

Short abstract
Improved techniques for defining disease-gene location and evaluating the biological candidacy of regional transcripts will hasten disease-gene discovery.
Abstract
Improved techniques for defining disease-gene location and evaluating the biological candidacy of regional transcripts will hasten disease-gene discovery.
Identifying the genes underlying susceptibility to human disease represents a major objective of current biomedical research. Increasingly, the focus of such gene-discovery efforts has shifted from rare, monogenic conditions towards the common conditions (such as diabetes, asthma, neuropsychiatric disorders and cancers) that account for the majority of human illness and mortality. These common conditions are termed 'multifactorial' because, in each case, susceptibility is attributed to the effects of genetic variation at a number of different genes and their interaction with relevant environmental exposures. The expectation is that identification and characterization of the genes that provide the inherited component of susceptibility will lead to substantial advances in our understanding of disease and, in turn, to improvements in diagnostic accuracy, prognostic precision, and the range and targeting of available therapeutic options.
The 'traditional' route to gene discovery - for the rarer, monogenic, Mendelian diseases at least - has been positional cloning. Typically, the gene responsible for a trait is first localized by linkage analysis to a small interval (ideally less than 1 centiMorgan, cM) by successive rounds of linkage mapping within families. Next, each of the handful of genes mapping to the interval so defined is assessed for their potential functional relevance to the disease and screened for etiological mutations. Well over 1,000 Mendelian diseases have been mapped using modifications of this procedure [1].
To date, the application of similar strategies to the identification of susceptibility genes underlying common, complex multifactorial traits has brought only limited success. The explanation for this pedestrian progress stems from the weak relationship between genotype - at any given locus - and phenotype that characterizes multifactorial traits. Not only does this mean that the correlation signals that we seek to detect by linkage analysis or population-based association studies are that much weaker in the first place, it also limits the capacity of these tools to provide precise estimates of disease-gene localization. The regions of interest defined through complex-trait linkage studies - even when analysis has involved thousands of families segregating the trait of interest - regularly exceed 30 cM in size, and contain many hundreds of genes. Large genomic intervals of interest can also be defined through the analysis of major chromosomal rearrangements, duplications and deletions, implicated in the development of cancers and certain multisystem syndromes.
Difficulties with the positional cloning approach have led many investigators to favor a strategy based primarily on identifying susceptibility variants through direct examination of biological candidates ('the candidate gene approach'). This strategy, too, has proven something of a disappointment [2], precisely because ignorance about the biology of complex diseases has typically frustrated efforts to define biological candidacy with any confidence.
The key to accelerating the discovery of susceptibility genes for multifactorial conditions clearly lies in developing improved strategies for refining both disease-gene location and assessments of biological candidacy (see Figure 1). This article describes some recent developments, with an emphasis on their impact on susceptibility-gene identification in man.
Figure 1.
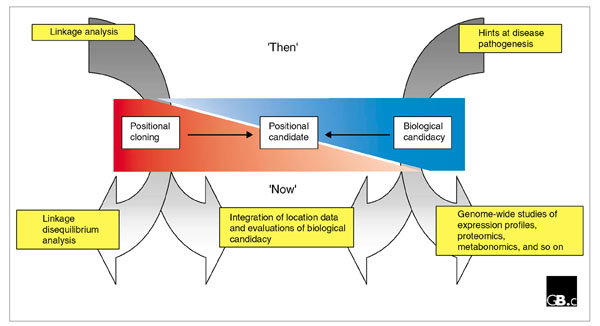
Candidate-gene identification past and present. In the past, the emphasis was on using linkage analysis to direct positional-cloning efforts, and on the application of our limited understanding of disease pathogenesis to select biologically relevant candidate genes. Now, additional techniques are providing new routes to the identification of disease-susceptibility genes. See text for further details.
New methods for defining susceptibility-gene location
Linkage analysis seeks to provide disease-gene localization through the direct observation of recombination events within families. Whilst it has proven an extremely effective tool for detecting and localizing the rare, penetrant variants that underlie most monogenic conditions (see above), it is all too often underpowered when it comes to the search for the common variants of modest penetrance that are generally held to influence susceptibility to common traits [3].
