

Background: CXCR2 plays an important role in various neutrophil-dominant inflammatory diseases.
Results: A macromolecular signaling complex containing CXCR2, NHERF1, and phospholipase C (PLC)-β2 regulates neutrophil calcium mobilization, chemotaxis, and transepithelial migration.
Conclusion: CXCR2·NHERF1·PLC-β2 macromolecular signaling complex is critical to neutrophil functions.
Significance: CXCR2 macromolecular complex might be a potential therapeutic target for neutrophil infiltration-associated inflammatory diseases.
Keywords: Chemotaxis, Inflammation, Neutrophil, Protein-Protein Interactions, Scaffold Proteins, Signal Transduction, CXCR2, PDZ
Abstract
Inflammation plays an important role in a wide range of human diseases such as ischemia-reperfusion injury, arteriosclerosis, cystic fibrosis, inflammatory bowel disease, etc. Neutrophilic accumulation in the inflamed tissues is an essential component of normal host defense against infection, but uncontrolled neutrophilic infiltration can cause progressive damage to the tissue epithelium. The CXC chemokine receptor CXCR2 and its specific ligands have been reported to play critical roles in the pathophysiology of various inflammatory diseases. However, it is unclear how CXCR2 is coupled specifically to its downstream signaling molecules and modulates cellular functions of neutrophils. Here we show that the PDZ scaffold protein NHERF1 couples CXCR2 to its downstream effector phospholipase C (PLC)-β2, forming a macromolecular complex, through a PDZ-based interaction. We assembled a macromolecular complex of CXCR2·NHERF1·PLC-β2 in vitro, and we also detected such a complex in neutrophils by co-immunoprecipitation. We further observed that the CXCR2-containing macromolecular complex is critical for the CXCR2-mediated intracellular calcium mobilization and the resultant migration and infiltration of neutrophils, as disrupting the complex with a cell permeant CXCR2-specific peptide (containing the PDZ motif) inhibited intracellular calcium mobilization, chemotaxis, and transepithelial migration of neutrophils. Taken together, our data demonstrate a critical role of the PDZ-dependent CXCR2 macromolecular signaling complex in regulating neutrophil functions and suggest that targeting the CXCR2 multiprotein complex may represent a novel therapeutic strategy for certain inflammatory diseases.
Introduction
Migration of leukocytes (such as monocytes and neutrophils) in response to various chemotactic stimuli is critical to maintaining host defense, but uncontrolled cellular infiltration of circulating leukocytes into tissues or organs can lead to a variety of chronic inflammatory conditions in the heart, vessels, lung, intestine, skin, etc. (1). Neutrophil-dominated inflammation is characterized by increased levels of interleukin-8 (IL-8/CXCL8) and neutrophil elastase (2). IL-8 is the prototypical member of the CXC subfamily of chemokines, which function as chemoattractants and activators of polymorphonuclear leukocytes (PMNs)4 (or neutrophils) (3). It is secreted by stimulated macrophages and other cells (such as endothelial or epithelial cells), and activates neutrophils by binding to two G-protein coupled receptors, CXCR1 (4) and CXCR2 (5). Both CXCR1 and CXCR2 are the major chemokine receptors on neutrophils, sharing 77% identity at the amino acid sequence level (3). CXCR2 appears to be the predominant receptor mediating IL-8 chemotactic response (6). Inhibition of CXCR2 is sufficient to prevent neutrophil margination and chemotaxis mediated by IL-8, suggesting that CXCR1 does not play a major role in neutrophil migration (7). CXCR2 couples to the pertussis toxin-sensitive Gi proteins to stimulate phosphatidylinositide-specific phospholipase C (PLC) activities (8). PLC molecules can be divided into six families of isozymes: β, γ, δ, ϵ, η, and ζ (9). The β family consists of four isoforms, PLC-β1 to PLC-β4. PLC-β2 has been detected primarily in hematopoietic cells, whereas PLC-β1 and PLC-β3 are found in a wide range of cells and tissues (10). PLC-β4 is predominantly expressed in certain neuronal cells (11, 12). Activation of PLC-β results in hydrolysis of the lipid phosphatidylinositol 4,5-bisphosphate, generating diacylglycerol, which activates PKC isoforms, and inositol 1,3,4-triphosphate, which releases calcium from intracellular stores. Stimulation of PMNs with chemoattractants (such as IL-8) results in increases in cytosolic calcium due to a combination of intracellular calcium release mediated by inositol 1,3,4-triphosphate and an influx of extracellular calcium. Both PLC-β2 and -β3 are expressed in PMNs and are the only isoforms responsible in murine PMNs for chemoattractant-stimulated PLC activity, but PLC-β2 is the major PLC isoform in neutrophils (13–15).
PDZ domains are modular protein interaction domains that form peptide-binding clefts and typically mediate interactions with the carboxyl termini of other proteins that terminate in consensus binding motifs (referred to as PDZ motif) (16–18). The PDZ domain-containing proteins (also referred to as PDZ scaffold proteins) that preferentially accumulate at the membranes include Na+/H+ exchanger regulatory factor-1 (NHERF1), NHERF2, and PDZ domain containing 1 (PDZK1), etc. NHERF1 and NHERF2 contain two PDZ domains and a carboxyl-terminal domain that mediates association with MERM proteins (merlin, ezrin, radixin, moesin), whereas PDZK1 contains four tandem PDZ domains (16, 17). PDZ scaffold proteins have been shown to be involved in the regulation of PLC-β isoforms (19, 20). All PLC-β isoforms contain consensus PDZ motifs, -X(S/T)X(L/V)-COOH (X represents any amino acid), at their carboxyl termini (21). The C terminus of PLC-β3, but not other PLC-β isoforms, was reported to specifically interact with the PDZ domains of NHERF2 in mouse small intestine (19), and Shank2, a PDZ protein present in the postsynaptic density in neuronal cells (22), whereas the PLC-β1 C-tail reportedly interacts with PAR-3, a PDZ scaffold protein in HeLa cells (23). Most recently, it was reported that in NHERF1 knock-out mice, PLC-β3 was down-regulated in mouse jejuna villus cells (24). Therefore, the specific interactions of different PLC-β isoforms with distinct PDZ proteins may be responsible for the specificity and diversity of agonist-induced intracellular signaling. Similar to PLC-β isoforms, both human and murine CXCR2 possess a consensus PDZ motif at their carboxyl termini, and the PDZ motif has been reported to modulate post-endocytic sorting and cellular chemotaxis in CXCR2-overexpressing HEK293 cells (25). A variety of PDZ scaffold proteins have been documented to nucleate the formation of compartmentalized multiprotein complexes that are critical for efficient and specific cellular signaling (26–32). Therefore, the PDZ motif of CXCR2 can, theoretically, mediate potential interactions with certain PDZ scaffold proteins. This may cluster CXCR2 with other relevant signaling molecules into multiprotein macromolecular signaling complexes. However, the molecular mechanisms that underlie the formation and/or regulation of the potential CXCR2 macromolecular complex and the functional significance of the CXCR2 complex in neutrophil mobilization, recruitment, and transmigration into various tissues during inflammatory diseases have not been determined.
In our present work, using a series of molecular and biochemical techniques and cellular functional studies, we sought to characterize a CXCR2 macromolecular signaling complex and define the critical role this complex might play in regulating neutrophil intracellular signaling and functional activities. Our data show that there is a physical coupling between CXCR2 and its downstream effector enzyme PLC-β2, which is mediated preferentially by the PDZ scaffold protein NHERF1. Moreover, we demonstrated that disturbing the CXCR2·NHERF1·PLC-β2 macromolecular complex attenuated CXCR2-mediated intracellular calcium signals in neutrophils and significantly suppressed neutrophilic chemotaxis and transepithelial migration, implicating a functional relevance of the CXCR2 macromolecular signaling complex in various neutrophil infiltration associated inflammatory diseases (such as inflammatory bowel diseases, chronic lung inflammation, atherosclerosis, etc.).
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Anti-human and murine CXCR2, PLC-β1, -β2, and -β3 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-NHERF1 polyclonal antibody was from Sigma, and mouse anti-NHERF1 monoclonal antibody was from Santa Cruz. Anti-HA HRP and anti-FLAG HRP were obtained from Sigma. Lipofectamine 2000, Hanks' buffered salt solution (HBSS), Fura-2, and the cell culture media and fetal bovine serum (FBS) were procured from Invitrogen. ChariotTM peptide/protein delivery reagent was purchased from Active Motif (Carlsbad, CA). Chemokines IL-8/CXCL8, growth-related oncogene α (GROα/CXCL1), macrophage inflammatory protein 2 (MIP-2/murine CXCL2), and N-formyl-methionine-leucine-phenylalanine (fMLP) were obtained from ProSpec (East Brunswick, NJ). The human and murine CXCR2 C-tail peptides (biotin-conjugate at N terminus): WT (biotin-FVGSSSGHTSTTL for human CXCR2 C-tail; and Biotin-FVSSSSANTSTTL for mouse CXCR2 C-tail), PDZ motif deletion, ΔTTL or PDZ motif mutant, AAA, were synthesized by Genemed Synthesis, Inc. (San Antonio, TX).
