

Abstract
Objectives
To examine the role of lipoprotein-associated phospholipase A2 (Lp-PLA2/PLA2G7) in human inflammation and coronary atherosclerosis.
Background
Lp-PLA2 has emerged as a potential therapeutic target in coronary heart disease (CHD). Data supporting Lp-PLA2 are indirect and confounded by species differences; whether Lp-PLA2 is causal in CHD remains in question.
Methods
We examined inflammatory regulation of Lp-PLA2 during experimental endotoxemia in human, probed the source of Lp-PLA2 in human leukocytes under inflammatory conditions, and assessed the relationship of variation in PLA2G7, the gene encoding Lp-PLA2, with coronary artery calcification (CAC).
Results
In contrast to circulating TNFα and CRP, blood and monocyte Lp-PLA2 mRNA decreased transiently, and plasma Lp-PLA2 mass declined modestly during endotoxemia. In vitro, Lp-PLA2 expression increased dramatically during human monocyte to macrophage differentiation and further in inflammatory macrophages and foam like-cells. Despite only a marginal association of SNPs in PLA2G7 with Lp-PLA2 activity or mass, numerous PLA2G7 SNPs were associated with CAC. In contrast, several SNPs in CRP were significantly associated with plasma CRP levels but had no relation with CAC.
Conclusions
Circulating Lp-PLA2 did not increase during acute phase response in human, while inflammatory macrophages and foam cells, but not circulating monocytes, are major leukocyte sources of Lp-PLA2. Common genetic variation in PLA2G7 is associated with sub-clinical coronary atherosclerosis. These data link Lp-PLA2 to atherosclerosis in human while highlighting the challenge in using circulating Lp-PLA2 as a biomarker of Lp-PLA2 actions in the vasculature.
Keywords: Lp-PLA2, PLA2G7, CAC
INTRODUCTION
Lipoprotein-associated phospholipase A2 (Lp-PLA2) has emerged as a potential therapeutic target in coronary heart disease (CHD) and phase III clinical trials are underway. Supporting evidence includes apparent atherogenic biochemical properties; Lp-PLA2 cleaves oxidized phosphatidylcholine on modified LDL producing inflammatory lysophosphatidylcholine and oxidized non-esterified fatty acids (1,2). In addition, enzymatic expression of Lp-PLA2 is up-regulated in human atherosclerosis (3), while circulating levels are associated with incident CHD (4). Promising proof-of-principle pre-clinical and clinical trials have been carried out (5–7). However, whether Lp-PLA2 is causal and whether its inhibition will prevent CHD events remain open questions.
Data for Lp-PLA2 in human atherosclerosis remain indirect and confounded by species differences in physiology and actions. Lp-PLA2 circulates in blood, bound to lipoproteins which modulate its actions. In rodents Lp-PLA2 is carried mostly on high-density lipoprotein (HDL) particles whereas in human the enzyme is bound to low-density lipoprotein (LDL) particles. Thus, confounding may be particularly marked for plasma Lp-PLA2 relative to other inflammatory markers, as regulation of atherogenic lipoproteins is a major influence on circulating Lp-PLA2 levels and activity (8). Indeed, whether circulating Lp-PLA2 is associated with CHD beyond a complete assessment of atherogenic lipoproteins remains uncertain (9).
Arterial Lp-PLA2 biosynthesis by macrophages and foam cells, rather than circulating levels or activity, may determine its atherogenicity (10). Lp-PLA2 expression within the necrotic core and surrounding macrophages of vulnerable and ruptured plaques is increased compared with less-advanced lesions (11), suggesting a potential role in promoting plaque instability. The extent to which human Lp-PLA2 is regulated in circulation by systemic inflammation, however, versus locally controlled in arterial macrophage-foam cells is uncertain. Further, lesion biosynthesis is difficult to measure in human limiting our ability to monitor Lp-PLA2 activity in disease-relevant tissue and to assess vascular efficacy of pharmacological inhibition.
