

Abstract
Non-technical summary
Activation of sympathetic nerves decreases blood flow to resting skeletal muscle, but this vasoconstrictor effect normally is blunted during exercise so that blood flow can increase to the working muscles. In rats and humans treated with nitroglycerin for 1 week, we show that overproduction of reactive oxygen species prevents the usual attenuation of sympathetic vasoconstriction in the working muscles, resulting in muscle ischaemia during exercise. Improved knowledge about the effect that reactive oxygen species has on muscle blood flow regulation may help us to better understand the decreased exercise tolerance that occurs with age as well as with chronic disease.
Abstract
Sympathetic vasoconstriction is normally attenuated in exercising muscle, but this functional sympatholysis is impaired in rats with hypertension or heart failure due to elevated levels of reactive oxygen species (ROS) in muscle. Whether ROS have a similar effect in the absence of cardiovascular disease or whether these findings extend to humans is not known. We therefore tested the hypothesis that chronic treatment with nitroglycerin (NTG) to induce nitrate tolerance, which is associated with excessive ROS production, impairs functional sympatholysis in healthy rats and humans. NTG treatment increased ethidium fluorescence in rat muscles and urinary F2-isoprostanes in humans, demonstrating oxidative stress. In vehicle-treated rats, sympathetic nerve stimulation (1 to 5 Hz) evoked decreases in femoral vascular conductance at rest (range, −30 to −63%) that were attenuated during hindlimb contraction (range, −2 to −31%; P < 0.05). In NTG-treated rats, vasoconstrictor responses were similar at rest, but were enhanced during contraction (range, −17 to −50%; P < 0.05 vs. vehicle). Infusion of the ROS scavenger tempol restored sympatholysis in these rats. In humans, reflex sympathetic activation during lower body negative pressure (LBNP) evoked decreases in muscle oxygenation in resting forearm (−12 ± 1%) that were attenuated during handgrip exercise (−3 ± 1%; P < 0.05). When these subjects became nitrate tolerant, LBNP-induced decreases in muscle oxygenation were unaffected at rest, but were enhanced during exercise (−9 ± 1%; P < 0.05 vs. before NTG). Collectively, these data indicate that functional sympatholysis is impaired in otherwise healthy nitrate-tolerant rats and humans by a mechanism probably involving muscle oxidative stress.
Introduction
Activation of the sympathetic nervous system plays an important role in the matching of muscle blood flow to metabolic demand during exercise by causing vasoconstriction in non-exercising muscles and visceral organs, which redirects cardiac output to the active muscles. At the same time, sympathetic vasoconstriction is attenuated in the active muscles in part due to an effect of muscle metabolites to reduce vascular responsiveness to α-adrenoreceptor activation (Ohyanagi et al. 1992; Hansen et al. 1996; Thomas & Victor, 1998; Dinenno & Joyner, 2004). Such modulation, termed functional sympatholysis (Remensnyder et al. 1962), may constitute a protective mechanism that blunts the otherwise deleterious effect of increased sympathetic vasoconstrictor drive to active muscle in order to optimize intramuscular blood flow distribution.
Many cardiovascular diseases are characterized by abnormal haemodynamic and sympathoexcitatory responses to exercise, which may compromise the close matching of muscle blood flow to metabolic demand, but the underlying mechanisms are incompletely understood. We have recently reported that functional sympatholysis is impaired in hypertensive humans, a finding that is congruent with our previous studies in rats with ischaemic heart failure or hypertension (Thomas et al. 2001; Zhao et al. 2006; Vongpatanasin et al. 2011). We and others have also shown that the production of reactive oxygen species (ROS) such as superoxide (O2−) and biomarkers of oxidative damage are elevated in muscles from rodents with heart failure or hypertension (Thomas et al. 2001; Tsutsui et al. 2001; Dalla Libera et al. 2005; Zhao et al. 2006). Furthermore, functional sympatholysis could be restored by infusion of the O2− scavengers tempol or tiron in our rat studies (Thomas et al. 2001; Zhao et al. 2006). Taken together, these findings suggest that muscle oxidative stress may be a common underlying mechanism that enhances sympathetic vasoconstriction during exercise in chronic cardiovascular disease.
Whether these findings have implications for sympathetic regulation of muscle blood flow in humans with cardiovascular disease is currently not known. Studying such patients can pose significant challenges due to the typical existence of co-morbidities and multiple drug therapies, variations in disease duration and severity, and the limitations inherent to a cross-sectional study design. To avoid these complications, we chose to begin to study the effect of oxidative stress on muscle blood flow regulation in healthy subjects. To induce oxidative stress in these subjects, we exploited the clinical phenomenon of nitrate tolerance. This reversible condition, in which the vasodilator and anti-ischaemic effects of organic nitrates such as nitroglycerin (NTG) wane with continuous exposure to the drugs, is thought to be mediated in part by increased production of O2− and the subsequent development of oxidative stress (Munzel et al. 1995b, 1999; Jurt et al. 2001; McGrath et al. 2002; Hink et al. 2007). We therefore hypothesized that sympathetic vasoconstriction would be markedly attenuated in the exercising muscles of subjects before exposure to NTG, but that this functional sympatholysis would be impaired after continuous treatment with NTG. Due to the lack of non-invasive techniques to directly assess muscle oxidative stress in humans, we also performed complementary experiments in rats to first establish that chronic NTG treatment could elevate muscle ROS sufficiently to impair functional sympatholysis.
Methods
Ethical approval
All procedures utilizing animals were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Centre and were conducted in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. All human subject protocols were approved by the University of Texas Southwestern Medical Centre Institutional Review Board and were conducted in accordance with the Declaration of Helsinki principles. Informed, written consent was obtained from each subject.
Animals
Forty-four Sprague–Dawley rats (220 ± 6 g) were anaesthetized with isoflurane (2.0–2.5%) and implanted with osmotic mini-pumps (Alzet model 2ML1, Alza Corporation) to deliver either vehicle (30% polyethylene glycol + 30% ethyl alcohol in water) or NTG (0.5 mg kg−1 h−1) subcutaneously for 7 days (n = 22 each group).
Experimental preparation
Anaesthesia was induced with ketamine (80 mg kg−1, i.p.) and maintained with α-chloralose (30 mg kg−1 h−1, i.v.). Rats were given atropine sulfate (0.5 mg kg−1, s.c.) to reduce tracheal secretions and were mechanically ventilated with room air and supplemental O2. Core temperature was maintained at 37°C using an external heat source. Catheters were inserted in a jugular vein and in a carotid artery, which was connected to a transducer (Statham P23 XL) to measure blood pressure (BP). A pulsed Doppler flow probe (Crystal Biotech, Matec Instrument Co.) was placed around the left femoral artery. Stimulating electrodes were affixed to the left sciatic nerve. The left hindlimb was fixed at the knee joint and connected to a force transducer (FT-10, Grass Instruments) via the calcaneal tendon. In a subset of rats (vehicle and NTG, n = 13 per group), a small non-occlusive catheter was inserted in the abdominal aorta proximal to the iliac bifurcation. In the remaining rats (vehicle and NTG, n = 9 per group), stimulating electrodes were affixed to the left lumbar sympathetic chain. The former group was used to evaluate muscle ROS production, while the latter group was used to evaluate sympathetic vasoconstriction as described in the following experimental protocols.
