

Abstract
Non-technical summary
The fetal cardiovascular defence to episodes of reduced oxygenation, or acute hypoxia, includes redistribution of the cardiac output away from peripheral and towards essential circulations, such as those perfusing the brain – the so called brain-sparing effect. The peripheral vasoconstriction is triggered by a chemoreflex and maintained by constrictor hormones. Nitric oxide (NO) synthesis during hypoxia opposes these mechanisms, but the balance of all effects favours constriction. Statins are drugs commonly used to lower cholesterol. Since women are delaying pregnancy until later in life, there is increasing clinical interest in treating pregnant women with statins. However, statins have other effects, including increasing NO levels, and their effects on the physiology of the fetus are completely unknown. Here, we show that fetal exposure to statins depresses the fetal peripheral constrictor response to acute hypoxia via increasing NO bioavailability. Use of statins in pregnancy should be viewed with caution.
Abstract
In addition to lowering cholesterol, statins increase nitric oxide (NO) bioavailability, improving endothelial function. In the fetus, enhanced NO during acute hypoxia opposes the fetal peripheral vasoconstrictor response, part of the brain-sparing defence. This study tested the hypothesis that treatment with statins depresses the fetal circulatory response to acute hypoxic stress via increasing NO bioavailability. Under anaesthesia, 12 fetal sheep at 118 ± 1 days of gestation (term ca 145 days) were instrumented with vascular catheters and a femoral artery Transonic flow probe for chronic recording. Five days later, all animals were subjected to 30 min of acute hypoxia (fetal arterial partial pressure of O2 (PaO2) reduced by ca 50%) before and 24 h after fetal treatment with pravastatin (25 mg i.v.). In half of the fetuses (n = 6), responses to hypoxia post-pravastatin were evaluated during NO synthesis blockade. Fetal exposure to pravastatin did not affect fetal basal cardiovascular function. Fetal PaO2 was similarly reduced in all acute hypoxia experiments from ca 21 to 10 mmHg. Fetal exposure to pravastatin markedly diminished the fetal femoral vasoconstrictor (5.1 ± 0.9 vs. 2.5 ± 0.5 mmHg (ml min−1)−1) and lactic acidaemic (4.4 ± 0.5 vs. 3.0 ± 0.3 mm) responses to acute hypoxia (both P < 0.05), without affecting plasma catecholamine responses. Post-pravastatin, the circulatory (5.8 ± 1.5 mmHg (ml min−1)−1) and metabolic (3.9 ± 0.3 mm) responses could be restored to control levels during fetal treatment with NO synthase blockade. Pravastatin depresses the fetal cardiovascular and metabolic defences to acute hypoxia via increasing NO bioavailability. The use of statins during pregnancy should be viewed with extreme caution.
Introduction
Statins are 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase inhibitors and they have become one of the most effective and widely prescribed drugs for the primary prevention of coronary heart disease (Steinberg, 2008). In addition to their lipid-lowering effects, several additional actions of statins have been shown, including decreases in arterial stiffness, reductions in platelet aggregation and improvements in vascular endothelial dysfunction (Gelosa et al. 2007; Maki-Petaja & Wilkinson, 2009). These benefits have been credited to increases in nitric oxide (NO) bioavailability and increased NO function (Blum & Shamburek, 2009). Considering the rising levels of obesity and associated lipid disorders in younger populations and that women are delaying childbirth until the fourth or fifth decades of life (Heffner, 2004; National-Centre-for-Health-Statistics, 2010), there is growing clinical interest in treating pregnant women with statins. Despite incidental reports that statins may have adverse effects on the fetus and mother, randomised clinical studies have reported no untoward, or serious side effects of statins in humans (Taguchi et al. 2008). Indeed, one multi-centre randomised controlled trial has begun recruiting patients in the United Kingdom to investigate if pravastatin could reduce circulating anti-angiogenic factors associated with pre-eclampsia (Ahmed, 2011). However, the effects of statins on the physiology of the fetus are completely unknown.
One of the most common challenges to the fetus during gestation is a reduction in oxygenation or acute hypoxia (Low, 2004). The fetal defence to acute hypoxia involves integrated cardiovascular, endocrine and metabolic responses, which facilitate fetal survival during the period of reduced oxygen availability (Rudolph, 1985). The cardiovascular defences include bradycardia and peripheral vasoconstriction, the latter significantly decreasing oxygen consumption in non-essential vascular beds (Cohn et al. 1974; Boyle et al. 1990). The peripheral vasoconstriction also contributes to the redistribution of the fetal cardiac output, shunting oxygenated blood away from non-essential vascular beds towards the brain, heart, adrenal and umbilical circulations (Cohn et al. 1974; Giussani et al. 1993). The physiology underlying this circulatory response involves activation of carotid chemoreflexes (Giussani et al. 1993) and the release of stress hormones into the fetal circulation, such as catecholamines (Jones & Robinson, 1975). The fetal metabolic responses to acute hypoxia involve an increase in the circulating concentrations of glucose and lactate (Jones, 1977). The fetal hyperglycaemia during acute hypoxia results from a decrease in glucose uptake and utilisation by peripheral tissues and an increase in hepatic glucose production (Hooper, 1995). The fetal lactic acidaemia results from anaerobic metabolism of glucose in hypoxic fetal tissues, particularly in the hind limb where blood flow and oxygen delivery are markedly declined (Boyle et al. 1990).
More recently, the contribution to these responses of local agents, in particular NO, independent of neuroendocrine control has gathered increasing attention (Morrison et al. 2003; Thakor & Giussani, 2009; Thakor et al. 2010b). Studies have shown that fetal NO bioavailability can increase during acute hypoxia, altering neuroendocrine constrictor influences and metabolic responses (Morrison et al. 2003). Further, increased generation of reactive oxygen species (ROS) during acute hypoxia can interact with NO, providing an oxidant tone to the fetal vasculature (Thakor & Giussani, 2009; Thakor et al. 2010b). Since statins increase NO bioavailability and they cross the placenta (Laufs et al. 1998; Edison & Muenke, 2004), it is likely that when given in pregnancy, they may alter the cardiovascular, endocrine and metabolic physiology of the fetus during basal and/or stressful intrauterine conditions, such as during acute hypoxia.
Therefore, this study investigated the in vivo effects of statins during basal and hypoxic conditions on cardiovascular, endocrine and metabolic functions in chronically instrumented late gestation fetal sheep. To determine the mechanisms via which statins affect cardiovascular, endocrine and metabolic functions in the fetus, fetal exposure to acute hypoxia after pravastatin was repeated in the presence of NO synthase blockade with the NO clamp. This technique permits in vivo blockade of de novo synthesis of NO during acute hypoxia without affecting basal cardiovascular function in the fetus (Gardner et al. 2001; Gardner & Giussani, 2003; Thakor & Giussani, 2005). Secondly, changes in fetal plasma catecholamine concentrations were measured during acute hypoxia before and after pravastatin. Finally, cardiac and vasomotor chemoreflex function was analysed during acute hypoxia before and after pravastatin.