Plasmids, Cloning, and Mutagenesis
The His-S-tagged fusion proteins for the full-length of CXCR2 or PLC-β2, or the C-terminal tail fragments of CXCR2 (last 45 amino acids; i.e. amino acids 316–360 for human CXCR2, and amino acids 315–359 for murine CXCR2) or human PLC-β2 (last 100 amino acids, i.e. amino acids 1086–1185) were generated by PCR cloning into pTriEx-4 or pET30 vectors (Novagen). The various C-tail mutants (PDZ motif mutation or deletion) for either CXCR2 or PLC-β2 were generated using the QuikChangeTM Site-directed Mutagenesis kit (Stratagene) and also cloned into pTriEx-4 or pET30 vectors. The fusion proteins were purified using Talon beads (binding to His tag), and eluted with 200 mm imidazole. The imidazole-eluted affinity-purified His-S-tagged CXCR2 or PLC-β2 fusion proteins (full-length and/or C-terminal tail fragments) were used in the subsequent biochemical assays (such as pulldown, pairwise binding, and macromolecular complex assembly).
Cell Culture and Transfection
The HL-60 cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA) and maintained in Iscove's modified Dulbecco's medium (Invitrogen) supplemented with 10% FBS, and penicillin/streptomycin at 37 ºC with 5% CO2. HL-60 cells were differentiated into the granulocyte lineage with 1.2% Me2SO in Iscove's modified Dulbecco's medium with 10% FBS for 5–7 days as described (33). The differentiated HL-60 (dHL-60) cells were transfected with pTriEx-4 vector encoding CXCR2 C-tail fragments (WT, PDZ motif mutation AAA, or PDZ motif deletion ΔTTL) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After transfection, the dHL-60 cells were used for Ca2+ mobilization, chemotaxis, or transmigration assays. The HEK293 cells and HT-29 human colonic epithelial cells were purchased from ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% FBS as described before (31). HEK293 cells were transfected using Lipofectamine 2000 with HA-tagged human CXCR2, murine CXCR2, and FLAG-tagged PLC-β1, -β2, -β3, and -β4, respectively, for various biochemical assays.
Human Neutrophil Isolation from Buffy Coats
Briefly, neutrophils from buffy coats (purchased from LifeBlood Inc.) of citrated human peripheral blood collected from healthy donors were isolated by dextran sedimentation followed by density gradient centrifugation in Histopaque (Sigma) as described previously (34). Contaminating red blood cells were lysed by hypotonic shock with 0.2% NaCl. The purity and viability of isolated neutrophils was assessed by trypan blue dye exclusion, and the viability of isolated neutrophils was routinely found to be >98%. Neutrophils were used immediately after isolation for all assays.
Murine Neutrophil Isolation from Mouse Bone Marrow
Isolation of murine neutrophils from bone marrow was carried out as reported previously (35). Briefly, the femurs and tibias were removed from euthanized mice and the bone marrow cells were flushed out of the bones with ice-cold Ca2+- and Mg2+-free HBSS (HBSS−). Bone marrow cells were collected by centrifugation at 800 × g for 5 min, and then resuspended in 3 ml of HBSS−. The cells were layered over a discontinuous 3-layer Percoll (Amersham Biosciences) gradient (75, 67, and 52%) and subjected to centrifugation at 1,060 × g for 30 min at 22–24 ºC. The lowest band between 75 and 67% (the 75/67% interface) was then collected as the neutrophil fraction and washed twice with HBSS−. Any remaining red blood cells were eliminated by hypotonic lysis, and the purity of the neutrophils was typically ≥90% as assessed by crystal violet staining.
Pulldown Assay
Freshly isolated human or murine neutrophils, dHL60 cells, or HEK293 cells overexpressing various constructs (3HA-tagged human CXCR2, murine CXCR2, or FLAG-tagged PLC-β1, -β2, -β3, -β4) were used for the GST pulldown assays as reported (30, 31). In brief, the cells were lysed in lysis buffer (PBS, 0.2% Triton) supplemented with a mixture of protease inhibitors (containing 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin, and 1 μg/ml of pepstatin) and phosphatase inhibitor mixture (Sigma), and the clear supernatant (16,000 × g, 15 min) was mixed with various GST-PDZ fusion proteins (GST-NHERF1, GST-NHERF2, or GST-PDZK1) or GST alone at 4 ºC for 3 h. The complex was pulled down by glutathione-agarose beads (BD Biosciences) at 4 ºC for 1 h, washed three times with lysis buffer, and eluted in Laemmli sample buffer containing β-mercaptoethanol. The eluents were separated by SDS-PAGE and immunoblotted with anti-HA, CXCR2, or PLC-β1, -β2, -β3, or anti-FLAG (for PLC-β4) antibodies, and the blots were visualized using a BioSpectrum 500 Imaging system (UVP).
Pairwise Binding
Purified GST-NHERF1 was mixed with purified His-S-CXCR2 C-tail fragments (WT, or various mutants on PDZ motif, ATA, AAA, ΔTTL), or His-S-PLC-β2 C-tail fragments (WT, or various mutants on PDZ motif, ARA, AAA, ΔSRL) in binding buffer (PBS, 0.2% Triton) supplemented with a mixture of protease inhibitors at 22–24 ºC for 1 h. Next the mixtures were incubated with S-agarose beads (Novagen) for 2 h. The beads were washed three times with binding buffer and eluted with Laemmli sample buffer. The eluents were separated by SDS-PAGE and immunoblotted with anti-NHERF1 antibody.
Macromolecular Complex Assembly
Purified His-S-tagged PLC-β2 C-tail (last 100 amino acids at C terminus containing the PDZ motif) or His-S-murine CXCR2 C-tail (last 45 amino acids at the C terminus containing the PDZ motif) was mixed with GST-PDZ scaffold proteins (or GST alone) in 200 μl of binding buffer (PBS, 0.2% Triton, supplemented with protease inhibitors), and the complex was pulled down with S-protein-agarose. This step is also referred to as pairwise binding as described above. The dimeric complex was then mixed with HEK293 cell lysates overexpressing 3HA-CXCR2 or full-length PLC-β2 for 3 h at 4 ºC, and washed extensively with lysis buffer. The bound proteins were then eluted and immunoblotted using anti-HA (for CXCR2) or PLC-β2 antibodies.
Co-immunoprecipitation
Fresh cells (dHL-60 or murine bone marrow neutrophils) were cross-linked with 1 mm dithiobis(succinimidyl propionate) as described before (36). Thereafter, the cells were solubilized in PBS, 0.2% Triton, and cleared lysates (16,000 × g, 15 min) were processed for co-immunoprecipitation and immunoblotting as described before (30, 31) using the respective antibodies against PLC-β2, CXCR2, or NHERF1. A Co-immunoprecipitation Kit (Pierce) was used to immobilize the anti-CXCR2 IgG to the resin and the co-precipitated protein complex was eluted with Laemmli sample buffer before being subjected to immunoblotting and probed for PLC-β2 and NHERF1. The same membrane was stripped using RestoreTM Plus Western blot Stripping Buffer (Thermo Scientific) and reprobed for CXCR2. In some experiments, anti-PLC-β2 IgG was used to immunoprecipitate the complex and the co-precipitated protein complex was resolved by SDS-PAGE and probed for CXCR2 and NHERF1. The signal was detected by ECL (Amersham Biosciences) or SuperSignal West Femto Substrate (Thermo Scientific) and images were captured using a BioSpectrum 500 Imaging system (UVP).
CXCR2 Peptide in Vitro Competitive Binding
In brief, affinity-purified HA-tagged human CXCR2 (37.5, 75, and 150 ng) was immobilized on the nitrocellulose membrane by spotting, and the membrane was blocked with TBS, 0.1% Tween supplemented with 1 μg/ml of BSA (TBST-BSA) for 1 h at 22 ºC. During this time, 10 μg of His-S-tagged NHERF1 was mixed with TBST-BSA in the presence or absence of 25 μg of human CXCR2 C-tail WT peptide for 1 h at 22–24 ºC, and then the mixture was added to the membrane and incubated for 12 h at 4 ºC. The membrane was washed extensively and immunoblotted with S-HRP (Novagen), which detects the S-tag within the His-S-NHERF1 fusion proteins on Western blot. For the murine CXCR2 peptide interference study, mouse CXCR2 C-tail WT peptide (0–1.8 μm) was mixed with GST-NHERF1 for 1 h at 22–24 ºC. Then His-S-tagged murine CXCR2 C-tail (last 45 amino acids) was added to the mixture and continued to incubate at 22–24 ºC for another 1 h. Next, S-agarose beads were added to the mixture for anadditional 2 h before the protein complex was eluted from the beads and immunoblotted with anti-NHERF1 antibody.