Experimental protocols
Evaluation of nitrate tolerance
At the beginning of each experiment, the BP responses to bolus injections of NTG (3 and 10 μg kg−1, i.v.) were measured in the vehicle- and NTG-treated rats.
In situ muscle ROS detection
Oxidation of the cell-permeable probe dihydroethidium (DHE) to yield the impermeant fluorescent product ethidium was used to evaluate intracellular O2− production in resting and contracting muscles (Supinski et al. 1999; Murrant & Reid, 2001; Zhao et al. 2006). DHE was freshly prepared and used for experiments in one vehicle- and one NTG-treated rat per day. As DHE is a light-sensitive dye, experiments were performed in a dimly lit room. DHE was infused via the aortic catheter alone (20 nmol min−1; vehicle and NTG, n = 7 per group) or in combination with the O2− scavenger tempol (4-hydroxy-2,2,6,6-tetramethyl-piperidine 1-oxyl; 3 μmol kg−1 min−1, i.v.; vehicle and NTG, n = 6 per group). Tempol infusion was begun 20 min prior to DHE. Twenty minutes after the start of the DHE infusion, the left sciatic nerve was stimulated at 2 to 3 times the motor threshold voltage with 100 ms trains of pulses at a rate of 30 trains min−1 to produce intermittent, tetanic contractions of the left hindlimb. After completion of a 15 min contraction period, the stimulator was turned off, and the plantaris muscles from the left and right hindlimbs were immediately excised, frozen in liquid nitrogen-cooled isopentane in OCT compound (Sakura Finetek), and stored at −80°C. Rats were killed with an overdose of sodium methohexital (150 mg kg−1, i.v.) followed by pneumothorax.
Sympathetic vasoconstriction in contracting hindlimb
In rats treated with vehicle or NTG (n = 9 per group), BP and femoral blood flow velocity (FBFV) responses to lumbar sympathetic nerve stimulation (LNS; 1 ms pulses of 5 V at randomized frequencies of 1, 2.5 or 5 Hz for 1 min) were evaluated at rest and during contraction of the left hindlimb as described above. After a 20 min recovery, infusion of tempol (3 μmol kg−1 min−1, i.v.) was begun and 30 min later sympathetic vasoconstrictor responses were re-evaluated in resting and contracting hindlimbs. Rats were killed with an overdose of sodium methohexital (150 mg kg−1, i.v.) followed by pneumothorax.
Human subjects
Nine healthy men with a mean age of 30 ± 2 years, height of 176 ± 2 cm and weight of 85 ± 7 kg participated in the study. Subjects were normotensive, had no history of cardiovascular disease and were not taking any medications. On experimental days, subjects were asked to abstain from caffeinated beverages for a minimum of 12 h and from strenuous physical activity for 24 h. Subjects were studied on two separate days to compare responses at baseline and after 6 days of continuous NTG treatment administered by transdermal patch (0.5 mg h−1). Subjects were instructed to change patches daily. A total of 18 subjects were initially enrolled in the study, but 9 subjects withdrew because they developed intolerable headaches and/or nausea during the first 24 h of NTG treatment (Christiansen et al. 2000).
Experimental measurements
Cardiovascular measurements
Heart rate (HR) was recorded continuously from the electrocardiogram and BP was measured by automated oscillometric sphygmomanometry (CE0050, Welch Allyn, Skaneateles Falls, NY, USA).
Skeletal muscle oxygenation
Near-infrared (NIR) spectroscopy (NIRO-500, Hamamatsu) was used to measure changes in tissue concentrations of oxygenated haemoglobin and myoglobin (HbO2+ MbO2) in the forearm, as previously described (Hansen et al. 1996; Chavoshan et al. 2002; Fadel et al. 2004). Briefly, two fibre optic bundles spaced 2 cm apart were placed over the flexor digitorum profundus muscle, which is the main muscle recruited during handgrip (Fleckenstein et al. 1992). NIR signals were sampled at a rate of 1 Hz, converted to chromophore concentrations using established algorithms, output to a personal computer, and digitally stored for later analysis. Changes in the NIR signals were quantified as a percentage of the total labile signal (TLS), which was defined in each experiment as the maximal decrease in HbO2+ MbO2 achieved during inflation of a pneumatic cuff on the upper arm to 220 mmHg just prior to the termination of exercise and sustained for 2 min. Compared to Doppler measures of forearm blood flow, changes in tissue oxygenation provide a reliable measure of sympathetic vasoconstriction in resting and exercising muscle (Fadel et al. 2004).
Reflex activation of sympathetic nerves
Lower body negative pressure (LBNP) was used to produce reflex sympathetic vasoconstriction in the forearm (Vissing et al. 1994). The subject's lower body was enclosed to the level of the iliac crest in a negative pressure chamber. LBNP at −20 mmHg was applied to evoke reproducible increases in muscle sympathetic nerve activity (MSNA) (Hansen et al. 1996). Multiunit recordings of MSNA were obtained with unipolar tungsten microelectrodes inserted into muscle fascicles of the peroneal nerve by microneurography (Vallbo et al. 1979). Neural signals were amplified, filtered (bandwidth 700–2000 Hz), rectified and integrated (time constant, 0.1 s) to obtain mean voltage neurograms.
Handgrip exercise
Maximal voluntary contraction (MVC) for each subject was designated as the greatest of at least three maximal squeezes of a handgrip dynamometer (Stoelting, Chicago, IL, USA). Subjects performed intermittent handgrip to the rhythm of a metronome (20 handgrips min−1; 50% duty cycle) at 30% MVC for 6 min. Force production was displayed on an oscilloscope to provide subjects with visual feedback.
Experimental protocols
Development of nitrate tolerance
Nitrate tolerance was assessed in each subject by measuring BP and HR during 5 min of quiet standing before, 3 h after and 6 days after initiating NTG treatment.
Systemic oxidative stress
Subjects provided spot urine samples after an overnight fast before and during nitrate tolerance. Samples were stored at –80°C for subsequent analysis of F2-isoprostanes using gas chromatography/mass spectrometry as previously described (Morrow & Roberts, 2002; Roberts & Morrow, 2002; Milne et al. 2007).
Sympathetic vasoconstriction in exercising forearm muscle
Subjects were studied in the supine position. BP, HR and forearm muscle oxygenation were measured in response to 2 min of LBNP at −20 mmHg applied at rest and during minutes 3–5 of handgrip exercise. In the majority of subjects (n = 6), the protocol was first performed at baseline (no treatment) and repeated on day 6 of NTG treatment. However, three randomly chosen subjects were studied in the reverse order. To evaluate the effect of NTG treatment on LBNP-induced sympathoexcitation, MSNA responses to LBNP were measured at rest and during handgrip before and during nitrate tolerance in a subset of subjects who consented to the microneurography procedure (n = 6).
Data analysis
Muscle DHE experiments
Plantaris muscles were sectioned transversely (10 μm) and random fields of each section were imaged at ×20 using an Optronics VI-470 CCD camera attached to a Leitz Laborlux-S epifluorescence microscope using Scion Image software. Images were taken by one of the investigators who was blinded to both the treatment (vehicle, NTG) and muscle status (rest, contraction). Muscles from paired vehicle- and NTG-treated rats were processed at the same time and imaged using the same settings. Red-fluorescent ethidium resulting from the oxidation of DHE was evaluated using NIH ImageJ software to quantify the ethidium-positive area of four to five non-overlapping fields, which were averaged to obtain a representative mean area in μm2 per muscle section.
