

Abstract
Spinal cord injury is associated with rapid bone loss and arrested long bone growth due to mechanisms that are poorly understood. In this study, we sought to determine the effects of severe T10 contusion spinal cord injury on the sublesional bone microenvironment in adolescent rats. A severe lower thoracic (vertebral T10) spinal cord injury was generated by weight drop (10 g×50 mm). Severely injured and body weight-matched uninjured male Sprague–Dawley rats were studied. At 3 and 5 days post-injury, we performed histological analysis of the distal femoral metaphysis, TUNEL assay, immunohistochemistry, real-time PCR, and western blot analysis compared to uninjured controls. We observed severe hindlimb functional deficits typical of this model. We detected uncoupled remodeling with increased osteoclast activity in the absence of osteoblast activity. We detected osteoblast, osteocyte, and chondrocyte apoptosis with suppressed osteoblast and chondrocyte proliferation and growth plate arrest due to spinal cord injury. We also detected altered gene expression in both whole bone extracts and bone marrow monocytes following spinal cord injury. We conclude that spinal cord injury results in altered gene expression of key regulators of osteoblast and chondrocyte activity. This leads to premature cellular apoptosis, suppressed cellular proliferation, growth plate arrest, and uncoupled bone remodeling in sublesional bone with unopposed osteoclastic resorption.
Keywords: Rehabilitation medicine, Spinal cord injury, Osteoporosis, Osteoblast, Chondrocyte
Introduction
Spinal cord injury (SCI) alters bone metabolism of the paralyzed limbs leading to osteoporosis and increased fracture risk. Studies indicate that 100% of individuals with SCI develop osteoporosis below the level of the injury [1]. This bone loss leads to fractures in up to 50% of individuals with complete SCI, with the most common occurrence at the metaphyses of the proximal tibia and distal femur [2–8]. In many cases, fractures are discovered after minimal trauma and are most commonly treated with bed rest and bracing. The combination of the injury and this treatment results in prolonged immobility, worsening disability, and serious medical complications including fracture non-union [9–11] and amputation [12, 13]. Long bone growth arrest [14, 15] has also been reported following SCI in children.
We recently reported growth plate abnormalities with chondrocyte disorganization at 10 days post-injury [16]. We also reported decreased bone formation and a mineralization defect in rats with severe SCI compared to the uninjured controls. Based on studies of markers of bone turnover in humans, it is known that spinal cord injury causes immediate suppression of bone formation [17, 18] with increased osteoclastic resorption resulting in uncoupled bone remodeling. The mechanisms responsible for osteoclast activation with osteoblast suppression remain poorly defined. There are few reports in the literature that examine cellular activity within the bone microenvironment following SCI. The immediate effects (within 3–5 days) of spinal cord injury on cellular activities at the osteochondrous junction are currently unknown. We hypothesized that SCI would result in early osteocyte and chondrocyte apoptosis leading to the profound growth plate abnormalities and bone loss previously observed at 10 days post-injury [16].
Methods
Animals and SCI
Adolescent male Sprague–Dawley (SD) rats (7 weeks old ) (200–225 g) were anesthetized with i.p. ketamine (87 mg/kg) and xylazine (13 mg/kg). A severe T10 contusion injury was produced utilizing the New York University (NYU) SCI impactor (10 g×50 mm) and we used the Basso, Beattie, Bresnahan (BBB) scale to confirm injury severity by rating hindlimb functional deficits on day 2 post-injury as previously described [16]. All injured animals demonstrated absence of motor function in the lower extremities. The control group consisted of uninjured, age-matched male Sprague–Dawley rats. A total of 11 animals were studied (control n=4, day 3 n=3, day 5 n=4). Animals were euthanized on day 3 or 5 post-injury for subsequent analyses. The Institutional Animal Care and Use Committee at the Forsyth Institute approved all animal procedures.