Measurement of Cytosolic Ca2+ Concentration
CXCR2 C-tail peptides (WT and ΔTTL) were mixed with the Chariot peptide delivery reagent (Active Motif; total volume of 400 μl) at 22 ºC for 30 min as reported (30, 31). The Chariot-peptide complex was then incubated with freshly isolated neutrophils (or dHL-60 cells) with gentle agitation at 37 ºC for 1 h. The peptide-delivered neutrophils (1 × 106) were labeled with 1 μm Fura-2 (Invitrogen) at 22 ºC for 30 min. The cells were incubated for another 30 min after being washed twice with HBSS− (Ca2+ and Mg2+ free) to allow complete de-esterification. Cells were resuspended in regular HBSS buffer at 106 cells/ml, and a total of 2 ml of the cell suspensions were placed in a continuously stirred cuvette at 37 ºC in a PTI fluorescence spectrophotometer (Photon Technology International). The fluorescence was monitored at excitation wavelengths of 340 and 380 nm, and an emission wavelength of 510 nm, and the intracellular calcium concentration ([Ca2+]i) was expressed as the fluorescence ratio at 340 and 380 nm (R). Fura-2 AM-loaded cells were stimulated with various chemoattractants (50–100 ng/ml of MIP-2 or IL-8, 0.5–1 μm fMLP). At the end of each experiment, cells were lysed with 0.1% Triton X-100 to obtain maximal fluorescence ratio (Rmax), and then Ca2+ was chelated with 4.5 mm EGTA (pH > 8.0) to determine the minimum fluorescence ratio (Rmin). The intracellular calcium levels (in nm) were calculated as described previously (37).
Neutrophil Chemotaxic Migration
Chemotaxic migration of neutrophil and dHL-60 cells was determined using 24-well Transwell chambers (Corning, Acton, MA) with 3-μm pore size membranes as reported (35). The Transwell filters were pre-coated with collagen overnight. Various chemoattractants (in 600 μl), including human IL-8, GROα, mouse MIP-2 (50–100 ng/ml), and fMLP (1 μm), were added to the lower chambers. Freshly isolated human and murine neutrophils were delivered with various CXCR2 C-tail peptides (WT or ΔTTL) using the Chariot peptide-delivery system as reported (30, 31), and then the cells (1 × 105 cells in 100 μl) were loaded to the upper compartment, and the chambers were incubated for 1 (for neutrophils) or 2 h (for dHL-60) at 37 ºC and 5% CO2. After incubation, the amount of migrated neutrophils in the bottom chamber of the Transwell were counted with a hemocytometer. The chemotaxic migration was expressed as the percentage of cells recovered from the bottom well to the cells applied in the top well.
Differentiated HL-60 Cell Transepithelial Migration
Each Transwell filter was coated with type I collagen and dried overnight (38). HT-29 cells (1 × 106) were seeded to the underside of each Transwell filter and incubated at 37 ºC overnight for adhesion as reported previously (38, 39). Thereafter, the filters were placed into a 24-well plate with fresh medium and the transepithelial resistance was monitored every day as HT-29 cells were becoming confluent. The dHL-60 cells were delivered with various CXCR2 C-tail peptides (WT, mutant AAA, and ΔTTL) using the Chariot peptide delivery system (30, 31). Then both sides of the Transwell filters were washed by regular HBSS and allowed to recover in HBSS at 37 ºC for 45 min. The peptide-delivered dHL-60 cells (3 × 106) were added to the upper well in 150 μl of PBS, and transmigration was initiated by applying various chemoattractants to the lower well at a concentration of 50–100 ng/ml (for IL-8 and GROα) or 1 μm (for fMLP) in 500 μl of HBSS. Cells were allowed to migrate for 2 h. Transmigrated dHL-60 cells were quantified indirectly by measuring the myeloperoxidase (MPO) activity as reported (38). We also monitored the effect of delivery of various CXCR2 peptides on MPO activity of dHL-60 cells and did not find a significant change of MPO activity caused by the different peptide delivery system (data not shown).
Statistical Analysis
Data are expressed as mean ± S.E. of at least three independent experiments. Statistical significance of the experimental data were determined by Student's t test and one-way analysis of variance. A value of p < 0.05 was considered to be significant.
RESULTS
CXCR2 Physically Interacts with PDZ Scaffold Proteins
Both human and murine CXCR2 possess consensus PDZ motifs (-STTL-COOH) at their C termini, which could potentially bind certain PDZ scaffold proteins through a PDZ-based interaction. Using a GST pulldown assay, we observed that CXCR2 interacted with several membrane-associated PDZ scaffold proteins (NHERF1, NHERF2, and/or PDZK1) (Fig. 1). The human CXCR2 overexpressed in HEK293 cells was pulled down by all three GST-PDZ scaffold proteins, but not by GST alone (Fig. 1A). However, the murine CXCR2 was shown to be pulled-down by NHERF1 and NHERF2, but not by PDZK1 (Fig. 1B).
FIGURE 1.

CXCR2 preferentially interacts with NHERF1 in neutrophils. A, HA-tagged human CXCR2 (overexpressed in HEK293 cells) was pulled down by PDZ scaffold proteins (NHERF1, NHERF2, and PDZK1). The membrane was blotted with anti-HA monoclonal antibody. B, His-S-tagged murine CXCR2 (overexpressed in HEK293 cells) was pulled down by the indicated PDZ scaffold proteins. The membrane was blotted with anti-mouse CXCR2 monoclonal antibody. C, NHERF1 and NHERF2 bound to endogenous CXCR2 from human neutrophils. The membrane was immunoblotted with anti-human CXCR2 monoclonal antibody. D, NHERF1 bound to endogenous CXCR2 from murine bone marrow neutrophils. Cell lysates of HEK293 cells that overexpressed 3HA-murine CXCR2 were loaded as a positive control. The membrane was immunoblotted with anti-murine CXCR2 monoclonal antibody. E, NHERF1 and NHERF2 bound to endogenous CXCR2 from neutrophil-like cells, dHL-60 cells. The membrane was immunoblotted with anti-human CXCR2 monoclonal antibody.
CXCR2 in Neutrophils Preferentially Interacts with NHERF1
To determine whether endogenous CXCR2 in neutrophils interacts with PDZ scaffold proteins, we performed a pulldown assay with the freshly isolated neutrophils. Human neutrophils were lysed with PBS, 0.2% Triton X-100, and the clear supernatant was mixed with various GST-PDZ scaffold proteins as described under “Experimental Procedures.” As shown in Fig. 1C, endogenous CXCR2 in human neutrophils binds to NHERF1 and NHERF2, but not to PDZK1, and human CXCR2 interacts with NHERF1 with a much higher affinity (Fig. 1C). In addition, we observed an interaction between NHERF1 and endogenous CXCR2 from mouse bone marrow-derived neutrophils; however, murine CXCR2 did not bind to NHERF2, nor to PDZK1 (Fig. 1D).
We also performed pulldown experiments with a human promyelocytic leukemia cell line, HL-60 (40). HL-60 cells were induced to differentiate into neutrophil-like cells by exposure to Me2SO for 5–7 days (41). These cells have proven to be useful for studying neutrophil functions such as oxidative burst, adhesion, chemotaxis, and migration. dHL-60 cells express CXCR2 and respond to CXCL8 in calcium mobilization (data not shown) and chemotaxic migration (see Fig. 7B) in our studies. Similar to what was observed with human neutrophils, CXCR2 preferentially binds to NHERF1 in dHL-60 cells (Fig. 1E).
FIGURE 7.

Disrupting the CXCR2 macromolecular complex attenuates the CXCR2 ligand-induced neutrophil chemotaxic migration. A, chemotaxic migration of mouse PMNs in response to CXCR2 ligands (MIP-2 and IL-8; both 50 ng/ml) pretreated with or without various CXCR2 C-tail peptides. *, p < 0.05; **, p < 0.01, compared with the control (no peptide). B, migration of dHL-60 cells (human PMN-like granulocytes) pre-delivered with various CXCR2 C-tail peptides when the cells were stimulated by IL-8 (CXCL8; 50 ng/ml), GROα (CXCL1; 50 ng/ml), or fMLP (1 μm). **, p < 0.01, compared with the control (no peptide).
PLC-β2 in Neutrophils Preferentially Interacts with NHERF1
CXCR2 couples to the pertussis toxin-sensitive Gi proteins to stimulate phosphatidylinositide-specific PLC activities. Similar to CXCR2, all human and murine PLC-β isoforms contain consensus PDZ motifs at their carboxyl termini (21). We therefore explored the potential interactions between β isoforms of PLC with the PDZ scaffold proteins that were shown to interact with CXCR2 in our studies. The constructs for all the four β isoforms of PLC (FLAG-tagged PLC-β1 to β4) were overexpressed in HEK293 cells, and we performed pulldown experiments using the various GST-PDZ fusion proteins used in above studies. We observed that PLC-β1, -β2, and -β3 overexpressed in HEK293 cells interact preferentially with NHERF1 (Fig. 2, A–C). There is also a very weak binding between NHERF2 and PLC-β1, -β2, and -β3 (Fig. 2, A–C). However, the overexpressed PLC-β4 in HEK293 cells was not pulled down by any of the three PDZ scaffold proteins (data not shown).
FIGURE 2.

PLC-β isoforms physically interact with PDZ scaffold proteins, whereas PLC-β2 in neutrophils preferentially interacts with NHERF1. A–C, HEK293 cells were overexpressed with FLAG-tagged PLC-β1 (A), PLC-β2 (B), and PLC-β3 (C), and the GST pulldown assays were performed with PDZ scaffold proteins as described under “Experimental Procedures.” The membranes were immunoblotted with anti-PLC-β1 (A), PLC-β2 (B), and PLC-β3 (C) monoclonal antibodies, respectively. Purified His-S-PLC-β1 (20 ng) was loaded as positive control for the PLC-β1 antibody (A). D and E, endogenous PLC-β2 from neutrophils freshly isolated from mouse bone marrow (D), or from neutrophil-like cells, dHL-60 cells (E), was pulled down by PDZ scaffold proteins. The membranes were immunoblotted with anti-PLC-β2 monoclonal antibody. Purified His-S-PLC-β2 was loaded as positive control for the PLC-β2 antibody.