Methods
All procedures were performed under the UK Animals (Scientific Procedures) Act 1986 and were approved by the Ethical Review Committee of the University of Cambridge. Twelve pregnant ewes at 118 ± 1 days of gestational age (term ca 145 days) and their fetuses were surgically instrumented under strict aseptic conditions as previously described in detail (Giussani et al. 2001; Gardner et al. 2002). In brief, food but not water, was withheld from the pregnant ewes for 24 h prior to surgery. Following induction with 20 mg kg−1 i.v. sodium thiopentone (Intraval Sodium; Merial Animal Health Ltd, Rhone Mérieux, Dublin, Ireland), general anaesthesia (1.5–2.0% isoflurane in 50:50 O2:N2O) was maintained using positive pressure ventilation. Midline abdominal and uterine incisions were made, the fetal hind limbs were exteriorised and, on one side, femoral arterial (i.d., 0.86 mm; o.d., 1.52 mm; Critchly Electrical Products, NSW, Australia) and venous (i.d., 0.56 mm; o.d., 0.96 mm) catheters were inserted. The catheter tips were advanced carefully to the descending aorta and inferior vena cava, respectively. Another catheter was anchored onto the fetal hind limb for recording of the reference amniotic pressure. A Transonic flow probe was positioned around the contralateral femoral artery (2R or 3S). The uterine incisions were closed in layers, the dead space of the catheters was filled with heparinised saline (80 i.u. heparin ml−1 in 0.9% NaCl) and the catheter ends were plugged with sterile brass pins. All catheters and the flow probe lead were exteriorised via a keyhole incision in the maternal flank and kept inside a plastic pouch sewn onto the maternal skin.
During recovery, ewes were housed in individual pens in rooms with a 12 h:12 h/light:dark cycle where they had free access to hay and water and were fed concentrates twice daily (100 g sheep nuts no. 6; H & C Beart Ltd, Kings Lynn, UK). Antibiotics were administered daily to the ewe (0.20–0.25 mg kg−1i.m. Depocillin; Mycofarm, Cambridge, UK) and fetus i.v. and into the amniotic cavity (600 mg in 2 ml 0.9% NaCl, benzylpenicillin; Crystapen, Schering-Plough, Animal Health Division, Welwyn Garden City, UK). Generally, normal feeding patterns were restored within 24–48 h of recovery. Following 48–72 h of post-operative recovery, ewes were transferred to metabolic crates where they were housed for the remainder of the protocol. The arterial and amniotic catheters were connected to sterile pressure transducers (COBE; Argon Division, Maxxim Medical, Athens, TX, USA). While on the metabolic crates, the patency of the fetal and maternal vascular catheters was maintained by a slow continuous infusion of heparinised saline (80 i.u. heparin ml−1 at 0.1 ml h−1 in 0.9% NaCl) containing antibiotic (1 mg ml−1 benzylpenicillin; Crystapen, Schering-Plough).
Following at least 5 days of post-operative recovery, fetuses were randomly assigned to one of the two experimental groups (Fig. 1). In Group 1 (n = 6), fetal cardiovascular, metabolic and endocrine functions were assessed during basal and stimulated conditions before and 24 h following fetal exposure to pravastatin. In Group 2 (n = 6), fetal cardiovascular, metabolic and endocrine functions were assessed during basal and stimulated conditions before and 24 h following fetal exposure to pravastatin during the NO clamp.
Figure 1. Experimental design and assignment to groups.
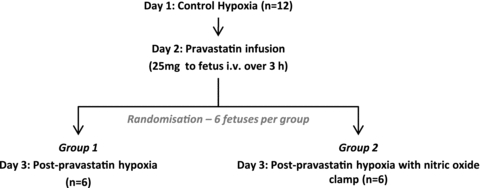
In Group 1, fetal cardiovascular, metabolic and endocrine functions were assessed during basal and stimulated conditions before and 24 h following fetal exposure to pravastatin. In Group 2, fetal cardiovascular, metabolic and endocrine functions were assessed during basal and stimulated conditions before and 24 h following fetal exposure to pravastatin in the presence of the NO clamp.
To avoid confounding influences due to maternal effects of pravastatin and/or differences in transplacental passage of pravastatin, pravastatin was infused directly to the fetus over 3 h (25 mg in 20 ml 0.9% NaCl solution i.v. at 6.6 ml h−1, pravastatin sodium, Sigma, UK; Fig. 2A). The dose chosen represents an intermediate level between clinical studies and doses used in previous animal experiments (Mondo et al. 2006; Torrens et al. 2009; StAmP, 2011). The ‘nitric oxide clamp’ combined i.v. treatment of the fetus with NG-nitro-l-arginine methyl ester (l-NAME, Sigma, 250 mg bolus dissolved in 1 ml saline) and continuous i.v. sodium nitroprusside (Sigma, UK) titrated to maintain basal arterial blood pressure (Gardner et al. 2001; Gardner & Giussani, 2003; Thakor & Giussani, 2005).
Figure 2. Pravastatin infusion and acute hypoxia protocols.
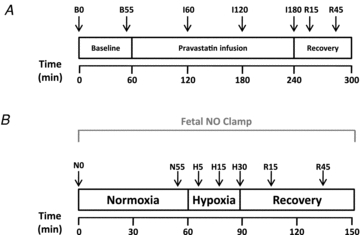
A, following a 1 h period of baseline, pravastatin was infused directly into the fetus for 3 h (25 mg in 20 ml 0.9% NaCl solution i.v. at 6.6 ml h−1, pravastatin sodium) followed by a 1 h recovery period. Maternal and fetal arterial samples for blood gas analysis (↓) were taken at 0 and 55 min of baseline, 60, 120 and 180 min of infusion and 15 and 45 min of recovery. Maternal and fetal cardiovascular variables were recorded continuously. B, each acute hypoxia protocol comprised 60 min normoxia, 30 min hypoxia and 60 min recovery. Maternal and fetal arterial samples for blood gas analysis (↓) were taken at 0 and 55 min of normoxia, 5, 15 and 30 min of hypoxia and 15 and 45 min of recovery. Additional blood samples for catecholamine analyses were taken at 0 and 55 min of normoxia, 15 and 30 min of hypoxia and 45 min of recovery.
