

Abstract
Inflammation is a multi-staged process whose expansive phase is thought to be driven by acutely released arachidonic acid (AA) and its metabolites. Inhibition of cyclooxygenase (COX), lipoxygenase (LOX), or soluble epoxide hydrolase (sEH) is known to be anti-inflammatory. Inhibition of sEH stabilizes the cytochrome P450 (CYP450) products epoxyeicosatrienoic acids (EETs). Here we used a non-selective COX inhibitor aspirin, a 5-lipoxygenase activation protein (FLAP) inhibitor MK886, and a sEH inhibitor t-AUCB to selectively modulate the branches of AA metabolism in a lipopolysaccharide (LPS)-challenged murine model. We used metabolomic profiling to simultaneously monitor representative AA metabolites of each branch. In addition to the significant crosstalk among branches of the AA cascade during selective modulation of COX, LOX, or sEH, we demonstrated that co-administration of t-AUCB enhanced the anti-inflammatory effects of aspirin or MK886, which was evidenced by the observations that co-administration resulted in favorable eicosanoid profiles and better control of LPS-mediated hypotension as well as hepatic protein expression of COX-2 and 5-LOX. Targeted disruption of the sEH gene displayed a parallel profile to that produced by t-AUCB. These observations demonstrate a significant level of crosstalk among the three major branches of the AA cascade and that they are not simply parallel pathways. These data illustrate that inhibition of sEH by both pharmacological intervention and gene knockout enhances the anti-inflammatory effects of aspirin and MK886, suggesting the possibility of modulating multiple branches to achieve better therapeutic effects.
Keywords: arachidonic acid, eicosanoid, interaction, metabolomics, synergism
1. Introduction
COXs, LOXs, and cytochrome P450s (CYP450s) are the key enzymes in the major branches of the arachidonate cascade [1]. COX enzymes catalyze the synthesis of PGs, prostacyclins, and thromboxanes (TXs). Decreasing the production of these pro-inflammatory AA metabolites using selective and non-selective COX inhibitors is currently the prominent therapeutic approach to relieve pain, fever, and inflammation [2]. Recent advances in understanding the role of 5-LOX and its associated activator FLAP in AA metabolism resulted in inhibitors of leukotriene (LT) and 5-HETE production [1, 3-5]. Both 5-LOX and FLAP are required for the cellular biosynthesis of LTs, prominent pro-inflammatory and chemotactic mediators [6]. FLAP binds free AA and presents it to 5-LOX, which catalyzes the formation of 5-(S)-hydroperoxy-6,8,11,14-eicosatetraenoic acid which is further metabolized to LTA4, 5-HETE and subsequent metabolites which among other effects mediate inflammation [5, 7-9]. In addition, simultaneous inhibition of both COX-2 and 5-LOX has been found to alleviate inflammation [10]. Approximately 20% of clinical pharmaceuticals were developed directly targeting at modulation of COX or 5-LOX branches of AA metabolism.
Besides the COX and LOX enzymes, some CYP450 epoxygenases catalyze the formation of anti-inflammatory epoxyeicosatrienoic acids (EETs) from AA [11]. EETs decrease inflammation, in part, by preventing the activation of nuclear factor-kappa B (NF-κB) [12-14]. EETs, in vivo, display multiple biological activities including regulation of blood pressure and cardioprotection [15-19]. However, EETs are rapidly metabolized to the more polar and less active DHETs in the presence of sEH [20, 21]. Inhibitors of sEH (sEHIs) in vivo reduce blood pressure during hypertension and normalize it during hypotension, although they appear devoid of effects under normotensive conditions [22-25]. sEHIs also attenuate pain and inflammation [26-28] as well as certain cardiovascular conditions [13, 15, 29-34] although they display no reported effects in normal subjects. The major effects of EETs could thus be viewed as modulating physiology towards a normal state. In addition, simultaneous inhibition of both COX-2 and sEH has been found to achieve additive results in alleviating pain, reducing COX-2 protein expression, and shifting oxylipin profiles towards resolution of inflammation [31].
Most previous studies [35-37] focused on the individual targeted branches which inevitably limited the understanding of the impact of therapy on the whole AA cascade by selective emphasis of just one branch of the cascade. This technical disadvantage unconsciously hindered a broader understanding of how individual therapies influence the entire cascade and the concept of informed use of drug combinations. To overcome such limitation, the new holistic omic technique- metabolomics has been developed as a promisingly comprehensive approach. Metabolomics has already been illustrated as a powerful tool in disease diagnosis [38], biomarker discovery [39], toxicity evaluation [40], gene function [41], and pathophysiological researches [42].
In this study, the metabolomic profiling approach targeting AA cascade, which can simultaneously measure many representative metabolites derived from AA (Figure 1A) [43], was employed. We applied this quantitative profiling method in a murine sepsis model with inhibitors of the COX and LOX pathways and a sEH inhibitor. These metabolomic data comprise a platform independent legacy database since they are quantitative. Our results demonstrate significant interactions among the COX, LOX and CYP450 branches of the AA cascade once thought to be discreet, parallel pathways, and suggest the possibility of therapeutic strategies for anti-inflammation by co-inhibition of sEH and FLAP or COX.
Fig. 1.
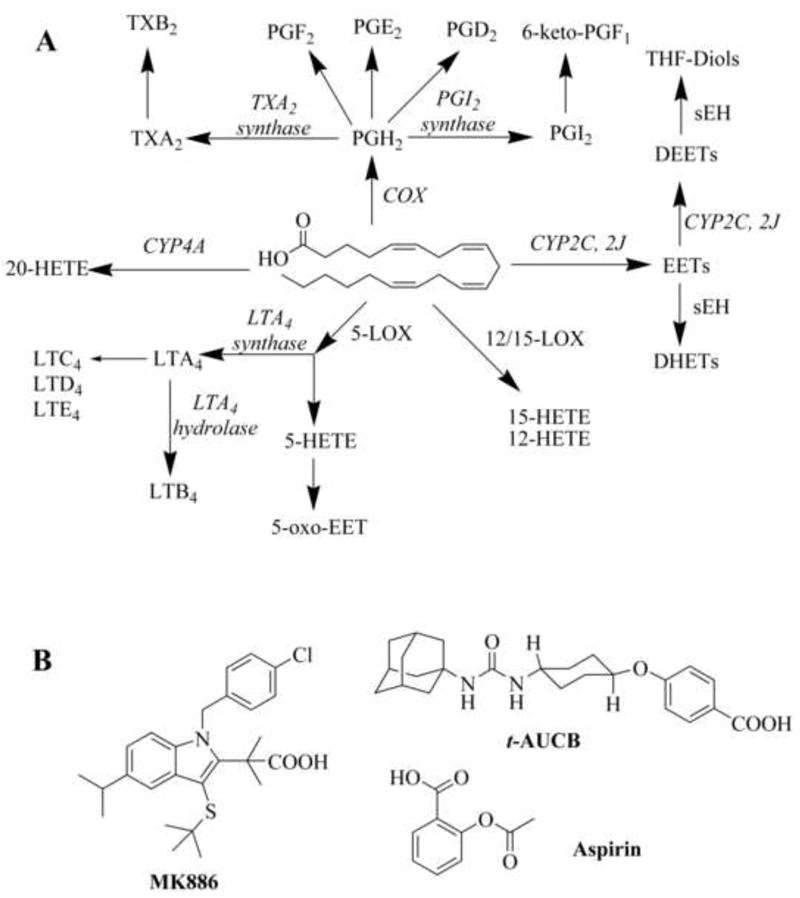
(A) A simplified schematic of the arachidonic acid cascade showing the target metabolite oxylipins that were simultaneously measured. The effects of inhibition of COX, FLAP, and/or sEH on the AA metabolism in LPS-challenged murine model are present in Figures 2, 3 and 4, respectively. (B) Structures of inhibitors used. Aspirin is a non-selective COX inhibitor. 3-[1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-2-yl]-2,2-dimethyl propanoic acid (MK 886) is a FLAP inhibitor, and trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) is a sEH inhibitor.