Both PLC-β2 and -β3 are expressed in neutrophils and are the only isoforms responsible for chemoattractant-stimulated PLC activity, but PLC-β2 is the major PLC isoform in neutrophils (13–15). Therefore, we further examined the binding features of endogenous PLC-β2 in neutrophils with PDZ scaffold proteins. Pulldown experiments demonstrated that endogenous PLC-β2 from murine bone marrow neutrophils interacts with NHERF1; however, neither NHERF2 nor PDZK1 was found to bind to PLC-β2 (Fig. 2D). In neutrophil-like dHL-60 cells, PLC-β2 preferentially binds to NHERF1, although it also binds to both NHERF2 and PDZK1, but with a relatively very low affinity (Fig. 2E). Endogenous PLC-β2 in human neutrophils also preferentially binds to NHERF1 (data not shown).
The Interaction between NHERF1 and CXCR2 or PLC-β2 Is PDZ Motif-dependent
The data resulting from the pulldown studies presented above (Figs. 1 and 2) does not provide information whether the interactions between CXCR2 or PLC-β2 and NHERF1 are direct, as cell lysates contain large numbers of other proteins as well. To test if CXCR2 or PLC-β2 binds NHERF1 directly or by other intermediary proteins, and to test the PDZ motif dependence, we performed a pairwise binding assay that detects a direct interaction between purified proteins in vitro (29, 30, 36, 42). Purified His-S fusion proteins containing the C-tail fragments (last 45 amino acids) of CXCR2 WT, C-tail ΔTTL (with PDZ motif TTL deleted), or C-tail AAA or ATA (with PDZ motif TTL mutated to AAA or ATA) were mixed with GST-NHERF1, and the mixtures were pulled down by S-protein-agarose (the S-protein can specifically bind to the S-tag within the fusion proteins). The protein complex was immunoblotted with anti-NHERF1 antibody. We observed that the CXCR2 C-tail directly interacts with NHERF1 in a PDZ motif-dependent manner, as the interaction between NHERF1 and CXCR2 C-tail lacking the PDZ motif (ΔTTL) or CXCR2 C-tail with PDZ motif mutations (AAA or ATA) was remarkably reduced (Fig. 3A). Similar findings were also observed between NHERF1 and murine CXCR2 (Fig. 3B). We also explored the binding features between the NHERF1 and PLC-β2 C-tail (last 100 amino acids), and found that the interaction between NHERF1 and PLC-β2 was also direct and PDZ motif-dependent, as the PLC-β2 C-tail mutants (-ARA, -AAA, -ΔSRL) demonstrated significantly reduced binding with NHERF1 (Fig. 3C).
FIGURE 3.

The interaction between NHERF1 and CXCR2 or PLC-β2 is PDZ motif-dependent. A, pairwise binding between GST-NHERF1 and His-S-tagged human CXCR2 C-tail (containing the last 45 amino acids) WT, PDZ motif deletion (ΔTTL), or PDZ motif mutants (ATA and AAA). The complex was pulled down by S-protein-agarose and immunoblotted with anti-NHERF1 IgG. Purified GST or GST-NHERF1 (20 ng) were loaded as negative or positive control for the anti-NHERF1 antibody, respectively. B, pairwise binding between GST-NHERF1 and His-S-tagged mouse CXCR2 C-tail (containing last 45 amino acids) with (WT) or without PDZ motif (ΔTTL). The complex was pulled down by the S-protein-agarose and immunoblotted with anti-NHERF1 IgG. C, pairwise binding between GST-NHERF1 and His-S-tagged PLC-β2 C-tail (containing last 100 amino acids) WT, PDZ motif deletion (ΔSRL), or mutation (ARA and AAA). The complex was pulled down by S-protein-agarose and immunoblotted with anti-NHERF1 IgG.
NHERF1 Couples CXCR2 and PLC-β2 into a Macromolecular Complex in Vitro
Results from the above studies demonstrated that CXCR2 in neutrophils preferentially interacts with the PDZ scaffold protein NHERF1 (Fig. 1, C and D), to which PLC-β2 in neutrophils also preferentially binds (Fig. 2, D and E), and the interactions between NHERF1 and CXCR2 or PLC-β2 are direct and PDZ motif-dependent (Fig. 3). An increasing number of PDZ scaffold proteins have been documented to nucleate the formation of compartmentalized multiprotein complexes that are critical for efficient and specific cellular signaling (26–30). Therefore, we hypothesized that the PDZ scaffold proteins (primarily NHERF1) might cluster CXCR2 and PLC-β2 into a macromolecular complex in a PDZ motif-dependent manner and this CXCR2 complex might play an essential role in CXCR2-mediated PLC-β2 signaling in neutrophil chemotaxis. To address this, we sought to see if we could first assemble a macromolecular complex of CXCR2, PDZ scaffold proteins, and PLC-β2 in vitro, which is represented schematically in Fig. 4A (upper panel). Using this in vitro macromolecular complex assembly assay (29, 30, 36, 42), we detected a complex consisting of CXCR2, PDZ scaffold proteins (NHERF1 or NHERF2), and PLC-β2 C-tail (Fig. 4A). NHERF1 and NHERF2 both were shown to be able to mediate macromolecular complex formation. However, it is to be noted that NHERF1 seems to be the most favored, as expected. The PDZK1 did not form a complex with the PLC-β2 C-tail and CXCR2 (Fig. 4A). His-S-tagged PLC-β2 C-tail does not bind directly to HA-tagged CXCR2 (see Fig. 4C, far left), nor does the complex form in the presence of GST alone (Fig. 4A). We observed a similar complex formation of murine CXCR2 with PLC-β2 mediated preferentially by NHERF1, although there was a relatively weak binding with NHERF2 and PDZK1 (Fig. 4B). We also performed a titration curve for the macromolecular complex assay by varying the concentration of NHERF1 to see if there was a linear increase in complex formation. Our data demonstrated the formation of a macromolecular complex (of His-S-tagged PLC-β2 C-tail, NHERF1, and HA-tagged CXCR2) in a dose-dependent manner (with GST-NHERF1 concentration range of 0–4 μm) (Fig. 4C).
FIGURE 4.

CXCR2, NHERF1, and PLC-β2 form a macromolecular complex in vitro and in neutrophils. A, schematic representation of in vitro macromolecular complex assembly (upper panel, refer to “Experimental Procedures” for details). Macromolecular complex of PLC-β2 C-tail, PDZ scaffold proteins, and human full-length CXCR2 (lower panel) are shown. B, macromolecular complex of PLC-β2 full-length, PDZ scaffold proteins, and mouse CXCR2 C-tail. C, dose-dependent (GST-NHERF1) macromolecular complex formation of His-S-tagged PLC-β2 C-tail, GST-NHERF1, and HA-tagged human CXCR2. D, endogenous PLC-β2 and NHERF1 were co-precipitated with CXCR2 from murine bone marrow neutrophils.
An Endogenous Macromolecular Complex Containing CXCR2, NHERF1, and PLC-β2 Exists in Neutrophils
Results from above demonstrated that there is a CXCR2 macromolecular complex in vitro, but did not indicate whether the macromolecular complex exists in neutrophils, which endogenously express these interacting proteins. To address this issue, freshly isolated murine bone marrow neutrophils were cross-linked as reported previously (36), and co-immunoprecipitation was performed using anti-murine CXCR2 antibody. We detected the endogenous PLC-β2 and NHERF1 that were co-precipitated with CXCR2 from freshly isolated neutrophils (Fig. 4D), indicating that a macromolecular complex consisting of endogenous CXCR2-PDZ scaffold proteins (NHERF1)-PLC-β2 is very likely to exist on the surface membranes of neutrophils. Similarly, PLC-β2 and NHERF1 were also co-immunoprecipitated with CXCR2 from dHL-60 cells (data not shown).
A CXCR2 C-tail-specific Peptide Disrupts the Physical Interaction between NHERF1 and CXCR2
From the findings described above, we established that the PDZ motif of CXCR2 is essential for the physical coupling of CXCR2 to PLC-β2 mediated by NHERF1 into a macromolecular signaling complex. Therefore, it is possible that a CXCR2 C-tail-specific peptide, which contains the PDZ motif, might compete with the endogenous CXCR2 for binding to NHERF1, thus disrupting the endogenous CXCR2 macromolecular complex. We used a human CXCR2 C-tail peptide comprising the last 13 amino acids at the extreme C terminus (biotin-conjugate; FVGSSSGHTSTTL-COOH) in Far Western blotting. We observed that the physical interaction between His-S-tagged NHERF1 and HA-tagged full-length human CXCR2 was disrupted in the presence of this human CXCR2 C-tail wild-type peptide (Fig. 5A), suggesting that the CXCR2 C-tail-specific peptide competed with full-length HA-tagged CXCR2 for binding to NHERF1 as expected. Similar results were also observed with a murine CXCR2 C-tail peptide (13 amino acids in length containing the C terminus PDZ motif) that was shown to disrupt the interaction between murine CXCR2 and NHERF1 in a dose-dependent manner by the pairwise binding assay (Fig. 5B).
