

Background: Preserving functional β-cell mass is essential for preventing and curing type 2 diabetes (T2D).
Results: Administration of FTY720 to db/db mice led to sustained normalization of hyperglycemia by stimulating in vivo β-cell regeneration through PI3K-dependent regulation of cyclin D3 and p57KIP2.
Conclusion: FTY720 is capable of promoting in vivo β-cell regeneration in obesity diabetes.
Significance: Sphingosine 1-phosphate signaling pathway potentially is a therapeutic target for treatment of T2D.
Keywords: Cell Proliferation, Diabetes, Pancreatic Islets, Regeneration, Sphingosine 1-Phosphate, FTY720, in Vivo β-Cell Regeneration
Abstract
Loss of insulin-producing β-cell mass is a hallmark of type 2 diabetes in humans and diabetic db/db mice. Pancreatic β-cells can modulate their mass in response to a variety of physiological and pathophysiological cues. There are currently few effective therapeutic approaches targeting β-cell regeneration although some anti-diabetic drugs may positively affect β-cell mass. Here we show that oral administration of FTY720, a sphingosine 1-phosphate (S1P) receptor modulator, to db/db mice normalizes fasting blood glucose by increasing β-cell mass and blood insulin levels without affecting insulin sensitivity. Fasting blood glucose remained normal in the mice even after the drug was withdrawn after 23 weeks of treatment. The islet area in the pancreases of the FTY720-treated db/db mice was more than 2-fold larger than that of the untreated mice after 6 weeks of treatment. Furthermore, BrdU incorporation assays and Ki67 staining demonstrated cell proliferation in the islets and pancreatic duct areas. Finally, islets from the treated mice exhibited a significant decrease in the level of cyclin-dependent kinase inhibitor p57KIP2 and an increase in the level of cyclin D3 as compared with those of untreated mice, which could be reversed by the inhibition of phosphatidylinositol 3-kinase (PI3K). Our findings reveal a novel network that controls β-cell regeneration in the obesity-diabetes setting by regulating cyclin D3 and p57KIP2 expression through the S1P signaling pathway. Therapeutic strategies targeting this network may promote in vivo regeneration of β-cells in patients and prevent and/or cure type 2 diabetes.
Introduction
Type 2 diabetes is one of the most prevalent human metabolic diseases. It is characterized by insulin resistance and the reduction of functional pancreatic β-cell mass (1). Although there is an initial compensatory increase of β-cell mass in response to insulin resistance, diabetes occurs when the functional β-cell mass fails to expand sufficiently (2, 3). Finding ways to preserve or increase the mass of functional β-cells in diabetic patients is therefore a key step in controlling or curing type 2 diabetes in humans (4, 5).
Pancreatic β-cells are plastic cells that modulate their mass in response to a variety of physiological (pregnancy) (6) and pathophysiological (obesity or insulin resistance) states (3). New β-cells may arise from the proliferation of pre-existing β-cells (7) or pancreatic progenitor cells (5, 8, 9), and the transdifferentiation of pancreatic non-β-cells to β-cells under certain conditions (10–13). Recent islet transplantation in diabetes patients suggest that diabetes may be cured by replenishing β-cell mass (14). Importantly, it has been shown that β-cell volume in obese humans without diabetes is 50% higher than that in normal lean subjects (2, 15) and increases in islet mass occur during pregnancy in humans (16, 17), suggesting that human islets are capable of expanding their mass in response to metabolic demands, although much lower compared with mice (15). Our goal, therefore, is to develop a pharmacological agent that can stimulate an increase in β-cell mass in vivo (4, 5).
Various nutrients and peptide hormones have been implicated as regulators of β-cell mass (18, 19). However, we are particularly interested in a group of membrane-derived bioactive lysophospholipids that have growth factor and hormone-like biological activities (20). Lysophospholipids, including lysophosphatidic acid and sphingosine 1-phosphate (S1P),2 regulate diverse biological processes including embryogenesis, vascular development, neurogenesis, uterine development, oocyte survival, immune cell trafficking, and inflammatory reactions through their receptors, a novel class of G protein-coupled receptors (GPCRs) (20, 21). Intriguingly, lysophospholipid levels are significantly increased during human pregnancy (22) and in obese mice (23). We have screened several lysophosphatidic acid and S1P analogs in a β-cell line and db/db mice, which exhibit severe depletion of insulin-producing β-cells (24), and identified that intraperitoneal injection of FTY720, a structural analog of sphingosine, can normalize hyperglycemia in db/db mice.
FTY720 (Fingolimod), a derivative of ISP-1 (myriocin), a fungal metabolite of the Chinese herb Iscaria sinclarii as well as a structural analog of sphingosine, is a potent immunosuppressant that was approved as a new treatment for multiple sclerosis (25, 26). FTY720 becomes active in vivo following phosphorylation by sphingosine kinase 2 (SphK2) to form FTY720(S)-phosphate (FTY720-P), which binds to four of the five S1P receptors (S1P1, S1P3, S1P4, and S1P5 but not S1P2) and prevents the release of lymphocytes from lymphoid tissue (27, 28). Here we report that oral administration of the FTY720 to db/db mice leads to normalization of hyperglycemia by stimulating β-cell in vivo regeneration through the PI3K-dependent regulation of cyclin D3 and p57KIP2.
EXPERIMENTAL PROCEDURES
Materials
FTY720 (2-amino-2-[4-octylphenyl]ethyl)-1,3-propanediol, hydrochloride, C19H33NO2·HC, 343.9), FTY720-P, S1P, SEW2871 (5-[4-phenyl-5-(trifluoromethyl)-2-thienyl]-3-[3-(trifluoromethyl)phenyl]-1,2,4-oxadiazole), CAY10444 (2-undecyl-thiazolidine-4-carboxylic acid), W123 (3-(2-(3-hexylphenylamino)-2-oxoethylamino)propanoic acid), and FTY720(R)-phosphate were purchased from Cayman Chemical (Ann Arbor, MI).
Animals and Procedures
Five-week-old female db/db mice (BKS.Cg-m+/+Leprdb) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed under controlled light (12 h light/12 h dark) and temperature conditions, and had free access to food (normal rodent chow) and water. All procedures were conducted in accordance with the guidelines on Animal Care and were approved by the Institutional Animal Care and Use Committee (IACUC) of Mount Sinai School of Medicine.
After 1 week, the fasting glucose levels of the 6-week-old mice were measured. Mice with normal glucose levels (<126 mg/dl) were randomly divided into control and FTY720 treatment groups. Mice in the FTY720-treated group were fed 10 mg/kg of FTY720 daily by feeding tube and their food intake and body weight were measured twice a week. Fasting glucose levels were measured at the end of each week. At 12 weeks of age (when the mice had been treated for 6 weeks), all mice were subjected to metabolic analysis. After metabolic analysis, the pancreases were removed from the mice at 13 and 16 weeks of age for immunohistochemical analysis, quantitation of islet area, or islet isolation.
Intraperitoneal Glucose Tolerance Test or Insulin Tolerance Test
For the glucose tolerance test, the mice were fasted for 16 h and intraperitoneally injected with 10% glucose (1 mg/g of body weight). Glucose levels were then measured after 0, 30, 60, 90, and 120 min by a Glucometer Elite (Bayer Corp., Elkhart, IN) (29). For the insulin tolerance test, the mice were fasted for 6 h and injected intraperitoneally with human regular insulin (0.55 units/kg) (Sigma). Tail blood samples were collected at 0, 30, 40, 60, 90, and 120 min for glucose measurement (30, 31).
Determination of Serum Insulin Levels
After overnight fasting, blood samples (50 μl) were collected in a heparinized microhematocrit tube for the determination of insulin concentration using a Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem Inc., Downers Grove, IL) (30).
