

SUMMARY
ICH is a disease with high rates of mortality and morbidity, with a substantial public health impact. Spontaneous ICH (sICH) has been extensively studied and a large body of data has been accumulated on its pathophysiology. However, the literature on traumatic ICH (tICH) is more limited, and there is a need for further investigations of this important topic. This review will highlight some of the cellular pathways in ICH with an emphasis on the mechanisms of secondary injury due to heme toxicity and to events in the coagulation process, which are common to both sICH and tICH.
Keywords: Intracranial hemorrhage, heme toxicity, iron toxicity, coagulation, inflammation, vascular response
BACKGROUND
ICH is a disease with high rates of mortality and morbidity, with a substantial public health impact. ICH can be classified as spontaneous or traumatic. Spontaneous ICH (sICH) has been extensively studied and a large body of data has been accumulated on its pathophysiology. However, the literature on traumatic ICH (tICH) is more limited. The need to investigate the specific mechanisms of tICH is underscored by the fact that ICH is a well known feature of severe TBI, and carries a high risk of morbidity and mortality. Progression of the hemorrhage is associated with poor clinical outcomes [1, 2]. This is true not only of large hemorrhages, but also of micro-bleeds detected only on susceptibility-weighted imaging (SWI) imaging and not on routine CT or MRI [3]. Moreover, these detrimental sequelae often extend beyond the area of the hemorrhage. Metabolic changes have been found in regions remote from focal hemorrhagic lesions, suggesting diffuse injury after human traumatic brain injury [4]. In a rat TBI model, severity of intracerebral hemorrhage correlates with degree of final cortical atrophy [5] In addition, TBI itself may induce coagulopathy, which further increases the extent of intracerebral hemorrhage and the incidence of poor outcome associated with such injuries [6].
The management of traumatic intracerebral hemorrhage (tICH) presents a paradox. On one hand, current management for severe TBI is directed towards preservation of adequate cerebral perfusion pressure (CPP). This approach frequently requires therapies that raise the arterial blood pressure when increased intracranial pressure (ICP) does not respond to efforts to return it to normal levels. On the other hand, increasing the blood pressure in traumatic injuries will likely increase blood loss. Since the progression of the hemorrhage is greatest in the first 24 hours, while the edema formation begins immediately after trauma and commonly peaks within 48-72 hours, the current CPP-driven management may be detrimental in terms of ICH progression. Ideally, the management to optimize CPP and to control ICH should be coordinated in the temporal progression of TBI. In addition to increasing the blood pressure pharmacologically to maintain adequate cerebral perfusion pressure, there is a need for strategies to reduce hemorrhage progression, and to address the harmful effects of the hemorrhage. To achieve this goal, an understanding of the pathophysiolgy of tICH is essential. Although there are significant differences between tICH and sICH, they share common processes and a review of the data in sICH could shed light on the mechanisms of injury in tICH.
This review will highlight some of the cellular pathways in ICH with an emphasis on the mechanisms of secondary injury due to heme toxicity and to events in the coagulation process, which are common to the different types of sICH and tICH.
Release of free heme
Heme is a major component of hemoproteins, including hemoglobin, myoglobin, cytochromes, guanylate cyclase, and nitric oxide synthase. Free heme is deposited in tissue only in pathological conditions. Hemorrhage, ischemia, edema, and mechanical injury damage are all processes that may result in the release of heme from hemoproteins [7]. Intracellular heme originates from cytoplasmic hemoproteins and from mitochondrial cytochromes located in neurons and glia [8]. Extracellular heme is released from dying cells and from extravasated hemoglobin from red blood cells [9]. The release of oxyhemoglobin (oxyHb) leads to superoxide anion (02•) and hydrogen peroxide (H202) release as oxyhemoglobin undergoes auto-oxidation to methemeglobin. Free heme is degraded by heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2) into Fe2+, CO, and one isomer of biliverdin, which rapidly reduces to free bilirubin. Free heme is lipophilic and enhances lipid peroxidation [10]. Free iron is also extremely toxic to cells (Huang et al, 2002; Kadoya et al, 1995; Panizzon et al, 1996). It reacts with H2O2 to form hydroxyl radicals, and degrades membrane lipid peroxides to yield alkoxy- and peroxy-radicals, which cause further chain reactions of free radical-induced damage [10, 11]. The result is oxidative damage to lipids, DNA, and proteins, leading to caspase activation and neuronal death [12]. Additionally, damage to endothelial cells causes BBB breakdown, resulting in vasogenic edema, increased ICP, and ischemia [13-15]. The effect of bilirubin formation after TBI is unclear. At low physiologic nanomolar concentrations in the healthy brain, bilirubin has potent anti-oxidative properties; but at high concentrations, it can act as a neurotoxin [7]. The level at which it is neuroprotective vs. neurotoxic is not clear, especially in the complex environment after TBI. The role of CO generation is controversial – it is beneficial by promoting relaxation of vascular smooth muscle and decreasing vasospasm [7].