Histology
Osteoclast, osteoblast, osteocyte, and chondrocyte numbers were studied in decalcified bone sections through the distal femoral metaphysis at 3 and 5 days post-injury and compared to uninjured controls. Femora with a small amount of adherent soft tissue were fixed in cold 4% paraformaldehyde, decalcified in cold 14% EDTA, embedded in paraffin, sectioned at 5 μ and placed on charged slides (Manco Inc., Avon, OH). Sections were stained with hematoxylin and eosin (H and E) for cellular identification (n=3 section/animal and 3–4 animals/condition). Adjacent sections were stained with Goldner’s Trichrome method for osteoblast identification or TRAP for mature osteoclast identification. Sections were analyzed under bright field microscopy (Axiovert; Carl Zeiss), and images were captured with a CCD camera (AxioCam MRc; Carl Zeiss). Osteoblasts were counted as cuboidal cells lining the trabecular bone surface and osteoclasts were counted as TRAP-positive multinucleated cells in the same region. Trabecular osteocytes were counted and reported as mean per high power field±SD. Proliferating and hypertrophic chondrocytes were counted per column and the growth plate width was determined. For growth plate width, two measurements were taken per section midway between the notch of the femoral condyle and the cortical edge using ImageJ software. All analyses were performed blinded by two independent individuals. Results are reported as average number of cells per column±SD or width in millimeters±SD.
Isolation of Rat Bone Marrow Monocytes
Bone marrow cells were obtained from the femora of rats at 3 and 5 days post-injury and uninjured controls. The femora were dissected and flushed with alpha-MEM supplemented with 10% fetal bovine serum (FBS). The isolated bone marrow monocytes were centrifuged at 2,000 RPM for 5 min and the pellet was resuspending in TRIZOL for RNA preparation.
Assessment of Chondrocyte Apoptosis by TUNEL Assay
TUNEL staining was performed on 5 mm deparaffinized bone sections through the distal femoral metaphysis using an in situ apoptosis kit (Roche) according to the manufacturer’s instructions. Negative controls were generated on adjacent sections by omitting TdT in the labeling solution. To preserve hypertrophic chondrocytes in the samples, sections were mounted on charged slides and then placed on a warming table at 55°C overnight. The slides were then heated at 60°C for 2 h prior to use. To visualize regions of bone and cartilage, Fast Green staining was also performed by standard histological methods. Sections were analyzed under bright field microscopy (Leica DMLS), and images were captured with a Leica DC 100 camera. To quantify apoptosis, positive cells per image (n=3 high powered field/section and 3–4 animals/condition) were counted by two independent individuals in a blinded fashion. Results are reported as average number of positive cells±SD.
Immunohistochemical Analysis of Osteocyte and Osteoblast Apoptosis by Caspase-3 Activation
Adjacent sections were stained for cleaved Caspase-3 (Cell Signaling Technology #9661, dilution 1:800) using the ABC method and visualized by light microscopy. Prior to primary antibody incubation slides were incubated in Retrieve-All Antigen Unmasking Solution (Signet) for 10 min at 90°C. Positive cells were counted and reported as mean per high power field±SD. All counts were performed by two independent individuals in a blinded fashion.
In Situ Cell Proliferation
Cellular proliferation was studied in decalcified femoral bone sections. Rats were injected with 50 mg/kg of 5-bromo-2-deoxyuridine (BrdU) solution 2 and 16 h prior to sacrifice. Femurs were excised and processed for histology as described above. Sections were stained with a mouse monoclonal anti-BrdU antibody (Sigma Aldrich B 8434, dilution 1:500) detected with the AEC chromagen kit (Sigma Aldrich, Stock Number AEC101) according to the manufacturer’s instructions, counter stained with Mayer’s hematoxylin solution (Sigma Aldrich, Catalogue Number MHS) for cellular identification, and visualized by light microscopy. Positive cells were counted and reported as mean per high power field±SD. All counts were performed by two independent individuals in a blinded fashion.
Western Blot Analysis
The distal femoral metaphyses of uninjured controls (n=3) and severely injured rats (day 3 n=3 and day 5 n=3) were resected, flash frozen in liquid nitrogen, and homogenized by hand in 1× RIPA buffer (Cell Signaling Technology) supplemented with 1 mM phenylmethylsulphonyl fluoride (PMSF) and protease inhibitor cocktail (Sigma). Protein lysates were then diluted 1:1 in Novex® Tris-Glycine SDS sample buffer (2×) (Invitrogen) and equivalent amounts of protein were loaded onto Novex Tris-Glycine SDS gels (Invitrogen), electrophoresed and transferred to nylon membranes following the manufacturer’s instructions. After blocking, membranes were incubated overnight at 4°C with the following primary antibodies (Cell Signaling Technology): cleaved caspase-3 (#9661), cleaved caspase-7 (#9491), and B-actin (#4970), washed to remove unbound antibody, and incubated with secondary antibody for 1 h at room temperature. Chemiluminescent signal was detected using the LUMIGLO reagents A and B (Cell Signaling Technology #7003) following the manufacturer’s instructions. Blots were then exposed to X-ray film. The cleaved caspase-3 antibody detects a 17/19-kDa cleavage product. The cleaved caspase-7 antibody detects a 20-kDa cleavage product. The B-actin antibody detects total B-actin at 45 kDa.