Immunohistochemical Analysis
Pancreases were removed from the db/db mice, fixed overnight in 4% formaldehyde solution, and embedded in paraffin. Paraffin sections (10 μm thick) were rehydrated, and antigen retrieval in 10 mm sodium citrate solution was performed using a microwave, followed by blocking endogenous peroxidase in 3% H2O2 solution. The following primary antibodies were used: guinea pig anti-swine insulin (1:300; DAKO Corp., Carpinteria, CA), rabbit anti-glucagon (1:200; Thermo Fisher Scientific, Fremont, CA), mouse anti-cyclin D3 (1:40; Vector Laboratory, Burlingame, CA), mouse anti-BrdU (1:10; BD Biosciences), rabbit anti-Ki67 (1:100; Abcam, Cambridge, MA), and rabbit anti-p57KIP2 (1:100; Abcam, Cambridge, MA). Goat anti-mouse/rabbit IgG (Vector Laboratory) and goat anti-guinea pig-mouse/rabbit IgG conjugated with the Alexa Fluor® dyes (Alexa Fluor® 488 and Alexa Fluor® 594; Invitrogen) were used for the secondary antibodies. All images were captured by a Zeiss Axioplan 2 microscope (32, 33).
Assessment of Islet Areas
To assess islet area in pancreas after 6 weeks of treatment with FTY720, six consecutive paraffin sections (10 μm) from a pancreas (10 pancreases for the control and 9 pancreases for the FTY720-treated group) were used. All islet images on a whole section were taken by a Axioplan 2 microscope at ×10 magnification, each islet area was measured by the Java-based image-processing program ImageJ (National Institutes of Health, Bethesda, MD), and the sum of all islet areas from a section was considered to be the islet area per pancreas.
Islet Isolation and Cell Culture
Islets were isolated from pancreases removed from the 13-week-old untreated and FTY720-treated db/db mice by Liberase (Roche Diagnostics) digestion, followed by discontinuous Ficoll gradient separation and manual stereomicroscopic selection to exclude contaminating tissues (30). Isolated islets (or INS-1 cells) were cultured in the medium (composed of RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 1% sodium pyruvate, 50 μm β-mercaptoethanol, 100 units/ml of penicillin, and 100 μg/ml of streptomycin). For treatment of INS-1 cells, the cells were plated in RPMI medium overnight and changed to Hanks' balanced salt solution containing 2 mm glucose and 0.5% FBS overnight. Then the cells were treated with various conditions in the same medium for 24 h.
Measurement of β-Cell Regeneration in Vivo
After overnight fasting, the mice were given 1 mg/ml of 5-bromo-2′-deoxyuridine (BrdU) in PBS intraperitoneally. Mice were sacrificed 24 h after BrdU injection and pancreases were removed and fixed with a 4% formaldehyde solution. The pancreases were then embedded in paraffin and sliced into 10-μm sections. Tissue sections were stained with the anti-BrdU monoclonal antibody provided in BrdU in Situ Detection Kit (BD Biosciences).
RT-PCR Analysis
Total RNA was extracted from islets isolated from treated and untreated mice using the RNeasy Mini Kit. The targeting cDNA sequences were obtained online (www.ncbi.nlm.nih.gov) and PCR primers were designed by PrimerQuestTM (IDT SciTools, Coralville, IA). Primers used in the conventional RT-PCR were listed in Table 1. RT-PCR was performed according to the manufacturer's instructions. Band intensity was quantified by the Java-based image-processing program ImageJ.
TABLE 1.
Sequences of primers and probes used for conventional and real-time RT-PCRs
| Gene | Sequences |
|---|---|
| Primers for conventional RT-PCRs (mouse) | |
| GAPDH | F: 5′-AACTTTGGCATTGTGGAAGGGCTC |
| R: 5′-ACCCTGTTGCTGTAGCCGTATTCA | |
| Cyclin D1 | F: 5′-TGCTGCAAATGGAACTGCTTCTGG |
| R: 5′-TACCATGGAGGGTGGGTTGGAAAT | |
| Cyclin D2 | F: 5′-AGGCCCAAGTTGAATGAGTCTGGA |
| R: 5′-ATCAGGTACCCAAGTTGCCCAAGA | |
| Cyclin D3 | F: 5′-TCCCACAGATGTCACAGCCATTCA |
| R: 5′-AAGCTGGTTGAGTGGGAAGGAAGA | |
| p21CIP1 | F: 5′-AATCCTGGTGATGTCCGACCTGTT |
| R: 5′-AGACCAATCTGCGCTTGGAGTGAT | |
| p27KIP1 | F: 5′-TGGCTCTGCTCCATTTGACTGTCT |
| R: 5′-ATGATTGACCGGGCCGAAGAGATT | |
| p57KIP2 | F: 5′-TGTACCATGTGCAAGGAGTACGCT |
| R: 5′-TTGGTATGGGCAGTACAGGAACCA | |
| S1P1 | F: 5′-TTCTCTTCTGCACCACCGTCTTCA |
| R: 5′-AGCAGGCAATGAAGACACTCAGGA | |
| S1P3 | F: 5′-TGGCTTCTCCATTTCCTCCTGTGT |
| R: 5′-ACGGATCCACTGGGCATACATCAA | |
| S1P4 | F: 5′-ATCTAGTGCTTGCCTCCCATCCAA |
| R: 5′-GGCCTGGGTTTCCTGTTGTTTGAA | |
| S1P5 | F: 5′-TTTCCAATAGCCGCTCTCCTGTGT |
| R: 5′-AGCTTGCCGGTGTAGTTGTAGTGA | |
| PI3Kr1 | F: 5′-CAGCAACCGAAACAAAGCGGAGAA |
| R: 5′-TGGCTTGAAGGTGAGGAACTGGAT | |
| PI3Kr2 | F: 5′-ATCAAAGTCTTCCACCGGGATGGT |
| R: 5′-TGAGGTCAGGTTTGAGGCTGTTCA | |
| PI3Kr3 | F: 5′-TGCTTTGGAAAGAAGCTGGCTTGG |
| R: 5′-AGGGCAAACAGGGTGATGAAGAGA | |
| Akt1 | F: 5′-ACGGGCACATCAAGATAACGGACT |
| R: 5′-AAGGTGGGCTCAGCTTCTTCTCAT | |
| Akt2 | F: 5′-TGGACTGCTGAAGAAGGACCCAAA |
| R: 5′-TTTCTGTACCACGTCCTGCCAGTT | |
| Primers and probes for quantitative real-time RT-PCRs (mouse) | |
| GAPDH | F: 5′-TCAACAGCAACTCCCACTCTTCCA |
| R: 5′-ACCCTGTTGCTGTAGCCGTATTCA | |
| Probe-/56-FAM/GGCTGGCAT/Zen/TGCTCTCAATGACAACT/3IABKFQ | |
| Cyclin D3 | F: 5′-TCT TCC TTC CCA CTC AAC CAG CTT |
| R: 5′-TTT GGC AAC TGA GAA GGT TGG AGC | |
| Probe-/56-FAM/TCCTGGGCC/Zen/ATGATGGTCAGAGAAAT/3IABKFQ | |
| p57KIP2 | F: 5′-ATG TAG CAG GAA CCG GAG ATG GTT |
| R: 5′-TTT ACA CCT TGG GAC CAG CGT ACT | |
| Probe-/56-FAM/TGAGAACAC/Zen/TCTGTACCATGTGCAAGG/3IABKFQ | |
| PI3Kr1 | F: 5′-AAT AGG TTA CAG TGC GGG CCG TAT |
| R: 5′-CAG TTT CCT TGG CTT TGC TCG GTT | |
| Probe-/56-FAM/CCAAGACAC/Zen/CATTACAAAGAAAGCCGGAC/3IABkFQ | |
| PI3Kr2 | F: 5′-TGC ATC CAG CAA GAT CCA AGG AGA |
| R: 5′-ACC ATC CCG GTG GAA GAC TTT GAT | |
| Probe-/56-FAM/TCAGGAAAG/Zen/GCGGGAACAACAAGTTG/3IABkFQ | |
| PI3Kr3 | F: 5′-TTC AGA CAT TGC TGT GCG GTT GTG |
| R: 5′-GCA AGT CTG CCA ACC ATT CCA AGT | |
| Probe-/56-FAM/GGGAAGGTA/Zen/AGGTGAGAACATTGTTGGG/3IABkFQ | |
| PDX-1 | F: 5′-ACT TAA CCT AGG CGT CGC ACA AGA |
| R: 5′-GGC ATC AGA AGC AGC CTC AAA GTT | |
| Probe-/56-FAM/AATTCTTGA/Zen/GGGCACGAGAGCCAGTT/3IABkFQ | |
| Bcl-2 | F: 5′-AAT TGT AAT TCA TCT GCC GCC GCC |
| R: 5′-ACA CTC CGG CTT CAC TGA GAA TGT | |
| Probe-/56-FAM/TGCGGTGCT/Zen/CTTGAGATCTCTGGTT/3IABkFQ | |
| Bcl-xL | F: 5′-TTG CTG GTC GCC GGA GAT AGA TTT |
| R: 5′-TGG GTC TGC TCT GTG TTT AGC GAT | |
| Probe-/56-FAM/ACTTATCTT/Zen/GGCTTTGGATCCTGGAAGAGA/3IABkFQ | |
| Primers and probes for quantitative real-time RT-PCRs (rat) | |
| GAPDH | F: 5′-TGA TGC TGG TGC TGA GTA TGT CGT |
| R: 5′-TCT CGT GGT TCA CAC CCA TCA CAA | |
| Probe-/56-FAM/AGTCTACTG/Zen/GCGTCTTCACCACCAT/3IABkFQ | |
| Cyclin D3 | F: 5′-TCA CTG CAT TTG GAT CTG GGT CCT |
| R: 5′-AGT TTA CGG GCG CTT GCT CTT CTA | |