Because of the potential harmful effects of hemoglobin (Hb) breakdown, HO-1 and HO-2 activity may be detrimental after TBI. HO inhibitors have been shown to reduce edema in models of ischemia, hemorrhage, and trauma [16-18]. However, there is also evidence that HO-1 is neuroprotective [16]. These discrepancies may result from the model used, and the brain region or even the cell type studied. In cell culture, for example, HO-1, induced in reactive astrocytes and microglia/macrophages, protects cortical astrocytes but not neurons from oxidative stress after exposure to Hb and H2O2 [17, 18]. Other investigations involve the anti-oxidant activity of bilirubin/biliverdin redox cycling [19], and the administration of deferoxamine, a scavenger for ferric iron which improved spatial memory in a rodent model of ICH [20].
Haptoglobin (Hp), a glycoprotein which binds free Hb almost irreversibly, is a potential endogenous neuroprotectant after ICH [13, 21, 22]. In the Hb-Hp complex, the iron moiety is situated within the hydrophobic pocket of Hb, preventing its oxidative and cytotoxic activities [23]. Hp binding also facilitates the clearance of free Hb by monocytes and macrophages, and promotes iron recycling [24]. In addition to preventing Hb-induced oxidative cellular damage to cells, Hp-Hb binding protects Hb itself from oxidative damage. This is especially important since oxidative changes to Hb render it unable to bind to Hp, or to the Hb scavenger receptor CD163 on macrophages [25], which mediate Hb endocytosis and clearance. Interestingly, the presence of free Hb alone could induce a state of hypohaptoglobinemia [11], making the induction of Hp expression even more crucial. Hp level in the brain is low at baseline [11], but its expression can be induced in the brain and in peripheral blood by intracerebral injection of blood [11]. Over-expression of Hp, induced pharmacologically with sulforaphane (an activator of nuclear factor-erythroid 2-related factor (Nrf2), reduces brain injury after experimental ICH [11].
Oligodendrocytes appear to be the major producer of Hp in the CNS [11]. Since oligodendrocytes are closely associated with axons, they are uniquely situated to decrease the effects of heme toxicity on white matter tracts, which are situated along the path of blood extravasation during aSAH. In fact, Hp-overexpressing mice show better axonal integrity than Hp-null mice [11]. This function of oligodendrocytes adds to their known activities in protecting axons from excitotoxic and oxidative stress. In addition to protecting neurons, Hp also protects oligodendrocytes themselves from heme toxicity [11], ensuring continued intracranial production of Hp. Thus, Hp is a promising endogenous agent that may be utilized to limit Hb-mediated toxicity in ICH.
Activation of coagulation
Coagulation disorders are common after TBI, including both hypercoagulability and coagulopathy. In the presence of coagulopathy, there is a ten-fold increase in the risk of mortality from TBI, and a thirty-fold increase in significant morbidity. Tissue factor (TF), a protein present in endothelial cells and leukocytes and the primary physiological initiator of the coagulation cascade, is released into the general circulation following injury to brain vessels [26]. Cerebral vessels have a rich store of TF and play a central role in inducing CNS and systemic coagulopathy [26, 27]. The large amount of TF released can overwhelm normal control mechanisms that prevent excessive coagulation. With significant hemorrhage, anti-thrombin, clotting factors, and platelets are consumed. At the same time, the expression of plasminogen activator-inhibitor (PAI-1) expression is increased, inhibiting the fibrinolytic system [28]. Tissue hypoperfusion and activation of the protein C pathway have also been implicated in the early coagulopathy seen in TBI [29]. These factors contribute to DIC, seen in as much as 25% of patients with severe TBI. DIC-induced coagulopathy may develop within hours of injury [30]. On the other hand, TF-dependent activation of coagulation can cause both micro-vascular and macro-vascular fibrin thrombi formation. Excessive micro-thrombi formation may obstruct flow in small vessels, and is proposed to be a cause of delayed ischemia in investigations of aSAH [31]. Because of the complexity of the coagulation system with its myriad factors, thrombin will be highlighted here as an example of the multiple secondary effects of activation of the coagulation system.