Muscle blood flow and oxygenation experiments
For both rat and humans studies, data were acquired and analysed using PowerLab hardware and software (ADInstruments, Milford, MA, USA). For the animal studies, femoral vascular conductance (FVC, kHz mmHg−1) was calculated online as the mean Doppler shift (kHz) divided by mean arterial pressure (MAP, mmHg). MAP, FBFV and FVC responses to LNS stimulation applied at rest and during hindlimb contraction were determined by calculating the differences between the average of 10 s of baseline immediately preceding LNS and the last 10 s during LNS. For the human studies, BP, HR and muscle oxygenation responses to LBNP applied at rest and during handgrip were determined by calculating the differences between the average of 20 s of baseline immediately preceding LBNP and the last 20 s during LBNP. MSNA was analysed by averaging 60 s of data at baseline and during LBNP and was expressed as burst frequency (bursts min−1) and total activity (burst frequency × mean burst amplitude).
Statistical analysis
All data are presented as means ± SEM. For the animal experiments, differences in baseline MAP and the MAP responses to intravenous NTG were analysed by unpaired Student's t tests, the muscle ethidium fluorescence data were analysed by ANOVA followed by Bonferonni's multiple comparison test, and all other data were analysed by repeated measures ANOVA followed by Bonferonni's multiple comparison test. For the human experiments, differences in F2-isoprostane levels were analysed by a paired Student's t test and all other data were analysed by repeated measures ANOVA followed by Bonferonni's multiple comparison test. P < 0.05 was considered significant.
Results
Development of nitrate tolerance in rats
When measured 7 days after osmotic pump implantation, MAP was similar among all of the vehicle- and NTG-treated rats (104 ± 4 vs. 103 ± 4 mmHg, P > 0.05). Acute administration of nitroglycerin (3 and 10 μg kg−1, i.v.) produced dose-dependent decreases in MAP in vehicle-treated rats (−31 ± 1 and −42 ± 1 mmHg, respectively) that were significantly attenuated in NTG-treated rats (−11 ± 1 and −19 ± 1 mmHg, P < 0.05 vs. vehicle), indicating the development of nitrate tolerance.
Skeletal muscle O2− is elevated in nitrate-tolerant rats
MAP, FBFV and FVC were similar in vehicle- and NTG-treated rats at rest (before infusion), after infusion of DHE alone or in combination with tempol, and during unilateral hindlimb contractions (Table 1). Peak forces generated by the contracting hindlimbs were similar in vehicle- and NTG-treated groups (1.91 ± 0.05 vs. 1.90 ± 0.05 kg, P > 0.05). In cryosections of resting muscle, ethidium fluorescence was 148% greater in the NTG-treated rats relative to the vehicle-treated controls (5245 ± 756 vs. 2118 ± 269 μm2, P < 0.05) (Fig. 1). In contracting muscle, ethidium fluorescence was increased relative to that of resting muscle in both groups, but remained significantly greater in the NTG- vs. vehicle-treated rats (9355 ± 834 vs. 4376 ± 768 μm2, P < 0.05). Treatment with tempol significantly reduced ethidium fluorescence in resting muscle of NTG-treated rats to a level that was similar to that of vehicle-treated rats. Tempol also prevented the contraction-induced increase in ethidium fluorescence in both groups of rats.
Table 1.
Haemodynamics in rats used to evaluate O2− production in resting and contralateral contracting plantaris muscles
| Vehicle (n = 7) | Vehicle + Tempol (n = 6) | NTG (n = 7) | NTG + Tempol (n = 6) | |
|---|---|---|---|---|
| MAP (mmHg) | ||||
| Rest | 106 ± 7 | 102 ± 7 | 98 ± 10 | 107 ± 6 |
| DHE infusion | 106 ± 6 | 93 ± 6 | 99 ± 10 | 97 ± 8 |
| Contraction | 114 ± 5 | 101 ± 6 | 104 ± 8 | 107 ± 5 |
| FBFV (kHz) | ||||
| Rest | 1.58 ± 0.32 | 1.44 ± 0.25 | 1.26 ± 0.22 | 1.00 ± 0.22 |
| DHE infusion | 2.60 ± 0.37 | 2.19 ± 0.19 | 1.84 ± 0.36 | 2.17 ± 0.30 |
| Contraction | 6.46 ± 0.73a,b | 5.72 ± 0.41a,b | 5.10 ± 0.60a,b | 5.67 ± 0.77a,b |
| FVC (kHz mmHg−1) | ||||
| Rest | 0.016 ± 0.004 | 0.014 ± 0.002 | 0.014 ± 0.003 | 0.009 ± 0.002 |
| DHE infusion | 0.026 ± 0.004 | 0.024 ± 0.002 | 0.019 ± 0.003 | 0.022 ± 0.002a |
| Contraction | 0.057 ± 0.006a,b | 0.057 ± 0.003a,b | 0.050 ± 0.006a,b | 0.052 ± 0.006a,b |
Values are means ± SEM. NTG, nitroglycerin; MAP, mean arterial pressure; FBFV, femoral blood flow velocity; FVC, femoral vascular conductance; DHE, dihydroethidium.
P < 0.05 vs. Rest
P < 0.05 vs. DHE infusion.
Figure 1. Muscle oxidative stress in nitrate-tolerant rats.
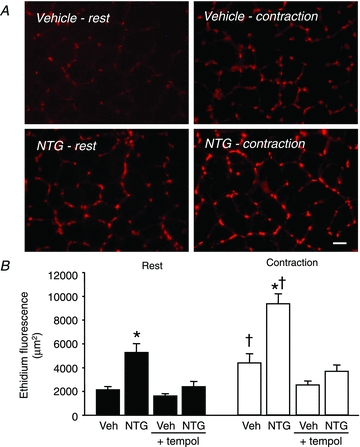
A, representative photomicrographs of ethidium fluorescence in resting and contracting muscles from a vehicle-treated rat and a NTG-treated rat. Scale bar, 40 μm. B, summary data showing increased ethidium fluorescence in resting and contracting muscles of NTG-treated rats compared to vehicle-treated rats (Veh). These increases were prevented by infusion of tempol. Veh and NTG, n = 7 per group; Veh + tempol and NTG + tempol, n = 6 per group. *P < 0.05 vs. vehicle; †P < 0.05 vs. rest.
