

Abstract
Abstract
The field of agonist-activated Ca2+ entry in non-excitable cells underwent a revolution some 5 years ago with the discovery of the Orai proteins as the essential pore-forming components of the low-conductance, highly Ca2+-selective CRAC channels whose activation is dependent on depletion of intracellular stores. Mammals possess three distinct Orai proteins (Orai1, 2 and 3) of which Orai3 is unique to this class, apparently evolving from Orai1. However, the sequence of Orai3 shows marked differences from that of Orai1, particularly in those regions of the protein outside of the essential pore-forming domains. Correspondingly, studies from several different groups have indicated that the inclusion of Orai3 is associated with the appearance of conductances that display unique features in their gating, selectivity, regulation and mode of activation. In this Topical Review, these features are discussed with the purpose of proposing that the evolutionary appearance of Orai3 in mammals, and the consequent development of conductances displaying novel properties – whether formed by Orai3 alone or in conjunction with the other Orai proteins – is associated with the specific role of this member of the Orai family in a unique range of distinct cellular activities.
Trevor Shuttleworth obtained his BSc from the University of London, and a PhD from the University of Otago in New Zealand, and was a faculty member in the Department of Biological Sciences at the University of Exeter before moving in 1988 to the University of Rochester Medical Center in Rochester, New York, where he is currently Professor of Pharmacology and Physiology. Although initially a comparative physiologist, investigating mechanisms of epithelial ion transport, his work gradually transitioned to become focused on cellular mechanisms of agonist-activated calcium entry in non-excitable cells, culminating in the discovery of a novel store-independent, arachidonic acid-regulated calcium channel (the ARC channel). Like the store-operated CRAC channels, ARC channels are formed by members of the recently discovered Orai protein family, but they display a unique heteropentameric stoichiometry comprising two Orai3 subunits and three Orai1 subunits, and their activation relies exclusively on the pool of STIM1 that is constitutively resident in the plasma membrane.
 |
From the Oxford English Dictionary –‘Exceptional (adj.) – of the nature of or forming an exception; out of the ordinary course, unusual, special.’
The discovery of Orai proteins and the evolution of Orai3
Although the essential biophysical properties of the Ca2+ release-activated Ca2+ (CRAC) channels had been known since the early 1990s (Lewis & Cahalan, 1989; Hoth & Penner, 1992, 1993; Zweifach & Lewis, 1993), the molecular identity of the key proteins involved in the formation of the channel, and its activation as a result of the depletion of the endoplasmic reticulum (ER) Ca2+ stores, remained unclear for almost another 15 years. The eventual breakthrough came as a result of studies using RNAi-based screens designed to reveal proteins involved in the store-operated entry of Ca2+ via the CRAC channel. First, STIM1 was identified as the protein that senses the depletion of Ca2+ from the ER, and induces the activation of the Ca2+ entry channels (Liou et al. 2005; Roos et al. 2005). Subsequently, similar RNAi-based screens resulted in the discovery of the Orai proteins as the molecular components that formed the channel itself (Feske et al. 2006; Vig et al. 2006b; Zhang et al. 2006). In one particularly compelling study, these screening approaches were coupled with parallel studies using linkage analysis of patients suffering from severe combined immunodeficiency (SCID) (Feske et al. 2006), on the basis that store-operated Ca2+ entry was known to represent a critical step in the activation of T lymphocytes during immune responses (Lewis, 2001). Both approaches independently converged on a single gene product that was named Orai1. Critically, expression of Orai1 in T cells obtained from the SCID patients fully restored normal store-operated Ca2+ entry and typical CRAC-like Ca2+ currents in these cells. Two additional homologues of this gene in humans were found, and their respective products named Orai2 and Orai3 (Feske et al. 2006). Although Orai1 appeared to be a plasma membrane protein containing four transmembrane domains, its sequence showed no obvious homology with any other known ion channel. Consequently, it was not certain whether Orai1 was the protein that actually formed the CRAC channel itself or was a regulator of that channel. Indeed, another group who identified the same protein around the same time, also using a Drosophila RNAi screening approach, named it CRACM1 for ‘CRAC modulator 1’ (Vig et al. 2006b). However, several groups subsequently showed that co-expression of Orai1 along with STIM1 resulted in the appearance of greatly increased CRAC-like store-operated currents (Mercer et al. 2006; Peinelt et al. 2006; Soboloff et al. 2006; Lis et al. 2007), and similar data were obtained with Drosophila Orai (Zhang et al. 2006). More specifically, studies involving the demonstration that certain point mutations in a predicted pore region of the Orai1 sequence markedly affected ion selectivity confirmed that it represented the genuine channel-forming protein (Prakriya et al. 2006; Vig et al. 2006a; Yeromin et al. 2006).
Although it soon became clear that genes encoding highly conserved homologues of Orai proteins were present in organisms ranging from Caenorhabditis elegans to humans (Zhang et al. 2006), the first really comprehensive phylogenetic analysis of the Orai protein family was done by Cai (2007) who screened available whole-genome and protein databases to identify a putative evolutionary pattern of Orai protein development. This analysis revealed the presence of just a single Orai in most invertebrates (C. elegans, Drosophila, Aedes). The same single Orai was also seen in those invertebrates closest to the vertebrate line, such as sea urchins and the invertebrate urochordates (tunicates). The transition to the vertebrate lineage was associated with the first appearance of two distinct Orai genes (Orai1 and Orai2), whilst Orai3 does not appear until the mammals, apparently evolving from Orai1 rather than Orai2 (Fig. 1). Consequently, in evolutionary terms, Orai3 is the ‘newest’ Orai family member. As noted by Cai (2007), ‘Lineage specific expansion of protein families is believed to be one of the primary means of adaptation and bringing new biological functions in organisms subject to diverse environmental selective pressures’. Consistent with this is the increasing realization that Orai3 possesses unique properties, and behaves in unique ways, that are distinct from either Orai1 or Orai2. Consequently, this ‘late arrival’ of Orai3 raises the possibility that its evolution might be associated with a critical involvement in distinct activities within the cell. It is this thesis that will be explored in this review.
Figure 1. The evolution of Orai proteins – based on Cai (2007).
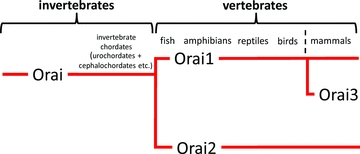
Invertebrates, including the invertebrate chordates, possess only a single Orai. The appearance of the Vertebrates is associated with the evolution of two Orai proteins – Orai1 and Orai2. Orai3 only appears in the mammals, apparently arising from Orai1. See text for details.
Structural distinctions
As noted above, the discovery of the Orai proteins, and the determination of their amino acid sequences, showed that they had no obvious relationship with any other known ion channel. This is perhaps not surprising, as it is now known that these proteins form channels with rather unique biophysical properties. Consistent with the shared fundamental features of the Orai channels – including their small overall whole-cell currents, very high selectivity for Ca2+, and predicted very small single channel conductance – the pore-forming transmembrane domains of all three Orai proteins show a high degree (∼82%) of conservation. However, simple examination of the overall sequences of these proteins reveals several regions where there are clear, often rather profound, differences. This is particularly so in the case of Orai3 (Fig. 2). Thus, compared to Orai1, major differences in sequence are seen in the cytosolic N- and C-termini (sequence identity of 34% and 46%, respectively), and in the extracellular loop between transmembrane domains 3 and 4 (21% sequence identity). As will be discussed later, of particular interest is the cytosolic N-terminus domain. In Orai1, this region is composed of ∼90 amino acids and contains clusters of prolines and arginines, particularly in the most distal parts of this domain. In contrast, the N-terminus of Orai3 comprises only ∼65 amino acids and has none of the clusters of prolines and arginines seen in Orai1. Only in the group of 22 residues immediately prior to the first transmembrane domain (H69–R91 in Orai1, H44–R66 in Orai3) is there obvious high sequence conservation between Orai1 and Orai3. Similarly, the extracellular loop linking transmembrane domains 3 and 4 is some 72 amino acids long in Orai3, compared to only 38 amino acids in Orai1. The net result is that the overall amino acid sequence identity between human Orai1 and Orai3 is approximately 57%. Together, the presence of obvious marked differences in the sequences of the ‘non-pore forming’ regions of the protein raises the possibility that they may impart unique properties to the respective channels, particularly in their modes of regulation and/or modulation. Of course, an obvious caveat to this is that simple sequence comparisons can often be a rather poor predictor of functional differences, as is clearly illustrated in those examples where a single point mutation in even large proteins can result in dramatic functional effects – such as the Δ508 mutation of the CFTR protein causing cystic fibrosis, and the E6V mutation in the haemoglobin sequence that gives rise to sickle-cell anaemia.
Figure 2. Structural comparison of the sequences of Orai1 and Orai3.
