

A colleague of mine once said ‘You know Fiona, this genetics is all very well but when we try and read a paper on genetics it's a bit like reading a car manual and then just when you get to the crucial bit, the language suddenly changes to Chinese and I can't make head nor tail of it’. I suspect he is not alone in his opinions and, if so, this is a great shame. It is well recognised in the genetics community that many health care professionals completed their training long before the great explosion in molecular genetics and so the role of the clinical geneticist includes that of being an educator. It is vital that we pass knowledge on to other clinicians so that they can make sense of all the new advances and use this to improve patient care.
Until recently cytogenetic and molecular genetics were two very separate disciplines. However as this review will show, molecular techniques are increasingly used in cytogenetics and the distinction between them is looking increasingly blurred and artificial.
Most of us are familiar with the standard karyotype obtained by looking at dividing cells under the microscope. For many years it has been a first line test in the investigation of learning disability, dysmorphic features and multiple congenital abnormalities. However, if you imagine your genetic material as being contained in a series of books then looking at a karyotype is a bit like looking at the bookshelf and seeing that all the books are there. It doesn't tell you if there are missing pages, missing chapters or indeed any spelling mistakes in any of the words on any of the pages. Newer molecular techniques are superseding it and its days as a front-line investigation are numbered.
Each of our cells should contain 46 chromosomes. Numbers 1 to 22 come in pairs and then there are the two sex chromosomes: XX in females and XY in males. Each chromosome is really a long tightly coiled string of DNA. Chromosomes are a method of organising the DNA into manageable units. It is estimated that each human cell contains about 2m of DNA. Donnai and Read1 illustrate this nicely: if you were to enlarge an average 10μm cell to the size of a lecture room 10m across, then the DNA would be like a piece of string 2000km long. If it were not carefully organised, there would be a hopeless tangle and protein synthesis would be almost impossible.
When chromosomes are analysed we look not only at the actual number but also to see if there are missing pieces or extra pieces, so-called deletions and duplications. There is a standard method of nomenclature which can be very confusing. The short arm of the chromosome is referred to as the P arm and the long arm as the q arm. Each arm is then divided into numbered bands. Thus if the cytogenetic laboratory identifies an abnormality they can describe its precise location. The problem is that the description can lead to very complex formulae e.g. 46,XX,dup(2)(pl3p22) which means duplication of part of short arm of Chromosome 2. This is why sometimes it can be hard for non-genetic clinicians to make sense of the report. It can be helpful to have a clarifying sentence on the report to make it clear what is being described.
The situation can become more problematic when a translocation is being described. A translocation is a chromosomal rearrangement caused by the exchange of segments between two or more chromosomes. Such a rearrangement is described as balanced if there is no loss or gain of genetic material: the amount of DNA is correct but not all in the right place. If there is a pathogenic loss or gain of genetic material i.e. too much of one chromosome and not enough of another, it is said to be unbalanced. The word pathogenic is important because there are some parts of the genome that can be missing or duplicated with no ill effect - often referred to as normal variants or more correctly large scale copy number polymorphisms. Most people with a balanced translocation are clinically normal (unless one of the breakpoints goes through a particular gene and affects the function of that gene) whereas most individuals with an unbalanced rearrangement will have abnormal clinical features. Individuals with balanced translocations are at increased risk of miscarriage and also are at risk of having a child with an unbalanced translocation.
The standard nomenclature for translocations is often very confusing e.g.45,XX,der(13;14)(pll;qll) This describes a common Robertsonian translocation where one copy of chromosome 13 is attached to one copy of chromosome 14. Individuals with this balanced translocation are usually clinically completely normal. If you do receive a report, the meaning of which is not entirely clear to you, then it may be helpful to call the genetics department and either the cytogenetic staff or one of the clinicians will be able to help.