Real-time PCR Analysis
We assessed the impact of SCI on genes controlling osteoblast (Runx2) and chrondrocyte (Indian hedgehog, Ihh) differentiation. Real-time reverse transcription-polymerase chain reaction (RT-PCR) was performed using the DyNAmo SYBR Green qPCR Kit according to the manufacturer’s instructions (Finnzymes, Espoo, Finland) in a 24-μl reaction volume containing 1 μl of a 1:4 dilution of first-strand reaction product and 1 μl of 10 μM gene-specific upstream and downstream primers mixture. Amplification and analysis of cDNA fragments was carried out using a 7,300 real-time PCR system (ABI, Foster City, CA). Cycling conditions were as follows: initial denaturation at 95°C for 10 min followed by 40 cycles consisting of a 15-s denaturation interval at 95°C and a 1 min annealing and extension interval at 60°C. Amplification of rat β-actin mRNA, the housekeeping gene, was used as an endogenous control to normalize results.
Statistical Analysis
For each outcome, an analysis of variance was performed to compare means among the experimental groups and to account for assays conducted in separate bone sections in the same animal (PROC MIXED; SAS, version 9.2; SAS Institute; Cary, NC). A Tukey test was used to adjust for multiple comparisons.
Results
Osteocyte Apoptosis and Uncoupled Remodeling in Sublesional Bone Following SCI
At 5 days post-injury, we detected a 37% reduction in osteocyte numbers in trabecular bone at the distal femoral metaphysis (Table 1 and Fig. 1) compared to uninjured controls. This corresponded to a 47% increase in caspase-3 positive osteocytes in the same region. In the uninjured animals, osteoclasts were seen in close proximity to osteoblasts along the trabecular bone surface, indicating (normal) coupled remodeling. At 5 days after injury, osteoclasts were seen lining the bone surface in the absence of osteoblasts, indicating uncoupled remodeling (Fig. 2). We detected a 257% increase in mature osteoclast numbers in trabecular bone at the distal femoral metaphysis at day 5 compared to uninjured controls. This corresponded to a 65% reduction in osteoblast numbers and a 75% decrease in osteoblast proliferation in the same region (Fig. 3, top panel). There was a 520% increase in cleaved caspase-3 positive osteoblasts at 5 days post-injury (Fig. 3, bottom panel). We detected an increase in cleaved caspase-3 (17/19 kDa cleavage product) and cleaved caspase-7 (20 kDa cleavage product) by Western blot analysis of protein samples derived from the distal femoral metaphysis at days 3 and 5 compared to uninjured controls (Fig. 4).
Table 1.
Effect of SCI on cell numbers and growth plate width at the distal femur
| Day 0 (n=4) (mean+/SD) | Day 3 (n=3) (mean+/SD) | Day 5 (n=4) (mean+/SD) | Percent change day 3, day 5 | |
|---|---|---|---|---|
| Osteocytes | 42.8±8.4 | 27.1±6.6 | 26.9±5.9 | 36%, 37% ↓ |
| Caspase+osteocytes | 6.0±2.1 | 6.2±2.0 | 9.0±3.8 | 3%, 47% ↑ |
| Osteoclasts | 4.4±2.1 | 5.3±4.6 | 15.7±4.2 | 20%, 257% ↑ |
| Osteoblast | 29.5±7.9 | 14.2±1.7 | 10.3±3.2 | 52%, 65% ↓ |
| Caspase+osteoblast | 1.0±0.8 | 4.0±1.4 | 6.2±1.0 | 300%, 520% ↑ |
| BrdU+osteoblast | 12.8±3.6 | 8.7±2.8 | 3.3±1.7 | 33%, 75% ↓ |
| Growth plate width (mm) | 0.37±0.03 | 0.26±0.02* | 0.25±0.07* | 31%, 32% ↓ |
| Proliferating chondrocytes per column | 15.81±2.07 | 13.83±1.99 | 11.00±2.25 | 12%, 30% ↓ |
| BrdU+proliferating chondrocyte | 56.1±5.5 | 31.3±10.3 | 20.1±6.8 | 45%, 64% ↓ |
| Hypertrophic chondrocytes per column | 10.38±1.63 | 5.08±1.73 | 5.06±1.50 | 51%, 51% ↓ |
| Tunel+proliferating chondrocyte | 0.21±0.30 | 2.77±2.40 | 5.91±5.13 | 1,219%, 2,714% ↑ |
| Tunel+hypertrophic chondrocyte | 0.66±0.52 | 4.06±3.62 | 17.00±8.02 | 511%, 2,459% ↑ |
p≤0.005 for all values except for
p≤0.05
Fig. 1.