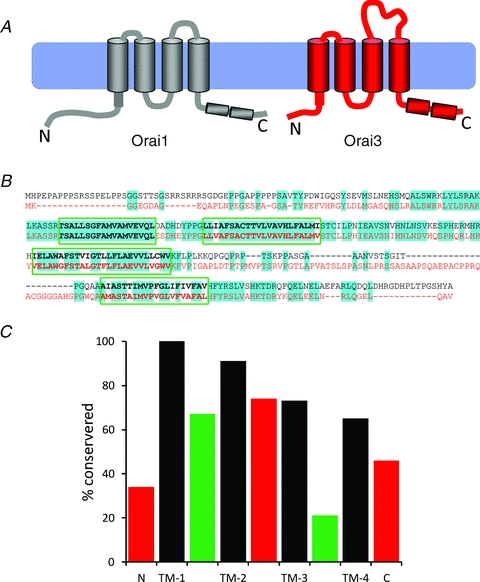
A, diagram illustrating the overall structural differences between Orai1 and Orai3, based on their respective protein sequences. Note the shorter N-terminus, the extended second extracellular loop, and the stronger probability of the two C-terminus coiled-coil domains in Orai3. B, protein sequence alignment of Orai1 (black) and Orai3 (red). Predicted conserved residues are highlighted (blue). C, graphical illustration of the degree of conservation in the protein sequences of the various regions of Orai1 and Orai3. Cytosolic domains are in red, transmembrane domains (TM-1-4) in black, and extracellular domains are in green.
Channel properties
As already noted, the discovery of Orai1 was based on its critical role in the store-operated entry of Ca2+. Consequently, the realization that Orai3 displayed properties that were distinct from those of Orai1 (and Orai2) began with studies showing that co-expression of Orai3 together with STIM1 failed to generate any measurable increase in either store-operated Ca2+ entry, or store-operated Ca2+-selective (CRAC-like) currents (Fig. 3A) (Mercer et al. 2006; DeHaven et al. 2007; Takahashi et al. 2007; Zhang et al. 2008). Similarly, whilst the siRNA-induced knockdown of Orai1 significantly reduced store-operated Ca2+ entry in cells expressing STIM1, the corresponding siRNA knockdown of Orai3 failed to affect such entry (Mercer et al. 2006). As a possible caveat to these knockdown data, it has been shown, at least in some cases, that the knockdown of Orai3 is accompanied by a significant increase in Orai1 expression (Gwack et al. 2007). Such an effect clearly could have obscured any Orai3-knockdown effect. Nevertheless, these early studies generally indicated that it was unlikely that Orai3 had a significant role in store-operated Ca2+ entry. However, contradicting these apparently negative results were studies showing that expression of Orai3 was able to rescue normal store-operated Ca2+ entry in cells in which such entry had been markedly reduced by the siRNA-induced knockdown of Orai1 (Mercer et al. 2006; DeHaven et al. 2007). Consistent with this, by using a more potent expression vector, Lis et al. (2007) were able to demonstrate that expression of Orai3 (CRACM3) in cells stably expressing STIM1 did indeed result in a modest increase in InsP3-induced store-operated CRAC-like currents (Fig. 3B). However, the magnitude of this effect was equivalent to only approximately 30% of that seen with a corresponding expression of Orai1 (CRACM1). Similar data were subsequently obtained by other groups (see, for example, DeHaven et al. 2007; 2008; Bogeski et al. 2010). Overall, the general consensus from these studies is that, like Orai1, the expression of Orai3 is capable of inducing a store-operated conductance, but its magnitude is considerably smaller than that seen with Orai1. As to the basis for this difference, it was known that the cytosolic N-terminal domain of Orai1 was essential for the activation of store-operated currents (Li et al. 2007; Takahashi et al. 2007; Muik et al. 2008; Park et al. 2009). Based on this, Lis et al. (2010), again using their more potent expression vector, showed that replacing the N-terminal cytosolic domain of Orai3 with the corresponding domain of Orai1 resulted in an approximate doubling in the magnitude of the measured store-operated Ca2+ currents, whilst the reverse exchange virtually eliminated all such currents. N-terminal deletion experiments narrowed the critical region down to amino acids 67–87 in Orai1 and 42–62 in Orai3. Although additional experiments showed that the appearance of significant store-operated currents depended on a single specific lysine residue in this domain (K85 in Orai1, and K60 in Orai3), the conservation of this residue in Orai1 and Orai3 cannot explain the differences in magnitude of store-operated Ca2+ currents between these two Orai family members. Alternatively, Bergsmann et al. (2011) have recently shown that N-terminal deletions to residues between W51 and Y55 significantly increase store-operated Orai3-dependent currents. The only sequence difference between Orai1 and Orai3 in this region is the substitution of a lysine in Orai1 for an arginine at position 53 in Orai3. Whether an R53K mutation in Orai3 would result in increased store-operated currents to levels similar to those seen with Orai1 has not been examined.
Figure 3. Examples of the biophysical differences between Orai1 and Orai3.
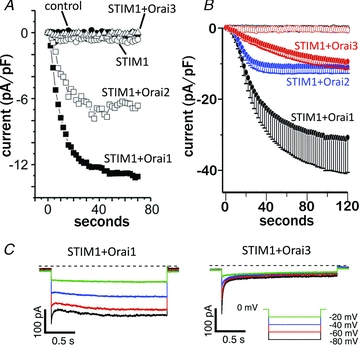
A, store-operated currents recorded in HEK293 cells following expression of DNA encoding the proteins indicated. Note that expression of STIM1 with either Orai1 or Orai2 results in greatly increased inward store-operated currents, whilst the corresponding expression of STIM1 with Orai3 fails to induce any significant store-operated currents. Data taken from DeHaven et al. (2007) with permission from The American Society for Biochemistry and Molecular Biology, © 2007. B, store-operated currents recorded in HEK293 cells following more extensive overexpression of STIM1 and the relevant Orai. Note that such overexpression now reveals store-operated currents with expression of STIM1+Orai3, but this still only represents approximately 30% of that seen with STIM1+Orai1. Data taken from Lis et al. (2007) with permission from Elsevier. C, fast inactivation of store-operated Ca2+ currents recorded during brief pulses to the negative potentials indicated, in STIM1-expressing cells transiently overexpressing Orai1 (left), or Orai3 (right). Note the much more pronounced and more rapid inactivation the cells expressing Orai3. Data taken from Lis et al. (2007) with permission from Elsevier.
Apart from their magnitude, the store-operated currents induced by the overexpression of Orai3 displayed additional differences from those seen with Orai1. One of the defining characteristics of native CRAC channels is their high selectivity for Ca2+ ions, and low permeability to monovalent cations such as Cs+ (Hoth & Penner, 1993; Zweifach & Lewis, 1996; Bakowski & Parekh, 2002; Kozak et al. 2002). Such selectivity properties are responsible for the marked inwardly rectifying current–voltage relationship, and very positive reversal potential, shown by these channels. Consistent with the evidence that native CRAC channels are formed by Orai1 subunits, the currents seen on overexpression of Orai1 together with STIM1 display a similar high selectivity for Ca2+ ions, and the consequent inwardly rectifying current–voltage relationship and very positive reversal potential (Prakriya et al. 2006; Vig et al. 2006a; Yeromin et al. 2006; Zhang et al. 2008). Essentially similar Ca2+ selectivity properties were indicated for the currents seen on expression of STIM1 with Orai3 (DeHaven et al. 2007; Lis et al. 2007). However, Lis et al. (2007) did report that, compared to those seen with Orai1, the Orai3 currents displayed a significantly increased permeability to Na+ when measured in the absence of external divalent cations, but no such difference was seen in the studies by DeHaven et al. (2007). This high degree of consistency in the Ca2+ selectivity of Orai1 and Orai3 is not surprising. Thus, earlier mutagenic studies in Orai1 suggested that a series of acidic residues in transmembrane domains 1 and 3 (E106, E190), and in the short extracellular loop between transmembrane domains 1 and 2 (D110, D112, D114) were critical determinants of a high Ca2+ selectivity (Vig et al. 2006a; Yeromin et al. 2006, 2007). The corresponding residues in Orai3 are E81 and E165 in the transmembrane domains, and E85, D87 and E89 in the extracellular 1–2 loop. Thus, the only difference is the substitution of glutamates for aspartates in two of the three residues in the 1–2 loop. Perhaps more critically, subsequent studies using a cysteine-scanning mutagenesis approach in Orai1 revealed that Ca2+ selectivity was exclusively determined by the E106 residue alone (McNally et al. 2009), and this is conserved (E81) in Orai3.
