vickers SJ, Bryon J, McMullan DJ, Griffiths MJ, Reilly JT, Vandenberghe EA, Wilson G, Watmore AE, Bown NP. Cytogenetically cryptic AML1-ETO and CBF beta-MYH11 gene rearrangements: incidence in 412 cases of acute myeloid leukaemia. Br J Haematol. 2000;111:1051–6. doi: 10.1046/j.1365-2141.2000.02474.x. [DOI] [PubMed] [Google Scholar]
- Mrózek K, Prior TW, Edwards C, Marcucci G, Carroll AJ, Snyder PJ, Koduru PR, Theil KS, Pettenati MJ, Archer KJ, Caligiuri MA, Vardiman JW, Kolitz JE, Larson RA, Bloomfield CD. Comparison of cytogenetic and molecular genetic detection of t(8;21) and inv(16) in a prospective series of adults with de novo acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol. 2001;19:2482–92. doi: 10.1200/JCO.2001.19.9.2482. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, Löwenberg B, Delwel R, Valk PJ. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94:131–4. doi: 10.3324/haematol.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Löwenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–28. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Bullinger L, Döhner K, Bair E, Fröhling S, Schlenk RF, Tibshirani R, Döhner H, Pollack JR. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–16. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné MC, De Vos J, Hernández JM, Hofmann WK, Mills KI, Gilkes A, Chiaretti S, Shurtleff SA, Kipps TJ, Rassenti LZ, Yeoh AE, Papenhausen PR, Liu WM, Williams PM, Foà R. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Kohlmann A, Schnittger S, Dugas M, Hiddemann W, Kern W, Hiddemann W, Kern W, Schoch C. Global approach to the diagnosis of leukemia using gene expression profiling. Blood. 2005;106:1189–98. doi: 10.1182/blood-2004-12-4938. [DOI] [PubMed] [Google Scholar]
- Schoch C, Kohlmann A, Schnittger S, Brors B, Dugas M, Mergenthaler S, Kern W, Hiddemann W, Eils R, Haferlach T. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc Natl Acad Sci USA. 2002;99:10008–13. doi: 10.1073/pnas.142103599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann A, Schoch C, Schnittger S, Dugas M, Hiddemann W, Kern W, Haferlach T. Molecular characterization of acute leukemias by use of microarray technology. Genes Chromosomes Cancer. 2003;37:396–405. doi: 10.1002/gcc.10225. [DOI] [PubMed] [Google Scholar]
- Gutiérrez NC, López-Pérez R, Hernández JM, Isidro I, González B, Delgado M, Fermiñán E, García JL, Vázquez L, González M, San Miguel JF. Gene expression profile reveals deregulation of genes with relevant functions in the different subclasses of acute myeloid leukemia. Leukemia. 2005;19:402–9. doi: 10.1038/sj.leu.2403625. [DOI] [PubMed] [Google Scholar]
- Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou X, Song G, Shurtleff SA, Pounds S, Cheng C, Ma J, Ribeiro RC, Rubnitz JE, Girtman K, Williams WK, Raimondi SC, Liang DC, Shih LY, Pui CH, Downing JR. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–87. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cavé H, Pane F, Aerts JL, De Micheli D, Thirion X, Pradel V, González M, Viehmann S, Malec M, Saglio G, van Dongen JJ. Standardization and quality control studies of 'real-time' quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003;17:2318–57. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- Boissel N, Renneville A, Biggio V, Philippe N, Thomas X, Cayuela JM, Terre C, Tigaud I, Castaigne S, Raffoux E, De Botton S, Fenaux P, Dombret H, Preudhomme C. Prevalence, clinical profile, and prognosis of NPM mutations in AML with normal karyotype. Blood. 2005;106:3618–20. doi: 10.1182/blood-2005-05-2174. [DOI] [PubMed] [Google Scholar]
- Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, Sonoda Y, Fujimoto T, Misawa S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8. [PubMed] [Google Scholar]
- Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol. 2001;113:983–8. doi: 10.1046/j.1365-2141.2001.02850.x. [DOI] [PubMed] [Google Scholar]
- Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–70. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- Falkenauer E. In: Genetic Algorithms and Grouping Problems. First. Falkenauer E, editor. Chichester: Wiley J & Sons Limited; 1998. The grouping genetic algorithm; pp. 81–110. [Google Scholar]
- de Ridder D, van der Linden CE, Schonewille T, Dik WA, Reinders MJ, van Dongen JJ, Staal FJ. Purity for clarity: the need for purification of tumor cells in DNA microarray studies. Leukemia. 2005;19:618–27. doi: 10.1038/sj.leu.2403685. [DOI] [PubMed] [Google Scholar]