2. Materials and Methods
2.1. Animals and Chemicals
All procedures and animal care were performed in accordance with the protocols approved by IACUC of the University of California, Davis. Male, 8-week-old C57BL/6 mice (Charles River Laboratories, MO) weighing 22-25g were used. For knockout studies male, 10- to 12-week-old mice on a 129X1/SvJ ×C57BL/6 background with targeted disruption of the Ephx2 gene (Ephx2-null mice) and their wild type conspecifics were used. These mice were originally obtained from Dr. C. Sinal, Dalhousie University, Halifax, NS, Canada under a National Cancer Institute Material Transfer Agreement 1-16268-04, and fostered at the University of California, Davis [44, 45]. The LPS (Escherichia coli serotype, L4130, 0111:B4) and aspirin were purchased from Sigma–Aldrich (St. Louis, NJ). The MK 886 (sodium salt) used was from Cayman Chemical Co (Ann Arbor, MI). t-AUCB was synthesized in house[46]. Aspirin and MK 886 doses were in the therapeutic range based on current literature [47, 48] and the dose of t-AUCB was chosen on basis of its potent IC50 (8 nM) and pharmacokinetic profiling [49].
2.2. Animal protocol
Aspirin (s.c.), t-AUCB (p.o.), or MK 886 (s.c.) were administrated alone or in combination immediately after i.p. injection of LPS (10 mg/kg) in saline. Mice receiving oral gavage of trans free trioleate and s.c. injection of saline immediately after i.p. injection of LPS or saline served as positive and negative controls, respectively. The Ephx2-null mice were administered aspirin (50 mg/kg, s.c.) or MK 886 (10 mg/kg, s.c.) immediately after receiving 10 mg/kg LPS (i. p.) in saline. Saline injected animals served as controls. Systolic blood pressure (BP) of mice was determined by a noninvasive tail-cuff method with a Visitech BP-2000 (Visitech Systems, Apex, NC) BP analysis system [26]. The lower limit of detection was set at 40 mmHg (1 mmHg= 133 Pa). All treatments were performed after 1-week training of BP determination for C57BL/6 mice and 2-week training of BP determination for the NIH colony mice, respectively. BP was recorded to characterize the development of septic shock 4 hours after LPS exposure and the mice was sacrificed 6 hours after LPS exposure. Blood was collected to separate plasma as the method detailed in [49]. Livers were excised following sacrifice and frozen immediately in liquid nitrogen. All samples were stored at -80 °C until analysis
2.3. Analysis of oxylipin mediators
Plasma was extracted for oxylipin analysis as the method reported in [49, 50] by Liu et al. Oxylipin concentrations of plasma were measured using an Agilent 1200 Series HPLC (Agilent Technologies, Inc. Santa Clara, CA) coupled with an Applied Biosystems 4000 QTRAP hybrid, triple-quadrupole MS instrument (Applied Biosystems, Foster City, CA) described in [43] by Yang et al.
2.4. Analysis of Data
Data were presented as mean ± sd. Statistic analyses were performed by ANOVA using the software SPSS 10.0 (SPSS Inc., Chicago, IL) with P < 0.05 as the significance level.
3. Results
3.1. Effect of selective modulation of COX, FLAP or sEH on AA metabolism
An LPS-challenged murine acute inflammation model was used to investigate the effects of reducing the flow of AA through the COX and LOX branches and stabilizing the EETs from the CYP450 branch of the AA cascade (Fig. 1A) with analysis by LC-MS/MS (Table S1). The structures of the inhibitors used are depicted in Figure 1B. In mice receiving LPS (10 mg/kg body weight, i.p.) dramatic increases in circulating plasma levels of the key metabolites PGE2, TXB2, 5-HETE, 15-HETE, and DHET regioisomers were observed (Fig. 2). As expected, inhibition of a single pathway significantly impacted the levels of the corresponding pathway metabolites (Fig. 2). Surprisingly, inhibition of a single pathway also had a profound effect on the production of metabolites from other pathways. Specifically, the sEHI t-AUCB (p.o) significantly decreased plasma DHET levels as would be expected from inhibition of sEH (Fig. 2A), but it also reduced the plasma levels of TXB2, 5-HETE, and 15-HETE (Figs. 2B and 2C). As seen before, these secondary effects were greater than the direct effects on the concentration of EETs. The non-selective COX inhibitor aspirin (s.c.) not only significantly inhibited the production of PGE2 and TXB2, as expected from inhibition of COX (Fig. 2B), but also significantly lowered plasma levels of DHETs, 5-HETE, and 15-HETE (Figs. 2A and 2C). The FLAP inhibitor MK 886 (s.c.) not only significantly suppressed the production of 5-HETE as anticipated (Fig. 2C), but also significantly decreased the plasma levels of 15-HETE, PGE2 and TXB2. It also dramatically increased the production of DHETs (especially 8,9-DHET, see Table S1) (Figs. 2A and 2B). These data suggest the presence of a complex cross-talk mechanism between and among distinct branches of the AA cascade.
Fig. 2.