A unique feature of the Orai3-induced store-operated currents first noted by Lis et al. (2007) was that activation of the currents was significantly slower than that seen with Orai1 (or Orai2), an effect that was suggested to indicate possible differences in the binding or interaction of the Orai3 with STIM1. Evidence suggests that the initial interaction of STIM1 with the channel involves the cytosolic C-terminal region of the Orai proteins (Li et al. 2007; Muik et al. 2008; Frischauf et al. 2009). In all three Orai subtypes, this region contains a predicted coiled-coil domain that is critical for interactions with STIM1 (Muik et al. 2008). However, bioinformatic analyses indicate that the probability of the coiled-coil nature of this region is significantly weaker for Orai1 than for Orai3 (Frischauf et al. 2009). Consistent with this, a single point mutation (L273S) within this coiled-coil domain is sufficient to result in the full inhibition of the interaction with STIM1 and subsequent activation of Orai1 currents, whilst the corresponding mutation in Orai3 (L285S) results in only a partial inhibition. Full inhibition of the Orai3-induced currents required the incorporation of an additional mutation (L292S) in the coiled-coil domain. Of course, precisely how this apparent ‘stronger’ interaction between STIM1 and Orai3 might result in the slower rate of activation noted above is difficult to envisage. Moreover, like Orai3, the predicted probability of the coiled-coil domain in Orai2 is notably much stronger than in Orai1 (Frischauf et al. 2009), yet Ca2+-selective store-operated currents in cells expressing STIM1 and Orai2 activate at a rate that is very similar to that seen with Orai1 (Lis et al. 2007). Together, it seems clear that the detailed interactions between STIM1 and the Orai proteins involved in the actual gating of the channel are complex – involving both stimulatory and inhibitory actions of multiple domains in both STIM1 and Orai proteins. Given this, it is perhaps not surprising that identifying precisely which features of the Orai proteins are responsible for such differences in the kinetics of activation on store depletion is problematic.
Another kinetic feature of the store-operated currents induced by Orai proteins, as well as by native CRAC channels, is a Ca2+-dependent fast inactivation seen during brief pulses to hyperpolarizing potentials. In this context, Lis et al. (2007) showed that the expression of Orai3 resulted in a significantly more pronounced, and more rapid, fast inactivation than that seen on expression of Orai1 (Fig. 3C). A subsequent study has suggested that this effect is dependent on the presence of three conserved glutamates (E281, E283, E284) in the C-terminal region of Orai3 (Lee et al. 2009). However, these glutamates are fully conserved in Orai2 (E233, E235, E236), and cells expressing STIM1 and Orai2 show a Ca2+-dependent fast inactivation that is significantly slower than that seen with Orai3. In contrast to these findings, Yamashita et al. (2007) have suggested that fast inactivation in Orai1 is determined by the same key acidic residues that were originally proposed to be involved in determining Ca2+ selectivity (see above). As already noted, the only difference in the corresponding residues in Orai3 is that the two aspartate residues D110 and D114 in Orai1 are glutamates in the corresponding Orai3 sequence (E85 and E89). Alternatively, based on an early study that had suggested that Ca2+-dependent fast inactivation may be calmodulin dependent (Litjens et al. 2004), a region in the cytosolic N-terminal domain of Orai1 (E68-R91) that binds calmodulin in a Ca2+-dependent manner was identified (Mullins et al. 2009). Mutations in this region that eliminate the ability of this domain to bind Ca2+–CaM also result in the inhibition of Ca2+-dependent fast inactivation. Comparison of the relevant critical CaM-binding sequence in Orai1 (S70-K87) with that in Orai3 (S45-K62) reveals just three differences – a Met–Leu change at position 2 in the sequence, a Gln–Arg at position 3, and a Lys–Arg at position 9. Whether these differences can account for the distinct rates of fast inactivation displayed by Orai1 and Orai3 remains to be examined. More recently, Srikanth et al. (2010) identified a short sequence in the intracellular loop connecting transmembrane domains 2 and 3 of Orai1 (151VSNV154) where mutation of the residues to alanines led to the complete loss of Ca2+-dependent fast inactivation. It was proposed that this loop acts as an ‘inactivation particle’ that may interact with the N-terminal CaM-binding region in Orai1 to modulate Ca2+-dependent fast inactivation. Once again, the only difference between this sequence in Orai1 and the corresponding sequence in Orai3 is that the final valine in Orai1 is an isoleucine in Orai3, theoretically representing only a modest increase in hydrophobicity. Finally, to make things even more complex, Scrimgeour et al. (2009) have shown that the rate and extent of fast inactivation is also highly dependent on the ratio of expressed STIM1 to Orai1 protein. Therefore simple differences in expression levels between transfected Orai1 and Orai3 constructs might significantly impact the rate of fast inactivation.
Channel pharmacology
Generally speaking, to date, there is no really specific pharmacology for Orai channels. Because they form highly Ca2+ selective conductances, they are subject to block by lanthanides such as La3+ (50–100 μm) and Gd3+ (1–5 μm), but these are essentially generic blockers of Ca2+ entry channels and certainly cannot be considered as diagnostic for the involvement of Orai proteins in such channels. Additional non-specific blockers include SKF96365, the myosin light chain kinase inhibitor ML-9 (Smyth et al. 2008), and the bistrifluoromethyl-pyrazole derivative BTP2 (Zitt et al. 2004). Undoubtedly, the one compound that has attracted most attention, and its effects most extensively studied, is 2-aminoethoxydiphenyl borate (2-APB). This compound was originally characterized as an inhibitor of InsP3 receptors (Maruyama et al. 1997; Bilmen & Michelangeli, 2002), but was subsequently shown to have multiple diverse effects including both the inhibition and activation of various different members of the TRP channel family (Voets et al. 2001; Trebak et al. 2002; Chung et al. 2004; Hu et al. 2004; Li et al. 2006; Juvin et al. 2007), and the inhibition of SERCA pumps (Missiaen et al. 2001; Peppiatt et al. 2003), as well as to effect store-operated Ca2+ entry via CRAC channels (Gregory et al. 2001; Iwasaki et al. 2001; Prakriya & Lewis, 2001). Regarding its effects on CRAC channels, early studies showed that it displayed a bifunctional effect that was dependent on the concentration used. Store-operated Ca2+ entry and native CRAC channel currents are enhanced by concentrations of 2-APB below 10–20 μm, but are profoundly inhibited at higher concentrations (above 30 μm) (Prakriya & Lewis, 2001). Essentially similar data were seen in cells expressing STIM1 and Orai1 (Peinelt et al. 2006; Lis et al. 2007; DeHaven et al. 2008; Peinelt et al. 2008). Importantly, all these stimulating and inhibitory effects on native CRAC channels and on expressed Orai1 did not involve any obvious changes in the ion selectivity of the channels, at least as evaluated by the current–voltage relationship. In marked contrast to this, in cells expressing STIM1 and Orai3, high concentrations of 2-APB were shown to induce a slowly developing, but ultimately rather profound, increase in store-operated currents (Lis et al. 2007; DeHaven et al. 2008; Peinelt et al. 2008; Schindl et al. 2008). In addition, this 2-APB induced enhancement of store-operated currents in the Orai3 expressing cells was accompanied by marked changes in ion selectivity resulting in a current–voltage relationship displaying clear double rectification, with large outward currents above +50 mV (DeHaven et al. 2008; Peinelt et al. 2008). In marked contrast, Zhang et al. (2008) reported that 50 μm 2-APB had no effect on store-operated Ca2+ entry in cells expressing both Orai3 and STIM1. The reason for this discrepancy is not clear.
Store-independent activation of Orai3 channels
The above data certainly indicate a potential for a unique response of store-operated Orai3 currents to the higher concentrations of 2-APB. Perhaps more surprisingly, several studies have demonstrated that Orai3 can be directly activated by similar concentrations of 2-APB, in a manner that is independent of any store depletion, and even of any involvement of STIM1 (DeHaven et al. 2008; Peinelt et al. 2008; Schindl et al. 2008; Zhang et al. 2008; Wang et al. 2009). These direct 2-APB induced currents display large inward and outward currents (i.e. they show double rectification) and a leftward shift in the reversal potential (Fig. 4) – features that indicate a marked reduction in Ca2+ selectivity, and an increased permeability to monovalent cations (DeHaven et al. 2008; Peinelt et al. 2008; Schindl et al. 2008; Zhang et al. 2008). Interestingly, both Peinelt et al. (2008) and DeHaven et al. (2008) noted that a similar, although very much smaller, direct effect of 2-APB could also be detected with Orai1 channels. The importance of this will become clear below.
Figure 4. Direct, store-independent activation of currents by 2-APB in cells expressing either Orai1 or Orai3.
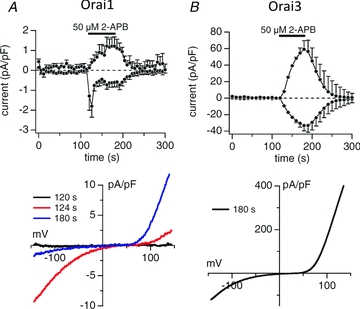
A, the effects of application of 2-APB (50 μm) on currents in HEK293 cells expressing Orai1. The top figure shows the inward (at −80 mV) and outward (at +80 mV) currents, whilst the bottom figure shows the relevant current–voltage relationships obtained at the time points indicated. B, the corresponding effects of 2-APB (50 μm) on inward (at −80 mV) and outward (at +80 mV) currents, and their current–voltage relationship in HEK293 cells expressing Orai3. Note that the scale for the measured currents in the Orai3-expressing cells is 40 times larger than for the Orai1 expressing cells. Data taken from Peinelt et al. (2008) with permission from John Willey and Sons.