FIGURE 5.

A CXCR2 C-tail-specific peptide disrupts the physical interaction between NHERF1 and CXCR2. A, binding of His-S-NHERF1 to 3HA-human CXCR2 in the presence of human CXCR2 C-tail WT peptide on a Far Western blot. The membrane was immunoblotted with HRP-conjugated S-protein, which detects S-tag within His-S-NHERF1. B, GST-NHERF1 binding to His-S-tagged mouse CXCR2 C-tail (WT) in the presence of mouse CXCR2 C-tail WT peptide in pairwise binding as described in the legend to Fig. 3B.
Disrupting the CXCR2 Macromolecular Complex Inhibits CXCR2 Ligand-induced Calcium Mobilization in Neutrophils
CXCR2 signaling plays an important role in the motility of various cells such as neutrophils, macrophages, and lymphocytes (43, 44), presumably through activating PLC-β2 to mobilize calcium from intracellular stores, and induce calcium influx, which is requisite for migration of leukocytes (including neutrophils) (45). As observed above, NHERF1 physically couples CXCR2 and PLC-β2 into a macromolecular complex (Fig. 4). This macromolecular complex may be essential for CXCR2 ligand (such as CXCL8/IL-8, CXCL1/GROα, or MIP-2/murine CXCL2)-induced calcium mobilization in neutrophils and the resultant neutrophilic chemotaxic migration observed in various inflammatory responses. We therefore set out to evaluate the functional significance of the macromolecular complex containing CXCR2, NHERF1, and PLC-β2 in the CXCR2 ligand-induced intracellular calcium mobilization.
As demonstrated in Fig. 5, CXCR2 C-tail PDZ motif-containing peptide disrupted the macromolecular complex of CXCR2, NHERF1, and PLC-β2; this peptide may have an inhibitory effect on the CXCR2 ligand-elicited calcium signal in neutrophils. To test the effect of the CXCR2 C-tail peptide on CXCR2 ligand-induced intracellular calcium mobilization, we delivered murine CXCR2 C-tail peptides into freshly isolated murine bone marrow neutrophils using the Chariot peptide delivery system (30), and then measured intracellular calcium mobilization induced by MIP-2 (murine CXCL2). We observed that neutrophils delivered with murine CXCR2 C-tail WT peptide showed a markedly decreased calcium signal compared with the control (no peptide) (Fig. 6). However, the CXCR2 C-tail ΔTTL (with PDZ motif deleted) peptide failed to block MIP-2-induced intracellular calcium mobilization in mouse neutrophils (Fig. 6). These results suggest that the CXCR2 C-tail WT peptide, when delivered into neutrophils, might disrupt the endogenous macromolecular complex (CXCR2·NHERF1·PLC-β2) by competing with the endogenous CXCR2 for binding to NHERF1, as observed in Fig. 5B. Therefore, neutrophils delivered with this peptide show a markedly decreased calcium signal compared with the control or PDZ motif deletion (ΔTTL) peptide. In addition, we also assessed the effects of the CXCR2 peptides on calcium signals induced by other chemoattractants, such as the bacterial derived peptide fMLP that stimulates calcium mobilization in neutrophils via activating fMLP receptor (46) (fMLP receptor does not possess any PDZ motif). We did not observe any significant difference in fMLP-induced calcium signals between neutrophils that were pre-delivered with the CXCR2 WT or ΔTTL peptide and the control (data not shown). Immunostaining of the delivered peptides was also performed to confirm delivery efficiency, and show the success of peptide delivery (data not shown) as reported (30, 31).
FIGURE 6.
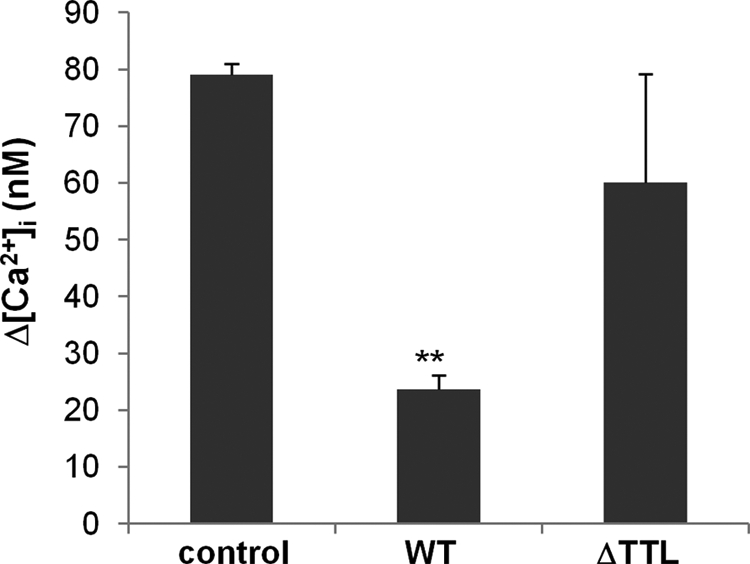
Disrupting the CXCR2 macromolecular complex inhibits CXCR2 ligand-induced calcium mobilization in neutrophils. Intracellular calcium mobilization induced by MIP-2 (100 ng/ml) in mouse bone marrow-derived neutrophils pre-delivered with mouse CXCR2 WT or ΔTTL peptides. **, p < 0.01 compared with the control (no peptide).
Disrupting the CXCR2 Macromolecular Complex Attenuates CXCR2 Ligand-induced Neutrophil Chemotaxic Migration
Chemotaxic migration of neutrophils in response to CXCR2 ligands is a downstream event of CXCR2-mediated intracellular calcium mobilization, which subsequently results in activation of the cytoskeletal machinery, leading to the directed migration of the cells up the chemoattractant gradient to the inflammatory site (47). To examine the functional significance of the macromolecular complex containing CXCR2, NHERF1, and PLC-β2 in CXCR2 ligand-induced neutrophilic chemotaxis, we delivered CXCR2 C-tail peptides (WT and ΔTTL) into neutrophils and monitored the chemotaxic migration across the cell-free collagen-coated Transwell filters in response to chemoattractants (48). As shown in Fig. 7A, CXCR2 C-tail WT peptide pre-treatment significantly suppressed MIP-2- and IL-8-induced neutrophilic chemotaxic migration compared with the controls (no peptide), however, the CXCR2 C-tail PDZ-deletion peptide (ΔTTL) failed to demonstrate a significant inhibitory effect on the chemotaxis of murine neutrophils (Fig. 7A). The CXCR2 C-tail WT peptide also demonstrated a similar inhibitory effect on chemotaxic migration of the dHL-60 cells induced by IL-8 and GROα, whereas the CXCR2 C-tail ΔTTL peptide did not cause a significant change for the chemotaxic migration (Fig. 7B). In addition, neither the WT peptide nor the ΔTTL peptide had any significant effect on fMLP-induced dHL-60 chemotaxic migration as compared with the control (Fig. 7B), suggesting that the CXCR2 C-tail PDZ motif-containing peptide does not affect fMLP-elicited signaling and cellular responses.
Disrupting the CXCR2 Macromolecular Complex Inhibits CXCR2 Ligand-induced Neutrophil Transepithelial Migration
Neutrophil transmigration across mucosal epithelia is the hallmark of many inflammatory diseases including inflammatory bowel disease. Neutrophil transepithelial migration has been shown to be deleterious to the epithelium, and hence, it is possible that blocking neutrophil transmigration might have an anti-inflammatory effect. To monitor neutrophil migration across epithelial monolayers in the physiologically relevant manner (basolateral-to-apical direction), we applied an in vitro system as reported (38) whereby neutrophils or neutrophil-like dHL-60 cells were added to the basal aspect of epithelial monolayers grown in an inverted fashion on the undersurface of permeable Transwell supports. The dHL-60 cells were delivered with human CXCR2 C-tail peptides (WT, AAA, or ΔTTL) and added to the basal sides of polarized colonic epithelial cell HT-29 monolayers. Our results showed that the CXCR2 WT peptide, but not the PDZ mutation (AAA) or PDZ deletion (ΔTTL) peptide, potently inhibited the basolateral-to-apical migration of dHL-60 cells across HT-29 monolayers induced by IL-8 (Fig. 8A) and GROα (data not shown). However, these CXCR2 peptides did not affect dHL-60 transepithelial migration in response to fMLP (Fig. 8B), suggesting the specificity of these peptides for CXCR2-mediated neutrophil transmigration. Similar results were also observed in the dHL-60 transmigration across other colonic epithelial monolayers, such as Caco2 and T84 cells (data not shown).
FIGURE 8.