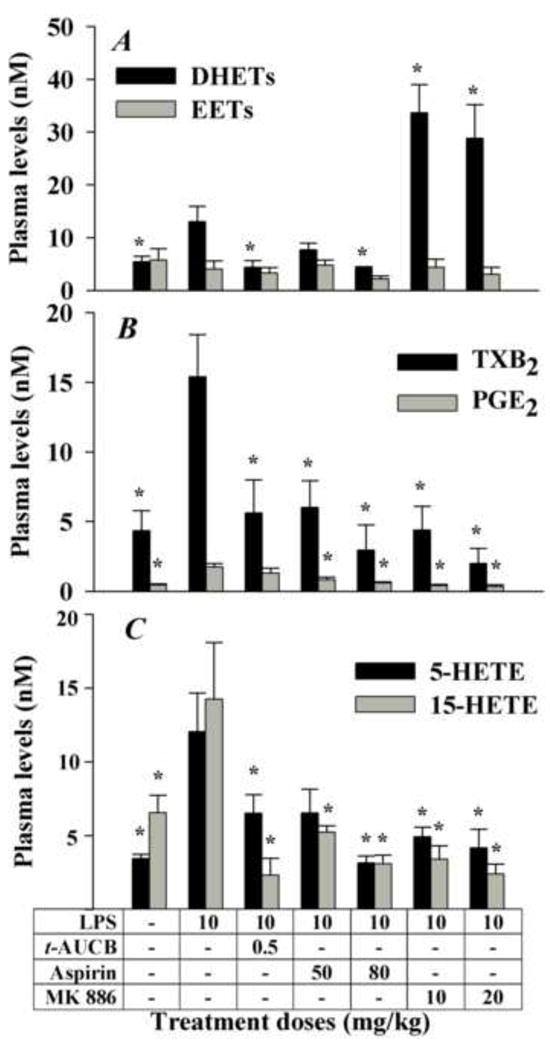
Selective modulation of three branches of the AA cascade in a murine model of inflammation reveals unexpected interactions. The COX and LOX branches were blocked by aspirin and MK 886, respectively. The CYP450 branch was impacted by preventing the degradation of EETs using a sEHI t-AUCB. (A) LPS administration led to a net increase in DHETs (black bars, sum of 5,6-, 8,9-, 11,12- and 14,15-DHETs) levels and to slight changes in EETs (gray bars, sum of 8,9-, 11,12- and 14,15-EETs) levels. A high dose of aspirin and t-AUCB decreased DHET levels. MK886 dramatically increased DHETs while displaying small changes in EETs levels. The data for 5,6-EET are excluded because of lactone formation and hydrolysis during sample preparation. (B) LPS administration led to a net increase in TXB2 (black bars) and PGE2 (gray bars) levels. A low dose of t-AUCB led to a decrease in TXB2 levels but not PGE2 levels. Aspirin and MK886 decreased the levels of both TXB2 and PGE2. (C) LPS administration led to a net increase in both 5-HETE (black bars) and 15-HETE (gray bars). High dose of aspirin, t-AUCB, and MK886 all led to decreases in 5-HETE and 15-HETE levels, while the lower dose of aspirin decreased only15-HETE levels significantly. The data in Figures 2-5 represent average ± SD (n = 4) unless otherwise noted. Animals were sacrificed 6 hours after treatments. * Significantly different from LPS control (P > 0.05) determined by ANOVA followed by Tukey’s posthoc comparison test.
3.2. Effect of co-administration of aspirin and t-AUCB on the plasma levels of TXB2 and PGE2
The above observations prompted us to investigate the interaction among COX, LOX and CYP450 products in shifting oxylipin mediator profiles. Firstly, the hypothesis that production of COX metabolites could be synergistically reduced by co-application of a therapeutic dose of COX inhibitor and a low dose of sEHI was tested. As shown in Fig. 3A a low dose of sEHI (t-AUCB) significantly decreased the plasma levels of TXB2 down to 37 ± 15% of LPS treatment without significantly reducing PGE2 levels. Aspirin (50 mg/kg, s.c.) on the other hand significantly reduced the plasma levels of both the COX-1 product TXB2 and the COX-1/2 product PGE2 down to 39 ± 12% and 48 ± 12%, respectively, compared to the LPS control (Fig. 3A). However when aspirin was co-administrated with t-AUCB (0.5 mg/kg, an ineffective dose in reducing PGE2 levels, p.o.) a synergistic reduction in PGE2 levels (to 21 ± 4%) was observed whereas an at least additive effect in reducing TXB2 (to 16 ± 9%) was evident (Fig. 3A). Inflammation created an imbalance in the TX to prostacyclin ratio as measured by plasma TXB2 and 6-keto-PGF1α levels. This pro-coagulatory ratio was shifted towards an anticoagulatory ratio by the high dose of aspirin. In some cases this change resulting from aspirin treatment may lead to gastrointestinal side effects. Co-administration of the sEH inhibitor with low dose of aspirin interestingly normalized the thromboxane to prostacyclin ratio (co-adminstration vs normal control, 0.64 ± 0.46 vs 0.78 ± 0.38, P = 0.70), despite the observation that the sEH inhibitor itself was less effective in decreasing the COX-2 product 6-keto-PGF1α. This may be beneficial to attenuate the side effect of high dose aspirin. Earlier, using a structurally different sEHI we demonstrated a decrease in hepatic COX-2 protein in inflamed mice [31]. Here this was confirmed using t-AUCB (Fig. S1). However aspirin did not prevent the upregulation of hepatic COX-2 (Fig. S1A), implying that aspirin and t-AUCB affect COX-2 dependent metabolites through independent mechanisms.
Fig. 3.
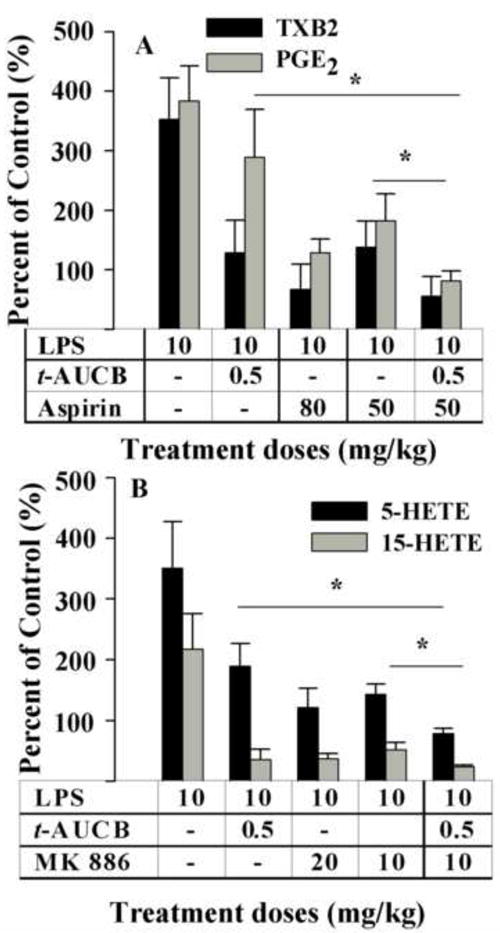
Dual inhibition of two branches of the AA acid cascade is synergistically effective in decreasing inflammatory mediators. (A) LPS administration led to significant increases in both PGE2 (gray bar) and TXB2 (black bar). t-AUCB is ineffective in significantly reducing PGE2 by itself but when co-administered with a low dose of aspirin led to a synergistic reduction in PGE2 (gray bar) levels. TXB2 (black bar) levels were also reduced by t-AUCB, aspirin, and co-administration of t-AUCB with a lower dose of aspirin. Aspirin decreased the production of TXB2 and PGE2 in a dose-related manner. (B) LPS administration led to significant increases in both 5-HETE (black bar) and 15-HETE (gray bar). t-AUCB, effective by itself, however, when co-administered with MK 886, led to a synergistic decrease in the production of both HETEs. MK 886 decreased the production of 5-HETE 15-HETE in a dose-related manner. These data indicate that co-administration of a low dose of t-AUCB with a therapeutic dose of MK 886 can further reduce the levels of proinflammatory molecules 5-HETE and 15-HETE. The data are depicted as percentage of control mice receiving vehicle without LPS. Control values are TXB2, 4.4 ± 1.4, and PGE2, 0.5 ± 0.1 nM, 5-HETE, 3.4 ± 0.3 nM; 15-HETE, 6.6 ± 1.2 nM. * Significantly different (P > 0.05) determined by ANOVA followed by Tukey’s or Games-Howell’s posthoc comparison test.