As to the molecular basis for these effects, in a series of experiments using chimeric constructs of Orai1 and Orai3, Zhang et al. (2008) concluded that transmembrane domains 2 and 3, together with the linking intracellular loop, were required for 2-APB to directly activate Orai3 channels to admit cations in a relatively non-selective way. Peinelt et al. (2008) used a rather different approach involving single-point mutations of putative critical amino acids in the selectivity filter of the Orai1 pore (E106D and E190A) to show that such mutations enabled 2-APB to directly gate the channels in a STIM1-independent manner. However, these residues are entirely conserved in the Orai3 sequence, so precisely why the effect of 2-APB should be so much more pronounced in the Orai3-induced currents was unclear. Nevertheless, these findings do suggest that 2-APB exerts its direct effects on Orai3 channels by inducing changes in the pore architecture. This was supported by direct evaluation of the pore size, as assayed by the permeation of methylated derivates of ammonium (Schindl et al. 2008). These authors showed that exposure to 2-APB increased the minimum pore size from a value of ∼3.8 Å to greater than 5.3 Å, an effect that was apparently dependent on the E165 residue of Orai3 that lies in the third transmembrane domain. However, as noted above, this residue is conserved in Orai1 (E190), so the basis for the different responses of Orai1 and Orai3 to 2-APB remained unresolved.
A more recent detailed analysis of the effects of 2-APB on Orai3 channel function by Yamashita et al. (2011) has, perhaps, helped to resolve these apparent differences. These authors showed that, like Orai1, store-operated Orai3 currents are potentiated by 2-APB at low concentrations (<10 μm) without affecting ion selectivity. This effect requires the presence of STIM1, and is strictly dependent on store depletion as it is not seen in cells whose store content is maintained. In addition, and contrary to the previous reports, they showed that 2-APB also inhibits the same store-operated currents at higher 2-APB concentrations (>20 μm). It was suggested that this inhibition of store-operated Orai3 currents by higher 2-APB concentrations may have been obscured in the earlier studies by the concomitant development of the 2-APB-induced store-independent mode of activity. Critically, this switch to a direct 2-APB-induced activity is associated with a corresponding reduction in ion selectivity, and is delayed in the presence of STIM1. Consistent with certain earlier reports noted above (DeHaven et al. 2008; Peinelt et al. 2008), the store-independent mode of activation by high concentrations of 2-APB in the absence of STIM1 was also seen in Orai1. However, the magnitude of the effect was equivalent to only about 5–6% of that seen with the Orai3 currents, and was lost in the presence of STIM1. The data indicated that both the store-dependent activation and the 2-APB-dependent potentiation seen at low 2-APB concentrations were associated with an increased binding of STIM1 to the channel. As for the direct store-independent gating of the Orai channels seen at higher concentrations of 2-APB, several lines of evidence pointed to this involving a functional uncoupling of STIM1 from the channel, with Orai1 being significantly more resistant than Orai3 to such uncoupling. The overall responses can therefore be explained by a mechanism in which 2-APB must first disengage STIM1 before it can directly gate the Orai3 channels. However, precisely why Orai1 should be more resistant than Orai3 to the uncoupling of STIM1 from the respective channel remains unclear.
Of course this store-independent activation of Orai3 channels by 2-APB represents a strictly pharmacological phenomenon. However, store-independent activation of channels containing Orai3 has also been shown under more physiologically relevant conditions. Thus, Orai3 has been shown to be an essential component of the store-independent, arachidonic acid activated, Ca2+-selective ARC channels (Mignen & Shuttleworth, 2000; Mignen et al. 2008a). These channels are found in a variety of different cell types, frequently co-existing with store-operated CRAC channels (Yoo et al. 2000; Mignen et al. 2003; Mignen et al. 2005; Li et al. 2008; Yeung-Yam-Wah et al. 2010), and display very similar basic biophysical properties. Interestingly, despite their insensitivity to store depletion, studies showed that activation of the ARC channels was absolutely dependent on the presence of STIM1, just like the CRAC channels (Mignen et al. 2007). However, although STIM1 was identified in 2005 as the sensor of ER Ca2+ store depletion, it was originally discovered almost 10 years earlier (Oritani & Kincade, 1996; Parker et al. 1996) as a plasma membrane protein with apparent functions in the suppression of tumour cell growth (Sabbioni et al. 1997; Manji et al. 2000; Williams et al. 2001, 2002). Evidence indicates that typically ∼15–25% of the total cell STIM1 is constitutively resident in the plasma membrane and, critically, it is this pool of STIM1 that is responsible for the activation of the ARC channels. The close biophysical similarities shown by CRAC and ARC channel currents, together with their co-dependence on STIM1 for activation, suggested that Orai proteins might also form these store-independent ARC channels. Subsequently, the specific involvement of Orai3 in these channels was revealed in studies with a HEK293 cell line stably expressing the m3 muscarinic receptor, where co-expression of STIM1 with either Orai1 or Orai3, but not Orai2, resulted in markedly increased (1.5- to 2-fold) arachidonic acid-activated store-independent ARC channel currents (Mignen et al. 2008a). Apart from their unique mode of activation, these currents could be readily distinguished from the co-existing CRAC channel currents by their complete additivity to the currents induced by store depletion, and the absence of any detectible fast inactivation. In cells stably expressing STIM1, the expression of Orai1 markedly increased both store-operated CRAC channel currents and store-independent arachidonic acid-activated ARC channel currents. However, whilst expression of a dominant-negative mutant of Orai3 (E81Q) had no effect on store-operated CRAC channel currents, the same expression reduced currents through the store-independent ARC channels to negligible levels (Mignen et al. 2008a). Together, these data indicated that the currents recorded for the arachidonic acid-activated ARC channels were likely to reflect the activity of heteromeric Orai1/Orai3 channels. The specific properties of these, and other potential heteromeric channels containing Orai3, will be discussed in the following section.
Orai3 in heteromeric channels
Essentially, the above reports describing the features of currents recorded following overexpression of just Orai3, either with or without STIM1, carry the implied assumption that the properties identified represent those of ‘Orai3 channels’. However, this essentially ignores the possibility of interactions and/or combinations with endogenous Orai proteins. The only exceptions to this general approach are those cases where the Orai3 construct was transfected into cells in which endogenous Orai1 had been reduced by siRNA treatment (Mercer et al. 2006; DeHaven et al. 2007). However, because early studies indicated that expression of Orai3, along with STIM1, failed to induce clearly detectible increases in store-operated currents (Mercer et al. 2006; Takahashi et al. 2007; Zhang et al. 2008), subsequent studies typically utilized fairly extensive overexpression of the protein, often resulting in currents that were up to 50–60 times larger than native store-operated currents in the same cell. Consequently, it is probably not unreasonable to assume that, under these conditions, monomeric Orai3 channels predominated in these studies. Nevertheless, the ability of Orai proteins to naturally form heteromeric assemblies was recognized even in some of the earliest studies on these proteins (Gwack et al. 2007; Lis et al. 2007; Zhang et al. 2008). Particularly convincing in this regard was the demonstration that overexpression of Orai3 together with an equal amount of a non-conducting pore mutant of Orai1 (E106Q), resulted in the complete abrogation of the increased store-operated currents seen on overexpression of Orai3 alone (Lis et al. 2007). Given the widespread expression of both Orai1 and Orai3 in most tissues (Gross et al. 2007; Gwack et al. 2007; Takahashi et al. 2007), naturally occurring Orai1/Orai3 heteromeric channels would certainly seem likely, raising the possibility that such combinations of Orai proteins might operate in unique ways in the cell (Mercer et al. 2006).
With regard to the potential properties of such heteromeric channels when activated by store depletion, Schindl et al. (2009) noted that, in contrast to the usual highly Ca2+ selective store-operated currents seen on expression of STIM1 with either Orai1 or Orai3, the currents seen on co-expression of STIM1 with both Orai1 and Orai3 together displayed small outward currents and a left-shifted reversal potential (∼+30 mV) – an effect apparently reflecting an increased permeability to Cs+ ions. As the authors noted, a problem of such a simple co-expression approach is that the resulting currents are likely to represent a mixture of those originating from homomeric and heteromeric channels. To address this, they examined the effect of expressing an Orai3–Orai1 dimer construct. As with the co-expression of the monomers, the store-operated currents resulting from expression of this Orai3–Orai1 dimer displayed marked inward rectification, along with significant outward currents and a reversal potential of ∼+26 mV. Again, the evidence indicated that this resulted from an increased permeability to Cs+. Interestingly, in the above study, FRET analysis following combined expression of differently tagged versions of the Orai3–Orai1 dimer indicated that they assembled to give functional tetrameric channels. Given this, it is curious that the properties reported above by Schindl et al. (2009) are very different from those seen on expression of the equivalent tetramers as concatenated constructs containing the appropriate pairs of Orai1 and Orai3 subunits, in either an Orai3–Orai1–Orai3–Orai1 conformation or an Orai3–Orai1–Orai1–Orai3 conformation (Mignen et al. 2009). As will be discussed below, these constructs gave store-operated currents displaying marked inward rectification, and no indication of any outward currents at least at voltages up to +60 mV. The reason for this obvious difference is not clear.