Thrombin has been shown to be a potent inducer of microparticle release from platelets and endothelial cells [35]. MPs are small vesicles which consist of a small amount of cytosol and bilayer plasma membrane, and which express surface antigens from the cell of origin. MP release occurs during many types of apoptogenic, procoagulant, or proinflammatory stimulation [32]. These stimuli trigger the migration of procoagulant phospholipids, such as phosphotidylserine, to the outer leaflet of the plasma membrane, with subsequent membrane budding which is released in the form of MPs. Phosphotidylserine creates an additional procoagulant surface for the assembly of clotting enzyme complexes on MP surfaces [28]. MPs also provide a reservoir of circulating TF, and enhance the catalytic efficiency of the TF/factor VIIa complex [28].
In addition to promoting fibrin clot formation, thrombin also mediates vasoconstriction through the thrombin receptor, PAR1, found on endothelial cells [33]. When cleaved by thrombin, activated PAR1 mediates vasoconstriction, and upregulates its own expression, increasing the vessel responsiveness to thrombin [33]. These processes potentiate vasoconstriction, which limits bleeding but can worsen ischemia after TBI. As is the case with many pathways reviewed, PAR1 activation also directly triggers inflammation [34] and increases BBB permeability, edema, and cell death [35].
Activation of Platelets
Beyond their crucial role in hemostasis, activated platelets have important secondary effects on the vasculature. The interaction of platelets with the exposed collagen of the blood vessel and with activated leukocytes contribute to platelet degranulation and to the formation of adhesion molecules in the endothelium [36]. These events trigger the release of eicosanoids and free radicals from granulocytes, increasing oxidative stress in the microenvironment of the hemorrhage. Platelets also directly contribute to vasoconstriction after ICH. Platelets and mast cells release 5-HT when activated. 5-HT has many actions, including increased vascular permeability [36], and vasoconstriction in large cerebral arteries but vasodilation in small cerebral arteries [37, 38]. It also stimulates the sensory fibers of the trigeminovascular system which release substance P (SP) antidromically [39], contributing to the effects of neurogenic inflammation. In experimental SAH, platelets also interact with perivascular nerves as follows: release of sensory fiber transmitters SP from these nerves stimulate the arachadonic acid cascade within platelets [40], contributing to inflammation through products of AA metabolism such as prostaglandins and leukotrienes.
Activation of Leukocytes
Leukocyte-endothelial cell interactions, mediated by intercellular adhesion molecule-1 (ICAM-1), lymphocyte function-associated antigen-1 (LFA-1), macrophage antigen-1 (Mac-1) and endothelial (E)-selectin, constitute another important element in the inflammatory response [41]. These interactions are believed to contribute to the development of vasospasm in aSAH. Infiltrating neutrophils may secrete TNFα and pro-inflammatory proteases, and generate ROS [42]. Dying leukocytes may stimulate macrophages to release pro-inflammatory mediators [43]. Mast cells migrate to vessel walls after SAH and release histamine when stimulated by bradykinin [44]. In studies of cerebral ischemia, leukocyte-endothelial cell adhesion helps to initiate and propagate reperfusion injury. Mice that have received neutralizing antibodies against leukocyte adhesion receptors and mutant mice that are genetically deficient in these adhesion receptors [45] experience less microvascular dysfunction and tissue injury following ischemia/reperfusion. If the same holds true for post-traumatic hemorrhage, then anti-leukocyte strategies should be explored as a therapeutic option.
Actions of Microglia, Astrocytes, and Oligodendrocytes
In studies of SAH, activated microglia have phagocytic activities which help to clear the hematoma. Thus, microglial activation and macrophage infiltration may be beneficial after hemorrhage. However, activated microglia also release cytokines [46-48], ROS [49, 50], and nitric oxide [51, 52], all of which also contribute to hemorrhage-induced brain injury. Use of anti-microglial strategies, such as tetracycline derivatives, in animal models of ICH have resulted in reduced injury size, edema, and improved neurologic function [53], but have not been substantiated in clinical studies. Importantly, these therapies must maintain the beneficial actions of microglia while decreasing their pro-inflammatory activities.