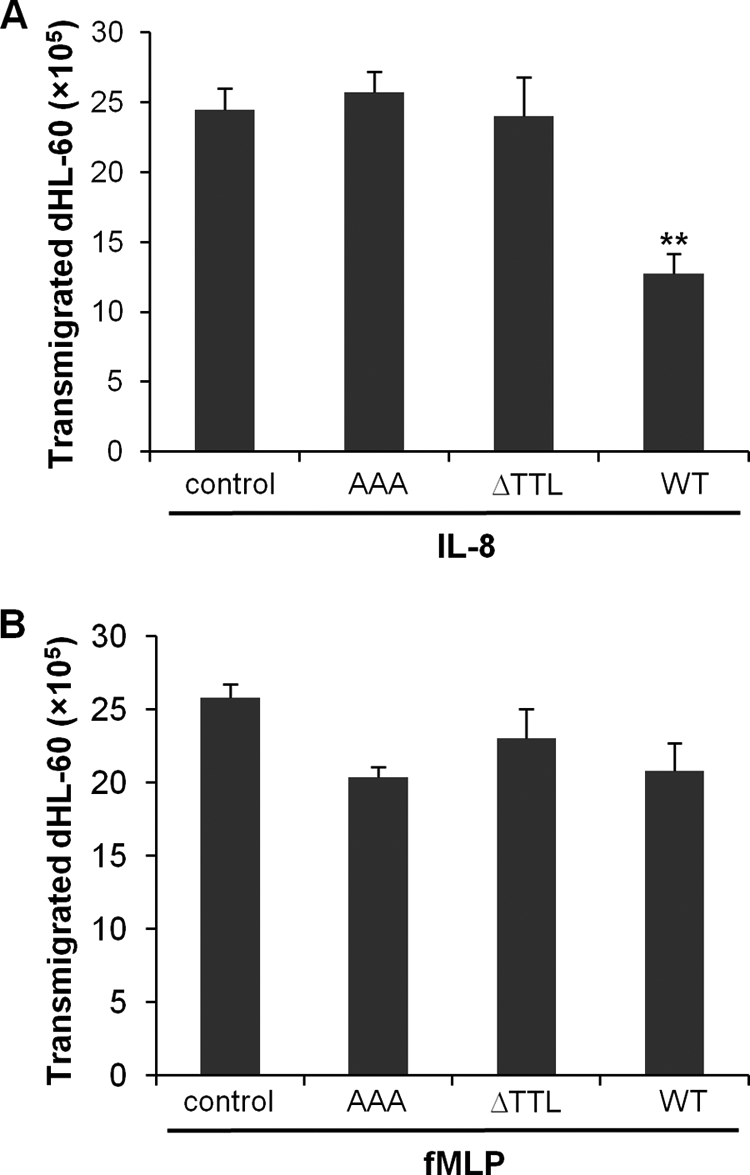
Disrupting the CXCR2 macromolecular complex inhibits CXCR2 ligand-induced neutrophil transepithelial migration. A, IL-8-induced dHL-60 cell transepithelial migration across HT-29 cell monolayers. dHL-60 cells (3 × 106) were pre-delivered with various human CXCR2 C-tail peptides as indicated, and the cells that migrated into the lower chamber at 2 h were quantified by a colorimetric MPO assay. **, p < 0.01 compared with control (no peptide). B, transepithelial migration of dHL-60 cells across HT-29 monolayers induced by fMLP. dHL-60 cells (3 × 106) were pre-delivered with various human CXCR2 C-tail peptides as indicated, and the transmigration was quantified by MPO activity assay.
DISCUSSION
The recruitment of circulating PMNs to sites of injury or infection is a fundamental event in the inflammatory response of the innate immune system, providing protection from invading bacteria. The process of neutrophil movement from the vasculature, through the interstitial matrix, and across mucosal surfaces requires sequential interactions and a complex series of signaling events between neutrophils and other cells within the tissues in response to a wide variety of chemotactic signals (e.g. cytokines, chemokines, lipopolysaccharide, fMLP, etc.) (49). However, excessive or unchecked neutrophil recruitment often leads to tissue dysfunction and damage, mainly resulting from the persistent release of harmful inflammatory cytokines, reactive oxygen species, and proteases by the infiltrated cells (50). Therefore, blocking neutrophil transmigration across cellular barriers such as vascular endothelium and mucosal epithelium represents an effective approach to prevent excessive inflammation in many diseases such as atherosclerosis, inflammatory bowel disease, cystic fibrosis, etc.
CXCR2 is a G protein-coupled receptor that is activated by ELR+ CXC chemokines, CXCL1–3 and CXCL5–8, including murine CXCL1 (keratinocyte-derived chemokine) and CXCL2 (MIP-2) (51). Activation of CXCR2 by its cognate ligands induces intracellular signals associated with chemotaxic migration, and recruitment and infiltration of leukocytes (especially PMNs) from the bloodstream during inflammation (52). It has been well documented that blocking the ELR+ CXC chemokines/CXCR2 biological axis via CXCR2 neutralizing antibodies or selective CXCR2 antagonists (such as SB225002) or neutralizing antibodies against cognate CXCR2 ligands (such as anti-CXCL1/keratinocyte-derived chemokine antibody) potently inhibited neutrophil migration and infiltration in vitro and in vivo, and significantly ameliorated ulcerative colitis in experimental animals (7, 35, 53–57). Genetic deletion of CXCR2 in colitic mice demonstrated reduced PMN infiltration, limited signs of mucosal damage, and reduced clinical symptoms (35, 58). Therefore, the ELR+ CXC chemokines/CXCR2 biological axis appears to represent an attractive potential therapeutic target (59). However, most of the studies have been conducted by systemic blockade of the functions of CXCR2 and its ligands in almost all cell types, including neutrophils. Thus, concern arises that these approaches might elicit global undesired effects on many other vital functions. This warrants the necessity of identifying highly selective and cell-specific therapeutic targets (i.e. merely in neutrophils).
Many studies have reported the association of the PDZ scaffold proteins with a wide variety of plasma membrane receptors, ion channels, and transporters, suggesting that PDZ scaffold proteins can nucleate the formation of multiprotein complexes that are critical for trafficking, transport, and signaling in cells (26–30). Several adaptor/scaffold proteins have been documented to bind the carboxyl-terminal domain of CXCR2 and are involved in chemokine-mediated signal transduction and cellular functions, such as adaptin 2 (60), VASP (61), LIM and SH3 protein-1 (62), and IQGAP1 (63). The carboxyl-terminal PDZ ligand motif was recently reported to modulate post-endocytic sorting and cellular chemotaxis in CXCR2-overexpressing HEK293 cells (25). Deletion of the PDZ motif was shown to impair cellular chemotaxis (25). However, the molecular identity of the potential PDZ scaffolding proteins that might interact with CXCR2 via PDZ-based interaction remains elusive. In our present study, we identified a novel interacting partner for CXCR2, i.e. the PDZ scaffold protein NHERF1, and NHERF1 interacts directly with CXCR2 via the C-tail PDZ-based interaction in both freshly isolated neutrophils and neutrophil-like differentiated HL-60 cells (physiologically more relevant cells than HEK293 cells). We applied multiple approaches to demonstrate the CXCR2-NHERF1 interaction, such as pulldown assay, pairwise binding, etc. Furthermore, our studies revealed that NHERF1 also preferentially binds to PLC-β2 in neutrophils, one of the downstream molecules in CXCR2 signaling (8). We then showed that NHERF1 cluster CXCR2 and PLC-β2 into a macromolecular signaling complex in vitro and also in neutrophils. We further demonstrated the functional relevance of this CXCR2·NHERF1·PLC-β2 macromolecular complex via delivery of a CXCR2 C-tail PDZ motif-containing peptide to disturb the endogenous CXCR2 complex, and we observed significantly inhibited cell signaling and functions induced by CXCR2 ligands. Our studies shed light on the cellular and molecular mechanism underlying the chemoattractant-induced neutrophil responses, the understanding of which provides unique targets for novel therapeutic approaches and agents that combat inflammatory conditions in various tissues and organs.
It is well documented that the activation of CXCR2 induces migration of various cell types (43, 44), and IL-8 and the other CXC chemokines act preferentially on neutrophils (43, 44). Given that CXCR2 and PLC-β2 are the primary players in neutrophils during the chemoattractant-induced response (6, 14), and our present studies demonstrated that both proteins can interact preferentially with NHERF1 (Figs. 1 and 2), and disrupting the complex (i.e. CXCR2-NHERF1-PLC-β2) using exogenous CXCR2 C-tail peptide (containing a PDZ motif) significantly attenuated the CXCR2-mediated intracellular calcium mobilization and the chemotaxic response of neutrophils to CXCR2 ligands (Figs. 6–8), we therefore propose a mechanism for the NHERF1-mediated coupling of CXCR2 to PLC-β2 signaling in neutrophils (Fig. 9). In brief, both CXCR2 and PLC-β2 directly interact with NHERF1. NHERF1 clusters CXCR2 and PLC-β2 in close proximity, thereby forming spatially compact signaling complexes beneath the plasma membrane. Consequently, the macromolecular signaling complex (CXCR2·NHERF1·PLC-β2) enables CXCR2 to transduce its signal to PLC-β2 with efficiency and specificity, and this complex is a prerequisite for the CXCR2 ligand-induced calcium signaling and the resultant neutrophil migration during various inflammatory diseases, such as atherosclerosis, cystic fibrosis, and inflammatory bowel disease. Therefore, our present study revealed a previously unidentified therapeutic target for the treatment of certain inflammatory diseases (proof-of-concept). In addition, compared with previous investigations that focused solely on CXCR2 and its ligands in controlling excessive inflammation, our present study introduces a new concept of the CXCR2 macromolecular complex mediated via PDZ-based interactions. By studying a network of protein complexes rather than CXCR2 alone in neutrophils, our results identified new potential therapeutic targets specifically for the modulation of neutrophil migration and infiltration in inflamed tissues or organs, and will lead to the near future investigations into the therapeutic efficacy of disrupting the CXCR2 PDZ motif-mediated protein interactions in inhibiting neutrophil infiltration in vivo. For instance, we can synthesize or screen certain small molecule inhibitors (serve to mimic the CXCR2 interacting partners) and develop novel types of nanoparticles as delivery systems for therapeutic small molecule inhibitors or siRNA to interrupt the PDZ motif-mediated CXCR2 macromolecular complex in neutrophils, and use them in future clinical trials.