3.3 Effects of co-administration of MK 886 and t-AUCB on plasma levels of 5-HETE and 15-HETE
LPS challenge led to a dramatic increase in the plasma levels of 5-HETE of 351 ± 77 % of that in the control vehicle treated mice (Fig. 3B). The sEHI t-AUCB (0.5 mg/kg, p.o), significantly reduced these plasma levels to 54 ± 10% of the LPS control level (Fig. 3B). As expected, the FLAP inhibitor MK 886 at 20 or 10 mg/kg (s.c.) significantly decreased the production of 5-HETE level to 40 ± 5 % of the LPS control level. When MK 886 was co-administrated with t-AUCB, the plasma level of 5-HETE was further decreased to 22 ± 2 % of LPS control level (Fig. 3B). Notably, the reduction in the 5-HETE level caused by the t-AUCB/MK 886 combination is well below the level of non-inflamed control mice.
Both t-AUCB and MK 886 also significantly decreased the production of 15-HETE, a 15-LOX and CYP450 mediated AA metabolite. LPS challenge led to a highly significant increase in the plasma levels of 15-HETE to 217 ± 58% of the control vehicle treated mice (Fig. 3B). Upon treatment with t-AUCB (0.5 mg/kg, p.o) or MK 886 (10 mg/kg, s.c.), the plasma levels of 15-HETE significantly decreased to 16 ± 8% and 24 ± 6% of the LPS control, respectively. However when MK 886 was co- administrated with t-AUCB the plasma level of 15-HETE was further decreased to 11 ± 1% of LPS control level. This is significantly different from that of MK 886 alone. Notably, the reduction in the 15-HETE level brought about by the MK 866 and the t-AUCB/MK 886 combination are both well below the levels in non-inflamed control mice. Thus t-AUCB has an unexpected additive effect with MK 886 on reducing the production of 15-HETE. Overall these results demonstrate that co-administration of t-AUCB with MK 886 additively decreases the production of both 5- and 15-HETE potentially through independent mechanisms.
3.4. Effect of targeted disruption of sEH gene on eicosanoid profiles
The effect of sEHI on modulation of the AA cascade was further supported using mice with a targeted disruption of the sEH gene. The LPS challenge led to a significant increase in plasma levels of both COX products PGE2 and PGD2 and the LOX product 5-HETE in both Ephx2-null mice and conspecific wild type mice (Figure 4). However, in Ephx2-null mice, the increase in PGE2 was significantly attenuated (by about two fold), although PGD2 remained equally high (Figs. 4A and 4B). Administration of aspirin (50 mg/kg, s.c.) to LPS treated animals also resulted in decreased plasma levels of PGD2 and PGE2 in both Ephx2-null (0.22 ± 0.02 and 0.07 ± 0.01 nM respectively) and conspecific wild type mice (0.31 ± 0.06 and 0.11 ± 0.03 nM, respectively). Interestingly, aspirin led to a further decrease in both PGE2 and PGD2 in the Ephx2-null mice over the wild type mice (Figs. 4A and 4B).
Fig.4.
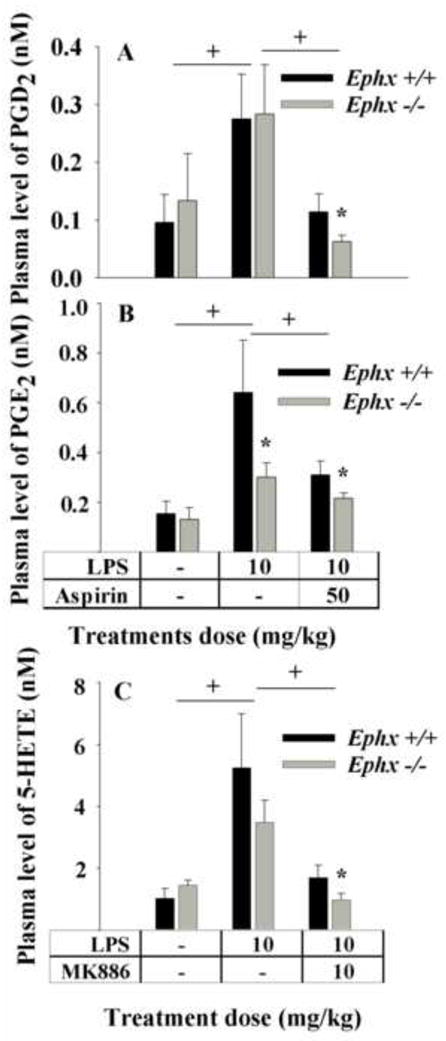
Disruption of the sEH gene in mice demonstrates an inflammatory mediator profile similar to that of a chemical knockout using a sEHI. LPS was administered to Ephx2-null mice (gray bar) and their wildtype conspecifics (black bar) and plasma eicosanoid profiles 6 h post-LPS were determined. (A) The plasma levels of PGD2 in Ephx2-null mice were not different than wild-type counterparts upon LPS administration. However whereas aspirin reduced PGD2 significantly in wild-type mice, a further synergistic reduction of PGD2 was detected in Ephx2-null mice receiving aspirin. (B) The plasma levels of PGE2 in Ephx2-null mice were significantly lower than wild-type mice upon inflammation, pointing out lower COX activity in the Ephx2-null animals. However whereas aspirin reduced PGE2 significantly in wild-type mice, a further reduction of PGE2 was detected in Ephx2-null mice receiving aspirin. (C) The plasma levels of 5-HETE in both Ephx2-null (n=4) and wild-type (n = 6) mice significantly increased after LPS administration. MK886 administration reduced 5-HETE levels in both strains. However, the Ephx2-null mice had still lower levels of 5-HETE after MK 886 administration. † denotes significant difference, * significant difference from wild mice with same treatment (P > 0.05) determined by ANOVA followed by Tukey’s posthoc comparison test.