Association (or more strictly, linkage disequilibrium) mapping, in contrast, seeks to localize susceptibility variants through the analysis of allele distributions in populations, relying on the observed consequences of unobserved recombination events during population history. Provided the susceptibility variant itself (or a variant highly correlated with it) is amongst the markers typed, association mapping is, generally, more powerful than linkage analysis [4]. Furthermore, because the extent of linkage disequilibrium (LD) within populations (typically tens of kilobases) is much less than that of linkage within families (typically tens of megabases), much tighter disease gene localization is possible.
One promising positional-cloning strategy for complex traits, therefore, starts with a preliminary linkage analysis to identify regions of interest. Following replication in other datasets, the strongest regions are then selected for LD mapping to refine susceptibility-gene location. Unless there is extensive allelic and locus heterogeneity, the association signal within such linked regions should be appreciable and, in principle, readily detected provided that a sufficiently dense set of markers is typed [5]. The markers that are used are increasingly likely to be single-nucleotide polymorphisms (SNPs). Although there have been several notable successes from this approach [6-8], widespread implementation has been stifled by the daunting scale associated with any exhaustive LD screen across a large genomic interval.
Several ongoing developments can be expected to facilitate such large-scale 'brute force' LD mapping efforts in the years to come. First, an increasingly comprehensive catalog of common variants [9] is providing the reagents for future LD studies. Second, a growing appreciation of the high-resolution structure of LD in human populations [10] indicates that most common variation within a region can be 'captured' by typing only a subset of the available variants (so-called 'haplotype-tagging' or htSNPs) [11]. The HapMap project is currently characterizing the haplotype structure of the genome in major ethnic groups, thereby defining htSNPs for use in future positional cloning endeavors. Third, improved genotyping platforms and strategies, using extensive multiplexing [12] and/or pooled DNA samples [13], promise to provide significant improvements in throughput combined with necessary reductions in cost and DNA consumption.
Significant issues remain unresolved, however. The density of SNPs required to provide a comprehensive survey of common disease variation remains unclear. On current evidence - except in population isolates, in which LD may be more extensive - a mean density exceeding one (haplotype-tagging) variant every five kilobases looks to be necessary, and considerably more in regions of high haplotypic diversity [14]. The relative importance of common versus rare variants in complex-trait susceptibility remains a contentious issue with enormous practical implications: a strategy based on common htSNPs will be poor at detecting rare penetrant mutations, and substantial allelic heterogeneity may seriously compromise the power of LD-mapping approaches [15]. The pros and cons of focusing the survey of a region on gene-related sequence [16] as opposed to an indiscriminate 'gene-blind' set of markers remain to be established. Finally, there remain significant analytical issues as to how best to analyze the large amounts of data that will arise. High-density LD surveys of several large genomic regions are now underway for a range of diseases. Whether successful or not in terms of disease-gene discovery, these 'pilot' studies, each generating several million genotypes, will help to address various unresolved issues, and inform future efforts.
Several groups have elected to bypass the linkage step and proceed directly to genome-wide LD analyses. Many of these efforts have been rather optimistic in terms of the density of markers typed, although interesting data have emerged from studies of population isolates [17]. In such populations - such as French Canadians and Finns - which have often grown from a limited number of recent founder individuals, LD may extend much further than in non-isolate populations, reducing the number of markers that need to be typed to obtain genome-wide coverage. Recently, a study of over 90,000 gene-based variants provided a valuable proof-of-principle by detecting a strong association between a variant in the LTA gene and myocardial infarction in Japanese [16].
These early studies indicate that it will be increasingly possible to translate the success of positional cloning (in the strict sense of the term, using positional mapping alone without reference to function) from rare, monogenic syndromes to common multifactorial traits. At the moment, the main obstacle remains the prodigious costs of implementing these strategies on the scale that is necessary.