Astrocytes modulate the neuronal response to brain injury through the production of angiogenic and neurotrophic factors. For instance, they influence neuronal sensitivity to glutamate toxicity by regulating the expression of the NMDA receptor subunit and the glutamate transporter excitatory amino acid carrier [54]. Interestingly, astrocytes could also modulate microglial ROS production [49] and thus play a role in limiting the harmful effects of micoglial ROS release, without compromising the other positive effects of microglia activation. Oligodendrocytes also play an important role. As discussed earlier, one of their responses to hemorrhage is the increased expression of haptoglobin [11], which binds free heme and limits heme-induced toxicity.
The vascular response
The different cells within the blood vessels respond differently to the presence of extravascular blood [55]. Cerebral arteries contain three structural layers: the external layer (the adventitia) contains axons of the perivascular nerves in a collagen sheath; the media contains smooth muscle; the internal layer (the intima) contains endothelial cells and the basement membrane. Oxyhemoglobin, released by lysed red blood cells, is pathogenic for all three layers [55]. In the adventitia, the loss of nerve fibers has been reported after sICH [56, 57]. This type of denervation could be expected to result in loss of neurogenic control of cerebral arteries [55] and impairment of autoregulation. In the media, myonecrosis occurs with loss of contractile protein in the smooth muscle [58, 59]; additionally, the amount of interstitial collagen increases [58]. Together these processes contribute to arterial narrowing which is not vasospasm-mediated [60]. In the intima layer, matrix metalloproteinases (MMPs) are released after hemorrhage. MMPs degrade the basement membrane and tight junction proteins, resulting in disruption of the BBB. Local substances released by multiple cell types also affect the permeability of the BBB [61]. Perivascular nerve fibers release CGRP, 5-HT, and SP which contribute to mast cell release of histamine [55], which increases inflammation.
Endothelial cells themselves release substances that may be detrimental in the setting of TBI. An example is endothelin, a potent vasoconstrictor, which helps to limit hemorrhage but may aggravate ischemic secondary injury after TBI. After ICH, endothelial cells also up-regulate the expression of receptors to SUR1 and to angiotensin I (AT1) [62]. These responses also contribute to vasoconstriction. SUR1 [63] and ET-1 [55]have the additional effect of increasing BBB permeability. Specific antagonists to SUR1, AT1 receptors, and ET-1 [55, 62, 63] have been shown to reduce cerebral edema and improve outcome in animal models of ICH. These are only a few examples of the diverse responses triggered in cerebral endothelial cells after ICH. These responses have been studied most extensively in the context of vasospasm after aSAH. It has been pointed out that vasospasm also plays a role after the different types of hemorrhage after TBI [64, 65], but it is unclear whether the treatment of post-traumatic vasospasm improves outcome.
Recent studies show that endothelial microparticles (MP) are released into the circulation at high proportions in TBI. In TBI, the proportion of endothelial MPs (compared to platelet MPs) was found to be higher than in many other disease entities, including spontaneous SAH, which is known to cause extensive cerebral endothelial damage [28]. This finding suggests that severe TBI and tICH result in more extensive endothelial activation than sICH, exacerbating the prothrombotic and proinflammatory signaling pathways in TBI.
Conclusion
Intracerebral hemorrhage initiates many cellular responses beyond those that restore hemostasis, and these responses may contribute to secondary brain injury (see Figure). ICH is often accompanied by increased ICP, ischemia, oxidative damage, vasogenic edema, and cytotoxic edema. These processes disrupt mitochondrial energetics, and lead to neuronal cell death. Heme-toxicity, iron-toxicity, and activation of coagulation are obviously key elements in both sICH and tICH. Thus, the data on cellular mechanisms in sICH may be applicable to the understanding of tICH. However, there are also significant differences among the different types of traumatic and spontaneous ICHs. These differences include: the distribution of the various hemorrhage subtypes (subdural, subarachnoid, intraparenchymal, or intraventricular) in spontaneous vs. traumatic bleeds, the extent and rate of hemorrhage progression, the degree and duration of increased intracranial pressure, the presence of blood beneath the subarachnoid layer vs. the dura. Additionally, the presence of concomitant injuries in TBI may influence the response to ICH. Thus, specific research of the pathophysiology of tICH and the recovery process is needed in order to identify specific therapeutic targets for these processes.