| Probe-/56-FAM/AG CCA ACC T/Zen/A GAT GGC TGC TGT GTA A/3IABkFQ | |
| p57KIP2 | F: 5′-TCG AAT TCG CCC TTA GCG ATG GAA |
| R: 5′-GCA CAT CCT GCT GGA AGT TGA AGT | |
| Probe-/56-FAM/TC CAG CGA C/Zen/A CCT TCC CAG TGA TA/3IABkFQ | |
| PDX-1 | F: 5′-AGT TGG GTA TAC CAG CGA GAT GCT |
| R: 5′-TTG TCC TCA GTT GGG AGC CTG ATT | |
| Probe-/56-FAM/TC TGG GAC T/Zen/C TTT CCT GGG ACC AAT T/3IABkFQ | |
Quantitative real-time RT-PCR was performed using an EXPRESS One-Step SuperScript quantitative RT-PCR Kit (from Invitrogen) according to the manufacturer's instructions of 7900HT Real-Time PCR Systems (Applied Biosystems, Foster City, CA). The mRNA levels were calculated by comparative CT methods (XTest/XGAPDH = 2ΔΔCT) with GAPDH as the endogenous reference gene. Primers and probes used in quantitative RT-PCR were designed by PrimerQuest (IDT SciTools, Coralville, IA) as listed in Table 1.
Western Blot Analysis
Islets isolated from 8-week-old C57/BL6 mice were treated with 0.1 or 0.2 μm FTY720 in the absence of FBS. After 24 or 48 h, islets were collected and lysed in RIPA lysis buffer. Aliquots containing 60 μg of protein were separated on 10% SDS-PAGE (34). After electrophoresis, proteins were blotted onto nitrocellulose membranes and probed with rabbit anti-p57KIP2 polyclonal antibody or mouse anti-cyclin D3 monoclonal antibody, followed by incubation with anti-rabbit or anti-mouse IgG conjugated with horseradish peroxidase. Proteins were detected by enhanced chemiluminescence.
Statistical Analysis
Data were expressed as mean ± S.E. Significant differences among groups were evaluated by one-way analysis of variance and Turkey's multiple comparisons test or by unpaired two-tailed Student's t test by using PRISM. Significance levels are described in individual figure legends.
RESULTS
Oral Administration of FTY720 to db/db Mice Normalizes Hyperglycemia without Affecting Insulin Sensitivity
We have prescreened several lysophosphatidic acid and S1P analogs in islet β-cells (supplemental Fig. S1). We found that intraperitoneal injection of FTY720 led to relatively normal fasting glucose levels in db/db mice (supplemental Fig. S2). To rigorously determine the effects of FTY720 in vivo, pre-diabetic (age of 6 weeks, fasting glucose <126 mg/dl) and diabetic (age of 8–9 weeks, fasting glucose = 430 mg/dl) female db/db mice were fed daily with FTY720 (+FTY720) and monitored by weekly fasting blood glucose measurements. Although fasting glucose levels increased significantly in the untreated group (−FTY720) by the age of 8 weeks and continued to increase over time (to about 500 mg/dl by the age of 12 weeks), levels in the FTY720-treated pre-diabetic db/db mice remained normal (∼126 mg/dl) and diabetic db/db mice also became normal after 6 weeks treatment with FTY720 (Fig. 1A). Importantly, this control of fasting glucose levels over time occurred despite the fact that FTY720 administration was changed from daily to weekly and ceased completely at the age of 29 weeks (Fig. 1A). In addition, the weight gain, a common side effect of insulin therapy, was significantly higher in the FTY720-treated group than in the untreated group (Fig. 1B).
FIGURE 1.

Metabolic features of the FTY720-treated db/db mice. A, fasting glucose levels. ○, FTY720-untreated group; ●, pre-diabetic db/db mice (FG < 126 mg/dl) at 6 weeks old treated with 10 mg/kg of FTY720; and ▴, diabetic db/db mice (FG ≅ 430 mg/dl) at 8–9 weeks old treated with 10 mg/kg of FTY720 (n = 4). I, fasting glucose of mice at the age of 6 to 12 weeks at a daily dosage of 10 mg/kg of FTY720 (n = 20 for the control and FTY720 treated pre-diabetic mice, respectively); II, fasting glucose levels of mice at the age of 12 to 20 weeks at the daily dosage of 10 mg/kg of FTY720 (n = 6 for the control and FTY720 treated mice, respectively); III, fasting glucose levels of mice at the age of 20 to 29 weeks at the weekly dosage of 10 mg/kg of FTY720 (n = 2 for control and FTY720-treated mice, respectively); IV, glucose levels from the mouse without FTY720 treatment after the age of 29 weeks (n = 2 for control and FTY720-treated mice, respectively). B, body weights. The mice were treated as in A and the body weights were measured weekly. C, the distribution of fasting glucose levels after 6 weeks treatment (*, p < 0.0001, one way analysis of variance test). D, fasting serum insulin levels of db/db mice with or without FTY720 treatment (*, p < 0.01). E, glucose tolerance test (1 g/kg 10% glucose) (*, p < 0.01) and F, insulin tolerance test (0.55 units/kg) (*, p < 0.05) were performed after 6 weeks treatment.
After 6 weeks treatment, the fasting glucose levels in all mice were normalized (Fig. 1C) and the fasting serum insulin levels were significantly elevated (Fig. 1D). Consistent with these findings, we found that glucose tolerance improved significantly in the FTY720-treated db/db mice (Fig. 1E) as compared with untreated mice but that insulin sensitivity was unaffected (Fig. 1F). Specifically, initial fasting glucose levels in the untreated group were about 500 mg/dl but declined rapidly in the first hour after insulin administration; glucose levels in the treated and untreated groups were similar after 70 min. These results indicated that administration of FTY720 to the db/db mice can normalize fasting blood glucose leading to prevention and reversal of diabetes without affecting insulin sensitivity.
FTY720 Treatment Increases β-Cell Mass in db/db Mice
Because FTY720 treatment increased fasting insulin levels in the db/db mice and did not affect the glucose-stimulated insulin secretion ex vivo (supplemental Fig. S3), we compared islet morphology, size, and insulin content in pancreases from the two groups of animals. Both H&E staining (Fig. 2A) and immunofluorescent staining for insulin (Fig. 2B) indicated that the untreated db/db mice experience a deterioration in islet morphology and a reduction in β-cell mass over time. In contrast, the FTY720-treated group exhibited normal islet morphology and large islet size (Fig. 2, A and B). Stereological quantification demonstrated that the insulin-positive cell area in these mice were significantly larger than those in the untreated group (Fig. 2C). Interestingly, it was recently reported that FTY720 does not impair human islets function and affect glucose-stimulated insulin secretion after 48 h treatment (35). Taken together, we conclude that the increase in insulin levels in the FTY720-treated db/db mice results from the increased β-cell mass in db/db mice.