The same authors also examined the effects of 2-APB (75 μm) on these store-operated Orai3–Orai1 currents, and showed that the initial action of the drug was to modestly increase inward currents in a STIM1-dependent fashion (Schindl et al. 2009). Subsequently, this inward current declined somewhat, with a simultaneous development of significant outward currents at membrane potentials above +40 mV, resulting in a current–voltage relationship displaying clear double-rectification. As such, these effects are very similar to those generated on expression of Orai3 alone and were thought to result from the replacement of two glutamates in the first extracellular loop of Orai1 with aspartates in Orai3 (Schindl et al. 2009). However, as already discussed, other studies have shown that this region of the protein is actually highly flexible and essentially non-selective (McNally et al. 2009).
As noted above, studies had shown that the store-independent arachidonic acid-activated ARC channel currents required the presence of both Orai1 and Orai3 (Mignen et al. 2008a), indicating that the functional ARC channel was heteromeric in nature. Given the evidence that the functional CRAC channel was formed by a tetrameric assembly of Orai1 subunits (Ji et al. 2008; Mignen et al. 2008b; Penna et al. 2008; Maruyama et al. 2009), it seemed reasonable to assume that the ARC channel might also be a tetramer, but one formed from both Orai1 and Orai3 subunits. Indeed, expression of concatenated tetrameric constructs containing either one or two Orai3 subunits along with the appropriate Orai1 subunits in cells stably expressing STIM1, all resulted in significant (3- to 4-fold) increases in arachidonic acid-activated currents (Mignen et al. 2009). Only a heterotetramer containing three Orai3 subunits failed to give significantly increased currents. However, further examination revealed that expression of these same concatenated heterotetramers resulted in a similar 3- to 4-fold increase in store-operated CRAC-like currents (Fig. 5). Because endogenous ARC channel currents are entirely independent of store depletion, it was clear that the channels formed on expression of these heterotetrameric constructs were not equivalent to those of the native ARC channels. Subsequent experiments eventually revealed that conductances whose properties matched, in every respect, those of the endogenous ARC channels were exclusively formed by pentameric constructs comprising two Orai3 subunits and three Orai1 subunits (Fig. 5). The essential ARC channel properties duplicated by expression of such constructs included the characteristic markedly inwardly rectifying current–voltage relationship, with an absence of any significant outward currents at voltages up to +100 mV, activation by the same low concentrations (2–8 μm) of arachidonic acid, insensitivity to 2-APB, and an absolute dependence on the pool of STIM1 residing in the plasma membrane for their activation (Mignen et al. 2009). Additional experiments involving co-expression of the concatenated pentamer with dominant-negative Orai1 or Orai3 monomers, or incorporation of such dominant-negative subunits into the concatenated construct, indicated that it was unlikely that either additional subunits were required to form the functional channel, or that individual subunits were being ‘excluded’ in the final channel assembly. Finally, the same specific arachidonic acid-activated currents could also be generated by the co-expression of appropriate combinations of either tetramers and monomers, or trimers and dimers, further demonstrating the validity of this admittedly unexpected heteropentameric conformation (Mignen et al. 2009).
Figure 5. Store-operated and arachidonic acid-activated currents in HEK293 cells stably expressing STIM1, and in the same cells expressing various concatenated heteromeric Orai1/Orai3 constructs.
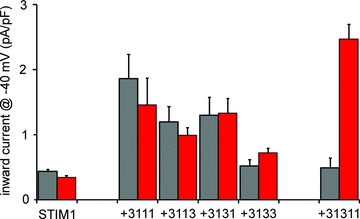
Expression of Orai1/Orai3 heterotetramers in various configurations and stoichiometries induces both increased store-operated (CRAC-like) currents (grey bars) and arachidonic acid-activated (ARC-like) currents (red bars). Only the 3–1–3–3 heterotetramer failed to induce any significantly increased currents. Expression of an Orai1/Orai3 heteropentameric construct containing two Orai3 subunits failed to increase store-operated currents, but induced large arachidonic acid-activated currents that displayed all the characteristic features of endogenous ARC channels. See text for details.
The specific requirement for Orai3 subunits in the ARC channel for it to be sensitive to activation by low concentrations of arachidonic acid was certainly intriguing. Examination of the basis for this effect showed that it required the presence of two, but no more than two, Orai3 subunits within the pentamer, and that the switch to a specific arachidonic acid sensitivity for channel activation was dependent exclusively on the cytosolic N-terminal domain of Orai3 (Thompson et al. 2010). However, it is important to note that this does not necessarily imply that this region of Orai3 is where arachidonic acid itself is acting, as it is not yet known whether the ARC channels are activated directly by arachidonic acid, or whether such activation involves some arachidonic acid-dependent regulator. Moreover, as already pointed out, activation of the ARC channels by arachidonic acid differs from store-operated Orai channels in that it specifically requires the presence of STIM1 in the plasma membrane (Mignen et al. 2007). Thus activation of the channel may involve an action of arachidonic acid on this pool of STIM1, either alone or in a pre-assembled complex with the channel, or it may drive the formation of such a complex. In addition, the data suggest that the acquisition of selective activation by arachidonic acid is not simply achieved by the presence of two Orai3 subunits alone. Thus, as noted above, tetrameric constructs containing two such subunits display an approximately equal sensitivity to activation by arachidonic acid and by store depletion (Mignen et al. 2009). Consequently, it would seem that the number of Orai1 subunits (i.e. three in the heteropentamer that forms the functional ARC channel), or the formation of Orai subunits into a pentameric channel structure, is also playing some role in determining the selectivity for activation by arachidonic acid.
Functional roles (Table 1)
Table 1.
Orai3-dependent physiological actions (see text for details)
| Physiological activity | Cell type | Channel involved |
|---|---|---|
| Store-operated Ca2+ entry | Breast cancer cells | Orai3 channels? |
| Store-operated Ca2+ entry (specifically under highly oxidizing conditions) | Effector T-helper cells | Orai3 channels? or Orai3/Orail channels? |
| Cell proliferation | Breast cancer cells | Orai3 channels? |
| Agonist-induced Ca2+ oscillations | HEK293 cells | ARC channels |
| Parotid and pancreatic acinar cells | (Oral + Orai3) | |
| Agonist-induced and glucose-induced insulin secretion | Pancreatic β cells | ARC channels (Orail +Orai3) |
To date, examination of the potential functional roles of Orai channels has been dominated by studies on Orai1, largely because of its critical role in CRAC channel function in several types of immune cells (Feske, 2009; Hogan et al. 2010). Yet, Orai3 expression displays a tissue distribution at least as wide as that of Orai1 (Gross et al. 2007; Gwack et al. 2007; Takahashi et al. 2007), suggesting the likelihood that important functional roles for this Orai family member exist, either alone or in combination with Orai1 (or Orai2). Unfortunately, in many studies, the simple demonstration that a particular cellular function or activity is impacted by the knockdown, or overexpression, of Orai1 is assumed to indicate that this implies the involvement of a classic store-operated CRAC-like conductance. Such an assumption clearly overlooks the possibility of any involvement of heteromeric Orai channels, such as those involving both Orai1 and Orai3.
Obviously, the demonstration of a role of Orai3 in any particular cellular function requires specific tools to identify such an activity. Examination of the simple overexpression of Orai3, either with or without STIM1, is an unreliable assay for identifying potential physiological roles of this protein as it is uncertain that the resulting channels will function, or be regulated, in the same way as they do when existing at endogenous expression levels. Much information about the important role of Orai1, particularly in immunological responses, has come from studies of patients carrying a critical mutation (R91W) in Orai1 (Feske, 2009), or from Orai1 knockout mice (Gwack et al. 2008; Vig et al. 2008; Braun et al. 2009). Unfortunately, the successful development of the corresponding Orai3 knockout mouse has yet to be achieved. Consequently, the most widely used approach to date has involved examination of the effect of 2-APB on the associated currents or entry of Ca2+, relying on the reported ability of higher concentrations of the drug (typically 50–100 μm) to increase such activity. However, the complex nature of the effects of 2-APB on STIM1 and Orai proteins (DeHaven et al. 2008), along with its obvious promiscuous effects on other molecules critical to cellular signalling activities, undoubtedly raises concerns as to the specificity and reliability of such an assay. A better approach might be the expression of a dominant-negative mutant of Orai3, such as the E81Q mutation, whose incorporation into the channel structure results in a non-conducting channel pore. Of course, the disadvantage of this approach is that it requires the transfection and expression of this mutant construct, something that it not always possible in native cells.
Orai3 and store-operated Ca2+ entry
Despite the well-established role of Orai1 in store-operated Ca2+ entry, there are indications that this feature may not be exclusive to the Orai1 protein. For example, McCarl et al. (2009) noted that, although clinically serious, the effects of deficiencies in Orai1 function in patients bearing various functionally relevant mutations in the gene were curiously limited in their range of targets – being essentially restricted to effects on T-cell activation, ectodermal dysplasia, and some skeletal muscle function and differentiation. As these authors noted, such a narrow range of effects is difficult to reconcile with the widespread expression of Orai1 throughout the body tissues, and the apparent universal presence of store-operated Ca2+ entry in cells. As an explanation, it was suggested that this might reflect an ability of other Orai family members (Orai2 and/or Orai3) to functionally replace Orai1 in store-operated Ca2+ entry in the unaffected tissues. However, as already noted, the original studies involving modest overexpression of Orai3 or siRNA-induced knockdown of endogenous Orai3 indicated that any such activity would likely be minimal, at best (Mercer et al. 2006; DeHaven et al. 2007; Takahashi et al. 2007; Zhang et al. 2008). Consistent with this are findings showing that, despite being expressed at normal levels and being efficiently delivered and inserted into the plasma membrane, expression of either Orai2 or Orai3 failed to rescue appreciable store-operated Ca2+ entry in T cells and fibroblasts obtained from SCID patients bearing the R91W mutant Orai1, which lacks any CRAC channel activity (Gwack et al. 2007). Clearly, at least for these cell types, store-operated Ca2+ entry is exclusively dependent on Orai1.