Acute hypoxia protocol
Animals were subjected to the acute hypoxia protocol before and 24 h following fetal exposure to pravastatin in the absence (Group 1, n = 6) or presence (Group 2, n = 6) of NO synthase blockade with the NO clamp (Fig. 2B). The acute hypoxia protocol consisted of 60 min normoxia, 30 min hypoxia and 60 min recovery (Fig. 2B). Acute hypoxia in the fetus was induced by decreasing the maternal inspired fraction of oxygen. A large transparent respiratory hood was placed over the ewe's head into which air was passed at a rate of ca 50 l min−1 for the period of normoxia. Following this normoxic baseline period, acute fetal hypoxia was induced for 30 min by changing the gas mixture breathed by the ewe to 15 l min−1 air, 35 l min−1 N2, 1.5–2.5 l min−1 CO2. This mixture was designed to reduce the fetal arterial partial pressure of oxygen (PaO2) to ca 10 mmHg while maintaining the maternal and fetal arterial partial pressure of CO2 (PaCO2). Following the 30 min period of acute hypoxia, the ewe was returned to breathing air for the 1 h recovery period.
During any acute hypoxia protocol, descending aortic blood samples (3 ml) were taken using sterile techniques from the mother and fetus at set time points (Fig. 2B) for the analysis of key fetal stress hormones (adrenaline and noradrenaline) and to determine arterial blood gas and acid–base status (ABL5 blood gas analyser; Radiometer, Copenhagen, Denmark; measurements corrected to 38°C for maternal and 39.5°C for fetal blood).
At the end of the experimental protocols, the effectiveness of NO synthase blockade by the NO clamp and the persistence of l-NAME in the system were tested by withdrawal of the sodium nitroprusside infusion. This unmasked the influence of fetal treatment with l-NAME alone and, if still acting, it should reveal fetal peripheral vasoconstriction and an increase in fetal arterial blood pressure. Following experimentation, the ewes and fetuses were humanely killed using a lethal dose of sodium pentobarbitone (200 mg kg−1i.v. Pentoject; Animal Ltd, York, UK). Post-mortem was carried out at 130 ± 1 days of gestation during which time the positions of the implanted catheters and flow probes were confirmed.
Adrenaline and noradrenaline measurements
Plasma adrenaline and noradrenaline were measured by a commercially available catecholamine radioimmunoassay previously validated for use with sheep plasma (2-CAT RIA, Diasourse; Poore et al. 2010). In an extraction plate, 10 μl of each standard or 300 μl of plasma were added to individual wells. Buffer and extraction buffer (50 μl of each) were then added and the mixture was incubated for 30 min at room temperature. The contents of the tray were then discarded and the plate washed with distilled water. Following this, 150 μl of acylation buffer and 25 μl of acylation reagent were added before a further 15 min incubation at room temperature. After washing the wells with water, 150 μl of 0.1 m HCl was added to each well to elute the amines from the extraction gel. Separately, two sets of 12 mm × 75 mm glass test tubes were labelled for adrenaline and noradrenaline quantification. The extracted samples, standards and controls were transferred to each adrenaline tube (100 μl of each) and noradrenaline tubes (25 μl of each). Enzyme solution (25 μl) was then added and the solution incubated at 37°C for 30 min. For adrenaline, 50 μl of radio-labelled 125I-adrenaline and 50 μl of adrenaline antiserum were added. For noradrenaline, 50 μl of radio-labelled 125I-noradrenaline and 50 μl of noradrenaline antiserum were added. Total counts were made with 50 μl of radio-labelled solution. The mixtures were then incubated overnight at 5°C. Chilled precipitating reagent (1 ml) was then added to all tubes (except the total count tubes) and the solutions mixed thoroughly before centrifuging for 15 min at 3000 g, at 5°C. The supernatant was decanted and the tubes blotted and left to dry upside-down for 2 min. All tubes were then counted for 2 min in a γ-counter (Cobra II Auto-Gamma; Packard Bioscience Co., Pangbourne, Berks, UK). For adrenaline, samples containing 17 and 58 ng ml−1, the inter-assay coefficients of variations were 5.6 and 6.1% and the intra-assay coefficients of variation were 14.0 and 4.6%. For noradrenaline, samples containing 17 and 58 ng ml−1, the inter-assay coefficients of variations were 10.0 and 6.1% and the intra-assay coefficients of variation were 4.0 and 4.6%.
Data and statistical analyses
All data are expressed as mean ± SEM. Data for fetal and maternal arterial blood pressure, amniotic pressure and fetal femoral blood flow were recorded continuously by a custom-built Data Acquisition System (MPAQ-Maastricht-Programmable AcQuisition system, Maastricht Instruments, The Netherlands, 500 Hz sample rate). Minute by minute averages were calculated during the pravastatin infusion protocol and the acute hypoxia protocol in each animal, and imported into an Excel spreadsheet for analysis. For statistical analysis, cardiovascular data were averaged over time as indicated in the Results and figure legends. Chemoreflex analysis was performed by calculating the slope of the relationship between fetal PaO2 and fetal heart rate or femoral vascular resistance within the first 15 min of hypoxia in individual fetuses, a period when the neural component of the chemoreflex is dominant (Morrison et al. 2003). Cardiovascular, metabolic and endocrine data were analysed comparing the effect of treatment, time and interactions between treatment and time by one-way or two-way repeated measures ANOVA as appropriate with post hoc Student–Newman–Keuls or Tukey's test (SigmaStat 3.5). Cardiovascular data before and after removal of the nitric oxide clamp were compared by Student's t test for paired data. For all comparisons, statistical significance was accepted when P < 0.05.
Results
Maternal blood gas and cardiovascular status
Basal values for maternal arterial blood gas, acid–base status and cardiovascular variables were similar in all animals and were within the normal range for pregnant Welsh Mountain ewes at this stage of gestation at the beginning of each protocol. At the start of the pravastatin infusion protocol, maternal pH, PaCO2 and PaO2 were 7.47 ± 0.01, 40.4 ± 1.2 mmHg and 103.6 ± 1.5 mmHg, respectively, in Group 1 (n = 6) and 7.48 ± 0.02, 40.1 ± 2.5 mmHg and 100.9 ± 4.6 mmHg, respectively, in Group 2 (n = 6). These variables were unaltered by fetal treatment with pravastatin. Similarly, maternal arterial blood pressure and heart rate were 99.9 ± 4.8 mmHg and 101.4 ± 5.5 beats min−1, respectively, in Group 1 (n = 6) and 95.0 ± 7.3 mmHg and 97.8 ± 7.9 beats min−1, respectively, in Group 2 (n = 6). These variables were also unaltered by fetal treatment with pravastatin.
Acute hypoxia did not affect maternal pH or PaCO2; however, it did lead to similar decreases in maternal PaO2 in all groups (Control hypoxia, from 99.9 ± 2.3 mmHg to 34.9 ± 1.2 mmHg vs. Pravastatin hypoxia, from 101.3 ± 1.3 mmHg to 35.6 ± 0.6 mmHg vs. Pravastatin hypoxia with NO clamp, from 96.0 ± 3.0 mmHg to 37.9 ± 2.1 mmHg; P < 0.05). Acute hypoxia led to similar increases in maternal arterial blood pressure and maternal heart rate in all groups (Δarterial blood pressure: Control +8.7 ± 1.9 mmHg vs. Pravastatin +9.5 ± 1.1 mmHg vs. Pravastatin with NO clamp +11.1 ± 2.0 mmHg; Δheart rate: Control +54.1 ± 5.6 beats min−1 vs. Pravastatin +41.4 ± 6.4 beats min−1 vs. Pravastatin with NO clamp +37.1 ± 3.7 beats min−1).