Figure 1.
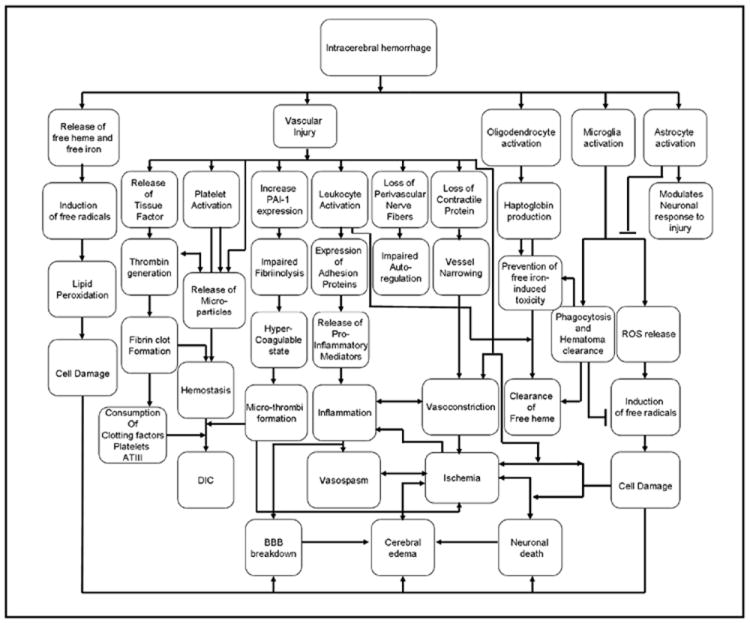
Representative pathways of secondary brain injury after ICH.
Acknowledgments
This study was supported by NIH grants (K08NS057339 to J.L., R01NS53560 and P01NS555104 to E.H.L.).
Footnotes
Conflict of Interest: The authors have no conflict of interest.
References
- 1.Narayan RK, Maas AI, Servadei F, Skolnick BE, Tillinger MN, Marshall LF. Progression of traumatic intracerebral hemorrhage: a prospective observational study. J Neurotrauma. 2008;25:629–639. doi: 10.1089/neu.2007.0385. [DOI] [PubMed] [Google Scholar]
- 2.White CL, Griffith S, Caron JL. Early progression of traumatic cerebral contusions: characterization and risk factors. J Trauma. 2009;67:508–514. doi: 10.1097/TA.0b013e3181b2519f. discussion 514-505. [DOI] [PubMed] [Google Scholar]
- 3.Park JH, Park SW, Kang SH, Nam TK, Min BK, Hwang SN. Detection of traumatic cerebral microbleeds by susceptibility-weighted image of MRI. J Korean Neurosurg Soc. 2009;46:365–369. doi: 10.3340/jkns.2009.46.4.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu HM, Huang SC, Hattori N, Glenn TC, Vespa PM, Hovda DA, Bergsneider M. Subcortical white matter metabolic changes remote from focal hemorrhagic lesions suggest diffuse injury after human traumatic brain injury. Neurosurgery. 2004;55:1306–1315. doi: 10.1227/01.neu.0000143028.08719.42. discussion 1316-1307. [DOI] [PubMed] [Google Scholar]
- 5.Immonen RJ, Kharatishvili I, Grohn H, Pitkanen A, Grohn OH. Quantitative MRI predicts long-term structural and functional outcome after experimental traumatic brain injury. Neuroimage. 2009;45:1–9. doi: 10.1016/j.neuroimage.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 6.Narayan RK, Maas AI, Marshall LF, Servadei F, Skolnick BE, Tillinger MN. Recombinant factor VIIA in traumatic intracerebral hemorrhage: results of a dose-escalation clinical trial. Neurosurgery. 2008;62:776–786. doi: 10.1227/01.neu.0000316898.78371.74. discussion 786-778. [DOI] [PubMed] [Google Scholar]
- 7.Chang EF, Claus CP, Vreman HJ, Wong RJ, Noble-Haeusslein LJ. Heme regulation in traumatic brain injury: relevance to the adult and developing brain. J Cereb Blood Flow Metab. 2005;25:1401–1417. doi: 10.1038/sj.jcbfm.9600147. [DOI] [PubMed] [Google Scholar]
- 8.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 9.Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien PJ, Little C. Intracellular mechanisms for the decomposition of a lipid peroxide. II. Decomposition of a lipid peroxide by subcellular fractions. Can J Biochem. 1969;47:493–499. doi: 10.1139/o69-077. [DOI] [PubMed] [Google Scholar]
- 11.Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, Aronowski J. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci. 2009;29:15819–15827. doi: 10.1523/JNEUROSCI.3776-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- 13.Keep RF, Xiang J, Ennis SR, Andjelkovic A, Hua Y, Xi G, Hoff JT. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:73–77. doi: 10.1007/978-3-211-09469-3_15. [DOI] [PubMed] [Google Scholar]
- 14.Thiex R, Tsirka SE. Brain edema after intracerebral hemorrhage: mechanisms, treatment options, management strategies, and operative indications. Neurosurg Focus. 2007;22:E6. doi: 10.3171/foc.2007.22.5.7. [DOI] [PubMed] [Google Scholar]
- 15.Bhasin RR, Xi G, Hua Y, Keep RF, Hoff JT. Experimental intracerebral hemorrhage: effect of lysed erythrocytes on brain edema and blood-brain barrier permeability. Acta Neurochir Suppl. 2002;81:249–251. doi: 10.1007/978-3-7091-6738-0_65. [DOI] [PubMed] [Google Scholar]