However, a recent study of certain cancerous and non-cancerous breast cell lines has demonstrated the existence of an endogenous store-operated Ca2+ entry pathway that is mediated by Orai3 (Motiani et al. 2010). This revealed that, in such cell lines that are negative for the oestrogen receptor (ER− lines), store-operated Ca2+ entry occurs via the ‘classic’ Orai1-dependent pathway. In contrast, store-operated Ca2+ entry in ER+ cell lines was mediated via an Orai3-dependent pathway. This specific involvement of Orai3 was demonstrated by the potentiating effect of 30–50 μm 2-APB on store-operated Ca2+ signals and on the associated currents, the full replication of the biophysical properties of such 2-APB-potentiated Orai3 channel currents (doubly rectifying current–voltage relationship and leftward shift in reversal potential), and the direct activation of currents by 2-APB in the absence of store depletion. In addition, whilst siRNA knockdown of Orai1, but not of either Orai2 or Orai3, virtually eliminated store-operated Ca2+ entry in the ER− cells, only the siRNA knockdown of Orai3 was effective in both markedly reducing store-operated Ca2+ entry and inhibiting the 2-APB induced potentiating effect on store-operated currents in the ER+ cells. These data suggest that, in these ER+ breast cancer cell lines, store-operated Ca2+ entry is being mediated by channels formed exclusively by Orai3. As such, this represents the first clear example of a native store-operated Ca2+ entry pathway mediated by Orai3. As to the functional role of this pathway, a recent study has shown that it probably plays a key role in the proliferation and survival of the breast cancer cells. Thus, siRNA knockdown of Orai3 results in the inhibition of cell proliferation, an arrested cell cycle progression, and an increase in apoptosis in such cells (Faouzi et al. 2011).
The siRNA data described above would seem to indicate that these channels are formed exclusively by Orai3 – i.e. they are genuine ‘Orai3 channels’. Whether similar such Orai3-mediated store-operated Ca2+ entry pathways exist in other cell types remains an interesting, and as yet largely unexplored, question. In this context, a comment in the report by Yamashita et al. (2011) is worth noting. In this, the authors point out that, because the ability of 2-APB to directly gate Orai3 channels is subject to inhibition by the level of functional STIM1 in the cell, an exclusive reliance on assays based on this feature as a ‘marker’ for such Orai3-dependent conductances could result in many false negatives, particularly when the endogenous STIM1 levels are high, or Orai3 levels are relatively low. In much the same way, the demonstration in some cell types that siRNA-induced knockdown of Orai3 is associated with a significant increase in Orai1 expression could also result in potential Orai3-dependent store-operated pathways being overlooked. Clearly, any comprehensive search for such conductances must involve the simultaneous application of multiple types of assay.
An additional novel role for Orai3 has recently been identified in the differentiation of naive T-helper (TH) cells into effector cells (Bogeski et al. 2010). This differentiation is associated with a reduced redox sensitivity, which is critical for allowing the cells to proliferate, differentiate, and function in the highly oxidizing environment typical of sites of inflammation. It was shown that, unlike Orai1 channels, store-operated channels formed on expression of Orai3 are not inhibited by oxidation by hydrogen peroxide – an effect associated with the absence of a critical reactive cysteine residue (C195) in Orai3. Correspondingly, it was shown that the decreased redox sensitivity of effector TH cells is associated with an increased expression of Orai3, along with increased levels of intracellular antioxidants, whilst siRNA knockdown of Orai3 in the effector TH cells rendered them significantly more redox sensitive. Presumably, this Orai3-dependent decreased sensitivity to reactive oxygen species will allow the Ca2+ signalling essential for effective cell proliferation and cytokine production to operate under conditions where the normal CRAC-like Orai1 channels would be unable to function (Bogeski et al. 2010). As the authors note, whether these redox-resistant Orai3-dependent channels seen in the effector TH cells represent monomeric Orai3 or heteromeric Orai1/Orai3 channels has yet to be resolved.
Orai3 and store-independent Ca2+ entry
Perhaps the most obvious unique property of channels involving Orai3 is their ability to be activated independently of store depletion, either pharmacologically by 2-APB or, more physiologically, by agonist-generated levels of intracellular arachidonic acid. With regards to the latter, the essential role of Orai3 along with Orai1 in forming the functional arachidonic acid-regulated Ca2+ selective ARC channel has already been discussed. As noted, the currents resulting from the activity of these channels, whilst superficially similar to those of the Orai1 CRAC channel, can be readily distinguished by several unique characteristics. These include the absence of any obvious Ca2+-dependent fast inactivation, a lack of inhibition by reductions in extracellular pH, insensitivity to either inhibitory or stimulatory effects of 2-APB, a specific dependence on STIM1 resident in the plasma membrane and, of course, their complete additivity with co-existing store-operated currents (Mignen & Shuttleworth, 2000; Mignen et al. 2003, 2007). In addition, it is now becoming increasingly clear that these ARC channels are found in a wide variety of different cell types, frequently coexisting with CRAC channels or other forms of store-operated channels (e.g. TRPC channels).
Of course, compared to the extensively studied CRAC channels, the store-independent ARC channels are relative ‘newcomers’ to the field, and their possible physiological roles have only recently begun to be explored. One particularly interesting feature is their apparent specific activation at low, physiologically relevant agonist concentrations, a property that has been demonstrated both in cell lines and in native cells (Mignen et al. 2001; Mignen et al. 2005). It has long been known that the Ca2+ signals induced by such agonist concentrations typically take the form of repetitive spikes or oscillations in cytosolic Ca2+ (Berridge & Galione, 1988; Berridge, 1990; Jacob, 1990; Thomas et al. 1991). Such oscillatory Ca2+ signals are thought to play a key role in providing a relatively low noise signal of almost infinite bandwidth, features that seem critical in the ability of the cell to selectively induce the appropriate response from the multitude of intracellular effectors that are known to respond to increases in cytosolic Ca2+– i.e. different effectors selectively respond to different frequencies of agonist-induced Ca2+ oscillations (Hajnoczky et al. 1995; De Koninck & Schulman, 1998; Dolmetsch et al. 1998). In addition to agonist concentration, a key determinant of the frequency of these oscillatory Ca2+ signals is the rate of agonist-activated Ca2+ entry (Rooney et al. 1989; Martin & Shuttleworth, 1994; Bootman et al. 1996). Exactly how the entering Ca2+ acts to modulate the frequency of the oscillations, which themselves reflect the repetitive release and re-uptake of Ca2+ from intracellular Ca2+ stores (essentially the ER), has long been a subject of debate. Effects on the refilling of the stores between each oscillation, and direct effects on the activity of the InsP3 receptors responsible for the release, have generally predominated in these discussions. However, we have recently described a novel mechanism by which Ca2+ entering specifically via the ARC channels can modulate oscillation frequency as a result of a direct activation of a phosopholipase C-δ (PLCδ) (Thompson & Shuttleworth, 2011). This activation occurs in a highly localized manner, and is entirely unaffected by a similar entry of Ca2+ via the co-existing CRAC channels. The assumption is that the InsP3 generated by this Ca2+ entry-dependent PLCδ activity will add to that resulting from the ‘classic’ receptor-stimulated PLCβ (or PLCγ) activity, to increase the frequency of Ca2+ oscillations. In confirmation of this, partial inhibition of this effect by the expression of either a dominant-negative Orai3 mutant, or a catalytically impaired PLCδ mutant, resulted in the clear reduction in agonist-induced Ca2+ oscillation frequency (Thompson & Shuttleworth, 2011).
Finally, a recent study has also indicated another, potentially clinically relevant, role of these ARC channels in insulin secretion by pancreatic β cells (Yeung-Yam-Wah et al. 2010). In this, it was shown that the known ability of glucose, as well as various insulin secretagogues including acetylcholine and cholecystokinin, to induce increases in cellular arachidonic acid results in the activation of ARC channels in the β cells, increasing cytosolic Ca2+ levels and enhancing the subsequent secretion of insulin.