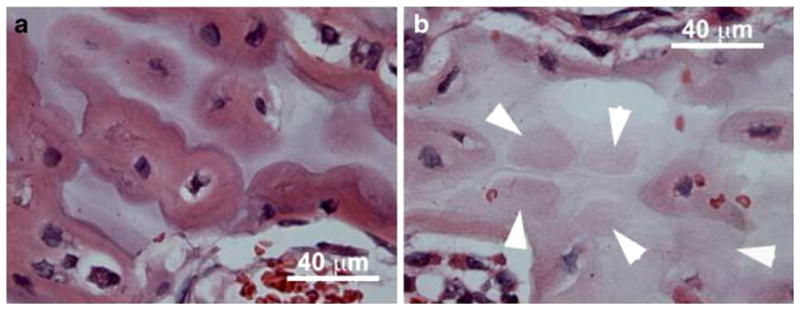
SCI stimulates immediate osteocyte cell death in sublesional trabecular bone. Osteocyte numbers were determined in trabecular bone of H and E stained sections through the distal femoral metaphysis in uninjured rats (a) and at 3 days post-injury (b). There was a marked reduction in the number of osteocytes following SCI (white arrows mark empty lacunae)
Fig. 2.

Uncoupled bone remodeling following SCI. Sections through the distal femoral metaphysis were stained with TRAP to identify mature osteoclasts in uninjured rats (a) and at 5 days post-injury (b). TRAP positive osteoclasts (black arrows, panel a) are seen in close proximity to osteoblasts (white arrows, panel a) in sections from uninjured animals. In contrast, TRAP positive osteoclasts (blue arrows, panel b) are seen without osteoblasts by 5 days post-injury
Fig. 3.

Accelerated osteoblast apoptosis and suppressed osteoblast proliferation following SCI. Sections through the distal femoral metaphysis were stained with cleaved caspase-3 (CC3) antibody to identify apoptotic osteoblasts in uninjured rats (top, panel a) and at 5 days post-injury (top, panel b). CC3 negative osteoblasts are seen in the uninjured rats (white arrows, panel a) and CC3 positive osteoblasts (red arrows, panel b) are seen at 5 days post-injury. Adjacent sections were stained with anti-BrdU antibody to identify proliferating osteoblasts in uninjured rats (bottom, panel c) and at 5 days post-injury (bottom, panel d). BrdU positive osteoblasts (white arrows, panel c) are seen only in the uninjured sections
Fig. 4.

SCI-induced apoptosis. Western blot analysis was performed on protein extracts derived from the distal femoral metaphysis of uninjured and injured rats. Cleaved caspase 3 (17/19 kDa) and cleaved caspase 7 (20 kDa) were detected in the day 3 and day 5 post-injury samples but not in the uninjured controls. β-actin (45 kDa) was used as a loading control
Decreased Growth Plate Width with Loss of Hypertrophic and Proliferating Chondrocytes Following SCI
H and E staining of adjacent sections through the chondro-osseous junction at the distal femur (Table 1) demonstrated a 51% reduction in the number of hypertrophic chondrocytes per column at day 5 compared to uninjured controls. There were 12.5% fewer proliferating chondrocytes at day 3 and 30% fewer BrdU positive cells at day 5. This loss of chondrocyte number resulted in a 31% reduction in growth plate width (Fig. 5 top) by 3 days post-injury that persisted at 5 days post-injury.