- 16.Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, Tysoe SA, Wolosker H, Baranano DE, Dore S, Poss KD, Snyder SH. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol. 1999;1:152–157. doi: 10.1038/11072. [DOI] [PubMed] [Google Scholar]
- 17.Regan RF, Chen J, Benvenisti-Zarom L. Heme oxygenase-2 gene deletion attenuates oxidative stress in neurons exposed to extracellular hemin. BMC Neurosci. 2004;5:34. doi: 10.1186/1471-2202-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Radic Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 19.Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci U S A. 2002;99:16093–16098. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100:672–678. doi: 10.3171/jns.2004.100.4.0672. [DOI] [PubMed] [Google Scholar]
- 21.Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- 22.Wada T, Oara H, Watanabe K, Kinoshita H, Yachi A. Autoradiographic study on the site of uptake of the haptoglobin-hemoglobin complex. J Reticuloendothel Soc. 1970;8:185–193. [PubMed] [Google Scholar]
- 23.Allison AC, Rees WA. The binding of haemoglobin by plasma proteins (haptoglobins); its bearing on the renal threshold for haemoglobin and the aetiology of haemoglobinuria. Br Med J. 1957;2:1137–1143. doi: 10.1136/bmj.2.5054.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Kinzie E, Berger FG, Lim SK, Baumann H. Haptoglobin, an inflammation-inducible plasma protein. Redox Rep. 2001;6:379–385. doi: 10.1179/135100001101536580. [DOI] [PubMed] [Google Scholar]
- 25.Buehler PW, Abraham B, Vallelian F, Linnemayr C, Pereira CP, Cipollo JF, Jia Y, Mikolajczyk M, Boretti FS, Schoedon G, Alayash AI, Schaer DJ. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood. 2009;113:2578–2586. doi: 10.1182/blood-2008-08-174466. [DOI] [PubMed] [Google Scholar]
- 26.Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479–488. doi: 10.1385/NCC:1:4:479. [DOI] [PubMed] [Google Scholar]
- 27.Keimowitz RM, Annis BL. Disseminated intravascular coagulation associated with massive brain injury. J Neurosurg. 1973;39:178–180. doi: 10.3171/jns.1973.39.2.0178. [DOI] [PubMed] [Google Scholar]
- 28.Morel N, Morel O, Petit L, Hugel B, Cochard JF, Freyssinet JM, Sztark F, Dabadie P. Generation of procoagulant microparticles in cerebrospinal fluid and peripheral blood after traumatic brain injury. J Trauma. 2008;64:698–704. doi: 10.1097/TA.0b013e31816493ad. [DOI] [PubMed] [Google Scholar]
- 29.Cohen MJ, Brohi K, Ganter MT, Manley GT, Mackersie RC, Pittet JF. Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the protein C pathway. J Trauma. 2007;63:1254–1261. doi: 10.1097/TA.0b013e318156ee4c. discussion 1261-1252. [DOI] [PubMed] [Google Scholar]
- 30.Halpern CH, Reilly PM, Turtz AR, Stein SC. Traumatic coagulopathy: the effect of brain injury. J Neurotrauma. 2008;25:997–1001. doi: 10.1089/neu.2008.0548. [DOI] [PubMed] [Google Scholar]
- 31.Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1761–1770. doi: 10.1038/jcbfm.2008.74. [DOI] [PubMed] [Google Scholar]
- 32.Morel O, Morel N, Freyssinet JM, Toti F. Platelet microparticles and vascular cells interactions: a checkpoint between the haemostatic and thrombotic responses. Platelets. 2008;19:9–23. doi: 10.1080/09537100701817232. [DOI] [PubMed] [Google Scholar]