The increase in 5-HETE in Ephx2-null (3.49 ± 0.70 nM) and wild type (5.25 ± 1.74 nM) mice was equally high upon LPS treatment (Fig. 4C). As expected, MK 886 treatment led to a decrease in the production of 5-HETE in Ephx2-null (0.98 ± 0.21 nM) and wild type (1.70 ± 0.40 nM) animals, respectively.
The EET and DHET levels in wild-type and Ephx2-null mice upon LPS, aspirin and MK886 administration were as expected (Table S2). The Ephx2-null mice had higher baseline levels of EETs compared to the wild-type counterparts. LPS administration raised DHET levels in wild-type but not Ephx2-null mice. Aspirin and MK886 decreased DHET levels only in wild-type mice. The use of t-AUCB in C57BL mice did not increase the plasma level of EETs while Ephx2-null mice have a significantly higher level of EET, which suggest that the accumulation of EETs may be a time- and/or dose-related process. This also cautions that the blood levels many not reflect the concentrations of eicosanoids in key tissues.
Notably, the non-inflamed Ephx2-null mice had about two fold lower baseline hepatic 5-LOX protein levels than wild type animals (Fig. S2). Inflammation in these animals led to a significant induction in hepatic 5-LOX protein. In addition, treatment with MK 886 significantly lowered baseline 5-LOX protein level to below non-inflamed levels in both sEH null and wild type mice (Figure S2). However, 5-HETE is not only a product of 5-LOX, but also a product of CYP 450 from AA [51]. This may explain the lack of reduction in 5-HETE in normal and LPS-challenged Ephx2-null mice although their 5-LOX protein levels are significantly lower than those of the conspecific widetype mice.
3.5. Anti-hypotensive and anti-inflammatory effects of aspirin, MK 886 and t-AUCB
Morbid hypotension is a hallmark of LPS-induced septic shock in mice [52]. Upon LPS challenge in current system, the systolic blood pressure (BP) of mice dropped under the detection limit (40 mmHg) (Fig. 5A). Therapeutic administration of aspirin (40 or 80 mg/kg, s.c.), or the FLAP inhibitor MK 886 (10 or 20 mg/kg, s.c) produced a dose-dependent anti-hypotensive effect (Fig. 5A). The sEHI t-AUCB (p.o) as well significantly reversed the LPS-induced hypotension in a dose-dependent manner (Fig. 5A). Co-administration of the low dose of t-AUCB (0.5 mg/kg, p.o.) with the low dose of aspirin (50 mg/kg, s.c.) or MK 886 (10 mg/kg, s.c.) produced further anti-hypotensive activity (Fig. 5A).
Fig. 5.
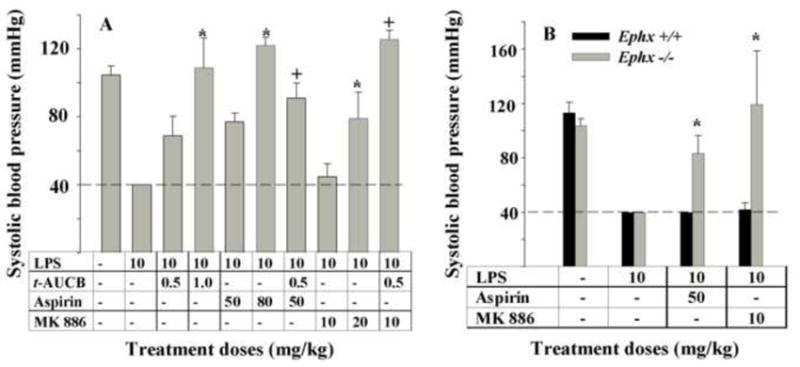
Selective modulation of branches of the AA cascade during a murine model of inflammation results in unexpected anti-hypotensive effects. Animals were treated as detailed in Fig.2 and systolic blood pressure was monitored 4 h after LPS administration. (A) LPS administration led to a profound decrease in blood pressure that was lower than the detection limit (40 mm Hg) of the instrument used (dashed line). Both doses of t-AUCB, and aspirin significantly reversed LPS-induced hypotension but MK 886 was effective only at a high dose. However, co-administration of the low doses of t-AUCB and MK 886 led to a synergistic reversal of blood pressure. Similarly, co-administration of t-AUCB and aspirin resulted in stronger reversal of hypotension. *Significantly different from its lower dose alone (P > 0.05) and † significantly different from the individual inhibitors alone as determined by ANOVA followed by Tukey’s posthoc comparison test. (B) The baseline systolic blood pressure of wild-type (black bar, n = 6) and Ephx2-null (gray bar, n = 4) mice were not different and LPS administration led to a similar decrease in both strains of mice. Aspirin or MK886 administration both led to reversal of hypotension only in the Ephx2-null mice but not in wild-type conspecifics. * Significantly different from Ephx2 wild mice control (P > 0.05) determined by by ANOVA followed by Tukey’s posthoc comparison test.
Plasma levels of interleukin-6 also reflect the inflammatory status in this model of inflammation [53]. Plasma concentrations of IL-6 are presented in Table S3. LPS administration significantly increased the release of IL-6 at both 6 and 24 hour. Administration of t-AUCB stimulated the release of IL-6 at 6 hour but significantly reduced IL-6 concentration at 24 h. Administration of aspirin significantly inhibited the production of IL-6 at 6 hours but had no effect on IL-6 production at 24 hour. Administration of MK 886 stimulated the production of IL-6 at both 6 and 24 hour. To our surprise, the beneficial effects of additively lowering the production of IL-6 were not observed during co-administration of t-AUCB with aspirin or MK 886 at 6 hour. However, the expected beneficial results were observed at 24 hour.
3.6. Anti-hypotensive effect of aspirin and MK886 in mice with targeted disruption of the sEH gene
The effect of t-AUCB on hypotension was supported using Ephx2-null mice. LPS treatment led to hypotension in both colonies of mice decreasing systolic blood pressure to under the detection limit (40 mmHg) (Fig. 5B). However, Ephx2-null mice recovered significantly better towards the normotensive state when given aspirin or MK 886 than wild type animals (Fig. 5B). These observations along with eicosanoid profiles of Ephx2-null mice demonstrate that genetic and pharmacological inhibition of sEH equally results in anti-hypotensive and anti-inflammatory effects that can be further enhanced with aspirin and MK 886.