Fetal blood gas and cardiovascular status
Basal values for fetal arterial blood gas, acid–base status and cardiovascular variables were similar in all animals and were within the normal range for the Welsh Mountain sheep fetus at this stage of gestation. At the start of the pravastatin infusion protocol, fetal pH, PaCO2 and PaO2 were 7.34 ± 0.01, 55.9 ± 1.9 mmHg and 21.9 ± 0.6 mmHg, respectively, in Group 1 (n = 6) and 7.33 ± 0.01, 53.4 ± 3.0 mmHg and 19.6 ± 0.6 mmHg, respectively, in Group 2 (n = 6). None of the variables were altered by fetal treatment with pravastatin. Similarly fetal arterial blood pressure, heart rate, femoral blood flow and femoral vascular resistance were 42.2 ± 4.0 mmHg, 180.1 ± 3.4 beats min−1, 33.0 ± 2.8 ml min−1 and 1.3 ± 0.2 mmHg (ml min−1)−1, respectively, in Group 1 (n = 6) and 46.1 ± 1.7 mmHg, 181.0 ± 10.0 beats min−1, 35.2 ± 10.0 ml min−1 and 1.3 ± 0.1 mmHg (ml min−1)−1, respectively, in Group 2 (n = 6). None of these variables were altered by fetal treatment with pravastatin.
In all fetuses, acute hypoxia induced significant falls in pH and PaO2 without any alteration to PaCO2 (Table 1). Fetal treatment with pravastatin or pravastatin with the NO clamp had no effect on the fetal arterial blood gas or acid–base status during basal or acute hypoxic conditions (Table 1). Acute hypoxia in the fetus led to a significant increase in arterial blood pressure and femoral vascular resistance, and significant falls in heart rate and femoral blood flow (Fig. 3). Fetal treatment with pravastatin significantly diminished the increase in fetal femoral vascular resistance during acute hypoxia, an effect which could be restored to control levels during fetal treatment with pravastatin in the presence of NO synthase blockade (Fig. 3). To address whether this effect of pravastatin was at the level of the fetal carotid body, the chemoreflex components of the cardiovascular responses were analysed post-pravastatin. The sensitivity of the femoral vasoconstrictor chemoreflex component was significantly diminished, while the cardiac chemoreflex response remained unaltered (Fig. 4). Again, the magnitude of the circulatory chemoreflex response could be restored to control levels during fetal treatment with pravastatin in the presence of NO synthase blockade (Fig. 4).
Table 1.
Fetal blood gases during the acute hypoxia protocol
| NO | N55 | H5 | H15 | H30 | R15 | R45 | |
|---|---|---|---|---|---|---|---|
| pH | |||||||
| Control | 7.34 ± 0.01 | 7.34 ± 0.01 | 7.34 ± 0.01 | 7.29 ± 0.02* | 7.25 ± 0.02* | 7.23 ± 0.02* | 7.29 ± 0.02 |
| Pravastatin | 7.34 ± 0.01 | 7.33 ± 0.01 | 7.34 ± 0.01 | 7.32 ± 0.01 | 7.29 ± 0.02* | 7.28 ± 0.02* | 7.30 ± 0.02 |
| Pravastatin + NO clamp | 7.30 ± 0.03 | 7.30 ± 0.03 | 7.30 ± 0.03 | 7.25 ± 0.03 | 7.21 ± 0.04* | 7.20 ± 0.04* | 7.24 ± 0.04 |
| PaCO2 (mmHg) | |||||||
| Control | 56.0 ± 1.0 | 55.6 ± 1.2 | 54.4 ± 1.3 | 56.9 ± 1.4 | 55.3 ± 1.2 | 54.4 ± 1.0 | 53.2 ± 1.2 |
| Pravastatin | 56.5 ± 1.7 | 53.0 ± 1.0 | 51.8 ± 1.1 | 52.5 ± 1.5 | 53.4 ± 1.4 | 51.9 ± 1.0 | 52.3 ± 1.1 |
| Pravastatin + NO clamp | 54.7 ± 1.6 | 55.1 ± 1.6 | 52.3 ± 1.8 | 55.4 ± 2.0 | 53.6 ± 2.5 | 51.9 ± 2.1 | 53.8 ± 1.7 |
| PaO2 (mmHg) | |||||||
| Control | 22.1 ± 0.9 | 21.2 ± 0.9 | 11.8 ± 0.5* | 9.9 ± 0.6* | 10.2 ± 0.7* | 25.6 ± 1.0 | 19.5 ± 1.8 |
| Pravastatin | 24.2 ± 1.2 | 23.5 ± 1.2 | 12.5 ± 0.6* | 10.8 ± 0.7* | 10.3 ± 0.8* | 25.7 ± 0.9 | 23.3 ± 1.5 |
| Pravastatin + NO clamp | 20.6 ± 1.4 | 20.7 ± 1.0 | 12.4 ± 1.3* | 11.1 ± 1.2* | 9.0 ± 0.4* | 23.4 ± 1.3 | 19.5 ± 1.8 |
Values represent the means ± SEM for arterial pH, PaCO2 and PaO2 during the acute hypoxia protocol at time points indicated in Fig. 2 for fetuses pre-pravastatin (n = 12), post-pravastatin (n = 6) or post-pravastatin with NO clamp (n = 6). Significant differences (P < 0.05)
vs. Normoxia. Two-way repeated measures ANOVA with post hoc Tukey's test.
Figure 3. Fetal cardiovascular function during the acute hypoxia protocol.

A, values represent the means ± SEM calculated every minute for fetal mean arterial blood pressure, fetal heart rate (bpm, beats min−1), femoral blood flow and femoral vascular resistance during 60 min of normoxia, 30 min of hypoxia (dashed box) and 60 min of recovery for fetuses pre-pravastatin (n = 12) or post-pravastatin (n = 6) or post-pravastatin treatment with NO clamp (n = 6). B, statistical summary of the fetal cardiovascular data showing mean ± SEM of the mean change from normoxic baseline for time periods in normoxia, hypoxia and recovery for fetuses pre-pravastatin (open circles, n = 12) or post-pravastatin (black circles, n = 6) or post-pravastatin treatment with NO clamp (grey squares, n = 6). Significant differences (P < 0.05): *, vs. Normoxia; †, vs. Control. Two-way repeated measures ANOVA with post hoc Tukey's test.
Figure 4. Chemoreflex sensitivity analysis.