- 33.Kai Y, Maeda Y, Sasaki T, Kanaide H, Hirano K. Basic and translational research on proteinase-activated receptors: the role of thrombin receptor in cerebral vasospasm in subarachnoid hemorrhage. J Pharmacol Sci. 2008;108:426–432. doi: 10.1254/jphs.08r11fm. [DOI] [PubMed] [Google Scholar]
- 34.Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x.1268 [DOI] [PubMed] [Google Scholar]
- 35.Sugawara T, Jadhav V, Ayer R, Chen W, Suzuki H, Zhang JH. Thrombin inhibition by argatroban ameliorates early brain injury and improves neurological outcomes after experimental subarachnoid hemorrhage in rats. Stroke. 2009;40:1530–1532. doi: 10.1161/STROKEAHA.108.531699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akopov S, Sercombe R, Seylaz J. Cerebrovascular reactivity: role of endothelium/platelet/leukocyte interactions. Cerebrovasc Brain Metab Rev. 1996;8:11–94. [PubMed] [Google Scholar]
- 37.Auer LM, Leber K, Sayama I. Effect of serotonin and its antagonist ketanserin on pial vessels. J Cereb Blood Flow Metab. 1985;5:517–522. doi: 10.1038/jcbfm.1985.78. [DOI] [PubMed] [Google Scholar]
- 38.Muhonen MG, Robertson SC, Gerdes JS, Loftus CM. Effects of serotonin on cerebral circulation after middle cerebral artery occlusion. J Neurosurg. 1997;87:301–306. doi: 10.3171/jns.1997.87.2.0301. [DOI] [PubMed] [Google Scholar]
- 39.Khalil Z, Helme RD. Serotonin modulates substance P-induced plasma extravasation and vasodilatation in rat skin by an action through capsaicin-sensitive primary afferent nerves. Brain Res. 1990;527:292–298. doi: 10.1016/0006-8993(90)91149-b.0006-8993(90)91149-B [DOI] [PubMed] [Google Scholar]
- 40.Gecse A, Kis B, Mezei Z, Telegdy G. Effects of inflammatory neuropeptides on the arachidonate cascade of platelets. Int Arch Allergy Immunol. 1999;118:166–170. doi: 10.1159/000024057. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 42.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 43.Stern M, Savill J, Haslett C. Human monocyte-derived macrophage phagocytosis of senescent eosinophils undergoing apoptosis Mediation by alpha v beta 3/CD36/thrombospondin recognition mechanism and lack of phlogistic response. Am J Pathol. 1996;149:911–921. [PMC free article] [PubMed] [Google Scholar]
- 44.Lee PY, Pearce FL. Histamine secretion from mast cells stimulated with bradykinin. Agents Actions. 1990;30:67–69. doi: 10.1007/BF01969000. [DOI] [PubMed] [Google Scholar]
- 45.Ishikawa M, Zhang JH, Nanda A, Granger DN. Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front Biosci. 2004;9:1339–1347. doi: 10.2741/1330. [DOI] [PubMed] [Google Scholar]
- 46.Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2000;20:53–65. doi: 10.1097/00004647-200001000-00009. [DOI] [PubMed] [Google Scholar]
- 47.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 48.Stoll G, Schroeter M, Jander S, Siebert H, Wollrath A, Kleinschnitz C, Bruck W. Lesion-associated expression of transforming growth factor-beta-2 in the rat nervous system: evidence for down-regulating the phagocytic activity of microglia and macrophages. Brain Pathol. 2004;14:51–58. doi: 10.1111/j.1750-3639.2004.tb00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J, Tsirka SE. Tuftsin fragment 1-3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2005;36:613–618. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- 51.Mander P, Borutaite V, Moncada S, Brown GC. Nitric oxide from inflammatory-activated glia synergizes with hypoxia to induce neuronal death. J Neurosci Res. 2005;79:208–215. doi: 10.1002/jnr.20285. [DOI] [PubMed] [Google Scholar]