A technique for looking at small specific microdeletions is known as Fluorescent in Situ Hybridisation (FISH). In this technique, a DNA probe that is known to bind to a specific sequence on the chromosome has a fluorescent dye attached. If the sequence of interest is present on the chromosome, a fluorescent spot will be seen under UV light. If the sequence is missing, there is a microdeletion, and there will be no visible fluorescent spot. Paediatricians and cardiologists will be very familiar with the use of this technique to detect 22ql 1 deletion syndrome.
Microdeletions can also be detected using multiplex ligation-dependent probe amplification (MLPA). This is a useful technique that can enable us to look for a number of microdeletions or duplications at once.
As mentioned earlier, these techniques are being superseded by genomic microarray techniques (array CGH). These arrays use comparative genome hybridisation (CGH) of a mixture of test DNA from a diagnostic sample and normal control DNA. This technique will show if there is more or less DNA in any particular region and so will identify very small deletions and duplications not detectable by any other means. This is a major advance and in some parts of the UK, karyotyping as a first-line investigation for learning disability; multiple congenital malformations and dysmorphism has already been replaced by array CGH. The potential drawback of array CGH is that it may show variations, the significance of which is sometimes unclear.
The basic principles of DNA structure are quite simple. Chromosomes are really long strings of DNA. The structure of DNA is that of a double helix i.e. two polynucleotide chains wrapped round each other. Each nucleotide has three modules: the base which can be adenine (A) guanine (G), cytosine (C) or thymine (T); the sugar which is deoxyribose and a phosphate. The deoxyribose has five carbon atoms and links to the phosphate either by the 5′ carbon or the 3′ to make the sugar phosphate backbone to which the bases are attached. The RNA is also made up of four types of nucleotide. The bases are also A, G and C but thymine is replaced by uracil (U). DNA and RNA sequences therefore have a 5′ end and a 3′ end.
Genes are the functional units of the DNA and the function of a gene is to specify the structure of a protein. Protein synthesis involves transcription of the coding DNA (cDNA) into messenger RNA (mRNA) and then translation of the mRNA into a polypeptide. A triplet of three nucleotides in the messenger RNA (mRNA) known as a codon, codes for a particular amino acid. For example AGU codes for serine. There are 61 of these codons, so almost all amino acids are represented by more than one codon. There are also 3 stop codons that tell the ribosome to dissociate and stop extending the protein. This part of the process from RNA to protein is usually referred to as translation.
A DNA sequence can therefore be written as a string of letters e.g. GTACACCTG. It is important to remember that sequences are written in a 5′ to 3′ direction. We also tend to use the term upstream to describe something that is closer to the 5′ end than a particular area of interest and the term downstream if it's closer to the 3′ end.
Chromosomes are a way of organising the DNA into manageable packages. Genes too are very organised. They consist of segments called exons, which code for proteins interrupted by non-coding sequences known as introns. Within the gene, there are also other regulatory elements e.g. promoters. It is estimated that the human genome contains approximately 21 000 genes (figure 1).
Fig 1.
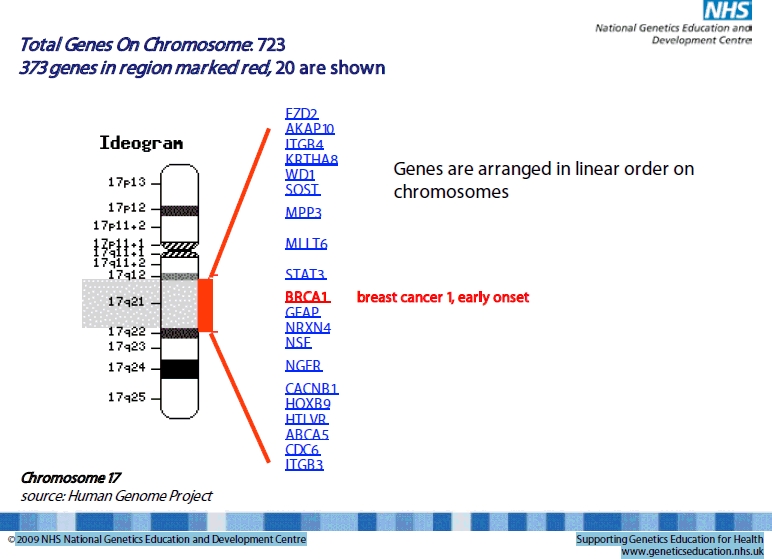
Illustration of the large number of genes contained within a small region of chromosome 17
When a molecular test is ordered for a particular clinical condition, the laboratory are looking at the sequence of a particular piece of DNA to see if it is different from what would be expected. A simple comparison is to consider it as proof reading a piece of reading material to see if there are any mistakes. In the case of DNA the reading material is composed of only 4 letters. So what ‘mistakes’ can occur in DNA and lead to diseases?