Fig. 5.

SCI-induced growth plate narrowing and chondrocyte apoptosis. H and E stained sections through the femoral metaphysis of uninjured rats (a) and at 3 days post-injury (b) demonstrates growth plate narrowing with fewer proliferating and hypertrophic chondrocytes per column (see Table 1). The growth plate is delineated by a white bracket (panels a and b). There are more TUNEL positive chondrocytes in the injured (d, white arrows) compared to uninjured rats (panel c, white arrow)
Accelerated Chondrocyte Apoptosis at the Femoral Growth Plate Following SCI
Staining of adjacent sections through the chondro-osseous junction at the distal femur (Fig. 5 bottom) demonstrated a dramatic increase in the number of TUNEL positive chondrocytes per column. At 3 days, there was a 511% increase and at 5 days there was a 2,459% increase in TUNEL positive hypertrophic chondrocytes compared to the uninjured controls. At 3 days, there was a 1,219% increase, and at 5 days, there was a 2,714% increase in TUNEL positive proliferating chondrocytes compared to the uninjured controls.
Altered Gene Expression in the Distal Femur Following SCI
Gene expression for both Runx2 (Fig. 6 top) and Indian hedgehog (Ihh, Fig. 6 bottom) was reduced in bone marrow monocytes at 3 and 5 days post-injury compared to uninjured controls.
Fig. 6.

SCI alters gene expression. Real-time PCR was performed to assess the impact of SCI on genes controlling osteoblast (Runx2) and chrondrocyte (Ihh) differentiation. Bone marrow monocytes (top) or whole bone extracts (bottom) were studied to compare gene expression in rats at 3 days post-injury (D3), 5 days post-injury (D5), and in uninjured controls (C). Following SCI there is a reduction in both Runx2 and Ihh expression
Discussion
In the current study, we assessed the impact of SCI on cellular activities at the chondro-osseus junction in rats. We report immediate osteocyte apoptosis in sublesional bone following SCI. This was followed by uncoupled bone remodeling with increased osteoclast numbers and decreased osteoblast numbers at 5 days post-injury. Osteoblast suppression was due to both inhibited proliferation and accelerated apoptosis. We also report fewer chondrocytes per column, reduced growth plate width, accelerated chondrocyte apoptosis, and suppressed chondrocyte proliferation with reduced Indian Hedge Hog gene expression due to SCI.
Normally, bone formation is tightly coupled to bone resorption. In SCI, bone remodeling becomes uncoupled with an initial decrease in bone formation and steadily increasing bone resorption [19]. In humans, bone formation rates normalize after 2 weeks post-injury; however, this immediate uncoupling leads to a 4% per month reduction in sublesional (below the neurologic level of injury) trabecular bone mineral content with a 40% reduction of sublesional BMD by 2 years post-injury [2]. We report immediate suppression of Runx2 expression in bone marrow monocytes. Runx2 is a master gene for osteoblast differentiation and activity. Our results therefore indicate suppression of osteoblastic differentiation due to spinal cord injury. In addition to this, suppressed osteoblast proliferation and viability contribute to the loss of osteoblasts in the sublesional trabecular bone and the resulting uncoupled remodeling. The physiologic signals that trigger uncoupled remodeling are poorly understood. Because we detect osteocyte apoptosis prior to osteoclast recruitment, we suspect osteocyte apoptosis may function as a cellular signal to trigger osteoclast recruitment to the sublesional bone. Osteocyte apoptosis has been reported in rodent models of hind-limb unloading [20] and was reported to trigger targeted remodeling of microdamage [21, 22]. Furthermore, osteocyte apoptotic bodies have been shown to have osteoclastogenic activity in vitro [23].