Conclusions
Together, the data presented indicate that Orai3 differs significantly in its sequence and structural features from other Orai proteins, and it displays several unique properties. Moreover, there is increasing evidence suggesting that the involvement of Orai3 is critical for certain specific, distinct roles in cells. As noted, an important contribution to our understanding of the role of Orai1, and indeed its initial identification, came from the study of patients carrying functionally critical mutations in this gene. To date, no equivalent identification of patients bearing similar mutations in Orai3 have been identified. Consequently, we are still at an early stage of essentially cataloguing the differences and unique properties and roles of the conductances in which Orai3 is involved and, in most cases, our understanding of their precise molecular bases is generally far from clear. This inevitably raises some problems for the appropriate interpretation of the physiological relevance of any such effects identified. Perhaps most notably in this context are the findings reporting the properties of the conductances induced by, often quite extensive, overexpression of Orai3. Generally, these properties are described as representing those of monomeric ‘Orai3 channels’ and, at the levels of expression often used, this is probably a valid characterization. However, whilst the examination of the biophysical properties of such channels may be revealing, any potential physiological implications would depend on whether these channels can be shown to exist endogenously in the appropriate native cells. Moreover, it is clear that the three Orai proteins have at least some level of affinity for each other, and are potentially capable of forming heteromeric channels. As already noted, this has significant implications for the appropriate interpretation of data obtained by experiments involving the simple knockdown of just a single member of the Orai family. It also raises problems for claims proposing the therapeutic potential of targeting just such a single Orai member. More immediately, given that most expression studies are done without deletion or knockdown of the endogenous Orai proteins in the cells, the formation of heteromeric channels of unknown, potentially multiple, configurations is clearly possible. Consequently, evaluation of genuine physiological relevance would seem to require the definitive identification, characterization and, ultimately, replication of the properties of those Orai channels that can be shown to form at endogenous levels of expression. An obvious technical difficulty in achieving this is that such endogenous conductances typically present as only very small whole-cell currents. Nevertheless, there are cases where the nature of endogenous Orai channels in various cell types has been established, the best example of which is obviously the store-operated CRAC channel which is formed exclusively by Orai1 subunits. Correspondingly, there are examples of endogenous channels that, at least, contain Orai3 subunits. Here, the store-independent ARC channels are the most obvious example, and these are formed by a heteropentamer composed of two Orai3 subunits and three Orai1 subunits. In addition, the recent evidence of conductances in certain breast epithelial cells and cell lines that appear to display properties consistent with those seen on overexpression of Orai3 alone is particularly interesting.
Analysis of genomic and proteomic sequences clearly indicates that Orai3 first appeared as a discrete entity in mammals, apparently evolving from Orai1 (Cai, 2007). However, it is always important to remember that such classifications are essentially a human invention, and that Nature typically does not partition things into such neat, distinct categories. Consequently, examination of the available sequences of various ‘pre-mammalian’ Orai1 proteins shows several, potentially critical, differences from that seen in mammalian Orai1. For example, the cytosolic N-terminus of chicken Orai1 is extremely short, comprising just the 28 residues immediately prior to the first transmembrane domain, and without any of the proline-rich and arginine-rich sequences seen in mammalian Orai1. The N-terminus of Xenopus Orai1 is intermediate in length between that of the chicken and the mammalian Orai1s, but again does not show any proline-rich and arginine-rich sequences. Such variations in sequence at least raise the possibility that some of these pre-mammalian Orai1 proteins might possess properties that display the beginnings of those ultimately more profoundly displayed in Orai3, and that Orai3 itself evolved in mammals to optimize, or maximize, these properties and their associated functions in cells.
In conclusion, although the study of Orai proteins, and the various channels that they can form, is still very much in its infancy, the evidence does suggest that Orai3, either alone or as part of a complex with other Orai family members, imparts certain unique features to the properties and regulation of the resulting conductances. This, alone, raises the possibility of its involvement in distinct roles within the cell, some of which are already being revealed (see Table 1). Consequently, it seems that Orai3 successfully fulfils the definition of an ‘exceptional’ Orai – one that is unusual, or special, and distinct from the perceived norm.
Acknowledgments
Data from the author's laboratory were largely obtained by Dr Olivier Mignen (present address: Université de Bretagne Occidentale, France) and Jill Thompson, and were supported by funds from National Institutes of Health grants GM040457 and DE019245 (to T.J.S.). The author thanks Dr Robert Dirksen for helpful comments on an earlier version of this manuscript.
Glossary
Abbreviations
- ARC channel
arachidonate-regulated Ca2+ channel
- CRAC channel
Ca2+ release-activated Ca2+ channel
- ER
endoplasmic reticulum
- PLC
phospholipase C
- SCID
severe combined immunodeficiency
- STIM1
stromal interacting molecule 1
References
- Bakowski D, Parekh AB. Monovalent cation permeability and Ca2+ block of the store-operated Ca2+ current ICRAC in rat basophilic leukemia cells. Pflugers Arch. 2002;443:892–902. doi: 10.1007/s00424-001-0775-8. [DOI] [PubMed] [Google Scholar]
- Bergsmann J, Derler I, Muik M, Frischauf I, Fahrner M, Plooheimer P, Schwarzinger C, Gruber HJ, Groschner K, Romanin C. Molecular determinants within N terminus of Orai3 protein that control channel activation and gating. J Biol Chem. 2011;286:31565–31575. doi: 10.1074/jbc.M111.227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Calcium oscillations. J Biol Chem. 1990;265:9583–9586. [PubMed] [Google Scholar]
- Berridge MJ, Galione A. Cytosolic calcium oscillators. FASEB J. 1988;2:3074–3082. doi: 10.1096/fasebj.2.15.2847949. [DOI] [PubMed] [Google Scholar]
- Bilmen JG, Michelangeli F. Inhibition of the type 1 inositol 1,4,5-trisphosphate receptor by 2-aminoethoxydiphenylborate. Cell Signal. 2002;14:955–960. doi: 10.1016/s0898-6568(02)00042-6. [DOI] [PubMed] [Google Scholar]
- Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, Takahashi N, Peinelt C, Griesemer D, Bozem M, Mori Y, Hoth M, Niemeyer BA. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. 2010;3:ra24. doi: 10.1126/scisignal.2000672. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Young KW, Young JM, Moreton RB, Berridge MJ. Extracellular calcium concentration controls the frequency of intracellular calcium spiking independently of inositol 1,4,5-trisphosphate production in HeLa cells. Biochem J. 1996;314:347–354. doi: 10.1042/bj3140347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, Bosl M, Stoll G, Nieswandt B. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113:2056–2063. doi: 10.1182/blood-2008-07-171611. [DOI] [PubMed] [Google Scholar]
- Cai X. Molecular evolution and structural analysis of the Ca2+ release-activated Ca2+ channel subunit, orai. J Mol Biol. 2007;368:1284–1291. doi: 10.1016/j.jmb.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. 2-Aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J Neurosci. 2004;24:5177–5182. doi: 10.1523/JNEUROSCI.0934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW., Jr Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem. 2008;283:19265–19273. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr Calcium inhibition and calcium potentiation of orai1, orai2, and orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–17556. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu KL, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol. 2011;226:542–551. doi: 10.1002/jcp.22363. [DOI] [PubMed] [Google Scholar]
- Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, Schindl R, Groschner K, Romanin C. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1–3 channels by a STIM1 coiled-coil mutant. J Biol Chem. 2009;284:21696–21706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RB, Rychkov G, Barritt GJ. Evidence that 2-aminoethyl diphenylborate is a novel inhibitor of store-operated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem J. 2001;354:285–290. doi: 10.1042/0264-6021:3540285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SA, Wissenbach U, Philipp SE, Freichel M, Cavalie A, Flockerzi V. Murine ORAI2 splice variants form functional CRAC channels. J Biol Chem. 2007;282:19375–19384. doi: 10.1074/jbc.M701962200. [DOI] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-Hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–5222. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX. 2-Aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Mori Y, Hara Y, Uchida K, Zhou H, Mikoshiba K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Receptors Channels. 2001;7:429–439. [PubMed] [Google Scholar]
- Jacob R. Calcium oscillations in electrically non-excitable cells. Biochim Biophys Acta. 1990;1052:427–438. doi: 10.1016/0167-4889(90)90152-4. [DOI] [PubMed] [Google Scholar]
- Ji W, Xu P, Li Z, Lu J, Liu L, Zhan Y, Chen Y, Hille B, Xu T, Chen L. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci U S A. 2008;105:13668–13673. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juvin V, Penna A, Chemin J, Lin YL, Rassendren FA. Pharmacological characterization and molecular determinants of the activation of transient receptor potential V2 channel orthologs by 2-aminoethoxydiphenyl borate. Mol Pharmacol. 2007;72:1258–1268. doi: 10.1124/mol.107.037044. [DOI] [PubMed] [Google Scholar]