FIGURE 2.

Effect of FTY720 on β-cell mass of db/db mice. A, H&E staining of pancreas from the untreated and FTY720-treated mice after 6 and 9 weeks treatment (scale bar, 50 μm). B, insulin immunostaining (green fluorescence) of pancreas after 6 weeks FTY720 treatment (scale bar, 50 μm). C, stereological quantification of the insulin positive areas in the control and FTY720-treated db/db mice. Six consecutive paraffin sections (10 μm) from each pancreas (10 pancreases from the untreated and 9 from the FTY720-treatment db/db mice) were used for islet area measurements. *, p < 0.01.
FTY720 Treatment Increases β-Cell Survival in db/db Mice
To determine the mechanism behind the increase in β-cell mass observed in the treated mice, first we examined β-cell survival because the S1P signal was previously reported to prevent cytokine-induced apoptosis in cultured islets (36). Although we detected few apoptotic β-cells in the pancreases of the untreated group, most likely due to the rapid clearance of apoptotic cells by phagocytes or adjacent cells in vivo (37), we found that levels of anti-apoptotic gene products, Bcl-2 (Fig. 3A) and Bcl-xL (Fig. 3B), were significantly increased in the islets from the FTY720-treated mice. Along with other report (38), these findings suggest that islet cells in these mice may have an increased survival ability through up-regulation of Bcl-2 and Bcl-xL.
FIGURE 3.
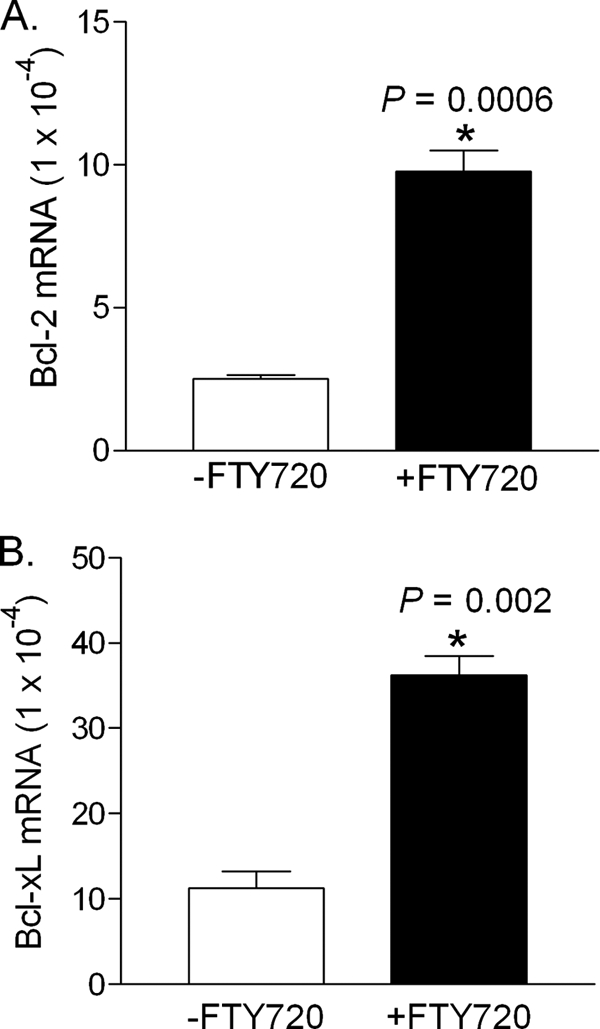
Effect of FTY720 on the expression of Bcl-2 and Bcl-xL in islets. Total RNAs were extracted from the islets isolated from the control and FTY720-treated mice at the age of 13 weeks and Bcl-2 (A) and Bcl-xL mRNA (B) in the db/db islets were quantitated by real time RT-PCT. *, p < 0.01.
FTY720 Treatment Increases β-Cell Proliferation in db/db Mice
Increases in islet mass occur during pregnancy in humans and animals (16, 17). Here we compared the degree of cell proliferation in islets from the two groups of animals by BrdU incorporation. In contrast to the untreated mice (Fig. 4A), BrdU-positive cells were easily observed in the islets of the FTY720-treated mice (Fig. 4, B–D).
FIGURE 4.

BrdU staining in pancreatic islets of the db/db pancreas. A, BrdU staining of islets in control db/db mice. B–D, BrdU staining of islets in FTY720-treated db/db mice. BrdU-positive cells (brown) were indicated by black arrows (scale bar, 100 μm).
We further examined β-cell proliferation in islets by staining of Ki67, a cellular marker for proliferation (39). Consistent with the results of BrdU incorporation, we did not observed any Ki67-positive cells within and outside islets in the untreated db/db mice. In contrast, Ki67 staining showed the proliferated cells in the islets, similar to BrdU incorporation in the islets of the FTY720-treated mice (Fig. 5). These data suggest that FTY720 induces β-cell proliferation in vivo.
FIGURE 5.

Ki67 staining in pancreatic islets of the db/db pancreas. The pancreatic sections were double-stained for insulin (green) and Ki67 (red). The Ki67-stained nucleus of the cell was identified within small and large islets of the FTY720-treated db/db mice (scale bar, 50 μm).
FTY720 Treatment Increases β-Cell Neogenesis in db/db Mice
New β-cells may arise from the proliferation of pancreatic progenitor cells located in the ductal lining (8, 9). We also examined BrdU incorporation in the pancreatic duct area. We were not able to detect any BrdU staining in the islets and ductal line of the untreated mice (Fig. 6A). As shown in Fig. 6, B–D, BrdU-positive cells were easily observed in the pancreatic duct lines of the FTY720-treated mice. Furthermore, the insulin-positive cells or small islets were also identified in the pancreatic duct area of the treated mice (Fig. 6, E and F). Our data suggested that FTY720 induces β-cell regeneration in vivo at least in the obese-diabetes setting.
FIGURE 6.

BrdU staining in the pancreatic duct areas of the db/db pancreas. A, BrdU staining of pancreas from control db/db mice. B–D, BrdU staining of pancreas from the FTY720-treated db/db mice. BrdU-positive cells (brown) in the duct lines are indicated by black arrows (scale bar, 100 μm). E and F, insulin immunostaining. Black arrows indicate the newly formed islets near the duct area (arrow) and insulin positive ductal line (scale bar, 100 μm).
FTY720 Treatment Down-regulates p57KIP2 and Up-regulates Cyclin D3 in db/db Islets
Cell proliferation is controlled by cyclin-dependent kinases, which are regulated positively by cyclins and negatively by inhibitors (CKIs). There are two families of CKIs: INK4 and CIP/KIP. Because p16INK4a is involved in the age-dependent decline in islet regenerative potential (40), we first examined p16INK4a levels in islets isolated from untreated and FTY720-treated db/db mice. We were unable to detect the expression of p16INK4a at 13 weeks of age, consistent with previous reports that the gene is repressed in young mice (41). Next we examined the CIP/KIP family, including p21CIP1, p27KIP1, and p57KIP2 (42). Of these, p57KIP2 was the most highly expressed in db/db islets from untreated mice (Fig. 7A); conversely, its expression was significantly down-regulated in the islets from the FTY720-treated db/db mice. In contrast, the expression of p21CIP1 and p27KIP1 did not vary significantly between the two groups. Interestingly, most of the p57KIP2-expressing cells are, like β-cells, terminally differentiated (42). Therefore, our results suggest that down-regulating p57KIP2 may allow these cells to re-enter the cell cycle from the differentiated state and begin proliferating.
FIGURE 7.