Functional sympatholysis is impaired in nitrate-tolerant rats
MAP, FBFV and FVC were similar in vehicle- and NTG-treated rats at rest and during unilateral hindlimb contraction (Table 2). Peak hindlimb forces during contraction also were similar in vehicle-and NTG-treated rats (2.00 ± 0.10 vs. 1.79 ± 0.10 kg, P > 0.05). In vehicle-treated rats, sympathetic nerve stimulation evoked frequency-dependent increases in MAP and decreases in FBFV and FVC at rest (Figs 2A and 3A). The robust vasoconstrictor responses evoked by 1, 2.5 and 5 Hz sympathetic stimulation at rest were significantly attenuated during hindlimb contraction (ΔFVC, −30 ± 4, −51 ± 3 and −63 ± 3% at rest vs. −2 ± 2, −16 ± 3 and −31 ± 6% during contraction; P < 0.05). NTG-treated rats exhibited a similar pattern of sympathetic vasoconstriction at rest (ΔFVC, −34 ± 2, −56 ± 2 and −69 ± 2%; P > 0.05 vs. vehicle controls), but significantly less attenuation of the vasoconstrictor responses during hindlimb contraction (ΔFVC, −17 ± 4, −32 ± 6 and −50 ± 6%; P < 0.05 vs. vehicle controls) (Figs 2B and 3A).
Table 2.
Haemodynamics in rats used to evaluate sympathetic vasoconstriction in hindlimb muscles at rest and during contraction
| Vehicle (n = 9) | NTG (n = 9) | |||
|---|---|---|---|---|
| − Tempol | + Tempol | − Tempol | + Tempol | |
| MAP (mmHg) | ||||
| Rest | 104 ± 6 | 93 ± 6b | 103 ± 5 | 83 ± 5b |
| Contraction | 106 ± 7 | 103 ± 7 | 108 ± 7 | 94 ± 7b |
| FBFV (kHz) | ||||
| Rest | 1.44 ± 0.27 | 1.63 ± 0.22 | 1.64 ± 0.12 | 1.98 ± 0.14 |
| Contraction | 4.84 ± 0.77a | 4.76 ± 0.78a | 6.41 ± 0.55a | 4.86 ± 0.45a,b |
| FVC (kHz mmHg−1) | ||||
| Rest | 0.014 ± 0.003 | 0.018 ± 0.003 | 0.016 ± 0.002 | 0.025 ± 0.002 b |
| Contraction | 0.046 ± 0.006a | 0.045 ± 0.005a | 0.059 ± 0.004a | 0.052 ± 0.003a |
Values are means ± SEM. NTG, nitroglycerin; MAP, mean arterial pressure; FBFV, femoral blood flow velocity; FVC, femoral vascular conductance.
P < 0.05 vs. Rest
P < 0.05 vs. − Tempol.
Figure 2. Representative original recordings of experiments in a vehicle-treated rat and a NTG-treated rat.
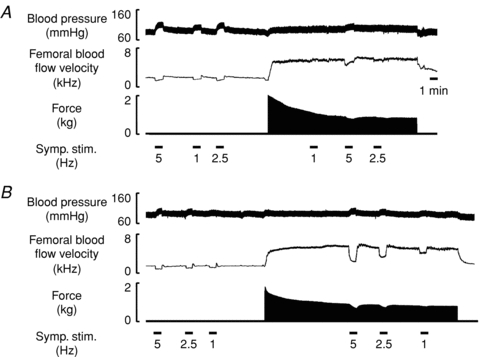
A, in the vehicle-treated rat, sympathetic nerve stimulation (symp. stim.) evoked frequency-dependent increases in arterial blood pressure and decreases in femoral blood flow velocity at rest, which were attenuated during hindlimb contraction. B, in the NTG-treated rat, sympathetic vasoconstrictor responses were abnormally preserved during hindlimb contraction.
Figure 3. Modulation of sympathetic vasoconstriction in rat hindlimb muscles.
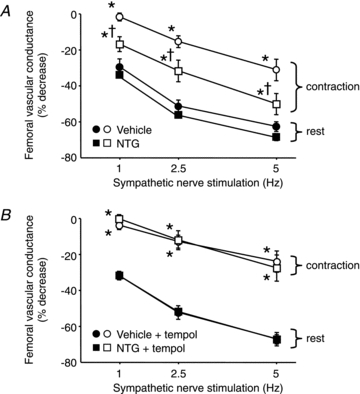
A, sympathetic stimulation evoked similar decreases in femoral vascular conductance in resting hindlimbs (filled symbols) of vehicle- and NTG-treated rats. These vasoconstrictor responses were attenuated during contraction in both groups (open symbols), but the degree of attenuation was less in the NTG-treated rats. B, in these same rats, infusion of tempol selectively attenuated sympathetic vasoconstriction only in the contracting hindlimbs of the NTG-treated rats. Vehicle and NTG, n = 9 per group. *P < 0.05 vs. rest; †P < 0.05 vs. vehicle.
Tempol normalizes functional sympatholysis in nitrate-tolerant rats
Infusion of tempol had modest effects on haemodynamics at rest, reducing MAP in both vehicle- and NTG-treated rats (−12 ± 6 and −20 ± 3 mmHg; P < 0.05 vs. before tempol) and increasing FVC in the NTG-treated rats (+0.009 ± 0.002 kHz mmHg−1; P < 0.05 vs. before tempol) (Table 2). Tempol had no effect on sympathetic vasoconstriction in the resting hindlimbs of either vehicle- or NTG-treated rats, or in the contracting hindlimbs of the vehicle-treated rats (Fig. 3B). In contrast, sympathetic vasoconstriction was attenuated to a greater extent in the contracting hindlimbs of the NTG-treated rats during tempol infusion (ΔFVC, −1 ± 2, −12 ± 5 and −28 ± 7%), such that the vasoconstrictor responses were no longer different from those of the vehicle-treated rats (ΔFVC, −4 ± 2, −13 ± 4 and −24 ± 6%; P > 0.05 vs. NTG).
Development of nitrate tolerance in humans
Three hours after initiating NTG treatment, MAP measured in the standing position was decreased (84 ± 3 vs. 95 ± 3 mmHg, pre-NTG; P < 0.05) and HR was increased (97 ± 3 vs. 78 ± 2 beats min−1, pre-NTG; P < 0.05). After 6 days of continuous NTG treatment, standing MAP (96 ± 3 mmHg) and HR (79 ± 3 beats min−1) had returned to the pre-NTG baseline, indicating the development of nitrate tolerance.
Systemic F2-isoprostanes are elevated in nitrate-tolerant humans
The level of urinary F2-isoprostanes, which is a sensitive biomarker of oxidative stress, was significantly increased by 38 ± 13% (2.15 ± 0.37 to 2.83 ± 0.46 ng (mg creatine)−1, P < 0.05) in subjects after the development of nitrate tolerance (Fig. 4). Urine samples were missing for two subjects due to failure to collect samples from one subject and loss of the NTG treatment sample in another subject.
Figure 4. Urinary F2-isoprostane levels in human subjects before and during nitrate tolerance.
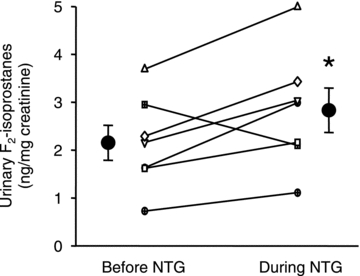
Individual (small symbols) and mean data (n = 7; large circles) are shown. *P < 0.05 vs. before NTG.