- 52.Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, Elango C, Singh AK, Singh I. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab. 2005;25:177–192. doi: 10.1038/sj.jcbfm.9600012. [DOI] [PubMed] [Google Scholar]
- 53.Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport. 2002;13:1067–1070. doi: 10.1097/00001756-200206120-00018. [DOI] [PubMed] [Google Scholar]
- 54.Canolle B, Masmejean F, Melon C, Nieoullon A, Pisano P, Lortet S. Glial soluble factors regulate the activity and expression of the neuronal glutamate transporter EAAC1: implication of cholesterol. J Neurochem. 2004;88:1521–1532. doi: 10.1046/j.1471-4159.2003.02301.x.2301 [DOI] [PubMed] [Google Scholar]
- 55.Sercombe R, Dinh YR, Gomis P. Cerebrovascular inflammation following subarachnoid hemorrhage. Jpn J Pharmacol. 2002;88:227–249. doi: 10.1254/jjp.88.227. [DOI] [PubMed] [Google Scholar]
- 56.Uemura Y, Sugimoto T, Okamoto S, Handa H, Mizuno N. Changes of neuropeptide immunoreactivity in cerebrovascular nerve fibers after experimentally produced SAH Immunohistochemical study in the dog. J Neurosurg. 1987;66:741–747. doi: 10.3171/jns.1987.66.5.0741. [DOI] [PubMed] [Google Scholar]
- 57.Fein JM, Flor WJ, Cohan SL, Parkhurst J. Sequential changes of vascular ultrastructure in experimental cerebral vasospasm Myonecrosis of subarachnoid arteries. J Neurosurg. 1974;41:49–58. doi: 10.3171/jns.1974.41.1.0049. [DOI] [PubMed] [Google Scholar]
- 58.Mayberg MR, Okada T, Bark DH. The significance of morphological changes in cerebral arteries after subarachnoid hemorrhage. J Neurosurg. 1990;72:626–633. doi: 10.3171/jns.1990.72.4.0626. [DOI] [PubMed] [Google Scholar]
- 59.Gomis P, Kacem K, Sercombe C, Seylaz J, Sercombe R. Confocal microscopic evidence of decreased alpha-actin expression within rabbit cerebral artery smooth muscle cells after subarachnoid haemorrhage. Histochem J. 2000;32:673–678. doi: 10.1023/a:1004115432660. [DOI] [PubMed] [Google Scholar]
- 60.Yamamoto Y, Bernanke DH, Smith RR. Accelerated non-muscle contraction after subarachnoid hemorrhage: cerebrospinal fluid testing in a culture model. Neurosurgery. 1990;27:921–928. doi: 10.1097/00006123-199012000-00010. [DOI] [PubMed] [Google Scholar]
- 61.Grossetete M, Phelps J, Arko L, Yonas H, Rosenberg GA. Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery. 2009;65:702–708. doi: 10.1227/01.NEU.0000351768.11363.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jung KH, Chu K, Lee ST, Kim SJ, Song EC, Kim EH, Park DK, Sinn DI, Kim JM, Kim M, Roh JK. Blockade of AT1 receptor reduces apoptosis, inflammation, and oxidative stress in normotensive rats with intracerebral hemorrhage. J Pharmacol Exp Ther. 2007;322:1051–1058. doi: 10.1124/jpet.107.120097. [DOI] [PubMed] [Google Scholar]
- 63.Simard JM, Geng Z, Woo SK, Ivanova S, Tosun C, Melnichenko L, Gerzanich V. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009;29:317. doi: 10.1038/jcbfm.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Armin SS, Colohan AR, Zhang JH. Traumatic subarachnoid hemorrhage: our current understanding and its evolution over the past half century. Neurol Res. 2006;28:445–452. doi: 10.1179/016164106X115053. [DOI] [PubMed] [Google Scholar]
- 65.Armin SS, Colohan AR, Zhang JH. Vasospasm in traumatic brain injury. Acta Neurochir Suppl. 2008;104:421–425. doi: 10.1007/978-3-211-75718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]