New approaches for defining susceptibility-gene biological candidacy
The candidate-gene approach relies on matching known or presumed gene functions to the known or presumed biology of the disease or phenotype under investigation. The aim is to identify genes with a strong prior claim to involvement in disease susceptibility and to prioritize these for analyses seeking evidence of disease-associated variation. Sadly, however, the history of candidate-gene studies, particularly for complex traits, has been characterized by a swathe of unsubstantiated, unreplicated claims and relatively few proven associations [1,2]. Amongst the many methodological and cultural reasons for this failure [18], two concern us here. First, because in general we know so little about the mechanisms responsible for complex diseases, any test of biological candidacy is necessarily speculative. Second, it is worth remembering that significant susceptibility effects are only likely to be found in a small minority of the set of genes whose products are directly involved in disease pathogenesis [19]. As a result, for most complex traits there are - even for genes with apparently robust biological credentials - relatively low prior odds (that is, odds estimated prior to the start of any genotyping) that variants within that gene play a major role in disease susceptibility. These low prior odds need to be borne in mind when interpreting the results of association studies [18].
The expectation is that application of the growing range of massively parallel genome-wide '-omics' technologies - from transcriptional profiling to metabonomics and beyond - will provide a basis for refining assessments of biological candidacy and identifying candidates with better prior odds. A growing number of examples attest to the potential of these strategies. Most to date have featured the use of expression data, with lists of promising candidates generated on the basis of both expression state (tissue expression profiles) and studies of differential expression. For some tissues, restricted or preferential tissue expression patterns have provided valuable clues to biological candidacy: for example, almost half of the 51 genes underlying monogenic retinal phenotypes are specifically or preferentially expressed in the retina [20]. For many complex traits, however, multisystem involvement and uncertainties over the most appropriate choice of tissues for study have limited the value of this approach. Even when the choice of target tissue is clear, sparse expression-state data in public repositories (for example, for the adipocyte) and/or inaccessibility of the tissues of interest (such as the pancreatic β cell) have often compromised progress.
Genome-wide studies of differential expression - whether undertaken by differential display, representational difference analysis or microarrays - have obvious potential to provide valuable clues to disease-associated pathways, thereby identifying candidates for genetic studies. Work on rodent models has predominated in this area; limitations on tissue accessibility, uncertainties over tissue selection, and concerns about the 'noise' arising from studies of 'free-range' subjects, have impeded equivalent studies in humans. But recent studies demonstrating that insulin resistance and type 2 diabetes are consistently and reproducibly associated with downregulation, in human skeletal muscle, of a coregulated set of oxidative phosphorylation genes [21,22] are reassuring and confirm the capacity to highlight candidates with key regulatory roles, in this case, PGC1-α/PPARGC1. Association studies of this gene have already generated positive, though as yet inconsistent, findings [23].
As with any transcript-profiling study, design is all-important. Comparisons between 'diseased' and 'control' tissue will favor direct detection of variants involved in gene regulation or mRNA stability, but not those that modify protein sequence (with some notable exceptions [24]). Where possible, there is considerable merit in complementing such 'disease versus control' candidate-mining efforts with additional transcriptional comparisons designed to detect genes regulated by environmental or pharmacological interventions pertinent to the disease phenotype. For example, in a study of the levels of high-density lipoprotein (HDL) cholesterol in baboons, transcript levels were studied in relation to both HDL levels and response to high-fat diet [25]. It is important to remember that candidacy mining using expression-profiling methods will perform poorly where trait susceptibility is governed by variation within genes with low-level and/or restricted expression (for example, limited to tight temporal and/or developmental windows).
Other genome-wide (-omics) technologies should, in time, provide equivalent power for defining novel etiological pathways and putative candidates. Studies aiming to define the protein markers of disease have often been limited by the scalability of conventional proteomic technologies [26], but recent advances (for example, the identification of putative plasma protein biomarkers for ovarian cancer [27]) should, as the relevant biomarkers are characterized, provide valuable biological insights and new candidates. The same is true of metabonomics-based analyses [28].