Values are the mean ± SEM for fetal femoral vascular resistance (FVR)/ΔPaO2 (A) and fetal heart rate (FHR)/ΔPaO2 (B) during the first 15 min of hypoxia in fetuses pre-pravastatin (open columns, n = 12), post-pravastatin (black columns, n = 6) or post-pravastatin with NO clamp (grey columns, n = 6). Significant differences (P < 0.05): *, vs. Control. One-way ANOVA with post hoc Student–Newman–Keuls test.
Fetal metabolic and endocrine responses to hypoxia
Basal values for fetal glucose and lactate were similar in all fetuses and were within the normal range for the Welsh Mountain sheep fetus at this stage of gestation. Acute hypoxia led to significant increases in fetal blood glucose and lactate (Fig. 5). Fetal treatment with pravastatin did not affect the fetal hyperglycaemic response to acute hypoxia but it did attenuate the increase in fetal blood lactate. Treatment of fetuses with pravastatin during the NO clamp prevented the suppression of the blood lactate response to acute hypoxia (Fig. 5).
Figure 5. Fetal metabolic and endocrine responses to acute hypoxia.
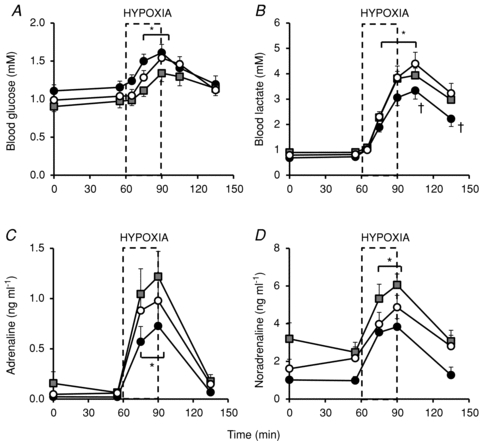
Values represent the mean ± SEM for fetal blood glucose and lactate (A and B) or plasma adrenaline and noradrenaline (C and D) during the acute hypoxia protocol in fetuses pre-pravastatin (open circles, n = 12), post-pravastatin (black circles, n = 6) or post-pravastatin with NO clamp (grey squares, n = 6). Significant differences (P < 0.05): *, vs. Normoxia; †, vs. Control. Dashed box represents the period of hypoxia.
Basal values for fetal plasma concentrations of adrenaline and noradrenaline were similar in all fetuses (0.04 ± 0.02 pg ml−1 and 1.60 ± 0.50 pg ml−1, respectively) and were not significantly different post-pravastatin with, or without, the NO clamp. Pre-pravastatin, acute hypoxia led to a significant increase in fetal plasma adrenaline and noradrenaline concentrations (0.97 ± 0.20 pg ml−1 and 4.86 ± 0.63 pg ml−1, respectively). Post-pravastatin, with or without the NO clamp, the increases in fetal plasma adrenaline and noradrenaline during acute hypoxia were not significantly altered (Fig. 5).
Removal of the NO clamp
Withdrawal of fetal infusion with sodium nitroprusside unmasked the well-known effects of NO synthase blockage by l-NAME in the fetus (Green et al. 1996), leading to increased arterial blood pressure, decreased heart rate, decreased femoral blood flow and increased femoral vascular resistance (Fig. 6). This demonstrated the effectiveness of NO synthesis blockade by the NO clamp and the persistence of the action of l-NAME within the system until the end of the experimental protocol.
Figure 6. Nitric oxide clamp removal.
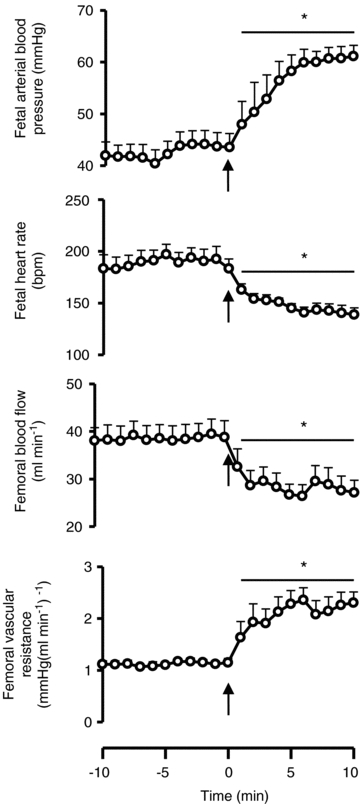
Values are the mean ± SEM for fetal mean arterial blood pressure, heart rate, femoral blood flow and femoral vascular resistance 10 min before and 10 min after the removal of the NO clamp (n = 6). Significant differences (P < 0.05): *, vs. before clamp removal. Student's t test for paired data. Arrow indicates time of clamp removal.
Discussion
The data show that fetal exposure to pravastatin significantly attenuated the fetal peripheral vasoconstrictor and lactic acidaemic responses, without affecting the cardiac chemoreflex or plasma catecholamine responses to acute hypoxic stress. Further, post-pravastatin in the presence of the NO clamp, the fetal peripheral vasoconstrictor response and lactic acidaemic responses could be restored to control levels. Therefore, the data support the hypothesis tested and show that the mechanism via which statins diminish the fetal circulatory response to hypoxia is via increasing NO bioavailability.
It is established that the gas NO generated from l-arginine by NO synthase, contributes to the maintenance of basal blood flow in the utero-placental (see Gardner et al. 2002) and several fetal vascular beds, including the cerebral (McCrabb & Harding, 1996), myocardial (Reller et al. 1995), femoral and carotid circulations (Green et al. 1996). NO opposes basal arterial tone in the fetus as systemic inhibition of NO synthase with l-NAME leads to generalised increases in peripheral vascular resistance and, thereby, an increase in arterial blood pressure (Green et al. 1996). Nitric oxide also plays a significant role in stimulated fetal cardiovascular function, for instance in response to hypoxic stress, as may occur during labour. In the fetus, as in the adult, the endothelium may act as a hypoxic sensor and effector system that releases vasoactive agents to act locally on the vascular smooth muscle, such as NO and endothelin (Green et al. 1998). The net fetal peripheral vasomotor response to acute hypoxia thus represents the balance between neural, endocrine and local paracrine vasoactive mechanisms. While endothelin does not appear to contribute significantly to the fetal cardiovascular response to acute hypoxia (Green et al. 1998), we have previously reported that fetal exposure to acute hypoxia during fetal NO synthase blockade leads to a significant enhancement of the femoral constrictor response (Morrison et al. 2003). In the fetus, hypoxia-induced increases in NO therefore limit the neural and endocrine constrictor influences on the fetal peripheral circulation (Morrison et al. 2003). More recently, it has become evident that the cellular oxidant milieu is also an important modulator of vascular resistance (Chen & Keaney, 2004). Vascular cells generate reactive oxygen species (ROS), such as the superoxide anion (•O2−; Dröge, 2002). Superoxide readily combines with NO, limiting its bioavailability (Dröge, 2002). Hence, under physiological conditions, manipulation of the vascular NO:•O2− ratio is also an important determinant of vascular tone. We have also shown that manipulation of the vascular NO:•O2− ratio with the antioxidants vitamin C or melatonin affects basal and stimulated cardiovascular function in the umbilical and fetal femoral circulations (Thakor et al. 2010a,b). Data in the present study show that statins oppose the fetal peripheral vasoconstrictor response to acute hypoxia via increasing NO bioavailability, further supporting the concept that enhanced NO limits chemoreflex and endocrine constrictor influences on the fetal peripheral circulations.