Effect of FTY720 on the expression of cyclin Ds and cyclin-dependent kinase (CDK) inhibitors in db/db islets. A, the mRNA levels of cyclin-dependent kinase inhibitors. *, p < 0.01. B, the mRNA levels of cyclin Ds. *, p < 0.01. Total RNAs were extracted from islets isolated from control and FTY720-treated mice at the age of 13 weeks, and the mRNA levels of interested targets were analyzed by RT-PCR. C, cyclin D3 immunostaining of pancreas after 6 weeks FTY720 treatment. The cells in the islet with cyclin D3-staining in their nucleus (brown) are indicated by black arrows (scale bar, 50 μm).
Furthermore, we examined the expression of the cyclin D family members cyclin D1, cyclin D2, and cyclin D3. Of these, cyclin D3 was expressed at the lowest levels in islets from untreated db/db mice (Fig. 7B). Although the expression of cyclin D1 and D2 was elevated in islets from the FTY720-treated db/db mice when compared with the untreated mice, the expression of cyclin D3 increased the most. Immunohistochemical analysis showed a dramatic increase in cyclin D3 expression in several islet cells (Fig. 7C) similar to that of BrdU incorporation (Fig. 4) and Ki67 staining (Fig. 5).
Finally, treatment of freshly isolated wild type islets with FTY720 or FTY720-P caused a dramatic down-regulation in p57KIP2 expression and a corresponding up-regulation in cyclin D3 expression in both proteins (Fig. 8A) and mRNAs (Fig. 8, B and C). These in vivo and ex vivo findings reveal an important role for p57KIP2 and cyclin D3 in regulation of the in vivo β-cell regeneration by FTY720.
FIGURE 8.

Effect of FTY720 on p57KIP2 and cyclin D3 expression in the wild type islets. A, Western blot analysis. Islets were isolated from 8-week-old wild type C57/BL6 mice and treated with 0.1 or 0.2 μm FTY720 in cell culture medium without FBS. After 24 and 48 h, protein was extracted and analyzed by Western blot analysis. B, quantitation of cyclin D3 mRNA and C, p57KIP2 mRNA levels. Isolated islets were treated with 0 (control), 0.1 or 0.2 μm FTY720 or FTY720-P for 48 h, and total RNAs were extracted and quantitative real-time RT-PCR was performed. *, p < 0.001, versus control.
FTY720 Treatment Up-regulates S1P Receptors in db/db Islets
It is well characterized that FTY720 is phosphorylated in vivo to form FTY720-P that binds S1P1, S1P3, S1P4, and S1P5 (25). We determined the expression of these receptors in the islets isolated from the untreated and FTY720-treated db/db mice. As shown in Fig. 9A, S1P1 and S1P3 were predominantly expressed in the db/db islets, whereas S1P4 and S1P5 were hardly detected. However, after FTY720 treatment of db/db mice, S1P1, S1P3, and S1P4 but not S1P5 were significantly elevated in the islets. Consistent with the findings in vivo, treatment of freshly isolated wild type islets with FTY720 caused a significant increase of S1P1, S1P3, and S1P4 but not S1P5 (Fig. 9B).
FIGURE 9.

Effect of FTY720 on the expression of S1P receptors in islet β-cells. A, expression of S1P receptors in vivo. The total RNAs were extracted from islets isolated from the untreated and FTY720-treated db/db mice at the age of 13 weeks, and the mRNA levels of S1P receptors were analyzed by RT-PCR and the band intensities were quantified by ImageJ (n = 5), *, p < 0.05. B, expression of S1P receptors ex vivo. Isolated wild type islets were treated with or without 0.2 μm FTY720 for 24 h and total RNAs were extracted and analyzed by RT-PCR for S1P receptors and the band intensities were quantified by ImageJ, *, p < 0.05. C, quantitation of cyclin D3 and D, p57KIP2 mRNA levels. INS-1 cells were treated with FTY720-P (FTY-P, 0.1 μm), FTY720(R)-P (FTY(R), 10 nm), FTY720 (FTY, 0.1 μm), FTY720 + Cay10444 (Cay, 10 μm), and FTY720 + W123 (20 μm) for 24 h and total RNAs were extracted and quantitative real-time RT-PCR was performed. The levels relative to the control were plotted. *, p < 0.05, versus control; #, p < 0.05, versus FTY720 or FTY720-P-treated groups.
We further evaluated the role of S1P receptors in regulation of cyclin D3 and p57KIP2 in INS-1 cells. Consistent with the findings in vivo and ex vivo, FTY720 or FTY720-P treatment significantly increased cyclin D3 expression in INS-1 cells, which was reversed significantly by CAY10444 (also known as BML-241), a selective antagonist of S1P3 (43), or by W123, a selective antagonist of S1P1 (44) (Fig. 9C). Furthermore, FTY720 or FTY720-P treatment significantly decreased p57KIP2 expression in INS-1 cells, which was reversed by CAY10444 or W123 (Fig. 9D). Interestingly, FTY720(R)-P, one of the stereoisomers of FTY720-P that is apparently not formed in vivo (45), did not affect cyclin D3 significantly but dramatically increased p57KIP2 expression (Fig. 9, C and D). These results suggest that at least S1P1 and S1P3 are involved in the FTY720-stimulated β-cell regeneration in vivo.
FTY720 Treatment Activates PI3K Signaling Pathway in db/db Islets
The PI3K/Akt pathway is known to play an important role in S1P/GPCR-mediated cellular responses, including survival and proliferation (46, 47). We determined the expression of three major PI3K regulatory subunits, PIK3r1 (encodes p85α, p55α, and p50α through differential promoter usage), PIK3r2 (encodes p85β), and PIK3r3 (encodes p55γ) (48), in the islets isolated from the untreated and FTY720-treated db/db mice. As shown in Fig. 10A, PI3Kr1 was moderately expressed and increased by FTY720 in vivo. PI3Kr2 and PI3Kr3, however, were undetectable in the islets from the untreated db/db mice and dramatically elevated in the islets from the FTY720-treated db/db mice, suggesting the involvement of PI3K. There was no difference in Akt1 and Akt2 between the treated and untreated groups because Akts are regulated by protein phosphorylation. It is known that expression of pancreatic and duodenal homeobox 1 (PDX-1), a transcription factor necessary for pancreatic development and β-cell maturation (49), is regulated by PI3K activation during pancreatic regeneration (50, 51). We found that the PDX-1 expression in the islets from the FTY720-treated db/db mice was 6-fold higher than that in the islets from the untreated mice (Fig. 10B), consistent with PI3K activation.
FIGURE 10.

Effect of FTY720 on the expression of PI3K regulatory subunits in the islet β-cells. Expression of PI3K regulatory subunits (A) and PDX-1 (B) in vivo. The total RNAs were extracted from the islets isolated from the untreated (−FTY720) and FTY720-treated db/db mice at the age of 13 weeks, and the mRNA levels of PI3K regulatory subunits, PI3Kr1, PI3Kr2, and PI3Kr3, as well as Akt1 and Akt2, were amplified by RT-PCR and visualized by agarose gel electrophoresis. The PDX-1 mRNA levels were quantified by quantitative RT-PCR. *, versus −FTY720 group. Expression of PI3Kr1 (C), PI3Kr2 (E), and PI3Kr3 in wild type islets. Isolated islets were treated with 0 (control), 0.1 or 0.2 μm FTY720 or FTY720-P for 48 h and total RNAs were extracted, and quantitative real time RT-PCR was performed. *, p < 0.001, versus control. Effects of wortmannin on the expression of (F) PDX-1, (G) cyclin D3, and (H) p57KIP2 in INS-1 cells. INS-1 cells were treated with FTY720 (FTY, 0.1 μm), FTY720-P (FTY-P, 0.1 μm), or S1P (0.1 μm) in the presence (+Wort) or absence (−Wort) of wortmannin (0.1 μm) for 24 h and total RNAs were extracted and quantitative real-time RT-PCR was performed. The levels relative to the control were plotted. *, p < 0.05, versus control, #, p < 0.05, versus the group without wortmannin.