Whilst considerable advances will arise from each platform in isolation, the key to a robust and comprehensive understanding of etiological pathways, and to a full description of disease pathogenesis that will maximally inform candidate selection, will be data integration. By synthesizing and modeling the changes that occur at the level of the genome, message, protein, post-translational modification, metabonome and physiome, such integration should lead to construction of a 'biological atlas' of each disease or phenotype of interest [29]. Within such a framework, data from 'distal' modalities, such as metabonomics and intermediate physiology, which more closely reflect the visible phenotype of interest, can inform the interpretation of 'proximal' modalities, such as expression data. This should help to distinguish, for example, those transcriptional changes that are directly related to disease etiology from those that are secondary or epiphenomenal. As an example, two large consortia - the UK-based Biological Atlas of Insulin Resistance (BAIR) [30] and the US-based Diabetes Genome Anatomy Project - have established complementary programs to reconstruct the development of type 2 diabetes and obesity, through coordinated transcriptional, proteomic, metabonomic, biochemical and physiological analyses in multiple tissue samples taken from rodent models at various stages of trait progression. One outcome of these studies will be an improved framework for the selection of functional candidates for genetic studies in man.
Integrating location and function
Despite the undoubted power of the approaches described above, we believe that the strongest routes to robust candidate identification will come from integrating the positional and functional routes to candidacy. Broadly speaking, there are two main ways by which this integration is being achieved. The first involves the use of functional genomic readouts - expression levels, protein levels, metabolite spectra, and so on - as 'endophenotypes' to be subjected to the same genome-wide quantitative trait linkage (and LD) strategies as have been traditionally performed on disease phenotypes. In the most comprehensive study published to date [31], genome-wide expression profiling on liver tissue from 111 F2 mice segregating obesity-related traits identified the only two transcripts that jointly met the following three criteria: first, physical location within a region on murine chromosome 2 harboring a locus for fat-pad mass, one of the obesity phenotypes of interest; second, linkage mapping of the variation in their expression levels to the same genomic location (indicating cis-acting expression effects); and third, transcriptional differences related to fat-pad mass trait values. Preliminary studies in human pedigrees have confirmed that familial aggregation of expression phenotypes remains detectable even under the less controlled circumstances inevitable in human studies [31,32], setting the stage for future linkage studies to map the loci that influence these expression levels, be they acting in cis (promoter variation within the genes themselves, for example) or in trans (via transcription factors).
The second way that integration is being achieved represents an updating of the 'positional candidate' strategy [33] to take account of contemporary informatics and functional genomics, and represents the intersection of positional cloning and functional candidate-selection approaches. As described above, the region of interest defined by a genome-wide linkage scan is generally around 30-40 megabases (about 1% of the genome) and contains several hundred genes. Given the costs currently associated with a comprehensive LD-based survey of such large regions, there is obvious interest in the use of biological candidacy as a means for evaluating and prioritizing regional transcripts for detailed mutation detection and association analysis. Looked at from another angle, one can view the genomic localization afforded by linkage studies (or physical mapping efforts) as providing a powerful basis for filtering the output of what tend to be relatively non-specific functional assessments of candidacy (such as descriptions of expression state and analyses of genome-wide differential expression).
It is of course, nowadays, an entirely straightforward matter to download the identities of all known genes mapping to a region of interest, and to skim through the list looking for obvious biological candidates. On occasion, this has proven extremely successful: inspection of the list highlights a single strong candidate, subsequently confirmed as the etiological locus [7]. In such circumstances, defining the overlap of function and location is a trivial matter. In general, however, a respectable prima facie case for disease involvement can be made for a substantial proportion of the genes mapping to the interval, making further prioritization essential if the list of genes to be studied is to be brought to manageable levels.