The sympathetic nervous system is critically involved in mediating cardiovascular and metabolic defences to acute hypoxia in the fetus. Acute hypoxia triggers a carotid chemoreflex, which activates the sympathetic chain and promotes femoral vasoconstriction via α-adrenergic receptors (Giussani et al. 1993). As the period of hypoxia progresses, the reflex-initiated cardiovascular responses are modified. Release of adrenaline and noradrenaline from the adrenal medulla into the fetal circulation maintains the peripheral vasoconstriction (Jones & Robinson, 1975). Blood glucose levels also increase during acute hypoxia as a result of the depression of insulin-dependent glucose uptake by the fetal tissues and/or activation of endogenous glucose production and/or stimulated glycogenolysis (Jones & Ritchie, 1983). Anaerobic metabolism of glucose in hypoxic fetal tissues, particularly in the ischaemic hind limb as a result of the femoral vasoconstriction, markedly contributes to the increase in blood lactate levels in the fetal circulation (Boyle et al. 1990). Since fetal treatment with phentolamine prevented the glycaemic response but enhanced insulin secretion during hypoxia (Jones & Ritchie, 1983), and since infusion of catecholamines increased glucose and lactate output in the sheep fetus (Apatu & Barnes, 1991), both the reduction in insulin-dependent glucose uptake and the increase in glucose and lactate production by the fetal tissues also involve neural and endocrine adrenergic pathways. In turn, it is known that statins can affect sympathetic outflow. Statins can depress sympathetic nerve activity and reduce catecholamine levels (Pliquett et al. 2003; Gomes et al. 2010). In the present study, fetal treatment with pravastatin suppressed the femoral vasoconstrictor and lactic acidaemic responses during acute hypoxia. Since sympathetic outflow is involved in mediating these responses, it could be argued that the inhibitory effects of pravastatin on the circulatory and metabolic fetal defence responses are secondary to suppression of sympathetic outflow rather than by increasing NO bioavailability at the peripheral vasculature. However, the lack of an effect of pravastatin on the increase in fetal plasma catecholamines and the fetal hyperglycaemia in the present study do not support a generalised effect of statins inhibiting sympathetic outflow as the mechanism underlying the suppressive effect on the femoral vasoconstrictor and blood lactate acidaemic responses in the fetus to acute hypoxia.
Similarly, it could be argued that the suppressive effect of pravastatin on the fetal peripheral vasoconstrictor response to acute hypoxia is due to an effect of statins at loci other than the peripheral vasculature. For example, at the level of the carotid body, NO and ROS have both been implicated in peripheral chemo-transduction mechanisms as enhanced NO can inhibit chemoreflex outflow, and increased ROS production sensitises the carotid chemoreflex (Li et al. 2010; Weir & Archer, 2010). However, two independent studies have shown that, in the fetus, both the bradycardia and the femoral vasoconstrictor responses to acute hypoxia are part of the same triggered carotid chemoreflex, since bilateral section of the carotid sinus nerves abolished both responses during acute hypoxia (Bartelds et al. 1993; Giussani et al. 1993). In the present study, fetal treatment with pravastatin diminished the fetal peripheral vasoconstrictor response to acute hypoxia without affecting the fetal bradycardia. Therefore, analysis of the chemoreflex component mediating the cardiovascular responses during the first 15 min of the hypoxic challenge revealed an effect of pravastatin on the vascular but not cardiac chemoreflex components. Dissociation of the cardiac and vascular arms of the carotid chemoreflex does not support an effect by pravastatin on NO at the level of the carotid body. Rather, the differential effect further supports an action of pravastatin at the level of the fetal peripheral vasculature.
There are several mechanisms via which pravastatin could increase vascular NO (Adam & Laufs, 2008). Statins inhibit the enzyme HMG-CoA reductase, ultimately preventing the generation of the intermediate mevalonate and, thereby, inhibiting cholesterol production (Buhaescu & Izzedine, 2007). Through lowering cholesterol levels, low density lipoproteins also decrease, an effect which has been shown to increase NO directly (Tamai et al. 1997). Acting as a precursor for cholesterol synthesis, mevalonate is also required for the synthesis of isoprenoid molecules which have important roles in normal cellular function. For instance, the isoprenoids farnesylpyrophosphate and geranylgeranylpyrophosphate facilitate membrane attachment of proteins such as the GTPase Rho. Normally Rho has direct negative effects on endothelial nitric oxide synthase (eNOS) mRNA activity or indirect inhibition of eNOS activity through inhibition of Akt which is known to activate eNOS through phosphorylation. Accordingly, statins up-regulate eNOS activity and eNOS mRNA stability secondary to enhanced eNOS phosphorylation by Akt (Kaesemeyer et al. 1999; Kureishi et al. 2000). Given that free radicals interact with NO to control the fetal peripheral circulation during basal and hypoxic conditions (Thakor et al. 2010b), changes in statin-induced ROS generation as well as statin-induced increases in NO bioavailability may have influenced the fetal defence responses to hypoxia. Indeed, statins have also been reported to prevent the activation of the GTP binding protein Rac1, which is needed to activate the pro-oxidant enzyme NAD(P)H oxidase (Adam & Laufs, 2008). This antioxidant effect extends beyond inhibiting ROS production by NAD(P)H oxidase as the ROS it generates can degrade BH4, a cofactor for NO synthase, which maintains NO bioavailability. Finally, statins have been reported to directly stimulate an increase in antioxidant enzyme production, such as catalase (Adam & Laufs, 2008).
The data presented not only provide an advance in our understanding of the physiological interactions between increased NO bioavailability and the neural, endocrine and metabolic defence responses to acute hypoxia in the fetus, but they have also significant clinical implications. The fetal peripheral vasoconstrictor response to acute hypoxia largely contributes to the redistribution of blood flow away from peripheral and towards essential vascular beds, such as those perfusing the brain (Cohn et al. 1974; Giussani et al. 1993). Therefore, in pregnant women taking statins, suppression of this fetal brain-sparing response may render the fetal central nervous system susceptible to hypoxic-ischaemic injury during periods of reduced fetal oxygenation, as may occur during complicated labour and delivery (Low, 2004). Alternatively, just as enhanced NO bioavailability induced by pravastatin may oppose an increase in vascular resistance in circulations which constrict such as the femoral vascular bed in the fetus, enhanced NO bioavailability induced by pravastatin may augment an increase in blood flow in circulations which vasodilate via NO-dependent mechanisms, such as the cerebral vascular bed (McCrabb & Harding, 1996). Whichever the case, there is clearly an urgent need to carry out further research into the mechanisms via which statins affect cardiovascular function in the fetus, before statins could be prescribed to pregnant women.