In this report, we examined inflammatory regulation of circulating Lp-PLA2 during experimental endotoxemia in human, probed the source of Lp-PLA2 in human leukocytes under inflammatory conditions, and determined the relationship of genetic variation in phospholipase A2, group VII (PLA2G7), the gene encoding Lp-PLA2, to coronary artery calcification (CAC) as well as plasma levels of Lp-PLA2 mass and activity. We found that, unlike blood tumor necrosis factor alpha (TNFα) and C-reactive protein (CRP), circulating Lp-PLA2 did not increase during the acute phase response in human, that inflammatory macrophages and foam cells, but not circulating or ex vivo monocytes, are primary leukocyte sources of Lp-PLA2, and that common genetic variation in PLA2G7 is associated with sub-clinical coronary atherosclerosis. These data link Lp-PLA2 to atherosclerosis in human while providing a human physiological context for the difficulty in using circulating Lp-PLA2 as a biomarker of disease or pharmacological efficacy in atherosclerosis.
METHODS
Clinical Studies
Human endotoxemia
Healthy volunteers on no medications and no significant medical history (N=32, 50% female; mean age 25.7±3.90) were studied as described previously (12,13) and in the supplement. Serial blood samples were collected before and after intravenous bolus infusion of 3 ng/kg US standard reference endotoxin and were prepared for plasma, whole-blood RNA and monocyte RNA (12).
Genetic association studies
The Penn Coronary Artery Calcification (PennCAC) resource included European-ancestry subjects recruited to three separate studies at U.Penn: the Study of Inherited Risk of Coronary Atherosclerosis (SIRCA; N=799), the Penn Diabetes Heart Study (PDHS; N=782), and the Philadelphia Area Metabolic Syndrome Network (PAMSyN; N=480). These studies are described in detail previously (14,15) and in the supplement. In each study, subjects with clinical atherosclerotic CVD were excluded. PLA2G7 SNPs were genotyped in all three studies. Plasma Lp-PLA2 mass and activity data were available in SIRCA and PDHS. Global CAC scores were determined by electron beam tomography (Imatron, San Francisco, CA) according to the method of Agatston(16). For all human studies described, the University of Pennsylvania (U.Penn) Institutional Review Board (IRB) approved each study and written informed consent was provided by all participants.
Laboratory Methods
Human monocyte, macrophage and foam cell studies
Human moncoyte isolation, macrophage (“M1” and “M2” phenotype) differentiation (17) and “foam cell” preparation was performed as described (12) and in supplement. Experiments were performed in batches using freshly-isolated monocytes, macrophages and foam cells derived from the same human volunteer.
Plasma LpPLA2, inflammatory and metabolic markers
Plasma and cell-media levels of Lp-PLA2 mass and activity, TNFα, and CRP, as well as lipid and biochemical markers were measured as described (13,14,18) and in supplement.
Real-time quantitative PCR and expression quantitative trait locus analysis
Whole-blood, isolated circulating monocyte, and human cultured monocyte, macrophage and foam cell mRNA was subjected to quantitative PCR (qPCR) using primers and probes (Applied Biosystems 7300 Real-Time PCR System, Foster City, CA) as described (12) for measurement of Lp-PLA2, TNFα, and β-actin mRNA (supplement). The relative quantitation 2−(ΔΔCt) method was used to determine fold-change from baseline (19). Exploratory expression Quantitative Trait Locus (eQTL) analysis is described in the supplement.
Genotyping
As described previously (20) and in the supplement, PennCAC participants were genotyped using the ITMAT Broad Care (IBC) CVD candidate gene array, which surveys ~50,000 SNPs in ~2,000 candidate genes (21). SNP data for PLA2G7 (N=19) and CRP (N=16) were selected for current analysis.