The challenge then lies in bringing together as many lines of evidence as possible about the biology of the genes mapping to the interval, and to relate these data to the biology of the disease or phenotype of interest. This is most definitely a non-trivial exercise, not least because it implies the need for an intimate dialogue between bioinformaticians on the one hand - as the people best placed to assemble information on gene biology - and biologists and clinicians on the other - as the people with expertise in the biology of the disease. The number of parameters that could potentially inform positional-candidate prioritization is substantial (see Table 1). For many of these, however, the datasets currently available are too sparse to offer much in the way of discriminant power, and expression data has been most widely used to date.
Table 1.
Attributes that can be used to define positional candidacy
| Attribute type | Attribute | Questions to be asked of each regional gene |
| Transcriptional | Expression state | Which genes are expressed in tissues relevant to the disease of interest? |
| Which genes show restricted or preferential expression in relevant tissues? | ||
| Which genes show expression within organelles (such as mitochondria) implicated in disease etiology? | ||
| Differential expression | Which genes show robust expression differences in comparison of pertinent tissues from 'disease' versus 'control'? | |
| Which genes show robust expression differences in response to relevant environmental, pharmacological or physiological manipulation? | ||
| Coregulation | Which genes appear to be coregulated with members of other pathways implicated in disease? | |
| Proteomics | Protein expression | Which genes code for proteins that show differences in expression and/or post-translational modification in diseased tissues, or response to relevant manipulation? |
| Interaction | Which genes code for proteins known to interact with other proteins implicated in disease etiology? | |
| Function | Which genes code for products that have functional (such as ion channels) or localization (for example, membrane proteins) properties that are pertinent to disease etiology? | |
| Metabonomics | Biomarkers of disease | Which genes code for proteins involved in regulation of metabolic pathways implicated in disease through metabonomic comparisons? |
| Pathway analysis | Which genes code for products involved in pathways implicated in disease? | |
| Comparative analysis | Homologies | Which genes have homologies (or other relationships) to genes implicated in equivalent phenotypes in other animals? |
| Ortholog mapping | Which genes are members of gene families for which other members are implicated in the disease or related diseases? | |
| Which genes are members of gene families for which other members map to other regions of linkage containing as yet unidentified susceptibility genes? |
Expression state has proven particularly useful in defining positional candidates for retinal [20,34,35] and cochlear [36] diseases; and de novo serial analysis of gene expression (SAGE) of the substantia nigra transcriptome was used as a tool for prioritizing positional candidates for Parkinson's disease [37]. Clearly, these types of data will be most valuable in circumstances where, as with Parkinson's, there are strong grounds for implicating specific tissues in disease pathogenesis.
Application of expression-state data is not limited to selection on the basis of predicted tissue distribution, but can be extended to incorporate knowledge regarding subcellular localization [38]. Prior linkage analysis in families with the French-Canadian subtype of Leigh syndrome had mapped the gene responsible to chromosome 2p, and the biology of the disease strongly implicated genes involved in mitochondrial function. Publicly available mRNA expression data were used to identify which of the regional transcripts had mRNA expression patterns resembling those of known mitochondrial genes. This analysis, together with data emerging from a proteomic survey of isolated mitochondria, pinpointed a single transcript, LRPPRC, which was confirmed as the disease gene by mutation screening in pedigrees segregating the disorder [38].
The most productive strategy to date, in animal models at least, has been the integration of information from positional localization with that from differential expression analysis. To identify genes underlying lipid phenotypes in baboons, a chromosomal-region-specific expression array - covering a region of interest defined by prior linkage - was used to detect regional transcripts with expression levels correlated with HDL-cholesterol levels and with the response to a high-fat diet [25]. In the NOD mouse model of autoimmune diabetes, Eaves and colleagues [39] used genome-wide transcriptional comparisons between NOD, NOD-derived congenic, and control strains to relate differential expression patterns to the locations of known susceptibility loci, and thereby to prioritize candidates. A similar congenic-based approach has been used successfully in rat models of hypertension [24,40]. The human homologs of these regional transcripts represent strong disease-susceptibility candidates. Issues of tissue availability will mean that, for many diseases, rodent models are likely to provide a more reliable source of such candidates than equivalent studies on human tissues.