In conclusion, treatment of late gestation fetal sheep with pravastatin did not alter fetal basal physiology, but it did significantly depress the fetal cardiovascular and metabolic responses to acute hypoxia. The mechanism underlying these suppressive effects of pravastatin is secondary to increased NO synthesis, since fetal treatment with NO synthase blockade with the NO clamp restored the responses to control levels. The use of statins during pregnancy should be viewed with extreme caution.
Acknowledgments
This work was supported by the British Heart Foundation, the BBSRC, the Frank Edward Elmore Fund and the James Baird Fund. D.A.G. is a Royal Society Wolfson Research Merit Award holder. We are thankful to Mr S. Gentle and Mrs S. Nicholls for their invaluable help with the maintenance of the animals. The authors have no conflicts of interest to declare.
Glossary
Abbreviations
- eNOS
endothelial nitric oxide synthase
- HMG-CoA
3-hydroxy-3-methyl-glutaryl-CoA
- NO
nitric oxide
- ROS
reactive oxygen species
Author contributions
The experiments were performed at The Barcroft Building, Department of Physiology, Development & Neuroscience, University of Cambridge. All authors were involved in the conception and design of the experiments, the collection, analysis and interpretation of data, and drafting the article or revising it critically for important intellectual content. We confirm that all authors approved the final version of the manuscript.
References
- Adam O, Laufs U. Antioxidant effects of statins. Arch Toxicol. 2008;82:885–892. doi: 10.1007/s00204-008-0344-4. [DOI] [PubMed] [Google Scholar]
- Ahmed A. New insights into the etiology of preeclampsia: identification of key elusive factors for the vascular complications. Thromb Res. 2011;127(Suppl. 3):S72–S75. doi: 10.1016/S0049-3848(11)70020-2. [DOI] [PubMed] [Google Scholar]
- Apatu RS, Barnes RJ. Release of glucose from the liver of fetal and postnatal sheep by portal vein infusion of catecholamines or glucagon. J Physiol. 1991;436:449–468. doi: 10.1113/jphysiol.1991.sp018560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelds B, van Bel F, Teitel D, Rudolph A. Carotid, not aortic, chemoreceptors mediate the fetal cardiovascular response to acute hypoxemia in lambs. Pediatr Res. 1993;34:51–55. doi: 10.1203/00006450-199307000-00013. [DOI] [PubMed] [Google Scholar]
- Blum A, Shamburek R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis. 2009;203:325–330. doi: 10.1016/j.atherosclerosis.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Boyle D, Hirst K, Zerbe G, Meschia G, Wilkening R. Fetal hind limb oxygen consumption and blood flow during acute graded hypoxia. Pediatr Res. 1990;28:94–100. doi: 10.1203/00006450-199008000-00004. [DOI] [PubMed] [Google Scholar]
- Buhaescu I, Izzedine H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin Biochem. 2007;40:575–584. doi: 10.1016/j.clinbiochem.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Chen K, Keaney JF. Reactive oxygen species-mediated signal transduction in the endothelium. Endothelium. 2004;11:109–121. doi: 10.1080/10623320490482655. [DOI] [PubMed] [Google Scholar]
- Cohn HE, Sacks EJ, Heymann MA, Rudolph AM. Cardiovascular responses to hypoxemia and acidemia in fetal lambs. Am J Obstet Gynecol. 1974;120:817–824. doi: 10.1016/0002-9378(74)90587-0. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Edison R, Muenke M. Mechanistic and epidemiologic considerations in the evaluation of adverse birth outcomes following gestational exposure to statins. Am J Med Genet A. 2004;131:287–298. doi: 10.1002/ajmg.a.30386. [DOI] [PubMed] [Google Scholar]
- Gardner DS, Fletcher AJ, Fowden AL, Giussani DA. A novel method for controlled and reversible long term compression of the umbilical cord in fetal sheep. J Physiol. 2001;535:217–229. doi: 10.1111/j.1469-7793.2001.00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DS, Fowden AL, Giussani DA. Adverse intrauterine conditions diminish the fetal defense against acute hypoxia by increasing nitric oxide activity. Circulation. 2002;106:2278–2283. doi: 10.1161/01.cir.0000033827.48974.c8. [DOI] [PubMed] [Google Scholar]
- Gardner DS, Giussani DA. Enhanced umbilical blood flow during acute hypoxemia after chronic umbilical cord compression: a role for nitric oxide. Circulation. 2003;108:331–335. doi: 10.1161/01.CIR.0000080323.40820.A1. [DOI] [PubMed] [Google Scholar]
- Gelosa P, Cimino M, Pignieri A, Tremoli E, Guerrini U, Sironi L. The role of HMG-CoA reductase inhibition in endothelial dysfunction and inflammation. Vasc Health Risk Manag. 2007;3:567–577. [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Gardner DS, Cox DT, Fletcher AJ. Purinergic contribution to circulatory, metabolic, and adrenergic responses to acute hypoxemia in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;280:R678–R685. doi: 10.1152/ajpregu.2001.280.3.R678. [DOI] [PubMed] [Google Scholar]
- Giussani DA, Spencer JA, Moore PJ, Bennet L, Hanson MA. Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. J Physiol. 1993;461:431–449. doi: 10.1113/jphysiol.1993.sp019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes ME, Tack CJ, Verheugt FW, Smits P, Lenders JWM. Sympathoinhibition by atorvastatin in hypertensive patients. Circ J. 2010;74:2622–2626. doi: 10.1253/circj.cj-10-0427. [DOI] [PubMed] [Google Scholar]
- Green LR, Bennet L, Hanson MA. The role of nitric oxide synthesis in cardiovascular responses to acute hypoxia in the late gestation sheep fetus. J Physiol. 1996;497:271–277. doi: 10.1113/jphysiol.1996.sp021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LR, McGarrigle HHG, Bennet L, Hanson MA. The role of endothelin-A receptors in cardiovascular responses to acute hypoxaemia in the late gestation sheep fetus. J Physiol. 1998;509:297–304. doi: 10.1111/j.1469-7793.1998.297bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner LJ. Advanced maternal age – how old is too old? N Engl J Med. 2004;351:1927–1929. doi: 10.1056/NEJMp048087. [DOI] [PubMed] [Google Scholar]