Functional sympatholysis is impaired in nitrate-tolerant humans
MAP and HR responses to LBNP or rhythmic handgrip, performed alone and in combination, were similar before and during nitrate tolerance (Table 3). Likewise, the decreases in muscle oxygenation in response to handgrip alone were similar before (−29 ± 5%) and during (−34 ± 9%; P > 0.05) nitrate tolerance. Prior to the development of nitrate tolerance, LBNP evoked decreases in muscle oxygenation in the resting forearm that were significantly attenuated during forearm exercise (−12 ± 1 vs. −3 ± 1%; P < 0.05) (Figs 5A and 6A). After the development of nitrate tolerance, LBNP evoked similar decreases in muscle oxygenation in the resting forearm (−13 ± 1%; P > 0.05 vs. before NTG), but larger decreases in oxygenation in the exercising forearm (−9 ± 1%; P < 0.05 vs. before NTG) (Figs 5B and 6A). The increments in MSNA evoked by LBNP were similar at rest and during handgrip exercise, both before and during nitrate tolerance (Table 3 and Fig. 6B).
Table 3.
Human subject cardiovascular and sympathoexcitatory responses to lower body negative pressure and handgrip exercise performed alone and in combination
| Before NTG | During NTG | |
|---|---|---|
| MAP (mmHg) | 84 ± 4 | 85 ± 4 |
| LBNP alone | −1 ± 1 | −1 ± 1 |
| HG alone | +5 ± 1 | +5 ± 1 |
| LBNP during HG | +3 ± 1 | +2 ± 1 |
| HR (beats min−1) | 60 ± 1 | 60 ± 3 |
| LBNP alone | +6 ± 1 | +6 ± 2 |
| HG alone | +9 ± 2 | +6 ± 1 |
| LBNP during HG | +5 ± 1 | +6 ± 2 |
| MSNA (bursts min−1) | 18 ± 5 | 17 ± 4 |
| LBNP alone | +11 ± 2 | +10 ± 2 |
| HG alone | +1 ± 1 | +0.4 ± 1 |
| LBNP during HG | +16 ± 3 | +12 ± 2 |
Values are means ± SEM. NTG, nitroglycerin; MAP, mean arterial pressure; HR, heart rate; MSNA, muscle sympathetic nerve activity; LBNP, lower body negative pressure; HG, handgrip. MAP and HR, n = 9; MSNA, n = 6.
Figure 5. Representative original recordings of an experiment in the same subject before and during nitrate tolerance.
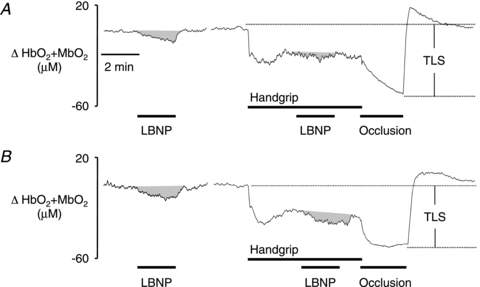
A, before NTG treatment, the decrease in muscle oxygenation (HbO2+ MbO2) evoked by lower body negative pressure (LBNP) at rest was significantly attenuated during handgrip. B, after the development of nitrate tolerance, the LBNP-induced decreases in muscle oxygenation were similar in resting and exercising muscles. TLS, total labile signal.
Figure 6. Modulation of sympathetic vasoconstriction in human forearm muscles.
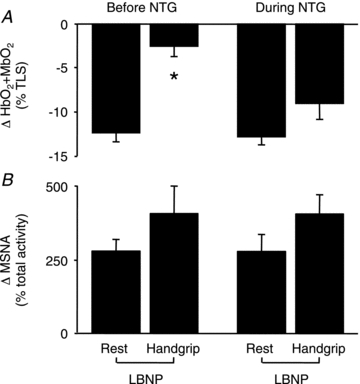
A, LBNP evoked decreases in oxygenation (HbO2+ MbO2) in resting forearm muscles that were attenuated during handgrip before, but not during, nitrate tolerance (n = 9). *P < 0.05 vs. rest. B, increases in muscle sympathetic nerve activity (MSNA) in response to LBNP were similar at rest and during handgrip, both before and during nitrate tolerance (n = 6).
Discussion
The major new finding of our study is that a week of continuous exposure to NTG in healthy rats and humans significantly impaired functional sympatholysis, resulting in enhanced sympathetic vasoconstriction in the exercising muscles. This impairment was accompanied by elevations in urinary F2-isoprostanes in NTG-treated humans and by increased DHE oxidation in the muscles of NTG-treated rats, both of which are sensitive indices of oxidative stress (Supinski et al. 1999; Murrant & Reid, 2001; Roberts & Morrow, 2002; Morrow, 2005; Milne et al. 2007). In the NTG-treated rats, the O2− scavenger tempol reduced muscle oxidative stress and restored functional sympatholysis, providing strong evidence that even a relatively brief period of oxidative stress in otherwise healthy subjects can markedly interfere with sympathetic vasoregulation in exercising muscle.
We previously reported that oxidative stress as reflected by elevated muscle O2− production or lipid peroxidation plays a causal role in impairing functional sympatholysis in rodent models of chronic cardiovascular disease, including ischaemic heart failure and hypertension (Thomas et al. 2001; Zhao et al. 2006). The present study extends this finding in two important ways. First, we have directly translated our findings from studies in anaesthetized rats undergoing electrically induced hindlimb contractions to conscious humans performing voluntary handgrip exercise. Our complementary experiments in nitrate-tolerant rats and humans provide evidence in support of the concept that oxidative stress shifts the finely tuned balance between vasodilator and vasoconstrictor influences in exercising muscle in favour of vasoconstriction. Second, we now have shown that the effect of oxidative stress to enhance sympathetic vasoconstriction in skeletal muscle is not a secondary consequence of chronic cardiovascular disease, but can occur in young, healthy subjects after only short-term exposure to NTG. Since oxidative stress may be present early in the course of cardiovascular disease (Armitage et al. 2009; Forstermann, 2010), it is tempting to speculate that the effect of ROS such as O2− to augment sympathetic vasoconstriction in skeletal muscle may contribute to disease progression and/or severity.
To induce oxidative stress in our human and animal subjects, we took advantage of a well-known clinically undesirable side-effect of NTG. While organic nitrates such as NTG produce beneficial haemodynamic effects when administered acutely, tolerance to these drugs develops upon continuous exposure (Munzel et al. 2011). The underlying mechanisms are not fully understood, but are thought to include reduced bioconversion of NTG to its active vasodilator metabolite by mitochondrial aldehyde dehydrogenase-2 (Chen et al. 2002), induction of oxidative stress due to both mitochondrial and cytosolic (NADPH oxidase-derived) O2− production (Munzel et al. 1995b; Sydow et al. 2004), and neurohormonal activation (increased angiotensin II, vasopressin, catecholamines) caused by the initial hypotensive effect of NTG (Parker et al. 1991). The latter may also contribute to cytosolic O2− production as angiotensin II is a potent activator of NADPH oxidase (Griendling et al. 1994; Rajagopalan et al. 1996).