We report loss of both proliferating and hypertrophic chondrocytes immediately following SCI due to accelerated apoptosis and suppressed proliferation. Chondrocyte apoptosis has been reported following hind-limb unloading and microgravity exposure in rats [24, 25]. Additionally, reduced chondrocyte numbers have been reported in the articular cartilage at the knee in rodents with SCI [26, 27]. Collectively, these findings suggest that mechanical loading is required for chondrogenesis. Chondrocyte differentiation within the growth plate normally proceeds through stages of proliferation, hypertrophy, and apoptosis. Stem cells enter the proliferating zone characterized by rapid cell division and subsequent columnar organization. Cells then stop dividing and begin to hypertrophy. Hypertrophic chondrocytes produce matrix components and the surrounding matrix calcifies as the chondrocytes undergo apoptosis and are replaced by bone [28]. The rate of chondrocyte differentiation is regulated by a locally acting feedback loop involving Indian hedgehog (Ihh) and parathyroid hormone-related protein (PTHrP). Ihh is expressed by chondrocytes transitioning from the proliferative to the hypertrophic zone, and it increases expression of PTHrP within the periarticular perichondrium. PTHrP binds to receptors on late proliferating chondrocytes and inhibits further differentiation [28]. In the current study, we found suppressed Ihh gene expression, suggesting an immediate role for this protein in SCI induced growth plate arrest.
From a clinical standpoint, normal chondrocyte activity has implications for both skeletal growth and fracture healing. Stunting of long bone skeletal growth has been reported in children with neurological impairment due to poliomyelitis [29, 30] as well as spinal cord injury [14, 15]. Delayed fracture union and non-union have also been reported following SCI [10, 11]. In one study, 53% of fractures occurring acutely after SCI that were treated non-operatively resulted in delayed union, mal-union, or nonunion [9]. Impaired fracture healing has great clinical relevance to the SCI population given the associated medical complications that arise including pressure ulcer formation and amputation [12].
Under normal circumstances, endochondral and intra-membranous ossification contribute to fracture healing. Endochondral ossification occurs at the fracture site where oxygen tension is lowest while intramembranous ossification proceeds in regions where the blood supply has been less disrupted. Vascular disruption at the bone fragments leads to decreased perfusion and hypoxia. This triggers commitment of mesenchymal stem cells to the chondrocyte lineage with subsequent chondrogenesis that recapitulates the developmental phases of proliferation, hypertrophy, and terminal differentiation seen within the growth plate. It has been shown that the same molecular mechanisms that control skeletal development also control fracture healing [31]. Impaired endochondral bone formation may contribute to fracture delayed union or nonunion post-SCI. Few studies have examined in detail the impact of SCI or other forms of skeletal unloading on long bone fracture healing. Incomplete maturation of woven bone and delayed calcium accumulation were seen in paraplegic rats with tibial fractures stabilized by intramedullary nailing [32].
Our findings suggest that osteocyte apoptosis precedes osteoclast recruitment to the region and may be a cellular signal stimulating uncoupled remodeling following SCI. We report inhibition of osteoblast differentiation via Runx2 suppression, decreased osteoblast proliferation, and accelerated osteoblast apoptosis in sublesional trabecular bone. Suppressed Ihh gene expression was seen with reduced chondrocyte proliferation and accelerated chondrocyte apoptosis resulting in growth plate arrest. These findings contribute to the understanding of uncoupled bone remodeling and long bone growth arrest that is seen clinically following SCI.
Acknowledgments
Support Grant sponsor: NIH/NICHD Grant number K12 HD001097-08 (L.M.)
We would like to thank Ms. Justine Dobeck for technical assistance and Dr. Paul Odgren for his valuable comments.
Footnotes
Conflicts of interest All authors have no conflicts of interest.
Contributor Information
Leslie R. Morse, Email: lmorse4@partners.org, Spaulding-Harvard Spinal Cord Injury Model System, Spaulding Rehabilitation Hospital, Boston, MA, USA. Department of Cytokine Biology, Forsyth Institute, Boston, MA, USA. Department of Physical Medicine and Rehabilitation, Harvard Medical School, Boston, MA, USA. Department of PMR, Harvard Medical School, The Forsyth Institute, 140 The Fenway, Boston, MA 02118, USA
Yan Xu, Department of Cytokine Biology, Forsyth Institute, Boston, MA, USA. Department of Medical Microbiology and Immunology, Kunming Medical University, Yunnan, China.
Bethlehem Solomon, Department of Cytokine Biology, Forsyth Institute, Boston, MA, USA.
Lara Boyle, Department of Cytokine Biology, Forsyth Institute, Boston, MA, USA.
Subbiah Yoganathan, Department of Cytokine Biology, Forsyth Institute, Boston, MA, USA.
Philip Stashenko, Department of Cytokine Biology, Forsyth Institute, Boston, MA, USA. Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, MA, USA.