FIGURE 9.

Proposed mechanism of the NHERF1-mediated coupling of CXCR2 to PLC-β2 signaling in neutrophils. Both CXCR2 and PLC-β2 directly interact with the PDZ scaffold protein NHERF1. NHERF1 clusters CXCR2 and PLC-β2 in close proximity via a PDZ-based interaction, thereby forming spatially compact signaling complexes beneath the plasma membrane. Consequently, the macromolecular signaling complex (CXCR2·NHERF1·PLC-β2) enables CXCR2 to transduce its signal to PLC-β2 with efficiency and specificity.
Acknowledgments
We sincerely thank Dr. Anjaparavanda P. Naren (University of Tennessee Health Science center) for constructive suggestions and generous assistance for this project, and Feng Zhou for formatting the references. We also thank Dr. P. G. Suh (Pohang University of Science and Technology, South Korea) for the FLAG-tagged PLC-β constructs.
This work was supported by American Heart Association (Greater SE Affiliate) Beginning Grant-in-aid 0765185B (to C. L.), an Elsa U. Pardee Foundation research grant (to C. L.), Wayne State University intramural startup fund and a Cardiovascular Research Institute Isis Initiative award (to C. L.), and National Institutes of Health Grant HL096800 (to F. S.).
- PMN
- polymorphonuclear leukocyte
- CXCR1/2
- CXC chemokine receptor 1/2
- CXCL
- CXC chemokine ligand
- MIP-2
- macrophage inflammatory protein 2
- PDZ
- PSD-95/SAP-90, Disc-large DLG, zonula occludens (ZO)-1
- NHERF
- Na/H exchanger regulatory factor
- PDZK1
- PDZ domain containing 1
- PLC
- phospholipase C
- GRO
- growth-related oncogene
- fMLP
- formyl-methionyl-leucyl-phenylalanine
- MPO
- myeloperoxidase
- dHL-60
- differentiated HL-60
- HBSS
- Hanks' buffered salt solution.
REFERENCES
- 1. Geng J. G. (2001) Directional migration of leukocytes. Their pathological roles in inflammation and strategies for development of anti-inflammatory therapies. Cell Res. 11, 85–88 [DOI] [PubMed] [Google Scholar]
- 2. Greene C. M., Carroll T. P., Smith S. G., Taggart C. C., Devaney J., Griffin S., O'Neill S. J., McElvaney N. G. (2005) TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 174, 1638–1646 [DOI] [PubMed] [Google Scholar]
- 3. Pease J. E., Sabroe I. (2002) The role of interleukin-8 and its receptors in inflammatory lung disease. Implications for therapy. Am. J. Respir. Med. 1, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Holmes W. E., Lee J., Kuang W. J., Rice G. C., Wood W. I. (1991) Structure and functional expression of a human interleukin-8 receptor. Science 253, 1278–1280 [DOI] [PubMed] [Google Scholar]
- 5. Murphy P. M., Tiffany H. L. (1991) Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 253, 1280–1283 [DOI] [PubMed] [Google Scholar]
- 6. Chuntharapai A., Kim K. J. (1995) Regulation of the expression of IL-8 receptor A/B by IL-8. Possible functions of each receptor. J. Immunol. 155, 2587–2594 [PubMed] [Google Scholar]
- 7. White J. R., Lee J. M., Young P. R., Hertzberg R. P., Jurewicz A. J., Chaikin M. A., Widdowson K., Foley J. J., Martin L. D., Griswold D. E., Sarau H. M. (1998) Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J. Biol. Chem. 273, 10095–10098 [DOI] [PubMed] [Google Scholar]
- 8. Wu D., LaRosa G. J., Simon M. I. (1993) G protein-coupled signal transduction pathways for interleukin-8. Science 261, 101–103 [DOI] [PubMed] [Google Scholar]
- 9. Harden T. K., Hicks S. N., Sondek J. (2009) Phospholipase C isozymes as effectors of Ras superfamily GTPases. J. Lipid Res. 50, S243–S248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhee S. G. (2001) Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70, 281–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang H., Lyubarsky A., Dodd R., Vardi N., Pugh E., Baylor D., Simon M. I., Wu D. (1996) Phospholipase C-β4 is involved in modulating the visual response in mice. Proc. Natl. Acad. Sci. U.S.A. 93, 14598–14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kano M., Hashimoto K., Watanabe M., Kurihara H., Offermanns S., Jiang H., Wu Y., Jun K., Shin H. S., Inoue Y., Simon M. I., Wu D. (1998) Phospholipase C-β4 is specifically involved in climbing fiber synapse elimination in the developing cerebellum. Proc. Natl. Acad. Sci. U.S.A. 95, 15724–15729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang H., Kuang Y., Wu Y., Xie W., Simon M. I., Wu D. (1997) Roles of phospholipase C-β2 in chemoattractant-elicited responses. Proc. Natl. Acad. Sci. U.S.A. 94, 7971–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Z., Jiang H., Xie W., Zhang Z., Smrcka A. V., Wu D. (2000) Roles of PLC-β2 and -β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science 287, 1046–1049 [DOI] [PubMed] [Google Scholar]
- 15. Wu D., Huang C. K., Jiang H. (2000) Roles of phospholipid signaling in chemoattractant-induced responses. J. Cell Sci. 113, 2935–2940 [DOI] [PubMed] [Google Scholar]
- 16. Fanning A. S., Anderson J. M. (1999) PDZ domains. Fundamental building blocks in the organization of protein complexes at the plasma membrane. J. Clin. Invest. 103, 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bezprozvanny I., Maximov A. (2001) PDZ domains. More than just a glue. Proc. Natl. Acad. Sci. U.S.A. 98, 787–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li C., Naren A. P. (2010) CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr. Biol. 2, 161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hwang J. I., Heo K., Shin K. J., Kim E., Yun C., Ryu S. H., Shin H. S., Suh P. G. (2000) Regulation of phospholipase C-β3 activity by Na+/H+ exchanger regulatory factor 2. J. Biol. Chem. 275, 16632–16637 [DOI] [PubMed] [Google Scholar]
- 20. Tang Y., Tang J., Chen Z., Trost C., Flockerzi V., Li M., Ramesh V., Zhu M. X. (2000) Association of mammalian trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J. Biol. Chem. 275, 37559–37564 [DOI] [PubMed] [Google Scholar]
- 21. Suh P. G., Hwang J. I., Ryu S. H., Donowitz M., Kim J. H. (2001) The roles of PDZ-containing proteins in PLC-β-mediated signaling. Biochem. Biophys. Res. Commun. 288, 1–7 [DOI] [PubMed] [Google Scholar]
- 22. Hwang J. I., Kim H. S., Lee J. R., Kim E., Ryu S. H., Suh P. G. (2005) The interaction of phospholipase C-β3 with Shank2 regulates mGluR-mediated calcium signal. J. Biol. Chem. 280, 12467–12473 [DOI] [PubMed] [Google Scholar]
- 23. Choi J. W., Lim S., Oh Y. S., Kim E. K., Kim S. H., Kim Y. H., Heo K., Kim J., Kim J. K., Yang Y. R., Ryu S. H., Suh P. G. (2010) Subtype-specific role of phospholipase C-β in bradykinin and LPA signaling through differential binding of different PDZ scaffold proteins. Cell Signal. 22, 1153–1161 [DOI] [PubMed] [Google Scholar]
- 24. Donowitz M., Singh S., Singh P., Salahuddin F. F., Chen Y., Chakraborty M., Murtazina R., Gucek M., Cole R. N., Zachos N. C., Kovbasnjuk O., Broere N., Smalley-Freed W. G., Reynolds A. B., Hubbard A. L., Seidler U., Weinman E., de Jonge H. R., Hogema B. M., Li X. (2010) Alterations in the proteome of the NHERF1 knock-out mouse jejunal brush-border membrane vesicles. Physiol. Genomics 42A, 200–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baugher P. J., Richmond A. (2008) The carboxyl-terminal PDZ ligand motif of chemokine receptor CXCR2 modulates post-endocytic sorting and cellular chemotaxis. J. Biol. Chem. 283, 30868–30878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang S., Yue H., Derin R. B., Guggino W. B., Li M. (2000) Accessory protein facilitated CFTR-CFTR interaction, a molecular mechanism to potentiate the chloride channel activity. Cell 103, 169–179 [DOI] [PubMed] [Google Scholar]
- 27. Hall R. A., Ostedgaard L. S., Premont R. T., Blitzer J. T., Rahman N., Welsh M. J., Lefkowitz R. J. (1998) A C-terminal motif found in the β2-adrenergic receptor, P2Y1 receptor and cystic fibrosis transmembrane conductance regulator determines binding to the Na+/H+ exchanger regulatory factor family of PDZ proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 8496–8501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Short D. B., Trotter K. W., Reczek D., Kreda S. M., Bretscher A., Boucher R. C., Stutts M. J., Milgram S. L. (1998) An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J. Biol. Chem. 273, 19797–19801 [DOI] [PubMed] [Google Scholar]