4. Discussion
The metabolomic profiling produced independent information that can be interpreted in terms of known biochemical pathways and physiological interactions [41, 54]. The above observations demonstrated that inhibition of sEH suppresses the action of COX and LOX pathways indicated both by the decrease in production of PGE2 and 5-HETE, and the repressed expression at the protein level. The inhibitors used do not affect the other arachidonic acid metabolizing enzymes investigated by in vitro bioassays. In the current and previous studies we extensively tested the hypothesis that the observed effects are not inhibitor specific but stem from inhibition of sEH [31]. The chemical knockout with sEHIs and genetic knockout of sEH gene resulted in similar metabolite patterns and biological effects indicating that the beneficial anti-hypotensive and anti-inflammatory effects are mostly due to the inhibition of the sEH pathway.
Interestingly, inhibition of COX or LOX pathways also had a significant impact on the other two pathways indicating that these pathways do not proceed in a parallel way but communicate in a dynamic manner. Inhibition of COX or LOX pathways had minimal effect on the levels of CYP450 produced EETs in this study (Figure 2). However, inhibition of the COX pathway significantly reduced the levels of the sEH produced DHETs, and the LOX products 5-HETE and 15-HETE potentially due to the feedback regulation of the anti-inflammatory effect from each individual inhibitor. Inhibition of the LOX pathway by MK 886 significantly decreased the production of COX products TXB2 and PGE2 while unexpectedly increasing the production of DHETs (Fig. 2). This increase in DHETs is under investigation. The differences in DHET levels between C57BL/6 and Ephx+/+ mice with the administration of LPS and MK 886 may due to the difference in strain and/or age of the mice used (Tables S1 and S2). Although FLAP inhibitors have not been clinically used to date, they have been demonstrated to be effective in inhibiting LT biosynthesis for the attenuation of atherosclerosis, adipose tissue inflammation [55, 56], liver injury [57], acute inflammation [58], pulmonary vascular reactivity, and pulmonary hypertension [59]. Here we demonstrated that co-administration of sEHI produced a beneficial anti-inflammatory effect observed as normalization of sepsis induced hypotension and reduced plasma level of 5-HETE. Parallel results were observed in Ephx2-null mice. Overall, reduced 5-HETE and 5-oxo-ETE (Fig. S3) concentration, the therapeutic anti-hypotensive and anti-inflammatory effects and the reduced 5-LOX protein expression in C57BL and Ephx2-null mice strongly suggest that simultaneous inhibition of sEH enhances the anti-inflammatory activity of FLAP inhibitors. These observations highlight a novel treatment for inflammation by simultaneous inhibition of sEH and the 5-LOX pathway.
One can presume that inhibition of the LOX pathway shifts the AA metabolism towards the other pathways by mass action in which case an increase in COX and CYP450 products should then be observed [31]. This has been predicted and shown in an in vitro cancer cell line system for COX and LOX products where inhibition of either COX-2 or 5-LOX alone resulted in activation of the other pathway in colon cancer cells [60]. This increase needs to be further investigated. In contrast, our data suggest that metabolism of AA by COX, LOX and CYP450 family enzymes does not only follow a simple mass flow rule in this sepsis model and each inhibitor used is anti-inflammatory due to a combination of effects on multiple pathways. Overall, these findings provide a rational justification for the common strategy of co-administration of pathway blockers to achieve better resolution of inflammation. Co-administration of nonsteroidal anti-inflammatory drugs (NSAIDs) is a popular therapeutic treatment of inflammation for the general purposes of maximizing the therapeutic effects and minimizing the adverse effects of the drugs [61]. Although the mechanisms by which aspirin asserts its many therapeutic effects are not totally clear, aspirin was used as a non-specific COX inhibitor different from the NSAIDs (indomethacin, rofecoxib and celebrex) previously tested [31]. The side effects of aspirin use in humans are well described and include clotting disorders, abdominal pain and other gastrointestinal problems [62, 63]. These effects are dose related [63]. Here we demonstrated in a murine model that the dose of aspirin can be reduced by co-administering sEHI while retaining the anti-inflammatory effect. We expect this may decrease at least some of the side effects attributed to inhibition of COX-1. Indeed, here the sEHI when combined with aspirin did not lead to a dramatic change in the ratio of PGI2 to TXA2, an indicator of blood clotting time (Table S1).
The fact that aspirin treatment had no effect in reducing the release of IL-6 at 24 h is likely due to its short half life. MK-886 did not repress the production of IL-6 at both 6 and 24 h, which could result from the low dose used because the co-dosing or t-AUCB and MK 886 had a significantly beneficial effect in reducing the production of IL-6. This suggests that the inflammation in this murine model is still exacerbating at 6 hour. This also indicates different anti-inflammatory actions and/or mechanisms of the three inhibitors used. In addition, the expected changes of oxylipin mediators (e.g. PGE2 and 5-HETE) and cytokines (e.g. IL-6) did not occur simultaneously, which suggests more complicated temporal roles of oxylipins and cytokines in regulation of inflammation.
In summary, by using the metabolomic profiling approach the current study demonstrates that inhibition of either COX, 5-LOX, or sEH enzymes has a profound impact on the global AA metabolism in an LPS-challenged murine model. In addition, inhibition of sEH by either pharmacological intervention with sEHI or gene disruption enhances the anti-inflammatory effects of COX and LOX inhibitors. It should also be noted that sEHIs are powerful anti-inflammatory agents on their own. A caution regarding these data is that both the propagation and resolution of the acute inflammatory response are dynamic as addressed in previous studies from this and many other laboratories. However, these measurements are made at two times post dosing based on previous temporal studies and monitoring the symptoms of treated animals. That said, these results demonstrate that sEHIs may have therapeutic utility not only as alonestand agents but also in combination with low doses of COX and LOX inhibitors for the treatment of inflammation. Furthermore in various other disease states, in which PG, LT, or epoxy fatty acid concentrations are altered in unfavorable ways, inhibitors of sEH have potential as therapeutic agents. Further exploration of the interactions among many pathways of AA metabolism may provide additional approaches to managing inflammatory diseases.
Supplementary Material
Acknowledgments
This work was supported in part by NIEHS R37 ES02710, NIEHS Superfund P42 ES04699, NIH R01 HL85727 and The America Asthma Association.