Statistical analysis
The effect of endotoxemia on plasma Lp-PLA2 mass and activity, TNFα, and CRP as well as whole-blood and monocyte mRNA was tested by repeated-measures analysis of variance (ANOVA). ANOVA was applied also to in vitro cell data. Post hoc t-tests were used to compare specific time-points and treatments. We observed heterogeneity of variance in several variables following LPS challenge, which was to be expected given the known variation in responses to endotoxin. We tested for homogeneity of variance using Levene’s test, and in cases where the assumption of homogeneity of variance was violated, we confirmed whether the group differences were significant using Tamhane’s post-hoc test.
In PennCAC, CAC scores were transformed by the natural log after adding 1 (Ln(CAC+1)), to correct for skewed distribution. This variable was used as the outcome in a linear regression model, with PLA2G7 and CRP SNPs, adjusting for age, gender and age-gender interaction. For linear regression analysis of SNP associations with plasma proteins, Lp-PLA2 mass and activity were normally distributed, and therefore used as outcomes while CRP was log-transformed. The linear regression model included adjustments for age, gender, and smoking. Analysis used PLINK v 1.06. Analyses of CAC and plasma proteins were performed separately in each sample and then subjected to meta-analysis. Meta-analysis applied a weighted Z-score method using METAL(22) (http://www.sph.umich.edu/csg/abecasis/Metal) as we applied in (23), described in supplement. In analysis of SNP data, we corrected for the number of independent tests within each gene (10 tests for 19 PLA2G7 SNPs, unadjusted P value threshold of 0.005, and 15 tests for 15 CRP SNPs, unadjusted P value threshold of 0.0033) using the method of Nyholt (24).
RESULTS
Lp-PLA2 is not induced in a human model of acute phase response
As we described (13,25) endotoxemia produced an acute, febrile illness associated with a marked, transient induction of plasma TNFα (P <0.001), followed by a delayed ~100-fold induction of plasma CRP at 24 hours (P<0.001) (Figure 1A). In contrast, plasma Lp-PLA2 mass and activity did not increase following LPS (Figure 1B). Indeed, levels of Lp-PLA2 mass tended to decline (by 18% at 6hours, P<0.01). The mRNA response to LPS in whole-blood for TNFα (Figure 1C) and Lp-PLA2 (Figure 1D) as well as in circulating monocytes for TNFα (Figure 1E) and Lp-PLA2 (Figure 1F) was similar to that of plasma proteins. The mRNA levels of Lp-PLA2 in circulating monocytes were low but detectable (baseline CTs~30, varying from CTs of 28–32 post-LPS).