To confirm NTG-induced oxidative stress in our human subjects, we measured the level of urinary F2-isoprostanes, which are stable isomers of PGF2α formed by ROS-mediated lipid peroxidation of arachidonic acid (Montuschi et al. 2004; Morrow, 2005). F2-isoprostanes in blood or urine have been shown to provide a specific, quantifiable index of systemic oxidative stress with little daily variability reported in most studies (Wang et al. 1995; Helmersson & Basu, 2001; Cracowski et al. 2002). The increase in urinary F2-isoprostanes (38%) that we observed in our study is comparable to increases previously reported in plasma (56%) and platelets (55%) of healthy, NTG-treated subjects (Jurt et al. 2001; McGrath et al. 2002). In rats, we extended this finding by using a more direct measure of intracellular O2− (oxidation of infused DHE) to capture its production in real time specifically in skeletal muscle (Supinski et al. 1999; Murrant & Reid, 2001; Zhao et al. 2006). These experiments yielded two important findings: chronic NTG treatment increased O2− more than 2-fold in both resting and contracting muscle, and these increases were markedly attenuated by infusion of the O2− scavenger tempol. Collectively, these findings indicate that continuous treatment with NTG is a viable and relatively rapid means to induce sustained oxidative stress in non-diseased muscle, thereby providing a compelling experimental model to use to test our main hypothesis that oxidative stress impairs functional sympatholysis.
Our finding that sympathetic vasoconstriction is enhanced in the contracting muscles of NTG-treated rats and humans could potentially be explained by an increase in either the sympathetic stimulus or in the vascular response to sympathetic activation. Chronic NTG treatment has been reported to increase central sympathetic outflow in pigs and to increase baroreflex-mediated sympathetic modulation of heart rate in humans (Zanzinger et al. 1998; Gori & Parker, 2002). In addition, circulating catecholamines have been reported to be elevated in some studies of nitrate-tolerant subjects (Parker et al. 1991). Despite the potential for excessive sympathetic/noradrenergic drive, we think that it is unlikely to explain the impaired functional sympatholysis in our study for several reasons. First, we directly measured MSNA in our human subjects and found no difference in baseline MSNA or in the LBNP-evoked increases in MSNA in the same subjects before and during NTG treatment, excluding a role for increases in central sympathetic drive or reflex-mediated sympathetic activation. Second, we circumvented this issue in the rats by directly controlling the vasoconstrictor stimulus via electrical stimulation of the lumbar sympathetic chain and saw clearly impaired sympatholysis in the NTG-treated rats. Third, we have previously shown that chronic infusion of a pressor dose of noradrenaline in rats does not impair functional sympatholysis, excluding a major role for circulating noradrenaline (Zhao et al. 2006).
It is more likely that the impaired functional sympatholysis that we observed in NTG-treated subjects is due to an enhanced vascular response to sympathetic activation. However, it is important to note that sympathetic vasoconstriction was increased only in the contracting muscles of nitrate-tolerant subjects – responses in the resting muscles were similar in vehicle- and NTG-treated rats, and in humans before and during NTG treatment. This indicates that the impaired sympatholysis is probably not due to a generalized supersensitivity to sympathetic stimulation, but is specifically related to events occurring in the contracting muscles. These could include changes in the production of vasoactive substances released either from the active muscle fibres or vascular endothelial cells, or changes in the downstream signalling pathways activated by these substances.
One key pathway that is markedly affected by chronic NTG treatment is nitric oxide (NO)-mediated vasodilatation, which is disrupted at multiple levels due to ROS-dependent uncoupling of NO synthase (NOS), scavenging of NO and inhibition of soluble guanylyl cyclase (Mulsch et al. 1997; Munzel et al. 2000; Warnholtz et al. 2002; Hink et al. 2003). We previously have shown that NOS-derived NO is one of the mediators of sympatholysis in both rodent and human skeletal muscle and that oxidative stress attenuates NO-dependent sympatholysis (Thomas et al. 1998, 2001; Thomas & Victor, 1998; Chavoshan et al. 2002; Zhao et al. 2006). We speculate that the impaired functional sympatholysis that we have observed in nitrate-tolerant subjects may therefore be mediated by an effect of O2− to reduce NO signalling in the muscle microcirculation. Consistent with this proposed mechanism, a previous study of NTG-treated rats showed that O2−-dependent attenuation of NO dilatation was greater in the smaller terminal arterioles than in the larger arcade arterioles of the microcirculation (Frame et al. 2002). As α-adrenergic vasoconstriction of the terminal arterioles is highly sensitive to attenuation by NO (Ohyanagi et al. 1992), loss of this dilator signal with prolonged NTG treatment would be expected to enhance sympathetic vasoconstriction of the terminal arterioles, thereby contributing to impaired sympatholysis. Although ROS generated during chronic NTG treatment inactivate mitochondrial aldehyde dehydrogenase-2 to reduce NTG bioactivation (Sydow et al. 2004), oxidative inhibition of this enzyme does not interfere with NOS-dependent NO production and therefore is unlikely to be responsible for impairing functional sympatholysis.
Our finding that tempol reduced O2− production in the contracting muscles of both the vehicle- and NTG-treated rats, but only improved functional sympatholysis in the NTG group suggests that a threshold level of O2− must be reached before sympatholysis is significantly impaired. The normal contraction-induced increase in O2− that occurs in non-diseased muscle apparently is not sufficient to interfere with the signalling pathways that attenuate sympathetic vasoconstriction, but probably modulates other redox-sensitive intracellular pathways such as activation of transcription factors and modulation of force production (Powers & Jackson, 2006). However, when O2− levels increase beyond the usual physiological range, detrimental effects such as NO scavenging become more pronounced and functional sympatholysis is impaired. In our previous study of hypertensive rats, we observed a similar differential effect of muscle O2− where low to moderate increases in contracting muscle had no impact on sympatholysis while moderate to high increases impaired sympatholysis (Zhao et al. 2006). The precise role of specific ROS (e.g. O2−, H2O2) to reduce NO signalling as well as other molecular changes induced by NTG treatment that could potentially reduce sympatholysis, such as decreased prostacyclin or increased endothelin-1 (Munzel et al. 1995a; Hink et al. 2003; Dinenno & Joyner, 2004; Wray et al. 2007), remain to be explored in future studies.
In summary, we found that continuous exposure of either rat or human subjects to NTG for 1 week induces a sustained oxidative stress and selectively enhances sympathetic vasoconstriction in active muscle without affecting the constrictor responses in resting muscle. The ability of a O2− scavenger to reverse this impaired functional sympatholysis in the NTG-treated rats strongly implicates a causal role for ROS. These findings are consistent with our previous studies showing impaired functional sympatholysis due to oxidative stress in rats with experimental hypertension or heart failure (Thomas et al. 2001; Zhao et al. 2006). We now, we believe for the first time, have extended this concept to humans and have shown that oxidative stress, even in the absence of chronic cardiovascular disease, can have a major negative influence on sympathetic regulation of muscle blood flow during exercise. These findings may provide a causal link between recent observations of vascular and muscle oxidative stress in healthy older individuals and previous reports of enhanced sympathetic vasoconstriction in exercising muscle in similarly aged individuals (Koch et al. 2003; Dinenno et al. 2005; Donato et al. 2007; Parker et al. 2007; Bailey et al. 2010; Kirby et al. 2011).
Acknowledgments
This work was supported by National Institutes of Health grant HL06296 (G.D.T.). The authors thank Jason Morrow (Vanderbilt University) for performing the F2-isoprostane analysis.