Abbreviations
- AA
arachidonic acid
- t-AUCB
trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid
- AUDA-BE
12-(3-adamantan-1-yl-ureido)-dodecanoic acid butyl ester
- COX
cyclooxygenase(s)
- CYP450
cytochrome P450
- DHET
dihydroxyeicosatrienoic acid
- EET
epoxyeicosatrienoic acids
- FLAP
5-lipoxygenase activation protein
- 5-HETE
5-hydroxyeicosatetrasanoic acid
- 5-HpETE
5-(S)-hydroperoxy-6,8,11,14-eicosatetraenoic acid
- LOX
lipoxygenase
- LPS
lipopolysaccharide
- LT
leukotriene
- MK 886
3-[1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-2-yl]-2,2-dimethyl propanoic acid
- NSAID
nonsteroidal anti-inflammatory drug
- PG
prostaglandin
- sEH
soluble epoxide hydrolase
- sEHI
sEH inhibitor
- TX
thromboaxane
Footnotes
Appendix Supplementary material.
Conflict of interest statement BDH is the founder of Arête Therapeutics. This company is moving sEHI through clinical trials for treating hypertension, pain, metabolic disease, inflammation, and other disorders. However, this study is independent from the company.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levick SP, Loch DC, Taylor SM, Janicki JS. Arachidonic acid metabolism as a potential mediator of cardiac fibrosis associated with inflammation. J Immunol. 2007;178:641–6. doi: 10.4049/jimmunol.178.2.641. [DOI] [PubMed] [Google Scholar]
- 2.Sciulli MG, Capone ML, Tacconelli S, Patrignani P. The future of traditional nonsteroidal antiinflammatory drugs and cyclooxygenase-2 inhibitors in the treatment of inflammation and pain. Pharmacol Rep. 2005;57(Suppl):66–85. [PubMed] [Google Scholar]
- 3.Claria J, Romano M. Pharmacological intervention of cyclooxygenase-2 and 5-lipoxygenase pathways. Impact on inflammation and cancer. Current pharmaceutical design. 2005;11:3431–47. doi: 10.2174/138161205774370753. [DOI] [PubMed] [Google Scholar]
- 4.Wickelgren I. Heart disease. Gene suggests asthma drugs may ease cardiovascular inflammation. Science. 2004;303:941. doi: 10.1126/science.303.5660.941a. [DOI] [PubMed] [Google Scholar]
- 5.Chen XX, Sood S, Yang CS, Li N, Sun Z. Five-lipoxygenase pathway of arachidonic acid metabolism in carcinogenesis and cancer chemoprevention. Curr Cancer Drug Tar. 2006;6:613–22. doi: 10.2174/156800906778742451. [DOI] [PubMed] [Google Scholar]
- 6.Evans JF, Ferguson AD, Mosley RT, Hutchinson JH. What’s all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci. 2008;29:72–8. doi: 10.1016/j.tips.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Peters-Golden M. Cell biology of the 5-lipoxygenase pathway. Am J Respir Crit Care Med. 1998;157:S227–32. [PubMed] [Google Scholar]
- 8.Ferguson AD, McKeever BM, Xu S, Wisniewski D, Miller DK, Yamin TT, et al. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science. 2007;317:510–2. doi: 10.1126/science.1144346. [DOI] [PubMed] [Google Scholar]
- 9.Ago H, Kanaoka Y, Irikura D, Lam BK, Shimamura T, Austen KF, et al. Crystal structure of a human membrane protein involved in cysteinyl leukotriene biosynthesis. Nature. 2007;448:609–12. doi: 10.1038/nature05936. [DOI] [PubMed] [Google Scholar]
- 10.Charlier C, Michaux C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur J Med Chem. 2003;38:645–59. doi: 10.1016/s0223-5234(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 11.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 12.Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol Sci. 2000;21:125–7. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- 13.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. P Natl Acad Sci USA. 2006;103:18733–8. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Node K, Huo YQ, Ruan XL, Yang BC, Spiecker M, Ley K, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol-Renal. 2005;289:F496–F503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 16.Sarkis A, Roman RJ. Role of cytochrome P450 metabolites of arachidonic acid in hypertension. Curr Drug Metab. 2004;5:245–56. doi: 10.2174/1389200043335603. [DOI] [PubMed] [Google Scholar]
- 17.Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ. Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostag Oth Lipid M. 2007;82:50–9. doi: 10.1016/j.prostaglandins.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elbekai RH, El-Kadi AOS. Cytochrome P450 enzymes: Central players in cardiovascular health and disease. Pharmacol Therapeut. 2006;112:564–87. doi: 10.1016/j.pharmthera.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Doggrell SA. Cytochrome P-450: a new target in the heart and coronary circulation. Drug Future. 2005;30:261–9. [Google Scholar]
- 20.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Morisseau C, Schmelzer K, Pedersen T, Eiserich J, Pinkerton K, Imig J, et al. Physiological roles of soluble epoxide hydrolase and therapeutic prospects of its inhibition. Faseb J. 2006;20:A669–A. [Google Scholar]
- 22.Imig JD, Zhao XY, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, et al. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46:975–81. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao XY, Inscho EW, Morisseau C, Hammock BD, Imig JD. Soluble epoxide hydrolase regulates renal microvascular angiotensin II reactivity in hypertension. Faseb J. 2002;16:A418–A. [Google Scholar]
- 24.Imig JD, Zhao XY, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–4. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 25.Imig JD. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc Drug Rev. 2006;24:169–88. doi: 10.1111/j.1527-3466.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 26.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. P Natl Acad Sci USA. 2005;102:9772–7. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. P Natl Acad Sci USA. 2005;102:2186–91. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olearczyk JJ, Quigley JE, Mitchell BC, Yamamoto T, Kim IH, Newman JW, et al. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond) 2009;116:61–70. doi: 10.1042/CS20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs) Prostag Oth Lipid M. 2007;82:42–9. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng VY, Morisseau C, Falck JR, Hammock BD, Kroetz DL. Inhibition of smooth muscle proliferation by urea-based alkanoic acids via peroxisome proliferator-activated receptor alpha-dependent repression of cyclin D1. Arterioscl Throm Vas. 2006;26:2462–8. doi: 10.1161/01.ATV.0000242013.29441.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, et al. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. P Natl Acad Sci USA. 2006;103:13646–51. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seubert JM, Sinal CJ, Graves J, DeGraff LM, Bradbury JA, Lee CR, et al. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–50. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis BB, Morisseau C, Newman JW, Pedersen TL, Hammock BD, Weiss RH. Attenuation of vascular smooth muscle cell proliferation by 1-cyclohexyl-3-dodecyl urea is independent of soluble epoxide hydrolase inhibition. J Pharmacol Exp Ther. 2006;316:815–21. doi: 10.1124/jpet.105.091876. [DOI] [PubMed] [Google Scholar]