Figure 1. Human endotoxemia does not induce circulating Lp-PLA2 protein or leukocyte Lp-PLA2 mRNA in vivo.



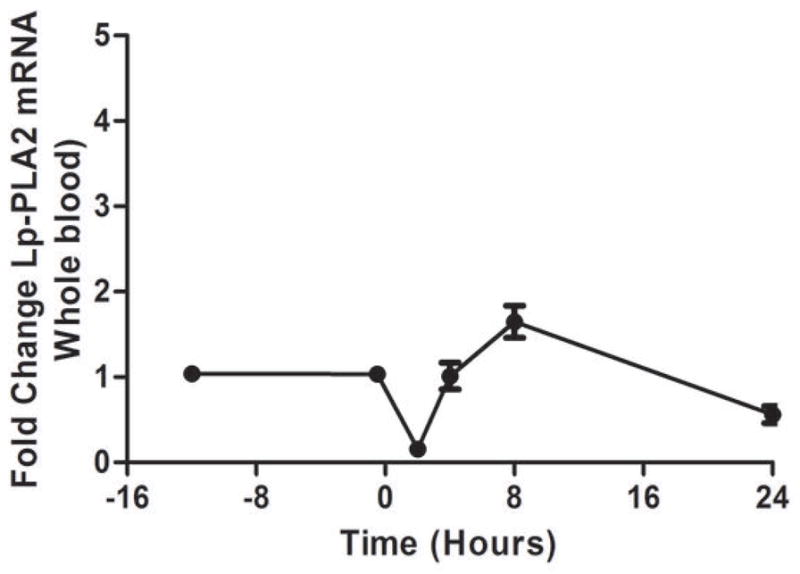
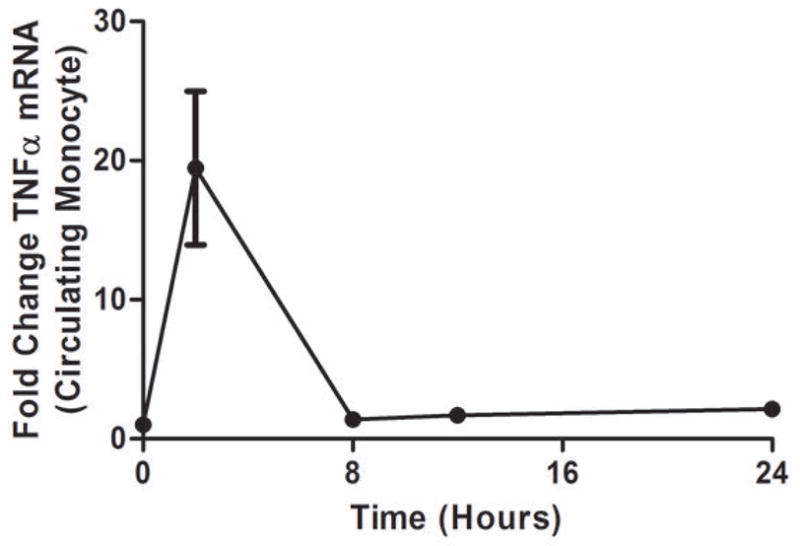

Endotoxemia (3ng/Kg LPS intravenously) markedly increased plasma levels of (A) TNFα and CRP (P<0.001) but not (B) Lp-PLA2 mass or activity which declined transiently (P<0.01). Following LPS, whole-blood (C) TNFα mRNA was markedly induced but (D) Lp-PLA2 mRNA is not. Similarly, LPS increased circulating monocyte mRNA levels of (E) TNFα but not (F) Lp-PLA2.
Lp-PLA2 expression is induced in inflammatory human macrophages and foam cells
Lp-PLA2 mRNA levels were low (CTs ~30) in freshly-isolated human monocytes but increased markedly (CTs ~20) following six-days of differentiation to mature macrophages (P<0.0001) (Figure 2A) and increased modestly during further polarization to M1 (P<0.0001) but not M2 macrophages (Figure 2B). Lp-PLA2 protein mass also was induced during differentiation to macrophages, with increases in both the cell-associated protein (P<0.0001) and the secreted protein (P=0.0004) (Figure 2C). Following loading of human monocyte-derived macrophages with acetylated LDL-C for 48hrs, cholesterol ester (128 vs. 0.6 ug CE/mg protein) and total cholesterol (422 vs. 316 ug chol/mg protein) were significantly higher in loaded versus unloaded cells consistent with findings for in vivo foam cells (26). Lp-PLA2 mRNA levels were significantly greater in foam cells compared with mature macrophages (P<0.01) (Figure 3A). Similarly, cell-associated (P=0.05) and secreted (P=0.008) Lp-PLA2 protein levels were higher in foam cells than in macrophages (Figure 3B). There was no Lp-PLA2 protein detectable in the media or acLDL used to treat cells. Overall, these data are consistent with lack of in vivo increase in plasma or monocyte levels of Lp-PLA2 during the acute phase and suggest that, in human atherosclerosis, Lp-PLA2 may be generated by macrophages and foam cells rather than circulating leukocytes.
Figure 2. Lp-PLA2 mRNA and protein increase during differentiation of human monocytes to macrophages in vitro.