For most of the studies described above, the expression data used were generated de novo, in part because of the patchy and disorganized nature of publicly available expression data. Certainly, expression-state data for many critical human tissues remain limited, and there is a clear need for a concerted effort to generate comprehensive transcript lists for a broad range of human tissues and cell types [41]. Such information, coupled with recent developments in setting standards for expression data [42,43] and the predicted expansion in usable, publicly available expression-profiling data [44], will hopefully reduce the need for costly de novo work and make expression-related data increasingly relevant to the evaluation of disease-gene candidacy.
As the breadth and depth of data available concerning each gene grows, two major challenges will need to be met, if these data are to be used optimally for assessments of disease-gene candidacy. The first challenge lies in displaying all these disparate data types - and related text-based information from the literature - to the user in such a way as to inform and assist decisions about positional-candidate prioritization. Through the EnsMart [45] component of the Ensembl database, it is already possible to combine information on genomic location, expression state (defined by a controlled expression vocabulary [42]), and the various functional attributes encoded by Gene Ontology terms [46,47]. The GeneSeeker program allows users to query on the basis of position, expression state and some phenotypic attributes [48]. Key to continued progress in this area is the construction of suitable controlled vocabularies or ontologies that permit uniform comparison of data from differing platforms, databases and species. These will allow additional attributes (see Table 1) to become increasingly accessible to the candidate-prioritization process.
The second challenge will be the development of, and validation of, heuristics for candidate-gene prioritization that take account of the range of data available and the domain expertise of the biologist as regards the disease or phenotype of interest. One interesting approach will be to consider the topography of expression, regulatory and protein-protein interaction networks; proteins that are known to be hubs of such networks show higher degrees of conservation [49] and may therefore be more likely disease candidates. Other current efforts have used Gene Ontology terms to group genes with similar properties, identifying potential candidates on the basis of potential functional overlap with known disease genes previously implicated in related diseases [50,51].
In conclusion, we have outlined some of the approaches currently being developed and applied to aid the identification of the genes that influence susceptibility to the major diseases that afflict mankind. These approaches are increasingly contingent on the integration of information from diverse data sources, and success will be dependent on overcoming the significant cultural and technical challenges that this imposes.
Acknowledgments
Acknowledgements
We thank the European Union (GIFT: QLRT-CT-1999-00546) for their support of D.S. and the Wellcome Trust (Collaborative Research Initiative Grant 006294/Z/01/Z) for their support of the collaboration between M.McC. and W.H.
References
- Glazier AM, Nadeau JH, Aitman TJ. Finding genes that underlie complex traits. Science. 2002;298:2345–2349. doi: 10.1126/science.1076641. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/S0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Wang WYS, Cordell HA, Todd JA. Association mapping of complex diseases in linked regions: estimation of genetic effects and feasibility of testing rare variants. Genet Epidemiol. 2003;24:36–43. doi: 10.1002/gepi.10216. [DOI] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TM, Mashima H, Schwarz PEH, et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet. 2000;26:163–175. doi: 10.1038/79876. [DOI] [PubMed] [Google Scholar]