- 34.Jin LM, Foss CE, Zhao XY, Mills TM, Wang MH, McCluskey LP, et al. Cytochrome P450 epoxygenases provide a novel mechanism for penile erection. Faseb J. 2006;20:539–41. doi: 10.1096/fj.05-4341fje. [DOI] [PubMed] [Google Scholar]
- 35.Borgdorff P, Tangelder GJ, Paulus WJ. Cyclooxygenase-2 inhibitors enhance shear stress-induced platelet aggregation. J Am Coll Cardiol. 2006;48:817–23. doi: 10.1016/j.jacc.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 36.Domoki F, Nagy K, Temesvari P, Bari F. Selective inhibitors differentially affect cyclooxygenase-dependent pial arteriolar responses in newborn pigs. Pediatr Res. 2005;57:853–7. doi: 10.1203/01.PDR.0000161415.62776.0A. [DOI] [PubMed] [Google Scholar]
- 37.Leach M, Hamilton LC, Olbrich A, Wray GM, Thiemermann C. Effects of inhibitors of the activity of cyclo-oxygenase-2 on the hypotension and multiple organ dysfunction caused by endotoxin: A comparison with dexamethasone. Brit J Pharmacol. 1998;124:586–92. doi: 10.1038/sj.bjp.0701869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HWL, et al. Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using H-1-NMR-based metabonomics. Nat Med. 2002;8:1439–44. doi: 10.1038/nm1202-802. [DOI] [PubMed] [Google Scholar]
- 39.Lewis GD, Wei R, Liu E, Yang E, Shi X, Martinovic M, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. J Clin Invest. 2008;118:3503–12. doi: 10.1172/JCI35111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindon JC, Keun HC, Ebbels TMD, Pearce JMT, Holmes E, Nicholson JK. The Consortium for Metabonomic Toxicology (COMET): aims, activities and achievements. Pharmacogenomics. 2005;6:691–9. doi: 10.2217/14622416.6.7.691. [DOI] [PubMed] [Google Scholar]
- 41.Fiehn O. Metabolomics - the link between genotypes and phenotypes. Plant Mol Biol. 2002;48:155–71. [PubMed] [Google Scholar]
- 42.Wikoff WR, Pendyala G, Siuzdak G, Fox HS. Metabolomic analysis of the cerebrospinal fluid reveals changes in phospholipase expression in the CNS of SIV-infected macaques. J Clin Invest. 2008;118:2661–9. doi: 10.1172/JCI34138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative Profiling Method for Oxylipin Metabolome by Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry. Anal Chem. 2009;81:8085–93. doi: 10.1021/ac901282n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–10. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 45.Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang HP, et al. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem. 2007;282:2891–8. doi: 10.1074/jbc.M608057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock B. Orally Bioavailable Potent Soluble Epoxide Hydrolase (sEH) Inhibitors. Journal of Medicinal Chemistry. 2007;50:3825–40. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cyrus T, Sung S, Zhao L, Funk CD, Tang S, Pratico D. Effect of low-dose aspirin on vascular inflammation, plaque stability, and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2002;106:1282–7. doi: 10.1161/01.cir.0000027816.54430.96. [DOI] [PubMed] [Google Scholar]
- 48.Paul L, Fraifeld V, Kaplanski J. Evidence supporting involvement of leukotrienes in LPS-induced hypothermia in mice. Am J Physiol-Reg I. 1999;276:R52–R8. doi: 10.1152/ajpregu.1999.276.1.R52. [DOI] [PubMed] [Google Scholar]
- 49.Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase (sEH) inhibitors for use in a murine model of inflammation. Brit J Pharmacol. 2009;156:284–96. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu JY, Park SH, Morisseau C, Hwang SH, Hammock BD, Weiss RH. Sorafenib has soluble epoxide hydrolase inhibitory activity, which contributes to its effect profile in vivo. Mol Cancer Ther. 2009;8:2193–203. doi: 10.1158/1535-7163.MCT-09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capdevila JH, Falck JR, Estabrook RW. Cytochrome-P450 and the Arachidonate Cascade. Faseb J. 1992;6:731–6. doi: 10.1096/fasebj.6.2.1537463. [DOI] [PubMed] [Google Scholar]
- 52.Weinberg JR, Boyle P, Meager A, Guz A. Lipopolysaccharide, Tumor-Necrosis-Factor, and Interleukin-1 Interact to Cause Hypotension. J Lab Clin Med. 1992;120:205–11. [PubMed] [Google Scholar]
- 53.Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–20. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dettmer K, Hammock BD. Metabolomics - A new exciting field within the “omics” sciences. Environ Health Persp. 2004;112:A396–A7. doi: 10.1289/ehp.112-1241997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Back M, Sultan A, Ovchinnikova O, Hansson GK. 5-Lipoxygenase-activating protein: a potential link between innate and adaptive immunity in atherosclerosis and adipose tissue inflammation. Circulation research. 2007;100:946–9. doi: 10.1161/01.RES.0000264498.60702.0d. [DOI] [PubMed] [Google Scholar]
- 56.Jawien J, Gajda M, Rudling M, Mateuszuk L, Olszanecki R, Guzik TJ, et al. Inhibition of five lipoxygenase activating protein (FLAP) by MK-886 decreases atherosclerosis in apoE/LDLR-double knockout mice. Eur J Clin Invest. 2006;36:141–6. doi: 10.1111/j.1365-2362.2006.01606.x. [DOI] [PubMed] [Google Scholar]
- 57.Titos E, Claria J, Planaguma A, Lopez-Parra M, Gonzalez-Periz A, Gaya J, et al. Inhibition of 5-lipoxygenase-activating protein abrogates experimental liver injury: role of Kupffer cells. J Leukocyte Biol. 2005;78:871–8. doi: 10.1189/jlb.1204747. [DOI] [PubMed] [Google Scholar]
- 58.Byrum RS, Goulet JL, Griffiths RJ, Koller BH. Role of the 5-lipoxygenase-activating protein (FLAP) in murine acute inflammatory responses. J Exp Med. 1997;185:1065–75. doi: 10.1084/jem.185.6.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Voelkel NF, Tuder RM, Wade K, Hoper M, Lepley R, Goulet JL, et al. Inhibition of 5-lipoxygenase-activating protein (FLAP) reduces pulmonary vascular reactivity and pulmonary hypertension in hypoxic rats. J Clin Invest. 1996;97:2491–8. doi: 10.1172/JCI118696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, Schiavone N, et al. Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther. 2006;5:2716–26. doi: 10.1158/1535-7163.MCT-06-0318. [DOI] [PubMed] [Google Scholar]
- 61.Picard P, Bazin JE, Conio N, Ruiz F, Schoeffler P. Ketorolac potentiates morphine in postoperative patient-controlled analgesia. Pain. 1997;73:401–6. doi: 10.1016/S0304-3959(97)00128-0. [DOI] [PubMed] [Google Scholar]
- 62.Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug-drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. Can Med Assoc J. 2007;177:347–51. doi: 10.1503/cmaj.070186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Graham DY, Smith JL. Aspirin and the stomach. Annals of internal medicine. 1986;104:390–8. doi: 10.7326/0003-4819-104-3-390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.