There may be a deletion or duplication in the DNA, i.e. a piece of the gene is missing. If the DNA is regarded as being the code or the instructions for making a particular protein, then it is clear that a deletion in the DNA will have a consequent effect on the protein. It will either be abnormal, reduced or sometimes non-existent. As a triplet of three nucleotides – a codon – codes for a particular amino acid, if one or more of those nucleotides is missing there will be a change in the reading frame. It is therefore referred to as a frameshift mutation.
There may be a change in which one nucleotide is changed to a different one. This is referred to as a point mutation. It will alter the codon and may therefore change the amino acid. Sometimes it can be hard for families to comprehend that the change in one particular ‘letter’ of the genetic code can cause such severe abnormalities. I sometimes use the example of a ‘gin and tonic’ versus a ‘gun and tonic’. One change in a letter, but two very different meanings ensue.
There are sometimes confusions about how mutations are described. A change that disturbs the reading frame is called a frameshift mutation, which is pretty straightforward. A point mutation that causes a change from one amino acid to another is called a missense mutation. If the mutation changes the codon for an amino acid to a stop codon, it is called a nonsense mutation. In the process of protein synthesis the non-coding introns are spliced out and the exons are joined together. If a mutation is likely to disrupt this process it is called a splice site mutation.
Rather like chromosome reports, molecular reports are written using internationally agreed standards. One of the most common tests carried out in any molecular genetics laboratory is mutation testing for cystic fibrosis. The commonest mutation is commonly known as F508. The symbol delta was used to denote a deletion so this description means a deletion of phenylalanine at amino acid 508 resulting from a three nucleotide deletion. The correct current nomenclature is p.F508del. The prefix ‘p.’ refers to a numbered amino acid in the protein product and the prefix ‘a’ refers to a numbered nucleotide in a gene sequence. If there is a point mutation or a substitution then the > symbol is used. Looking at another cystic fibrosis example; c.621+1G>T means that there is a change from a G to a T at position 621.
So, having ploughed through all this theory - how does genetic testing work in clinical practice?
Clinicians are aware that the time taken to get a molecular result varies widely. Why is this? Recalling the ‘reading material’ analogy, there is a huge variation in the gene size with some resembling a tabloid newspaper and others War and Peace or indeed all 24 volumes of the Encyclopaedia Britannica!
The other major time factor is whether the condition relates to common mutations that can be confirmed first, or whether the majority are private mutations. At the extremes of this spectrum, we could be looking for a spelling mistake that is always on line 6 of page 3 in your favourite tabloid or anywhere at all in the encyclopaedia. This can help account for the variation in reporting time and also the variation in cost. For some large genes that have a lot of private mutations genetic testing is rarely performed. The diagnosis should be clinically obvious and the cost to the NHS of large-scale testing would be prohibitive.
Let's look at the example of Duchenne Muscular Dystrophy. This is a life-limiting X-linked disorder. The diagnosis is suspected in boys who have muscle weakness and a significantly raised creatine kinase (CK). The dystrophin gene is found at Xp21, i.e. band 21 on the short arm of the X chromosome. It is an enormous gene comprising over 2 million DNA base pairs. Over 99% of the gene comprises introns which are non-coding. However this still leaves 79 exons. Looking for a mutation anywhere is this massive gene would be a very onerous undertaking. Approximately 75% of males will give a positive result on a dystrophin deletion/duplication analysis. If deletion/duplication analysis is negative, then further testing is carried out to look for point mutations. It can be a source of frustration for families and clinicians alike to go through this process, but this is the most logical and resource-efficient way to do it.