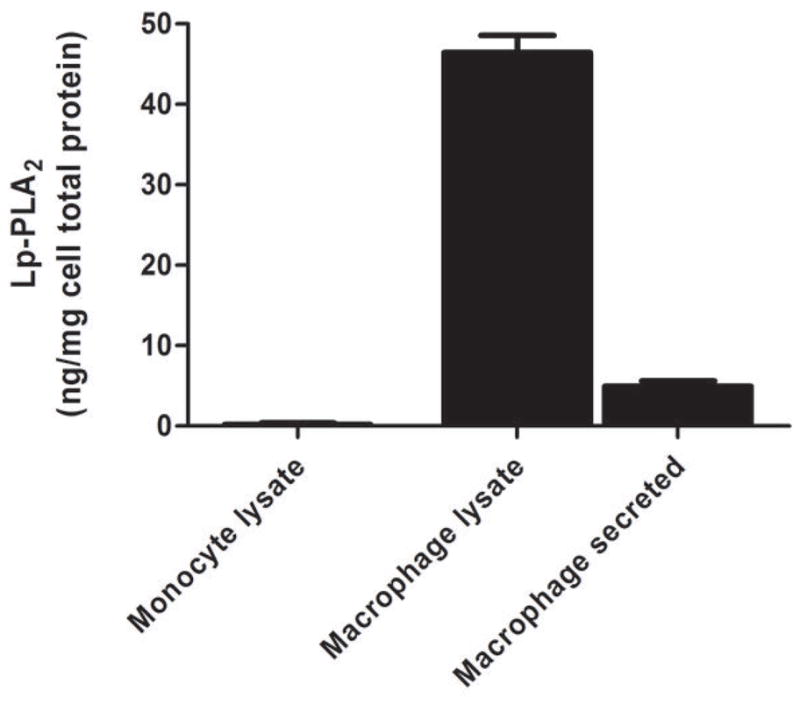
Lp-PLA2 mRNA levels increased markedly during differentiation from monocytes to mature macrophages (P<0.0001) (A) and increased modestly during further polarization to M1 macrophages (P<0.001) but fell during M2-polarization (P<0.001) (B). Lp-PLA2 protein mass also increased significantly during differentiation to macrophages, with increases in both the cell-associated protein (P<0.0001) and the secreted protein (P=0.0004) † (C). (ANOVA and Bonferroni post-hoc tests).
† As monocytes were grown in suspension, protein levels were measured in monocyte cell lysates but could not be measured in media.
Figure 3. Lp-PLA2 mRNA and protein are upregulated in human foam-like cells in vitro.
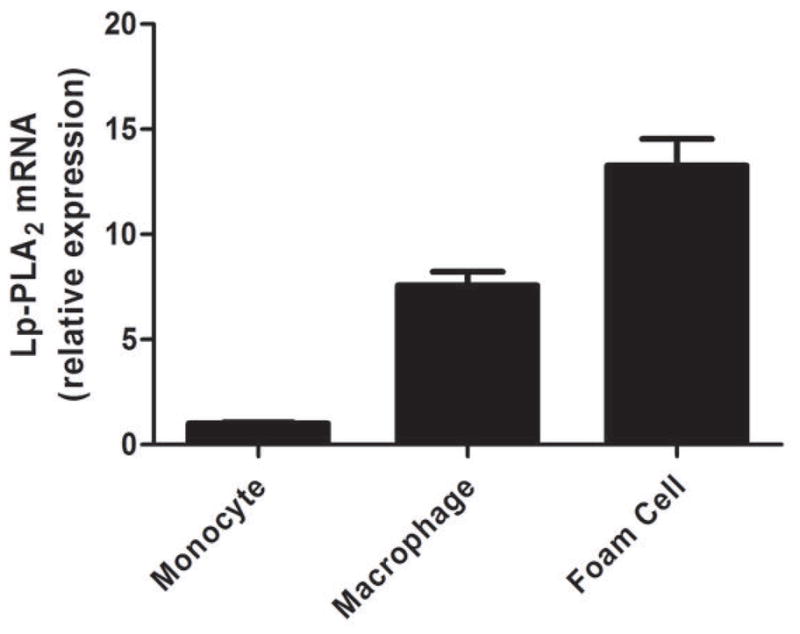
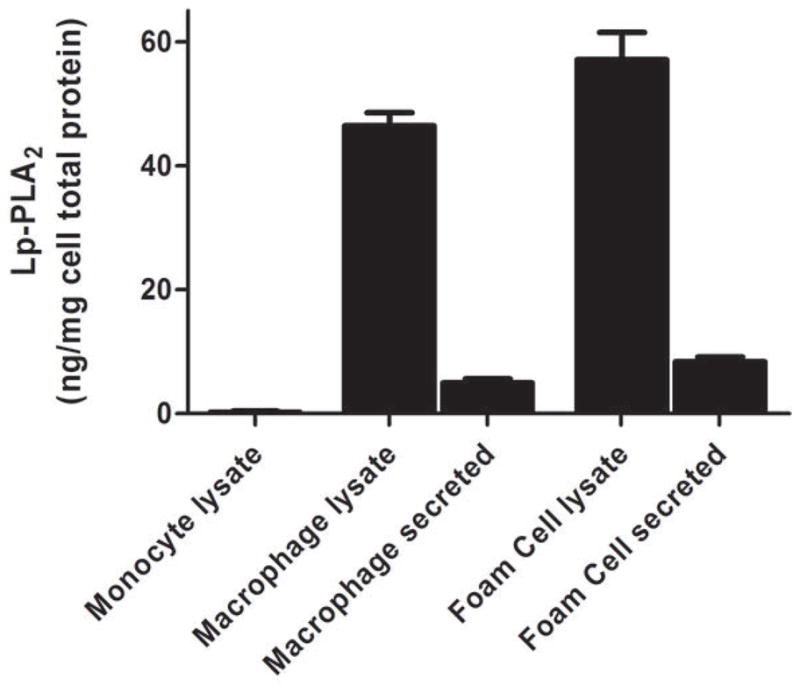
Lp-PLA2 mRNA was significantly greater (P<0.01) in foam cells than in mature macrophages (A). Similarly, cell-associated (P=0.05) and secreted (P=0.008) Lp-PLA2 protein levels were higher in foam cells than in macrophages † (B). (ANOVA and Bonferroni post-hoc tests). † As monocytes were grown in suspension, protein levels were measured in monocyte cell lysates but could not be measured in media.