- 29. Naren A. P., Cobb B., Li C., Roy K., Nelson D., Heda G. D., Liao J., Kirk K. L., Sorscher E. J., Hanrahan J., Clancy J. P. (2003) A macromolecular complex of β2-adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. U.S.A. 100, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li C., Dandridge K. S., Di A., Marrs K. L., Harris E. L., Roy K., Jackson J. S., Makarova N. V., Fujiwara Y., Farrar P. L., Nelson D. J., Tigyi G. J., Naren A. P. (2005) Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J. Exp. Med. 202, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li C., Krishnamurthy P. C., Penmatsa H., Marrs K. L., Wang X. Q., Zaccolo M., Jalink K., Li M., Nelson D. J., Schuetz J. D., Naren A. P. (2007) Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 131, 940–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li C., Schuetz J. D., Naren A. P. (2010) Tobacco carcinogen NNK transporter MRP2 regulates CFTR function in lung epithelia. Implications for lung cancer. Cancer Lett. 292, 246–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carrigan S. O., Weppler A. L., Issekutz A. C., Stadnyk A. W. (2005) Neutrophil-differentiated HL-60 cells model Mac-1 (CD11b/CD18)-independent neutrophil transepithelial migration. Immunology 115, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pizurki L., Morris M. A., Chanson M., Solomon M., Pavirani A., Bouchardy I., Suter S. (2000) Cystic fibrosis transmembrane conductance regulator does not affect neutrophil migration across cystic fibrosis airway epithelial monolayers. Am. J. Pathol. 156, 1407–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Farooq S. M., Stillie R., Svensson M., Svanborg C., Strieter R. M., Stadnyk A. W. (2009) Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J. Pharmacol. Exp. Ther. 329, 123–129 [DOI] [PubMed] [Google Scholar]
- 36. Li C., Roy K., Dandridge K., Naren A. P. (2004) Molecular assembly of cystic fibrosis transmembrane conductance regulator in plasma membrane. J. Biol. Chem. 279, 24673–24684 [DOI] [PubMed] [Google Scholar]
- 37. Hirst R. A., Harrison C., Hirota K., Lambert D. G. (2006) Measurement of [Ca2+]i in whole cell suspensions using fura-2. Methods Mol. Biol. 312, 37–45 [PubMed] [Google Scholar]
- 38. Lee W. Y., Chin A. C., Voss S., Parkos C. A. (2006) In vitro neutrophil transepithelial migration. Methods Mol. Biol. 341, 205–215 [DOI] [PubMed] [Google Scholar]
- 39. Hess D. J., Henry-Stanley M. J., Erickson E. A., Wells C. L. (2002) Effect of tumor necrosis factor α, interferon γ, and interleukin-4 on bacteria-enterocyte interactions. J. Surg. Res. 104, 88–94 [DOI] [PubMed] [Google Scholar]
- 40. Collins S. J. (1987) The HL-60 promyelocytic leukemia cell line. Proliferation, differentiation, and cellular oncogene expression. Blood 70, 1233–1244 [PubMed] [Google Scholar]
- 41. Sai J., Walker G., Wikswo J., Richmond A. (2006) The IL sequence in the LLKIL motif in CXCR2 is required for full ligand-induced activation of Erk, Akt, and chemotaxis in HL60 cells. J. Biol. Chem. 281, 35931–35941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li C., Naren A. P. (2011) Analysis of CFTR interactome in the macromolecular complexes. Methods Mol. Med. 741, 255–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huber A. R., Kunkel S. L., Todd R. F., 3rd, Weiss S. J. (1991) Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science 254, 99–102 [DOI] [PubMed] [Google Scholar]
- 44. Baggiolini M., Loetscher P., Moser B. (1995) Interleukin-8 and the chemokine family. Int. J. Immunopharmacol. 17, 103–108 [DOI] [PubMed] [Google Scholar]
- 45. Rah S. Y., Park K. H., Han M. K., Im M. J., Kim U. H. (2005) Activation of CD38 by interleukin-8 signaling regulates intracellular Ca2+ level and motility of lymphokine-activated killer cells. J. Biol. Chem. 280, 2888–2895 [DOI] [PubMed] [Google Scholar]
- 46. Thomas K. M., Pyun H. Y., Navarro J. (1990) Molecular cloning of the fMet-Leu-Phe receptor from neutrophils. J. Biol. Chem. 265, 20061–20064 [PubMed] [Google Scholar]
- 47. Cicchetti G., Allen P. G., Glogauer M. (2002) Chemotactic signaling pathways in neutrophils. From receptor to actin assembly. Crit. Rev. Oral Biol. Med. 13, 220–228 [DOI] [PubMed] [Google Scholar]
- 48. Xu L., Tong R., Cochran D. M., Jain R. K. (2005) Blocking platelet-derived growth factor-D/platelet-derived growth factor receptor β signaling inhibits human renal cell carcinoma progression in an orthotopic mouse model. Cancer Res. 65, 5711–5719 [DOI] [PubMed] [Google Scholar]
- 49. Liu Y., Shaw S. K., Ma S., Yang L., Luscinskas F. W., Parkos C. A. (2004) Regulation of leukocyte transmigration. Cell surface interactions and signaling events. J. Immunol. 172, 7–13 [DOI] [PubMed] [Google Scholar]
- 50. Nathan C. (2006) Neutrophils and immunity. Challenges and opportunities. Nat. Rev. Immunol. 6, 173–182 [DOI] [PubMed] [Google Scholar]
- 51. Olson T. S., Ley K. (2002) Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R7–28 [DOI] [PubMed] [Google Scholar]
- 52. Niggli V. (2003) Signaling to migration in neutrophils. Importance of localized pathways. Int. J. Biochem. Cell Biol. 35, 1619–1638 [DOI] [PubMed] [Google Scholar]
- 53. Matzer S. P., Zombou J., Sarau H. M., Röllinghoff M., Beuscher H. U. (2004) A synthetic, non-peptide CXCR2 antagonist blocks MIP-2-induced neutrophil migration in mice. Immunobiology 209, 225–233 [DOI] [PubMed] [Google Scholar]
- 54. Bento A. F., Leite D. F., Claudino R. F., Hara D. B., Leal P. C., Calixto J. B. (2008) The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. J. Leukocyte Biol. 84, 1213–1221 [DOI] [PubMed] [Google Scholar]
- 55. McColl S. R., Clark-Lewis I. (1999) Inhibition of murine neutrophil recruitment in vivo by CXC chemokine receptor antagonists. J. Immunol. 163, 2829–2835 [PubMed] [Google Scholar]
- 56. Murphy C. T., Moloney G., Hall L. J., Quinlan A., Faivre E., Casey P., Shanahan F., Melgar S., Nally K. (2010) Use of bioluminescence imaging to track neutrophil migration and its inhibition in experimental colitis. Clin. Exp. Immunol. 162, 188–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ajuebor M. N., Zagorski J., Kunkel S. L., Strieter R. M., Hogaboam C. M. (2004) Contrasting rolls for CXCR2 during experimental colitis. Exp. Mol. Pathol. 76, 1–8 [DOI] [PubMed] [Google Scholar]
- 58. Buanne P., Di Carlo E., Caputi L., Brandolini L., Mosca M., Cattani F., Pellegrini L., Biordi L., Coletti G., Sorrentino C., Fedele G., Colotta F., Melillo G., Bertini R. (2007) Crucial pathophysiological role of CXCR2 in experimental ulcerative colitis in mice. J. Leukocyte Biol. 82, 1239–1246 [DOI] [PubMed] [Google Scholar]
- 59. Bizzarri C., Beccari A. R., Bertini R., Cavicchia M. R., Giorgini S., Allegretti M. (2006) ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Ther. 112, 139–149 [DOI] [PubMed] [Google Scholar]
- 60. Fan G. H., Yang W., Wang X. J., Qian Q., Richmond A. (2001) Identification of a motif in the carboxyl terminus of CXCR2 that is involved in adaptin 2 binding and receptor internalization. Biochemistry 40, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Neel N. F., Barzik M., Raman D., Sobolik-Delmaire T., Sai J., Ham A. J., Mernaugh R. L., Gertler F. B., Richmond A. (2009) VASP is a CXCR2-interacting protein that regulates CXCR2-mediated polarization and chemotaxis. J. Cell Sci. 122, 1882–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raman D., Sai J., Neel N. F., Chew C. S., Richmond A. (2010) LIM and SH3 protein-1 modulates CXCR2-mediated cell migration. PLoS One 5, e10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Neel N. F., Sai J., Ham A. J., Sobolik-Delmaire T., Mernaugh R. L., Richmond A. (2011) IQGAP1 is a novel CXCR2-interacting protein and essential component of the “chemosynapse.” PLoS One 6, e23813. [DOI] [PMC free article] [PubMed] [Google Scholar]