- Lemon WJ, Palatini JJ, Krahe R, Wright FA. Theoretical and experimental comparisons of gene expression indexes for oligonucleotide arrays. Bioinformatics. 2002;18:1470–6. doi: 10.1093/bioinformatics/18.11.1470. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Kennedy A, Zhou X, Radtke I, Phillips LA, Shurtleff SA, Downing JR. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia. 2007;21:2000–9. doi: 10.1038/sj.leu.2404808. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Goudswaard CS, van Putten W, Bijl MA, Sanders MA, Hugens W, Uitterlinden AG, Erpelinck CA, Delwel R, Löwenberg B, Valk PJ. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood. 2005;106:3747–54. doi: 10.1182/blood-2005-05-2168. [DOI] [PubMed] [Google Scholar]
- Alcalay M, Tiacci E, Bergomas R, Bigerna B, Venturini E, Minardi SP, Meani N, Diverio D, Bernard L, Tizzoni L, Volorio S, Luzi L, Colombo E, Lo Coco F, Mecucci C, Falini B, Pelicci PG. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood. 2005;106:899–902. doi: 10.1182/blood-2005-02-0560. [DOI] [PubMed] [Google Scholar]
- Kohlmann A, Bullinger L, Thiede C, Schaich M, Schnittger S, Döhner K, Dugas M, Klein HU, Döhner H, Ehninger G, Haferlach T. Gene expression profiling in AML with normal karyotype can predict mutations for molecular markers and allows novel insights into perturbed biological pathways. Leukemia. 2010;24:1216–20. doi: 10.1038/leu.2010.73. [DOI] [PubMed] [Google Scholar]
- van Beers E, de Best L, van Vliet M, Brand J, Burgmer P, de Quartel L, Dumee B, Provoost A, Valk P, Delwel H, Büllinger L, van der Spek P, Viëtor H, Döhner H, Löwenberg B. A validated diagnostic microarray for newly diagnosed adult AML patients. Oral Communication at European Hematology Association annual meeting London 2011. Abstract O469.
- Kohlmann A, Schoch C, Dugas M, Rauhut S, Weninger F, Schnittger S, Kern W, Haferlach T. Pattern robustness of diagnostic gene expression signatures in leukemia. Genes Chromosomes Cancer. 2005;42:299–307. doi: 10.1002/gcc.20126. [DOI] [PubMed] [Google Scholar]
- Opitz L, Salinas-Riester G, Grade M, Jung K, Jo P, Emons G, Ghadimi BM, Beissbarth T, Gaedcke J. Impact of RNA degradation on gene expression profiling. BMC Med Genomics. 2010;3:36. doi: 10.1186/1755-8794-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sample preparation for gene expression profiling. - Generation of biotinylated, amplified cRNA using The Illumina Total Prep RNA Amplification Kit (Applied Biosystems/Ambion, Austin, USA). - Generation of low leukemic blast load AML samples by a "dilution/mixture" approach. - Hybridization on Illumina HumanHT-12 v3 Expression BeadChips, staining and detection of cRNAs on microarrays using an I-Scan system. Additional table S1 - Citations in previous studies of the identified markers. Additional figures S1 to S7. Figure S1. Three-dimensional projection of the 3 principal components in a principal-components analysis of APL, t(8;21)-AML, inv(16)-AML, NK-AML, and Normal Bone Marrow samples belonging to the Training Set, with the use of the 10-marker classifiers. Figure S2. Per class median confidence level of the assignment for APL, t(8;21)-AML, inv(16)-AML, NK-AML and normal bone marrow samples belonging to the Training Set according to the number of markers per class (from 1 to 100 markers per class). Figure S3. Median confidence level of the class assignments for the NK-AML samples belonging to the Training Set according to FLT3 and NPM1 mutational status and the number of markers per class (from 1 to 100 markers per class). Figure S4. Relationship between the percentage of leukemic blasts within the 101 Test Set samples (X axis) and the confidence level of their class assignments (Y axis) (samples with poor quality control criteria and AML cell lines were excluded). Figure S5. Confidence level of the class assignments according to the percentage of leukemic blasts including comparisons between diluted and not diluted samples. Figure S6. Results of the class assignment for the 10 AML Test Set samples with suboptimal quality control criteria based on the 10-marker classifiers characterizing the APL, t(8;21)-AML, inv(16)-AML and NK-AML classes. Figure S7. Sensitivity, specificity, negative and positive predictive values of the prediction model for the class assignment of the 111 Test Set samples (all AML samples with or without optimal quality control criteria - AML cell line samples excluded) to the APL, t(8;21)-AML, inv(16)-AML and NK-AML classes with 10-marker classifiers.