Exploratory interrogation of PLA2G7 SNP eQTLs for Lp-PLA2 mRNA expression in publicly-available data revealed nominal associations of several SNPs in the PLA2G7 region with exon probe levels in PBMCs (best P=0.0059, rs12181971) and brain (best P=0.008, rs12195701) (27), as well as skin (best P=0.021, rs16874962), fat (best P=0.019, rs16874962) and lymphoblastoid cells (best P=0.037, rs7745519) (MuTHER twin2 study resource (28)). These modest associations, however, were not significant after correction for multiple testing (Supplement). Macrophage and foam cell expression datasets were not available for testing a more atherosclerosis-relevant cell type.
Common polymorphisms in PLA2G7 are associated with coronary calcification but only weakly with plasma Lp-PLA2 mass or activity
Individually in SIRCA or PDHS samples, there were no significant associations between PLA2G7 SNPs and Lp-PLA2 mass or activity. In the combined meta-analysis, only one SNP (rs1805017) had nominal association with Lp-PLA2 mass (P=0.02; P=0.2 after Bonferroni correction) (Table 1A). As a positive control, we performed similar analysis of plasma CRP using common CRP SNPs. In contrast to findings for PLA2G7-Lp-PLA2, there were significant associations between numerous CRP SNPs and plasma CRP in the SIRCA and PDHS samples and in the overall meta-analysis (Table 1B). Nine of sixteen CRP SNPs had nominal (P<0.05) associations with CRP levels and eight of these SNPs had significant associations after Bonferroni correction.
Table 1.