Glossary
Abbreviations
- BP
blood pressure
- DHE
dihydroethidium
- FBFV
femoral blood flow velocity
- FVC
femoral vascular conductance
- HbO2+ MbO2
oxygenated haemoglobin + myoglobin
- HR
heart rate
- LBNP
lower body negative pressure
- LNS
lumbar nerve stimulation
- MAP
mean arterial pressure
- MSNA
muscle sympathetic nerve activity
- MVC
maximal voluntary contraction
- NIR
near-infrared
- NO
nitric oxide
- NOS
nitric oxide synthase
- NTG
nitroglycerin
- O2−
superoxide
- ROS
reactive oxygen species
- TLS
total labile signal
Author contributions
The experiments were performed in the laboratories of the Hypertension Division at the University of Texas Southwestern Medical Centre. P.J.F. contributed to the design of the experiments, the collection, analysis and interpretation of the data, and drafting the article. M.F. contributed to the design of the experiments and the collection, analysis and interpretation of the data. K.M.G. and Z.W. contributed to the collection and analysis of the data. G.D.T. contributed to the conception and design of the experiments, the analysis and interpretation of the data, and revising the article critically for important intellectual content. All authors approved the final version.
References
- Armitage ME, Wingler K, Schmidt HH, La M. Translating the oxidative stress hypothesis into the clinic: NOX versus NOS. J Mol Med. 2009;87:1071–1076. doi: 10.1007/s00109-009-0544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, McEneny J, Mathieu-Costello O, Henry RR, James PE, McCord JM, Pietri S, Young IS, Richardson RS. Sedentary aging increases resting and exercise-induced intramuscular free radical formation. J Appl Physiol. 2010;109:449–456. doi: 10.1152/japplphysiol.00354.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavoshan B, Sander M, Sybert TE, Hansen J, Victor RG, Thomas GD. Nitric oxide-dependent modulation of sympathetic neural control of oxygenation in exercising human skeletal muscle. J Physiol. 2002;540:377–386. doi: 10.1113/jphysiol.2001.013153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen I, Iversen HK, Olesen J. Headache characteristics during the development of tolerance to nitrates: pathophysiological implications. Cephalalgia. 2000;20:437–444. doi: 10.1046/j.1468-2982.2000.00064.x. [DOI] [PubMed] [Google Scholar]
- Cracowski JL, Durand T, Bessard G. Isoprostanes as a biomarker of lipid peroxidation in humans: physiology, pharmacology and clinical implications. Trends Pharmacol Sci. 2002;23:360–366. doi: 10.1016/s0165-6147(02)02053-9. [DOI] [PubMed] [Google Scholar]
- Dalla Libera L, Ravara B, Gobbo V, Danieli Betto D, Germinario E, Angelini A, Vescovo G. Skeletal muscle myofibrillar protein oxidation in heart failure and the protective effect of Carvedilol. J Mol Cell Cardiol. 2005;38:803–807. doi: 10.1016/j.yjmcc.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ. Combined NO and PG inhibition augments α-adrenergic vasoconstriction in contracting human skeletal muscle. Am J Physiol Heart Circ Physiol. 2004;287:H2576–H2584. doi: 10.1152/ajpheart.00621.2004. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Masuki S, Joyner MJ. Impaired modulation of sympathetic α-adrenergic vasoconstriction in contracting forearm muscle of ageing men. J Physiol. 2005;567:311–321. doi: 10.1113/jphysiol.2005.087668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, Seals DR. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circ Res. 2007;100:1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- Fadel PJ, Keller DM, Watanabe H, Raven PB, Thomas GD. Noninvasive assessment of sympathetic vasoconstriction in human and rodent skeletal muscle using near-infrared spectroscopy and Doppler ultrasound. J Appl Physiol. 2004;96:1323–1330. doi: 10.1152/japplphysiol.01041.2003. [DOI] [PubMed] [Google Scholar]
- Fleckenstein JL, Watumull D, Bertocci LA, Parkey RW, Peshock RM. Finger-specific flexor recruitment in humans: depiction by exercise-enhanced MRI. J Appl Physiol. 1992;72:1974–1977. doi: 10.1152/jappl.1992.72.5.1974. [DOI] [PubMed] [Google Scholar]
- Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459:923–939. doi: 10.1007/s00424-010-0808-2. [DOI] [PubMed] [Google Scholar]
- Frame MD, Fox RJ, Kim D, Mohan A, Berk BC, Yan C. Diminished arteriolar responses in nitrate tolerance involve ROS and angiotensin II. Am J Physiol Heart Circ Physiol. 2002;282:H2377–H2385. doi: 10.1152/ajpheart.00429.2001. [DOI] [PubMed] [Google Scholar]
- Gori T, Parker JD. Nitrate tolerance: a unifying hypothesis. Circulation. 2002;106:2510–2513. doi: 10.1161/01.cir.0000036743.07406.53. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- Hansen J, Thomas GD, Harris SA, Parsons WJ, Victor RG. Differential sympathetic neural control of oxygenation in resting and exercising human skeletal muscle. J Clin Invest. 1996;98:584–596. doi: 10.1172/JCI118826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmersson J, Basu S. F2-isoprostane and prostaglandin F2αmetabolite excretion rate and day to day variation in healthy humans. Prostaglandins Leukot Essent Fatty Acids. 2001;65:99–102. doi: 10.1054/plef.2001.0295. [DOI] [PubMed] [Google Scholar]
- Hink U, Daiber A, Kayhan N, Trischler J, Kraatz C, Oelze M, Mollnau H, Wenzel P, Vahl CF, Ho KK, Weiner H, Munzel T. Oxidative inhibition of the mitochondrial aldehyde dehydrogenase promotes nitroglycerin tolerance in human blood vessels. J Am Coll Cardiol. 2007;50:2226–2232. doi: 10.1016/j.jacc.2007.08.031. [DOI] [PubMed] [Google Scholar]
- Hink U, Oelze M, Kolb P, Bachschmid M, Zou MH, Daiber A, Mollnau H, August M, Baldus S, Tsilimingas N, Walter U, Ullrich V, Munzel T. Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J Am Coll Cardiol. 2003;42:1826–1834. doi: 10.1016/j.jacc.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Jurt U, Gori T, Ravandi A, Babaei S, Zeman P, Parker JD. Differential effects of pentaerythritol tetranitrate and nitroglycerin on the development of tolerance and evidence of lipid peroxidation: a human in vivo study. J Am Coll Cardiol. 2001;38:854–859. doi: 10.1016/s0735-1097(01)01414-0. [DOI] [PubMed] [Google Scholar]
- Kirby BS, Crecelius AR, Voyles WF, Dinenno FA. Modulation of postjunctional α-adrenergic vasoconstriction during exercise and exogenous ATP infusions in ageing humans. J Physiol. 2011;589:2641–2653. doi: 10.1113/jphysiol.2010.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch DW, Leuenberger UA, Proctor DN. Augmented leg vasoconstriction in dynamically exercising older men during acute sympathetic stimulation. J Physiol. 2003;551:337–344. doi: 10.1113/jphysiol.2003.042747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath LT, Dixon L, Morgan DR, McVeigh GE. Production of 8-epi prostaglandin F2α in human platelets during administration of organic nitrates. J Am Coll Cardiol. 2002;40:820–825. doi: 10.1016/s0735-1097(02)02037-5. [DOI] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- Montuschi P, Barnes PJ, Roberts LJ., 2nd Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- Morrow JD. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler Thromb Vasc Biol. 2005;25:279–286. doi: 10.1161/01.ATV.0000152605.64964.c0. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ., 2nd Mass spectrometric quantification of F2-isoprostanes as indicators of oxidant stress. Methods Mol Biol. 2002;186:57–66. doi: 10.1385/1-59259-173-6:57. [DOI] [PubMed] [Google Scholar]