Furthermore, treatment of the wild type islets with FTY720 or FTY720-P dramatically increased the expression of PI3Kr1 and PI3Kr3 (Fig. 10, C and E) but not PI3Kr2, probably due to its high expression (about 100-fold higher than PI3Kr1 and PI3Kr3) in the wild type islets (Fig. 10D). These results suggest that S1P signaling may mediate PI3K function partly through regulating expression of the PI3K regulatory subunits.
To substantiate the role of PI3K in the FTY720-stimulated β-cell regeneration, INS-1 cells were treated with FTY720, FTY720-P, or S1P, respectively, in the presence or absence of wortmannin, a specific PI3K inhibitor (52). We found that wortmannin completely blocked the increase of PDX-1 by FTY720, FTY720-P, or S1P (Fig. 10F). Moreover, wortmannin essentially reversed the effects of FTY720, FTY720-P, and S1P on cyclin D3 (Fig. 10G) and p57KIP2 (Fig. 10H). Our data strongly suggest that FTY720-stimulated in vivo β-cell regeneration is PI3K-dependent.
DISCUSSION
In this study, we showed that orally administered FTY720 can normalize blood glucose levels of db/db mice by preserving β-cell mass in vivo through increasing in survival ability and regeneration of β-cells, which in turn contributes to the well controlled fasting blood glucose levels observed in FTY720-treated db/db mice. Moreover, we demonstrated that FTY720 treatment of db/db mice up-regulates the expression of PDX-1 and cyclin D3 and down-regulates p57KIP2 in the islets through S1P receptors in a PI3K-dependent manner.
FTY720 is a structural analog of sphingosine and becomes active in vivo following phosphorylation by sphingosine kinase 2 to form FTY720(S)-P, which binds to four of the five S1P receptors (25, 53). We found that expression of S1P1, S1P3, and S1P4 was significantly increased in the islets from the FTY720-treated db/db mice and the effects of FTY720 are attenuated by S1P receptor antagonists, suggesting the involvement of S1P signaling in the db/db islets.
Intriguingly, it was recently reported that S1P signaling plays a key role in adiponectin-mediated survival of pancreatic β-cells, nutrient uptake, nutrient utilization, and mitochondrial proliferation (54, 55). Our findings suggest that FTY720 when administered to db/db mice acts on an intrinsic pathway that is physiologically important for β-cell survival and regeneration under metabolic stress.
Our findings identify a novel way to control β-cell regeneration by down-regulating p57KIP2 and up-regulating cyclin D3 expression through S1P/GPCR signaling. p57KIP2 is an imprinted gene that is conserved between rodents and humans and is required for normal development and differentiation (42). It has been reported that p57KIP2 is paternally imprinted and highly expressed in human pancreatic β-cells (56), and controls both self-renewal and exit from the cell cycle of pancreatic progenitors during pancreatic development (57). p57KIP2 expression, but not p21CIP1 or p27KIP1, is induced by transcription factor E47, which is involved in the transition from proliferation to cell cycle exit (58). Down-regulation of p57KIP2 by FTY720 may reverse this transition in vivo. Interestingly, cyclin D3, which is required for normal expansion of immature T lymphocytes (59), is a target of p57KIP2 inhibition (42). Our findings suggest that orchestrating p57KIP2 and cyclin D3 expression may be critical to inducing β-cell regeneration in vivo.
S1P receptors are a group of GPCRs, and S1P/GPCR signaling has been reported to control cell proliferation and survival through activation of AMP-activated protein kinase or PI3K/Akt pathways (46, 60). We found that PI3Kr2 and PI3Kr3, which are highly expressed in normal islets, were hardly detected in db/db islets and dramatically increased after FTY720 treatment, which is accompanied with a 6-fold increase of PDX-1. Inhibition of PI3K essentially reverses the effect of FTY720 on PDX-1, cyclin D3, and p57KIP2, suggesting that PI3K is required for FTY720-stimulated β-cell regeneration in vivo.
Based on these findings and others (54, 55), we propose the mechanism for S1P signaling in β-cell regeneration and survival in vivo as elucidated in Fig. 11. In this model, S1P signaling is a core component of the adiponectin-mediated pleiotropic actions (54) and FTY720 acts on this core, which is physiologically important for β-cell regeneration and survival under metabolic stress.
FIGURE 11.

Proposed mechanism for FTY720-stimulated β-cell regeneration in vivo. S1P signaling is a core component of the adiponectin-mediated pleiotropic actions (54). Binding of adiponectin to its receptors enhances deacylation of ceramide to sphingosine, which can then be phosphorylated to form S1P (54). FTY720, a structural analog of sphingosine, is phosphorylated in vivo to form FTY720-P (25). Inside-out signaling of S1P/FTY720-P via S1P receptors activates the PI3K/Akt pathway, which leads to up-regulation of PDX-1 and cyclin D3 and down-regulation of p57KIP2, and subsequently β-cell regeneration in vivo. FTY720 also promotes cell survival through activation of PI3K/Akt (38) and expression of Bcl-2 and Bcl-xL. In addition, S1P receptor signaling plays a key role in adiponectin-mediated AMP-activated protein kinase activation, which is important for mitochondrial biogenesis and β-cell survival (54). Thus, FTY720 acts on an intrinsic pathway that is physiologically important for β-cell regeneration and survival under metabolic stress. Sph, sphingosine; S1PRs, S1P receptors; AdipoR, adiponectin receptors.
Although FTY720 promotes β-cell regeneration, the risk of tumorigenesis is low. Concentrations of FTY720 identical to those used in this study (10 mg/kg) reportedly inhibit growth, migration, and invasion of pancreatic cancer cells (61). The compound has also been used in phase III clinical trials in patients with relapsing multiple sclerosis with a few reported cancer formation (26).
Is the FTY720-induced β-cell regeneration observed in db/db mice relevant to humans? β-Cell volume in obese humans with non-diabetes is 50% higher than that in normal lean subjects (2, 15) and increases in islet mass occur during pregnancy in humans (16, 17), suggesting that the mechanism in humans for β-cell mass expansion may be triggered by increased metabolic demand. Surprisingly, in contrast to the adult mouse, in which β-cell progenitors can be activated by pancreatic injury (8), partial pancreatectomy in adult humans does not provoke β-cell regeneration (62). These findings suggest that metabolic demand, but not pancreatic injury, can trigger human β-cell regeneration. It has been reported that p57KIP2 is highly expressed in human pancreatic β-cells (56) and controls both self-renewal and exit from the cell cycle of pancreatic progenitors during pancreatic development (57). High expression of p57KIP2 may inhibit β-cell regeneration in humans, unless p57KIP2 expression is reduced by some means and allows cells to re-enter the cell cycle (57).
In addition, human islets contain abundant cyclin D3, but a variable amount of cyclin D1 and little cyclin D2 (63). In contrast to mice in which cyclin D2 is required for β-cell expansion (64), overexpression of cyclin D3 and cdk6 effectively drives human β-cell replication (63), suggesting that increasing in cyclin D3, but not cyclin D1 and D2, is essential for human β-cell replication. It is worth noting that FTY720 does not have any toxic effects on human islets in vitro and in vivo after human islets were transplanted into the chemically induced diabetic immunodeficient mice (35). Therefore, it is possible that FTY720 that down-regulates p57KIP2 and up-regulates cyclin D3 expression in db/db islets may also increase islet mass in humans with metabolic demand, such as obesity and type 2 diabetes patients. Further study in human islets will validate the potential role of FTY720 in stimulating β-cell proliferation in type 2 diabetes.
Supplementary Material
Acknowledgment
We greatly thank Dr. Susan Bonner-Weir, Joslin Diabetes Center, Harvard University Medical School, for analyzing BrdU incorporation data and reviewing manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS063962 from the NINDS and the Iacocca Family foundation.

This article contains supplemental Figs. S1–S3.
- S1P
- sphingosine 1-phosphate
- GPCR
- G protein-coupled receptor
- FTY720
- (2-amino-2-[4-octylphenyl]ethyl)-1,3-propanediol
- BrdU
- 5-bromo-2′deoxyuridine
- PDX-1
- pancreatic and duodenal homeobox 1.