The other question that needs to be asked when ordering genetic testing is: ‘why am I doing this?’. Professor Peter Farndon of the UKGTN (United Kingdom Genetic Testing Network) and NGEDC (National Genetics and Educational Centre) has described five good reasons for doing a genetic test:
Diagnosis
Treatment
Prognosis and Management
Pre-symptomatic Diagnostic Screening
Genetic Risk Assessment
When is genetic testing helpful in making a diagnosis? Well the good news is that history and examination is still the mainstay of clinical diagnosis, even for genetic disorders. A very good example of when genetic testing may or may not be helpful is to consider the two types of neurofibromatosis. Neurofibromatosis type I is an obvious clinical diagnosis and genetic testing is only rarely required. However neurofibromatosis type II is a very different condition. Skin changes are not obvious and there is a high risk of intracranial tumours. Genetic testing is very helpful for the at-risk relatives of an affected individual and will enable them to be appropriately investigated and followed up.
Genetic testing may not help make the diagnosis on an individual but may assist with genetic risk assessments for the wider family. In the example of the Duchenne family, identification of the mutation in the boy will enable us to see if his mother is a carrier (which would give a risk of ½ for future sons) or whether she is just at risk of gonadal mosaicism which give a significantly lower, though not zero, risk. Identification of mutations in other conditions can give an opportunity for prenatal diagnosis or even pre-implantation genetic diagnosis.
As technology advances, identification of particular mutations may have a greater influence on choices of treatment. Mucopolysaccharidosis type I is a lysosomal storage disorder caused by a deficiency of alpha iduronidase. Certain mutations such as W402X will lead to the Hurler form of the disease for which the treatment of choice is a bone marrow transplant. Other mutations cause the Scheie or Hurler Scheie forms which are treated with enzyme replacement therapy.
Genetic testing in the UK is organised through the UK Genetic Testing Network. If a laboratory wishes to offer a genetic test for other UK genetic centres the test undergoes a vigorous assessment to see what the benefits of performing the test might be for the patient and or the wider family. Technical methods, sensitivity and specificity are also examined. For any new test, a set of testing criteria is drawn up to ensure that these often expensive tests are being used on the appropriate target population. Other key questions are what impact performing a test will have on the NHS and also what impact not doing the test would have? If the test is approved, then a recommendation is made that commissioners should fund the test, although this is not mandatory. There is also a realisation that some genetic testing will move into mainstream medicine as not everyone with a genetic disorder will always see a clinical geneticist. Having this clear process in place should ensure that genetic testing is provided on an equitable basis throughout the whole of the UK and that the tests provided are fit for purpose.
This review is by no means comprehensive. Some of the more complex issues such as genomic imprinting or mitochondrial inheritance are beyond its scope. Further source material can be found at www.geneticseducation.nhs.uk and www.ukgtn.nhs.uk.
So, where does clinical genetics go next? We hope to see the widespread introduction of CGH microarray, although in these difficult financial times it may not be viewed as a priority by some. That said, failing to move to CGH microarray and persisting with conventional karyotyping, could be viewed as similar to the rejection of penicillin and continuing with poultices and other such treatments! Improvements in technology mean that whole genome sequencing will not be prohibitively expensive and its introduction into clinical practice may not be too far away. Such a development would be very exciting but would mean that we need to ensure that those ordering and using such tests have a thorough understanding of their uses and limitations.
The author has no conflict of interest.
REFERENCES
- 1.Read A, Donnai D. New Clinical Genetics. Oxford: Scion Publishing Ltd; 2007. [Google Scholar]