- Hooper SB. Fetal metabolic responses to hypoxia. Reprod Fertil Dev. 1995;7:527–538. doi: 10.1071/rd9950527. [DOI] [PubMed] [Google Scholar]
- Jones CT. The development of some metabolic responses to hypoxia in the foetal sheep. J Physiol. 1977;265:743–762. doi: 10.1113/jphysiol.1977.sp011741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CT, Ritchie JW. The effects of adrenergic blockade on fetal response to hypoxia. J Dev Physiol. 1983;5:211–222. [PubMed] [Google Scholar]
- Jones CT, Robinson RO. Plasma catecholamines in foetal and adult sheep. J Physiol. 1975;248:15–33. doi: 10.1113/jphysiol.1975.sp010960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaesemeyer WH, Caldwell RB, Huang J, Caldwell RW. Pravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol-lowering actions. J Am Coll Cardiol. 1999;33:234–241. doi: 10.1016/s0735-1097(98)00514-2. [DOI] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- Li Y-L, Zheng H, Ding Y, Schultz HD. Expression of neuronal nitric oxide synthase in rabbit carotid body glomus cells regulates large-conductance Ca2+-activated potassium currents. J Neurophysiol. 2010;103:3027–3033. doi: 10.1152/jn.01138.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JA. Reflections on the occurrence and significance of antepartum fetal asphyxia. Best Pract Res Clin Obstet Gynaecol. 2004;18:375–382. doi: 10.1016/j.bpobgyn.2004.02.002. [DOI] [PubMed] [Google Scholar]
- McCrabb G, Harding R. Role of nitric oxide in the regulation of cerebral blood flow in the ovine foetus. Clin Exp Pharmacol Physiol. 1996;23:855–860. doi: 10.1111/j.1440-1681.1996.tb01133.x. [DOI] [PubMed] [Google Scholar]
- Maki-Petaja K, Wilkinson I. Anti-inflammatory drugs and statins for arterial stiffness reduction. Curr Pharm Des. 2009;15:290–303. doi: 10.2174/138161209787354221. [DOI] [PubMed] [Google Scholar]
- Mondo CK, Yang W-S, Zhang N, Huang T-G. Anti-oxidant effects of atorvastatin in dexamethasone-induced hypertension in the rat. Clin Exp Pharmacol Physiol. 2006;33:1029–1034. doi: 10.1111/j.1440-1681.2006.04482.x. [DOI] [PubMed] [Google Scholar]
- Morrison S, Gardner DS, Fletcher AJ, Bloomfield MR, Giussani DA. Enhanced nitric oxide activity offsets peripheral vasoconstriction during acute hypoxaemia via chemoreflex and adrenomedullary actions in the sheep fetus. J Physiol. 2003;547:283–291. doi: 10.1113/jphysiol.2002.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National-Center-for-Health-Statistics. Health, United States, 2010: With Special Feature on Death and Dying. Hyattsville, Maryland: 2010. [PubMed] [Google Scholar]
- Pliquett RU, Cornish KG, Zucker IH. Statin therapy restores sympathovagal balance in experimental heart failure. J Appl Physiol. 2003;95:700–704. doi: 10.1152/japplphysiol.00265.2003. [DOI] [PubMed] [Google Scholar]
- Poore KR, Boullin JP, Cleal JK, Newman JP, Noakes DE, Hanson MA, Green LR. Sex- and age-specific effects of nutrition in early gestation and early postnatal life on hypothalamo-pituitary-adrenal axis and sympathoadrenal function in adult sheep. J Physiol. 2010;588:2219–2237. doi: 10.1113/jphysiol.2010.187682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reller MD, Burson MA, Lohr JL, Morton MJ, Thornburg KL. Nitric oxide is an important determinant of coronary flow at rest and during hypoxemic stress in fetal lambs. Am J Physiol Heart Circ Physiol. 1995;269:H2074–H2081. doi: 10.1152/ajpheart.1995.269.6.H2074. [DOI] [PubMed] [Google Scholar]
- Rudolph A. Distribution and regulation of blood flow in the fetal and neonatal lamb. Circ Res. 1985;57:811–821. doi: 10.1161/01.res.57.6.811. [DOI] [PubMed] [Google Scholar]
- StAmP. 2011. The StAmP trial: A proof of principle, double-blind, randomised placebo-controlled, multi centre trial of pravastatin to ameliorate early onset pre-eclampsia. http://www.stamp.bham.ac.uk/investigations/docs/StAmP_Protocol_Version_3.1_05Jan2011.pdf.
- Steinberg D. The statins in preventive cardiology. N Engl J Med. 2008;359:1426–1427. doi: 10.1056/NEJMp0806479. [DOI] [PubMed] [Google Scholar]
- Taguchi N, Rubin ET, Hosokawa A, Choi J, Ying AY, Moretti ME, Koren G, Ito S. Prenatal exposure to HMG-CoA reductase inhibitors: Effects on fetal and neonatal outcomes. Reprod Toxicol. 2008;26:175–177. doi: 10.1016/j.reprotox.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Tamai O, Matsuoka H, Itabe H, Wada Y, Kohno K, Imaizumi T. Single LDL apheresis improves endothelium-dependent vasodilatation in hypercholesterolemic humans. Circulation. 1997;95:76–82. doi: 10.1161/01.cir.95.1.76. [DOI] [PubMed] [Google Scholar]
- Thakor A, Giussani D. Nitric oxide reduces vagal baroreflex sensitivity in the late gestation fetus. Pediatr Res. 2009;65:269–273. doi: 10.1203/PDR.0b013e318193f134. [DOI] [PubMed] [Google Scholar]
- Thakor AS, Giussani DA. Role of nitric oxide in mediating in vivo vascular responses to calcitonin gene-related peptide in essential and peripheral circulations in the fetus. Circulation. 2005;112:2510–2516. doi: 10.1161/CIRCULATIONAHA.105.562546. [DOI] [PubMed] [Google Scholar]
- Thakor AS, Herrera EA, Serón-Ferré M, Giussani DA. Melatonin and vitamin C increase umbilical blood flow via nitric oxide-dependent mechanisms. J Pineal Res. 2010a;49:399–406. doi: 10.1111/j.1600-079X.2010.00813.x. [DOI] [PubMed] [Google Scholar]
- Thakor AS, Richter HG, Kane AD, Dunster C, Kelly FJ, Poston L, Giussani DA. Redox modulation of the fetal cardiovascular defence to hypoxaemia. J Physiol. 2010b;588:4235–4247. doi: 10.1113/jphysiol.2010.196402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrens C, Kelsall CJ, Hopkins LA, Anthony FW, Curzen NP, Hanson MA. Atorvastatin restores endothelial function in offspring of protein-restricted rats in a cholesterol-independent manner. Hypertension. 2009;53:661–667. doi: 10.1161/HYPERTENSIONAHA.108.122820. [DOI] [PubMed] [Google Scholar]
- Weir EK, Archer SL. The role of redox changes in oxygen sensing. Respir Physiol Neurobiol. 2010;174:182–191. doi: 10.1016/j.resp.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]