REFERENCES
- 1. Porte D., Jr. (1991) Banting lecture 1990. β-Cells in type II diabetes mellitus. Diabetes 40, 166–180 [DOI] [PubMed] [Google Scholar]
- 2. Butler A. E., Janson J., Bonner-Weir S., Ritzel R., Rizza R. A., Butler P. C. (2003) β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 [DOI] [PubMed] [Google Scholar]
- 3. Rhodes C. J. (2005) Type 2 diabetes, a matter of β-cell life and death? Science 307, 380–384 [DOI] [PubMed] [Google Scholar]
- 4. Leahy J. L., Hirsch I. B., Peterson K. A., Schneider D. (2010) Targeting β-cell function early in the course of therapy for type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 95, 4206–4216 [DOI] [PubMed] [Google Scholar]
- 5. Bonner-Weir S., Li W. C., Ouziel-Yahalom L., Guo L., Weir G. C., Sharma A. (2010) β-Cell growth and regeneration. Replication is only part of the story. Diabetes 59, 2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sorenson R. L., Brelje T. C. (1997) Adaptation of islets of Langerhans to pregnancy. β-Cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm. Metab. Res. 29, 301–307 [DOI] [PubMed] [Google Scholar]
- 7. Dor Y., Brown J., Martinez O. I., Melton D. A. (2004) Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 [DOI] [PubMed] [Google Scholar]
- 8. Xu X., D'Hoker J., Stangé G., Bonné S., De Leu N., Xiao X., Van de Casteele M., Mellitzer G., Ling Z., Pipeleers D., Bouwens L., Scharfmann R., Gradwohl G., Heimberg H. (2008) β-Cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132, 197–207 [DOI] [PubMed] [Google Scholar]
- 9. Li W. C., Rukstalis J. M., Nishimura W., Tchipashvili V., Habener J. F., Sharma A., Bonner-Weir S. (2010) Activation of pancreatic duct-derived progenitor cells during pancreas regeneration in adult rats. J. Cell Sci. 123, 2792–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lipsett M., Finegood D. T. (2002) β-Cell neogenesis during prolonged hyperglycemia in rats. Diabetes 51, 1834–1841 [DOI] [PubMed] [Google Scholar]
- 11. Minami K., Okuno M., Miyawaki K., Okumachi A., Ishizaki K., Oyama K., Kawaguchi M., Ishizuka N., Iwanaga T., Seino S. (2005) Lineage tracing and characterization of insulin-secreting cells generated from adult pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 102, 15116–15121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Desai B. M., Oliver-Krasinski J., De Leon D. D., Farzad C., Hong N., Leach S. D., Stoffers D. A. (2007) Preexisting pancreatic acinar cells contribute to acinar cell, but not islet β-cell, regeneration. J. Clin. Invest. 117, 971–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thorel F., Népote V., Avril I., Kohno K., Desgraz R., Chera S., Herrera P. L. (2010) Conversion of adult pancreatic α-cells to β-cells after extreme β-cell loss. Nature 464, 1149–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shapiro A. M., Lakey J. R., Ryan E. A., Korbutt G. S., Toth E., Warnock G. L., Kneteman N. M., Rajotte R. V. (2000) Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 343, 230–238 [DOI] [PubMed] [Google Scholar]
- 15. Butler P. C., Meier J. J., Butler A. E., Bhushan A. (2007) The replication of beta cells in normal physiology, in disease and for therapy. Nat. Clin. Pract. Endocrinol. Metab. 3, 758–768 [DOI] [PubMed] [Google Scholar]
- 16. Van Assche F. A., Aerts L., De Prins F. (1978) A morphological study of the endocrine pancreas in human pregnancy. Br. J. Obstet. Gynaecol. 85, 818–820 [DOI] [PubMed] [Google Scholar]
- 17. Rieck S., Kaestner K. H. (2010) Expansion of β-cell mass in response to pregnancy. Trends Endocrinol. Metab. 21, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bouwens L., Rooman I. (2005) Regulation of pancreatic β-cell mass. Physiol. Rev. 85, 1255–1270 [DOI] [PubMed] [Google Scholar]
- 19. Assmann A., Hinault C., Kulkarni R. N. (2009) Growth factor control of pancreatic islet regeneration and function. Pediatr. Diabetes 10, 14–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishii I., Fukushima N., Ye X., Chun J. (2004) Lysophospholipid receptors. Signaling and biology. Annu. Rev. Biochem. 73, 321–354 [DOI] [PubMed] [Google Scholar]
- 21. Gardell S. E., Dubin A. E., Chun J. (2006) Emerging medicinal roles for lysophospholipid signaling. Trends Mol. Med. 12, 65–75 [DOI] [PubMed] [Google Scholar]
- 22. Tokumura A., Kanaya Y., Miyake M., Yamano S., Irahara M., Fukuzawa K. (2002) Increased production of bioactive lysophosphatidic acid by serum lysophospholipase D in human pregnancy. Biol. Reprod. 67, 1386–1392 [DOI] [PubMed] [Google Scholar]
- 23. Samad F., Hester K. D., Yang G., Hannun Y. A., Bielawski J. (2006) Altered adipose and plasma sphingolipid metabolism in obesity. A potential mechanism for cardiovascular and metabolic risk. Diabetes 55, 2579–2587 [DOI] [PubMed] [Google Scholar]
- 24. Leiter E. H., Chapman H. D., Coleman D. L. (1989) The influence of genetic background on the expression of mutations at the diabetes locus in the mouse. V. Interaction between the db gene and hepatic sex steroid sulfotransferases correlates with gender-dependent susceptibility to hyperglycemia. Endocrinology 124, 912–922 [DOI] [PubMed] [Google Scholar]
- 25. Brinkmann V., Billich A., Baumruker T., Heining P., Schmouder R., Francis G., Aradhye S., Burtin P. (2010) Fingolimod (FTY720), discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 9, 883–897 [DOI] [PubMed] [Google Scholar]
- 26. Cohen J. A., Barkhof F., Comi G., Hartung H. P., Khatri B. O., Montalban X., Pelletier J., Capra R., Gallo P., Izquierdo G., Tiel-Wilck K., de Vera A., Jin J., Stites T., Wu S., Aradhye S., Kappos L. (2010) Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 362, 402–415 [DOI] [PubMed] [Google Scholar]
- 27. Brinkmann V., Davis M. D., Heise C. E., Albert R., Cottens S., Hof R., Bruns C., Prieschl E., Baumruker T., Hiestand P., Foster C. A., Zollinger M., Lynch K. R. (2002) The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 277, 21453–21457 [DOI] [PubMed] [Google Scholar]
- 28. Verzijl D., Peters S. L., Alewijnse A. E. (2010) Sphingosine 1-phosphate receptors. Zooming in on ligand-induced intracellular trafficking and its functional implications. Mol. Cells 29, 99–104 [DOI] [PubMed] [Google Scholar]
- 29. Song K., Zhang X., Zhao C., Ang N. T., Ma Z. A. (2005) Inhibition of Ca2+-independent phospholipase A2 results in insufficient insulin secretion and impaired glucose tolerance. Mol. Endocrinol. 19, 504–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao Z., Zhao C., Zhang X. H., Zheng F., Cai W., Vlassara H., Ma Z. A. (2009) Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 150, 2569–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao Z., Zhang X., Zhao C., Choi J., Shi J., Song K., Turk J., Ma Z. A. (2010) Protection of pancreatic β-cells by group VIA phospholipase A2-mediated repair of mitochondrial membrane peroxidation. Endocrinology 151, 3038–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang X. H., Zhao C., Ma Z. A. (2007) The increase of cell membranous phosphatidylcholines containing polyunsaturated fatty acid residues induces phosphorylation of p53 through activation of ATR. J. Cell Sci. 120, 4134–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang X. H., Zhao C., Seleznev K., Song K., Manfredi J. J., Ma Z. A. (2006) Disruption of G1 phase phospholipid turnover by inhibition of Ca2+-independent phospholipase A2 induces a p53-dependent cell-cycle arrest in G1 phase. J. Cell Sci. 119, 1005–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seleznev K., Zhao C., Zhang X. H., Song K., Ma Z. A. (2006) Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J. Biol. Chem. 281, 22275–22288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Truong W., Emamaullee J. A., Merani S., Anderson C. C., James Shapiro A. M. (2007) Human islet function is not impaired by the sphingosine 1-phosphate receptor modulator FTY720. Am. J. Transplant 7, 2031–2038 [DOI] [PubMed] [Google Scholar]