- Hugot J-P, Chamaillard M, Zouali H, Lesage S, Cézard J-P, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Leaves NI, Anderson GG, Ponting CP, Broxholme J, Holt R, Edser P, Bhattacharyya S, Dunham A, Adcock IM, et al. Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma. Nat Genet. 2003;34:181–186. doi: 10.1038/ng1166. [DOI] [PubMed] [Google Scholar]
- Reich DE, Gabriel SB, Altshuler D. Quality and completeness of SNP databases. Nat Genet. 2003;33:457–458. doi: 10.1038/ng1133. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Johnson GCL, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, et al. Haplotype tagging for the identification of common disease genes. Nat Genet. 2001;29:233–237. doi: 10.1038/ng1001-233. [DOI] [PubMed] [Google Scholar]
- Oliphant A, Barker DL, Stuelpnagel JR, Chee MS. BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques. 2002;Suppl:56–58. [PubMed] [Google Scholar]
- Sham P, Bader KS, Craig I, O'Donovan M, Owen M. DNA pooling: a tool for large-scale association studies. Nat Rev Genet. 2002;3:862–871. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- Dawson E, Abecasis GR, Bumpstead S, Chen Y, Hunt S, Beare DM, Pabial J, Dibling T, Tinsley E, Kirby S, et al. A first-generation linkage disequilibrium map of human chromosome 22. Nature. 2002;418:544–548. doi: 10.1038/nature00864. [DOI] [PubMed] [Google Scholar]
- Wright AF, Hastie ND. Complex genetic diseases: controversy over the Croesus code. Genome Biology. 2001;2:comment2007.1–2007.8. doi: 10.1186/gb-2001-2-8-comment2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda R, Sato H, Sato H, Hori M, Nakamura Y, et al. Functional SNPs in the lymphotoxin-3 gene that are associated with susceptibility to myocardial infarction. Nat Genet. 2002;32:650–654. doi: 10.1038/ng1047. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Escamilla MA, Service SK, Spesny M, Meshi DB, Poon W, Molina J, Fournier E, Gallegos A, Mathews C, et al. Genomewide linkage disequilibrium mapping of severe bipolar disorder in a population isolate. Am J Hum Genet. 2002;71:565–574. doi: 10.1086/342291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Bell JI. Association study designs for complex disease. Nat Rev Genet. 2001;2:91–99. doi: 10.1038/35052543. [DOI] [PubMed] [Google Scholar]
- Miller RD, Kwok P-Y. The birth and death of human single-nucleotide polymorphisms: new experimental evidence and implications for human history and medicine. Hum Mol Genet. 2001;10:2195–2198. doi: 10.1093/hmg/10.20.2195. [DOI] [PubMed] [Google Scholar]
- Katsanis N, Worley KC, Gonzalez G, Ansley SJ, Lupski JR. A computational/functional genomics approach for the enrichment of the retinal transcriptome and the identification of positional candidate retinopathy genes. Proc Natl Acad Sci USA. 2002;99:14326–14331. doi: 10.1073/pnas.222409099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek J, Andersen G, Urhammer SA, Gaede PH, Drivsholm T, Borch-Johnsen K, Hansen T, Pedersen O. Mutation analysis of peroxisome proliferator-activated receptor-γ coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to type II diabetes mellitus. Diabetologia. 2001;44:2220–2226. doi: 10.1007/s001250100032. [DOI] [PubMed] [Google Scholar]
- Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, Wahid FN, Al-Majali KM, Trembling PM, Mann CJ, Shoulders CC, et al. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21:76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- Cox LA, Birnbaum S, VandeBerg JL. Identification of candidate genes regulating HDL cholesterol using a chromosomal region expression array. Genome Res. 2002;12:1693–1702. doi: 10.1101/gr.333502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers TA, Yates JR. Review of proteomics with applications to genetic epidemiology. Genet Epidemiol. 2003;24:83–98. doi: 10.1002/gepi.10226. [DOI] [PubMed] [Google Scholar]
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–577. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HWL, Clarke S, Schofield PM, McKilligin E, Mosedale DE, Grainger DJ. Rapid and non-invasive diagnosis of the presence and severity of coronary heart disease using 1H-NMR-based metabonomics. Nat Med. 2002;8:1439–1444. doi: 10.1038/nm802. [DOI] [PubMed] [Google Scholar]
- Vidal M. A biological atlas of functional maps. Cell. 2001;104:333–339. doi: 10.1016/s0092-8674(01)00221-5. [DOI] [PubMed] [Google Scholar]