(A) Lack of association between SNPs in PLA2G7 and circulating Lp-PLA2 protein mass or activity but (B) significant relation of multiple SNPs in CRP with circulating CRP levels*.
| (A) | Lp-PLA2 Mass Meta-analysis (n=1723) | Lp-PLA2 Activity Meta-analysis (n=1683) | ||||
|---|---|---|---|---|---|---|
| †PLA2G7 SNP (Allele: Minor/Major) | Type of SNP | MAF | Z | P | Z | P |
| rs1805017 (A/G) | Non-synonymous Coding | 0.26 | 2.34 | 0.02 | 0.23 | 0.82 |
| rs2216465 (C/G) | Intronic | 0.34 | 1.54 | 0.12 | 0.66 | 0.51 |
| rs1421378 (G/A) | Upstream | 0.41 | 1.37 | 0.17 | 0.37 | 0.71 |
| rs17288905 (G/A) | Intronic | 0.08 | 1.74 | 0.08 | 0.79 | 0.43 |
| rs10948300 (T/A) | Intronic | 0.21 | 1.77 | 0.08 | 0.31 | 0.76 |
| rs12195701 (A/G) | Intronic | 0.21 | 1.46 | 0.14 | 0.77 | 0.44 |
| rs1051931 (A/G) | Non-synonymous Coding | 0.21 | 1.53 | 0.13 | 0.07 | 0.95 |
| rs7756935 (C/A) | Intronic | 0.21 | 1.53 | 0.13 | 0.07 | 0.95 |
| rs12528807 (C/A) | Intronic | 0.08 | −0.36 | 0.72 | 0.44 | 0.66 |
| rs1362931 (A/C) | Intronic | 0.20 | 1.15 | 0.25 | 0.06 | 0.96 |
| rs1421372 (A/G) | Intronic | 0.20 | 1.15 | 0.25 | 0.06 | 0.96 |
| rs3799861 (A/G) | Intronic | 0.20 | 1.15 | 0.25 | 0.06 | 0.96 |
| rs9472830 (A/G) | Intronic | 0.20 | 1.07 | 0.29 | 0.08 | 0.94 |
| rs16874962 (A/G) | Intronic | 0.07 | −1.12 | 0.26 | −0.46 | 0.64 |
| rs3799863 (A/T) | Intronic | 0.05 | −0.71 | 0.48 | 1.01 | 0.31 |
| rs1421368 (G/A) | Intronic | 0.10 | 0.42 | 0.68 | 0.02 | 0.99 |
| rs9349373 (A/G) | Upstream | 0.20 | 0.23 | 0.82 | −0.49 | 0.63 |
| rs16874967 (A/G) | Intronic | 0.06 | −0.34 | 0.74 | 0.68 | 0.49 |
| rs1805018 (G/A) | Non-synonymous Coding | 0.06 | −0.34 | 0.74 | 0.68 | 0.49 |
| (B) | Meta-analysis (n=2026) | |||
|---|---|---|---|---|
| †CRP SNP (Allele: Minor/Major) | Type of SNP | MAF | Z | P |
| rs1205 (A/G) | 3′UTR | 0.34 | −5.63 | 1.8E-08 |
| rs1800947 (G/C) | Synonymous coding | 0.07 | −5.41 | 6.4E-08 |
| rs3091244 (A/G) | 5′ Upstream | 0.37 | 4.40 | 1.1E-05 |
| rs12084589 (A/C) | 5′ Upstream | 0.07 | 3.23 | 1.0E-03 |
| rs12068753 (A/T) | 5′ Upstream | 0.07 | 3.18 | 1.0E-03 |
| rs3093059 (G/A) | 5′ Upstream | 0.07 | 3.14 | 2.0E-03 |
| rs3093068 (G/C) | 3′ Downstream | 0.06 | 2.94 | 3.0E-03 |
| rs2794521 (G/A) | 5′ Upstream | 0.25 | 0.29 | 0.77 |
| rs3093066 (A/C) | 3′UTR | 0.001 | 1.99 | 0.05 |
| rs2808634 (A/G) | 5′ Upstream | 0.27 | 0.39 | 0.69 |
| rs1130864 (A/G) | 3′UTR | 0.30 | 2.94 | 0.003 |
| rs2808631 (G/A) | 3′ Downstream | 0.002 | −1.49 | 0.14 |
| rs3093070 (C/A) | 3′ Downstream | 0.02 | −0.82 | 0.41 |
| rs3093069 (C/A) | 3′ Downstream | 0.001 | 1.37 | 0.17 |
| rs3093071 (A/C) | 3′ Downstream | 0.01 | 1.23 | 0.22 |
Meta-analysis of data from the Study of Inherited Risk of Coronary Atherosclerosis (SIRCA) and the Penn Diabetes Heart Study (PDHS).