- 36. Laychock S. G., Sessanna S. M., Lin M. H., Mastrandrea L. D. (2006) Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet β-cells. Endocrinology 147, 4705–4712 [DOI] [PubMed] [Google Scholar]
- 37. Lauber K., Blumenthal S. G., Waibel M., Wesselborg S. (2004) Clearance of apoptotic cells. Getting rid of the corpses. Mol. Cell 14, 277–287 [DOI] [PubMed] [Google Scholar]
- 38. Potteck H., Nieuwenhuis B., Lüth A., van der Giet M., Kleuser B. (2010) Phosphorylation of the immunomodulator FTY720 inhibits programmed cell death of fibroblasts via the S1P3 receptor subtype and Bcl-2 activation. Cell Physiol. Biochem. 26, 67–78 [DOI] [PubMed] [Google Scholar]
- 39. Scholzen T., Gerdes J. (2000) The Ki67 protein. From the known and the unknown. J. Cell Physiol. 182, 311–322 [DOI] [PubMed] [Google Scholar]
- 40. Krishnamurthy J., Ramsey M. R., Ligon K. L., Torrice C., Koh A., Bonner-Weir S., Sharpless N. E. (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443, 453–457 [DOI] [PubMed] [Google Scholar]
- 41. Dhawan S., Tschen S. I., Bhushan A. (2009) Bmi-1 regulates the Ink4a/Arf locus to control pancreatic β-cell proliferation. Genes Dev. 23, 906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pateras I. S., Apostolopoulou K., Niforou K., Kotsinas A., Gorgoulis V. G. (2009) p57KIP2, “Kip”ing the cell under control. Mol. Cancer Res. 7, 1902–1919 [DOI] [PubMed] [Google Scholar]
- 43. Jongsma M., Hendriks-Balk M. C., Michel M. C., Peters S. L., Alewijnse A. E. (2006) BML-241 fails to display selective antagonism at the sphingosine 1-phosphate receptor, S1P(3). Br. J. Pharmacol. 149, 277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huwiler A., Pfeilschifter J. (2008) New players on the center stage. Sphingosine 1-phosphate and its receptors as drug targets. Biochem. Pharmacol. 75, 1893–1900 [DOI] [PubMed] [Google Scholar]
- 45. Albert R., Hinterding K., Brinkmann V., Guerini D., Müller-Hartwieg C., Knecht H., Simeon C., Streiff M., Wagner T., Welzenbach K., Zécri F., Zollinger M., Cooke N., Francotte E. (2005) Novel immunomodulator FTY720 is phosphorylated in rats and humans to form a single stereoisomer. Identification, chemical proof, and biological characterization of the biologically active species and its enantiomer. J. Med. Chem. 48, 5373–5377 [DOI] [PubMed] [Google Scholar]
- 46. Radeff-Huang J., Seasholtz T. M., Matteo R. G., Brown J. H. (2004) G protein-mediated signaling pathways in lysophospholipid induced cell proliferation and survival. J. Cell Biochem. 92, 949–966 [DOI] [PubMed] [Google Scholar]
- 47. Guillermet-Guibert J., Bjorklof K., Salpekar A., Gonella C., Ramadani F., Bilancio A., Meek S., Smith A. J., Okkenhaug K., Vanhaesebroeck B. (2008) The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. U.S.A. 105, 8292–8297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vanhaesebroeck B., Guillermet-Guibert J., Graupera M., Bilanges B. (2010) The emerging mechanisms of isoform-specific PI3K signaling. Nat. Rev. Mol. Cell Biol. 11, 329–341 [DOI] [PubMed] [Google Scholar]
- 49. Ashizawa S., Brunicardi F. C., Wang X. P. (2004) PDX-1 and the pancreas. Pancreas 28, 109–120 [DOI] [PubMed] [Google Scholar]
- 50. Watanabe H., Saito H., Nishimura H., Ueda J., Evers B. M. (2008) Activation of phosphatidylinositol 3-kinase regulates pancreatic duodenal homeobox-1 in duct cells during pancreatic regeneration. Pancreas 36, 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Watanabe H., Saito H., Ueda J., Evers B. M. (2008) Regulation of pancreatic duct cell differentiation by phosphatidylinositol 3-kinase. Biochem. Biophys. Res. Commun. 370, 33–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wymann M. P., Bulgarelli-Leva G., Zvelebil M. J., Pirola L., Vanhaesebroeck B., Waterfield M. D., Panayotou G. (1996) Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol. 16, 1722–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Strader C. R., Pearce C. J., Oberlies N. H. (2011) Fingolimod (FTY720), a recently approved multiple sclerosis drug based on a fungal secondary metabolite. J. Nat. Prod. 74, 900–907 [DOI] [PubMed] [Google Scholar]
- 54. Holland W. L., Miller R. A., Wang Z. V., Sun K., Barth B. M., Bui H. H., Davis K. E., Bikman B. T., Halberg N., Rutkowski J. M., Wade M. R., Tenorio V. M., Kuo M. S., Brozinick J. T., Zhang B. B., Birnbaum M. J., Summers S. A., Scherer P. E. (2011) Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 17, 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maceyka M., Harikumar K. B., Milstien S., Spiegel S. (2012) Trends Cell Biol. 22, 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kassem S. A., Ariel I., Thornton P. S., Hussain K., Smith V., Lindley K. J., Aynsley-Green A., Glaser B. (2001) p57(KIP2) expression in normal islet cells and in hyperinsulinism of infancy. Diabetes 50, 2763–2769 [DOI] [PubMed] [Google Scholar]
- 57. Georgia S., Soliz R., Li M., Zhang P., Bhushan A. (2006) p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev. Biol. 298, 22–31 [DOI] [PubMed] [Google Scholar]
- 58. Rothschild G., Zhao X., Iavarone A., Lasorella A. (2006) E proteins and Id2 converge on p57Kip2 to regulate cell cycle in neural cells. Mol. Cell Biol. 26, 4351–4361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sicinska E., Aifantis I., Le Cam L., Swat W., Borowski C., Yu Q., Ferrando A. A., Levin S. D., Geng Y., von Boehmer H., Sicinski P. (2003) Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 4, 451–461 [DOI] [PubMed] [Google Scholar]
- 60. Levine Y. C., Li G. K., Michel T. (2007) Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J. Biol. Chem. 282, 20351–20364 [DOI] [PubMed] [Google Scholar]
- 61. Shen Y., Cai M., Xia W., Liu J., Zhang Q., Xie H., Wang C., Wang X., Zheng S. (2007) FTY720, a synthetic compound from Isaria sinclairii, inhibits proliferation and induces apoptosis in pancreatic cancer cells. Cancer Lett. 254, 288–297 [DOI] [PubMed] [Google Scholar]
- 62. Menge B. A., Tannapfel A., Belyaev O., Drescher R., Müller C., Uhl W., Schmidt W. E., Meier J. J. (2008) Partial pancreatectomy in adult humans does not provoke β-cell regeneration. Diabetes 57, 142–149 [DOI] [PubMed] [Google Scholar]
- 63. Fiaschi-Taesch N. M., Salim F., Kleinberger J., Troxell R., Cozar-Castellano I., Selk K., Cherok E., Takane K. K., Scott D. K., Stewart A. F. (2010) Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 59, 1926–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Georgia S., Bhushan A. (2004) Beta cell replication is the primary mechanism for maintaining postnatal β-cell mass. J. Clin. Invest. 114, 963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.