Ricardo A. Battaglino, Department of Cytokine Biology, Forsyth Institute, Boston, MA, USA. Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, MA, USA
References
- 1.Kocina P. Body composition of spinal cord injured adults. Sports Med. 1997;23:48–60. doi: 10.2165/00007256-199723010-00005. [DOI] [PubMed] [Google Scholar]
- 2.Dauty M, Perrouin VB, Maugars Y, Dubois C, Mathe JF. Supralesional and sublesional bone mineral density in spinal cord-injured patients. Bone. 2000;27:305–9. doi: 10.1016/s8756-3282(00)00326-4. [DOI] [PubMed] [Google Scholar]
- 3.Garland DE, Stewart CA, Adkins RH, Hu SS, Rosen C, Liotta FJ, et al. Osteoporosis after spinal cord injury. J Orthop Res. 1992;10:371–8. doi: 10.1002/jor.1100100309. [DOI] [PubMed] [Google Scholar]
- 4.Garland DE, Adkins RH, Kushwaha V, Stewart C. Risk factors for osteoporosis at the knee in the spinal cord injury population. J Spinal Cord Med. 2004;27:202–6. doi: 10.1080/10790268.2004.11753748. [DOI] [PubMed] [Google Scholar]
- 5.Demirel G, Yilmaz H, Paker N, Onel S. Osteoporosis after spinal cord injury. Spinal Cord. 1998;36:822–5. doi: 10.1038/sj.sc.3100704. [DOI] [PubMed] [Google Scholar]
- 6.Eser P, Schiessl H, Willnecker J. Bone loss and steady state after spinal cord injury: a cross-sectional study using pQCT. J Musculoskelet Neuronal Interact. 2004;4:197–8. [PubMed] [Google Scholar]
- 7.Szollar SM, Martin EM, Parthemore JG, Sartoris DJ, Deftos LJ. Demineralization in tetraplegic and paraplegic man over time. Spinal Cord. 1997;35:223–8. doi: 10.1038/sj.sc.3100401. [DOI] [PubMed] [Google Scholar]
- 8.Wilmet E, Ismail AA, Heilporn A, Welraeds D, Bergmann P. Longitudinal study of the bone mineral content and of soft tissue composition after spinal cord section. Paraplegia. 1995;33:674–7. doi: 10.1038/sc.1995.141. [DOI] [PubMed] [Google Scholar]
- 9.Garland DE, Saucedo T, Reiser TV. The management of tibial fractures in acute spinal cord injury patients. Clin Orthop Relat Res. 1986;213:237–40. [PubMed] [Google Scholar]
- 10.McMaster WC, Stauffer ES. The management of long bone fracture in the spinal cord injured patient. Clin Orthop Relat Res. 1975;112:44–52. [PubMed] [Google Scholar]
- 11.Nottage WM. A review of long-bone fractures in patients with spinal cord injuries. Clin Orthop Relat Res. 1981;155:65–70. [PubMed] [Google Scholar]
- 12.Morse LR, Giangregori L, Battaglino RA, Holland R, Craven BC, Stolzmann K, et al. VA-based survey of osteoporosis management in spinal cord injury. PMR. 2009 Mar 1;1:240–4. doi: 10.1016/j.pmrj.2008.10.008. Ref Type: Journal (Full) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morse LR, Battaglino RA, Stolzmann KL, Hallett LD, Waddimba A, Gagnon D, et al. Osteoporotic fractures and hospitalization risk in chronic spinal cord injury. Osteoporos Int. 2009;20:385–92. doi: 10.1007/s00198-008-0671-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell J, Bonnett C. Spinal cord injury in children. Clin Orthop Relat Res. 1975;112:114–23. [PubMed] [Google Scholar]
- 15.Duval-Beaupere G, Lougovoy J, Trocellier L, Lacert P. Trunk and leg growth in children with paraplegia caused by spinal cord injury. Paraplegia. 1983;21:339–50. doi: 10.1038/sc.1983.58. [DOI] [PubMed] [Google Scholar]
- 16.Morse L, Teng YD, Pham L, Newton K, Yu D, Liao WL, et al. Spinal cord injury causes rapid osteoclastic resorption and growth plate abnormalities in growing rats (SCI-induced bone loss in growing rats) Osteoporos Int. 2008;19:645–52. doi: 10.1007/s00198-007-0494-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts D, Lee W, Cuneo RC, Wittmann J, Ward G, Flatman R, et al. Longitudinal study of bone turnover after acute spinal cord injury. J Clin Endocrinol Metab. 1998;83:415–22. doi: 10.1210/jcem.83.2.4581. [DOI] [PubMed] [Google Scholar]