- Kozak JA, Kerschbaum HH, Cahalan MD. Distinct properties of CRAC and MIC channels in RBL cells. J Gen Physiol. 2002;120:221–235. doi: 10.1085/jgp.20028601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Yuan JP, Zeng W, So I, Worley PF, Muallem S. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci U S A. 2009;106:14687–14692. doi: 10.1073/pnas.0904664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Jiang J, Yue L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol. 2006;127:525–537. doi: 10.1085/jgp.200609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YS, Wu P, Zhou XY, Chen JG, Cai L, Wang F, Xu LM, Zhang XL, Chen Y, Liu SJ, Huang YP, Ye DY. Formyl-peptide receptor like 1: a potent mediator of the Ca2+ release-activated Ca2+ current ICRAC. Arch Biochem Biophys. 2008;478:110–118. doi: 10.1016/j.abb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282:29448–29456. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, Penner R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Zierler S, Peinelt C, Fleig A, Penner R. A single lysine in the N-terminal region of store-operated channels is critical for STIM1-mediated gating. J Gen Physiol. 2010;136:673–686. doi: 10.1085/jgp.201010484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litjens T, Harland ML, Roberts ML, Barritt GJ, Rychkov GY. Fast Ca2+-dependent inactivation of the store-operated Ca2+ current (ISOC) in liver cells: a role for calmodulin. J Physiol. 2004;558:85–97. doi: 10.1113/jphysiol.2004.065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji SS, Parker NJ, Williams RT, Van Stekelenburg L, Pearson RB, Dziadek M, Smith PJ. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 2000;1481:147–155. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- Martin SC, Shuttleworth TJ. Ca2+ influx drives agonist-activated [Ca2+]i oscillations in an exocrine cell. FEBS Lett. 1994;352:32–36. doi: 10.1016/0014-5793(94)00913-9. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem (Tokyo) 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Ogura T, Mio K, Kato K, Kaneko T, Kiyonaka S, Mori Y, Sato C. Tetrameric Orai1 is a teardrop-shaped molecule with a long, tapered cytoplasmic domain. J Biol Chem. 2009;284:13676–13685. doi: 10.1074/jbc.M900812200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, Kutok J, Hivroz C, Ledeist F, Plogmann K, Ehl S, Notheis G, Albert MH, Belohradsky BH, Kirschner J, Rao A, Fischer A, Feske S. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. 2009;124:1311–1318. doi: 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Yamashita M, Engh A, Prakriya M. Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci U S A. 2009;106:22516–22521. doi: 10.1073/pnas.0909574106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer JC, DeHaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Shuttleworth TJ. IARC, a novel arachidonate-regulated, noncapacitative Ca2+ entry channel. J Biol Chem. 2000;275:9114–9119. doi: 10.1074/jbc.275.13.9114. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Reciprocal regulation of capacitative and arachidonate-regulated noncapacitative Ca2+ entry pathways. J Biol Chem. 2001;276:35676–35683. doi: 10.1074/jbc.M105626200. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Ca2+ selectivity and fatty acid specificity of the noncapacitative, arachidonate-regulated Ca2+ (ARC) channels. J Biol Chem. 2003;278:10174–10181. doi: 10.1074/jbc.M212536200. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol. 2007;579:703–715. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J Physiol. 2008a;586:185–195. doi: 10.1113/jphysiol.2007.146258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008b;586:419–425. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. The molecular architecture of the arachidonate-regulated Ca2+-selective ARC channel is a pentameric assembly of Orai1 and Orai3 subunits. J Physiol. 2009;587:4181–4197. doi: 10.1113/jphysiol.2009.174193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Yule DI, Shuttleworth TJ. Agonist activation of arachidonate-regulated Ca2+-selective (ARC) channels in murine parotid and pancreatic acinar cells. J Physiol. 2005;564:791–801. doi: 10.1113/jphysiol.2005.085704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaen L, Callewaert G, De Smedt H, Parys JB. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium. 2001;29:111–116. doi: 10.1054/ceca.2000.0163. [DOI] [PubMed] [Google Scholar]
- Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285:19173–19183. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K, Romanin C. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- Mullins FM, Park CY, Dolmetsch RE, Lewis RS. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci U S A. 2009;106:15495–15500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oritani K, Kincade PW. Identification of stromal cell products that interact with pre-B cells. J Cell Biol. 1996;134:771–782. doi: 10.1083/jcb.134.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker NJ, Begley CG, Smith PJ, Fox RM. Molecular cloning of a novel human gene (D11S4896E) at chromosomal region 11p15.5. Genomics. 1996;37:253–256. doi: 10.1006/geno.1996.0553. [DOI] [PubMed] [Google Scholar]
- Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol. 2008;586:3061–3073. doi: 10.1113/jphysiol.2008.151365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT, Roderick HL. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium. 2003;34:97–108. doi: 10.1016/s0143-4160(03)00026-5. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney TA, Sass EJ, Thomas AP. Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single fura-2-loaded hepatocytes. J Biol Chem. 1989;264:17131–17141. [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbioni S, Barbanti-Brodano G, Croce CM, Negrini M. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997;57:4493–4497. [PubMed] [Google Scholar]
- Schindl R, Bergsmann J, Frischauf I, Derler I, Fahrner M, Muik M, Fritsch R, Groschner K, Romanin C. 2-Aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J Biol Chem. 2008;283:20261–20267. doi: 10.1074/jbc.M803101200. [DOI] [PubMed] [Google Scholar]
- Schindl R, Frischauf I, Bergsmann J, Muik M, Derler I, Lackner B, Groschner K, Romanin C. Plasticity in Ca2+ selectivity of Orai1/Orai3 heteromeric channel. Proc Natl Acad Sci U S A. 2009;106:19623–19628. doi: 10.1073/pnas.0907714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrimgeour N, Litjens T, Ma L, Barritt GJ, Rychkov GY. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J Physiol. 2009;587:2903–2918. doi: 10.1113/jphysiol.2009.170662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, DeHaven WI, Bird GS, Putney JW., Jr Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci. 2008;121:762–772. doi: 10.1242/jcs.023903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- Srikanth S, Jung HJ, Ribalet B, Gwack Y. The intracellular loop of Orai1 plays a central role in fast inactivation of Ca2+ release-activated Ca2+ channels. J Biol Chem. 2010;285:5066–5075. doi: 10.1074/jbc.M109.072736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Murakami M, Watanabe H, Hasegawa H, Ohba T, Munehisa Y, Nobori K, Ono K, Iijima T, Ito H. Essential role of the N-terminus of murine Orai1 in store-operated Ca2+ entry. Biochem Biophys Res Commun. 2007;356:45–52. doi: 10.1016/j.bbrc.2007.02.107. [DOI] [PubMed] [Google Scholar]
- Thomas AP, Renard DC, Rooney TA. Spatial and temporal organization of calcium signalling in hepatocytes. Cell Calcium. 1991;12:111–126. doi: 10.1016/0143-4160(91)90013-5. [DOI] [PubMed] [Google Scholar]
- Thompson JL, Mignen O, Shuttleworth TJ. The N-terminal domain of Orai3 determines selectivity for activation of the store-independent ARC channel by arachidonic acid. Channels (Austin) 2010;4:398–410. doi: 10.4161/chan.4.5.13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL, Shuttleworth TJ. Orai channel-dependent activation of phospholipase C-δ: a novel mechanism for the effects of calcium entry on calcium oscillations. J Physiol. 2011;589:5057–5069. doi: 10.1113/jphysiol.2011.214437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, Bird GSJ, McKay RR, Putney JW., Jr Comparison of human TRPC3 channels in receptor-activated and store-operated modes – Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J Biol Chem. 2002;277:21617–21623. doi: 10.1074/jbc.M202549200. [DOI] [PubMed] [Google Scholar]
- Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006a;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006b;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Prenen J, Fleig A, Vennekens R, Watanabe H, Hoenderop JG, Bindels RJ, Droogmans G, Penner R, Nilius B. CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. J Biol Chem. 2001;276:47767–47770. doi: 10.1074/jbc.C100607200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Deng X, Zhou Y, Hendron E, Mancarella S, Ritchie MF, Tang XD, Baba Y, Kurosaki T, Mori Y, Soboloff J, Gill DL. STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci U S A. 2009;106:7391–7396. doi: 10.1073/pnas.0900293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ, Dziadek MA. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673–685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, Senior PV, Van Stekelenburg L, Layton JE, Smith PJ, Dziadek MA. Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim Biophys Acta. 2002;1596:131–137. doi: 10.1016/s0167-4838(02)00211-x. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Navarro-Borelly L, McNally BA, Prakriya M. Orai1 mutations alter ion permeation and Ca2+-dependent fast inactivation of CRAC channels: evidence for coupling of permeation and gating. J Gen Physiol. 2007;130:525–540. doi: 10.1085/jgp.200709872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Somasundaram A, Prakriya M. Competitive modulation of Ca2+ release-activated Ca2+ channel gating by STIM1 and 2-aminoethyldiphenyl borate. J Biol Chem. 2011;286:9429–9442. doi: 10.1074/jbc.M110.189035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung-Yam-Wah V, Lee AK, Tse FW, Tse A. Arachidonic acid stimulates extracellular Ca2+ entry in rat pancreatic beta cells via activation of the noncapacitative arachidonate-regulated Ca2+ (ARC) channels. Cell Calcium. 2010;47:77–83. doi: 10.1016/j.ceca.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia WM, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Zhang SL, Kozak JA, Jiang W, Yeromin AV, Chen J, Yu Y, Penna A, Shen W, Chi V, Cahalan MD. Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem. 2008;283:17662–17671. doi: 10.1074/jbc.M801536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci U S A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitt C, Strauss B, Schwarz EC, Spaeth N, Rast G, Hatzelmann A, Hoth M. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem. 2004;279:12427–12437. doi: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]