- Mulsch A, Bauersachs J, Schafer A, Stasch JP, Kast R, Busse R. Effect of YC-1, an NO-independent, superoxide-sensitive stimulator of soluble guanylyl cyclase, on smooth muscle responsiveness to nitrovasodilators. Br J Pharmacol. 1997;120:681–689. doi: 10.1038/sj.bjp.0700982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel T, Daiber A, Gori T. Nitrate therapy: new aspects concerning molecular action and tolerance. Circulation. 2011;123:2132–2144. doi: 10.1161/CIRCULATIONAHA.110.981407. [DOI] [PubMed] [Google Scholar]
- Munzel T, Giaid A, Kurz S, Stewart DJ, Harrison DG. Evidence for a role of endothelin 1 and protein kinase C in nitroglycerin tolerance. Proc Natl Acad Sci U S A. 1995a;92:5244–5248. doi: 10.1073/pnas.92.11.5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel T, Hink U, Yigit H, Macharzina R, Harrison DG, Mulsch A. Role of superoxide dismutase in in vivo and in vitro nitrate tolerance. Br J Pharmacol. 1999;127:1224–1230. doi: 10.1038/sj.bjp.0702622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel T, Li H, Mollnau H, Hink U, Matheis E, Hartmann M, Oelze M, Skatchkov M, Warnholtz A, Duncker L, Meinertz T, Forstermann U. Effects of long-term nitroglycerin treatment on endothelial nitric oxide synthase (NOS III) gene expression, NOS III-mediated superoxide production, and vascular NO bioavailability. Circ Res. 2000;86:E7–E12. doi: 10.1161/01.res.86.1.e7. [DOI] [PubMed] [Google Scholar]
- Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995b;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant CL, Reid MB. Detection of reactive oxygen and reactive nitrogen species in skeletal muscle. Microsc Res Tech. 2001;55:236–248. doi: 10.1002/jemt.1173. [DOI] [PubMed] [Google Scholar]
- Ohyanagi M, Nishigaki K, Faber JE. Interaction between microvascular α1- and α2-adrenoceptors and endothelium-derived relaxing factor. Circ Res. 1992;71:188–200. doi: 10.1161/01.res.71.1.188. [DOI] [PubMed] [Google Scholar]
- Parker BA, Smithmyer SL, Jarvis SS, Ridout SJ, Pawelczyk JA, Proctor DN. Evidence for reduced sympatholysis in leg resistance vasculature of healthy older women. Am J Physiol Heart Circ Physiol. 2007;292:H1148–H1156. doi: 10.1152/ajpheart.00729.2006. [DOI] [PubMed] [Google Scholar]
- Parker JD, Farrell B, Fenton T, Cohanim M, Parker JO. Counter-regulatory responses to continuous and intermittent therapy with nitroglycerin. Circulation. 1991;84:2336–2345. doi: 10.1161/01.cir.84.6.2336. [DOI] [PubMed] [Google Scholar]
- Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2006;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remensnyder JP, Mitchell JH, Sarnoff SJ. Functional sympatholysis during muscular activity. Observations on influence of carotid sinus on oxygen uptake. Circ Res. 1962;11:370–380. doi: 10.1161/01.res.11.3.370. [DOI] [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supinski G, Nethery D, Stofan D, DiMarco A. Extracellular calcium modulates generation of reactive oxygen species by the contracting diaphragm. J Appl Physiol. 1999;87:2177–2185. doi: 10.1152/jappl.1999.87.6.2177. [DOI] [PubMed] [Google Scholar]
- Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, Ullrich V, Mulsch A, Schulz E, Keaney JF, Jr, Stamler JS, Munzel T. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG. Impaired metabolic modulation of α-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci U S A. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Victor RG. Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol. 1998;506:817–826. doi: 10.1111/j.1469-7793.1998.817bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Zhang W, Victor RG. Impaired modulation of sympathetic vasoconstriction in contracting skeletal muscle of rats with chronic myocardial infarctions: role of oxidative stress. Circ Res. 2001;88:816–823. doi: 10.1161/hh0801.089341. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Ide T, Hayashidani S, Suematsu N, Shiomi T, Wen J, Nakamura K, Ichikawa K, Utsumi H, Takeshita A. Enhanced generation of reactive oxygen species in the limb skeletal muscles from a murine infarct model of heart failure. Circulation. 2001;104:134–136. doi: 10.1161/01.cir.104.2.134. [DOI] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjork HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Vissing SF, Scherrer U, Victor RG. Increase of sympathetic discharge to skeletal muscle but not to skin during mild lower body negative pressure in humans. J Physiol. 1994;481:233–241. doi: 10.1113/jphysiol.1994.sp020434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongpatanasin W, Wang Z, Arbique D, Arbique G, Adams-Huet B, Mitchell JH, Victor RG, Thomas GD. Functional sympatholysis is impaired in hypertensive humans. J Physiol. 2011;589:1209–1220. doi: 10.1113/jphysiol.2010.203026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ciabattoni G, Creminon C, Lawson J, Fitzgerald GA, Patrono C, Maclouf J. Immunological characterization of urinary 8-epi-prostaglandin F2α excretion in man. J Pharmacol Exp Ther. 1995;275:94–100. [PubMed] [Google Scholar]
- Warnholtz A, Mollnau H, Heitzer T, Kontush A, Moller-Bertram T, Lavall D, Giaid A, Beisiegel U, Marklund SL, Walter U, Meinertz T, Munzel T. Adverse effects of nitroglycerin treatment on endothelial function, vascular nitrotyrosine levels and cGMP-dependent protein kinase activity in hyperlipidemic Watanabe rabbits. J Am Coll Cardiol. 2002;40:1356–1363. doi: 10.1016/s0735-1097(02)02133-2. [DOI] [PubMed] [Google Scholar]
- Wray DW, Nishiyama SK, Donato AJ, Sander M, Wagner PD, Richardson RS. Endothelin-1-mediated vasoconstriction at rest and during dynamic exercise in healthy humans. Am J Physiol Heart Circ Physiol. 2007;293:H2550–H2556. doi: 10.1152/ajpheart.00867.2007. [DOI] [PubMed] [Google Scholar]
- Zanzinger J, Czachurski J, Seller H. Impaired modulation of sympathetic excitability by nitric oxide after long-term administration of organic nitrates in pigs. Circulation. 1998;97:2352–2358. doi: 10.1161/01.cir.97.23.2352. [DOI] [PubMed] [Google Scholar]
- Zhao W, Swanson SA, Ye J, Li X, Shelton JM, Zhang W, Thomas GD. Reactive oxygen species impair sympathetic vasoregulation in skeletal muscle in angiotensin II-dependent hypertension. Hypertension. 2006;48:637–643. doi: 10.1161/01.HYP.0000240347.51386.ea. [DOI] [PubMed] [Google Scholar]