- 18.Szollar SM, Martin EM, Sartoris DJ, Parthemore JG, Deftos LJ. Bone mineral density and indexes of bone metabolism in spinal cord injury. Am J Phys Med Rehabil. 1998;77:28–35. doi: 10.1097/00002060-199801000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Uebelhart D, Hartmann D, Vuagnat H, Castanier M, Hachen HJ, Chantraine A. Early modifications of biochemical markers of bone metabolism in spinal cord injury patients. A preliminary study. Scand J Rehabil Med. 1994;26:197–202. [PubMed] [Google Scholar]
- 20.Basso N, Heersche JN. Effects of hind limb unloading and reloading on nitric oxide synthase expression and apoptosis of osteocytes and chondrocytes. Bone. 2006;39:807–14. doi: 10.1016/j.bone.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Vashishth D, Verborgt O, Divine G, Schaffler MB, Fyhrie DP. Decline in osteocyte lacunar density in human cortical bone is associated with accumulation of microcracks with age. Bone. 2000;26:375–80. doi: 10.1016/S8756-3282(00)00236-2. [DOI] [PubMed] [Google Scholar]
- 22.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000;15:60–7. doi: 10.1359/jbmr.2000.15.1.60. [DOI] [PubMed] [Google Scholar]
- 23.Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23:915–27. doi: 10.1359/jbmr.080207. [DOI] [PubMed] [Google Scholar]
- 24.Hecht JT, Makitie O, Hayes E, Haynes R, Susic M, Montufar-Solis D, et al. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J Orthop Res. 2004;22:759–67. doi: 10.1016/j.orthres.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Montufar-Solis D, Duke PJ, Durnova G. Spaceflight and age affect tibial epiphyseal growth plate histomorphometry. J Appl Physiol. 1992;73:19S–25. doi: 10.1152/jappl.1992.73.2.S19. [DOI] [PubMed] [Google Scholar]
- 26.Moriyama H, Yoshimura O, Kawamata S, Takayanagi K, Kurose T, Kubota A, et al. Alteration in articular cartilage of rat knee joints after spinal cord injury. Osteoarthr Cartil. 2008;16:392–8. doi: 10.1016/j.joca.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Moriyama H, Nishihara K, Hosoda M, Saka Y, Kanemura N, Takayanagi K, et al. Contrasting alteration patterns of different cartilage plates in knee articular cartilage after spinal cord injury in rats. Spinal Cord. 2009;47:218–24. doi: 10.1038/sc.2008.96. [DOI] [PubMed] [Google Scholar]
- 28.van der Eerden BC, Karperien M, Gevers EF, Lowik CW, Wit JM. Expression of Indian hedgehog, parathyroid hormone-related protein, and their receptors in the postnatal growth plate of the rat: evidence for a locally acting growth restraining feedback loop after birth. J Bone Miner Res. 2000;15:1045–55. doi: 10.1359/jbmr.2000.15.6.1045. [DOI] [PubMed] [Google Scholar]
- 29.Gullickson G, Jr, Olson M, Kottke FJ. The effect of paralysis of one lower-extremity on bone growth. Arch Phys Med Rehabil. 1950;31:392–400. [PubMed] [Google Scholar]
- 30.Ring PA. Shortening and paralysis in poliomyelitis. Lancet. 1957;273:980–3. doi: 10.1016/s0140-6736(57)92003-2. [DOI] [PubMed] [Google Scholar]
- 31.Jepsen KJ, Price C, Silkman LJ, Nicholls FH, Nasser P, Hu B, et al. Genetic variation in the patterns of skeletal progenitor cell differentiation and progression during endochondral bone formation affects the rate of fracture healing. J Bone Miner Res. 2008;23:1204–16. doi: 10.1359/JBMR.080317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aro H, Eerola E, Aho AJ. Fracture healing in paraplegic rats. Acta Orthop Scand. 1985;56:228–32. doi: 10.3109/17453678508993001. [DOI] [PubMed] [Google Scholar]