- The Biological Atlas of Insulin Resistance (BAIR) Consortium http://www.icggri.ic.ac.uk/bair/
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen K-Y, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003;33:422–425. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- Collins FS. Positional cloning moves from the perditional to traditional. Nat Genet. 1995;9:347–350. doi: 10.1038/ng0495-347. [DOI] [PubMed] [Google Scholar]
- Sullivan LS, Heckenlively JR, Bowne SJ, Zuo J, Hide WA, Gal A, Denton M, Inglehearn CF, Blanton SH, Daiger SP. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohocki MM, Malone KA, Sullivan LS, Daiger SP. Localization of retina/pineal-expressed sequences: identification of novel candidate genes for inherited retinal disorders. Genomics. 1999;58:29–33. doi: 10.1006/geno.1999.5810. [DOI] [PubMed] [Google Scholar]
- Skvorak AB, Weng Z, Yee AJ, Robertson NG, Morton CC. Human cochlear expressed sequence tags provide insight into cochlear gene expression and identify candidate genes for deafness. Hum Mol Genet. 1999;8:439–452. doi: 10.1093/hmg/8.3.439. [DOI] [PubMed] [Google Scholar]
- Hauser MA, Li Y-J, Takeuchi S, Walters R, Noureddine M, Maready M, Darden T, Hulette C, Martin E, Hauser E, et al. Genomic convergence: identifying candidate genes for Parkinson's disease by combining serial analysis of gene expression and genetic linkage. Hum Mol Genet. 2003;12:671–676. doi: 10.1093/hmg/12.6.671. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaves IA, Wicker LS, Ghandour G, Lyons PA, Peterson LB, Todd JA, Glynne RJ. Combining mouse congenic strains and microarray gene expression analyses to study a complex trait: the NOD model of type 1 diabetes. Genome Res. 2002;12:232–243. 10.1101/gr.214102. Article published online before print in January 2002. [PubMed] [Google Scholar]
- McBride WM, Carr FJ, Graham D, Anderson NH, Clark JS, Lee WK, Charchar FJ, Brosnan MJ, Dominiczak AF. Microarray analysis of rat chromosome 2 congenic strains. Hypertension. 2003;41:847–853. doi: 10.1161/01.HYP.0000047103.07205.03. [DOI] [PubMed] [Google Scholar]
- Jongeneeel CV, Iseli C, Stevenson BJ, Riggins GJ, Lal A, Mackay A, Harris RA, O'Hare MJ, Neville AM, Simpson AJG, Strausberg RL. Comprehensive sampling of gene expression in human cell lines with massively parallel signature sequencing. Proc Natl Acad Sci USA. 2003;100:4702–4705. doi: 10.1073/pnas.0831040100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso J, Visagie J, Theiler G, Christoffels A, Bardien-Kruger S, Smedley D, Otgaar D, Greyling G, Jongeneel V, McCarthy MI, et al. eVOC: a controlled vocabulary for unifying gene expression data. Genome Res. 2003;13:1222–1230. doi: 10.1101/gr.985203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, et al. Minimum information about a microarray experiment (MIAME) - toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, Abeygunawardena N, Holloway E, Kapushesky M, Kemmeren P, Lara GG, et al. ArrayExpress - a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003;31:68–71. doi: 10.1093/nar/gkg091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EnsMart http://www.ensembl.org/EnsMart/
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology http://www.geneontology.org/
- Van Driel MA, Cuelenaere K, Kemmeren PP, Leunissen JA, Brunner HG. A new web-based data mining tool for the identification of candidate genes for human genetic disorders. Eur J Hum Genet. 2003;11:57–63. doi: 10.1038/sj.ejhg.5200918. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Wolf YI, Koonin EV. No simple dependence between protein evolution rate and the number of protein-protein interactions: only the most prolific interactors tend to evolve slowly. BMC Evol Biol. 2003;3:1. doi: 10.1186/1471-2148-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Iratxeta C, Bork P, Andrade MA. Association of genes to genetically inherited diseases using data mining. Nat Genet. 2002;31:316–319. doi: 10.1038/ng895. [DOI] [PubMed] [Google Scholar]
- Freudenberg J, Propping P. A similarity-based method for genome-wide prediction of disease-relevant human genes. Bioinformatics. 2002;18(suppl 2):S110–S115. doi: 10.1093/bioinformatics/18.suppl_2.s110. [DOI] [PubMed] [Google Scholar]