For all analyses, the effect allele corresponds to the minor allele. MAF=minor allele frequency. The 19 SNPs in PLA2G7 fall into 10 LD blocks (r2>0.8), therefore we used Bonferroni multiple testing correction for 10 effective tests, giving a P value threshold of significance of 0.005. The 15 SNPs in CRP fall into 15 LD blocks (r2>0.8), therefore we used Bonferroni multiple testing correction for 15 effective tests, giving a P value threshold of significance of 0.0033.
Association of PLA2G7 SNPs with CAC was assessed initially in SIRCA&PAMSyN together with follow-up in PDHS. Multiple PLA2G7 SNPs had nominal associations with CAC in SIRCA&PAMSYN (eleven with P<0.05; lowest P<0.0001 for rs1421378). Replication signals in PDHS were modest (strongest rs10948300 P=0.02) likely due to limited power; however, in PDHS sixteen of nineteen SNPs had effects in the same direction as in SIRCA/PAMSYN (χ2=8.9, P=0.003). Meta-analysis of the combined sample found several SNP associations with CAC (rs9349373, P=0.002; rs2216465, P=0.002; rs12195701, P=0.004) that were significant after Bonferroni correction (Table 2A). Including plasma Lp-PLA2 mass or activity in the model did not attenuate the association between PLA2G7 SNPs and CAC. These findings support recent associations of variation in PLA2G7 with CHD (29–31).
Table 3.
Association of SNPs in (A) PLA2G7 but not in (B) CRP with Coronary Artery Calcification.
| (A) | *SIRCA&PAMSYN (n=1279) | *PDHS (n=782) | Meta-analysis (n=2061) | |||||
|---|---|---|---|---|---|---|---|---|
| †PLA2G7 SNP (Allele: Minor/Major) | Type of SNP | MAF | Beta | P | Beta | P | Z | P |
| rs1421378 (G/A) | Upstream | 0.41 | 0.31 | 9.8E-05 | 0.05 | 0.65 | 3.35 | 8.1E-04 |
| rs2216465 (C/G) | Intronic | 0.34 | 0.29 | 3.9E-04 | 0.05 | 0.67 | 3.06 | 0.002 |
| rs12528807 (C/A) | Intronic | 0.08 | 0.39 | 0.004 | 0.40 | 0.07 | 3.36 | 7.7E-04 |
| rs9349373 (A/G) | Upstream | 0.20 | 0.26 | 0.008 | 0.22 | 0.13 | 3.04 | 0.002 |
| rs10948300 (T/A) | Intronic | 0.21 | 0.24 | 0.013 | 0.32 | 0.02 | 3.36 | 7.8E-04 |
| rs1805017 (A/G) | Non-synonymous Coding | 0.26 | 0.22 | 0.014 | 0.09 | 0.49 | 2.37 | 0.018 |
| rs3799863 (A/T) | Intronic | 0.05 | 0.46 | 0.016 | −0.23 | 0.39 | 1.37 | 0.17 |
| rs16874967 (A/G) | Intronic | 0.06 | 0.39 | 0.02 | −0.14 | 0.56 | 1.41 | 0.16 |
| rs1805018 (G/A) | Non-synonymous Coding | 0.06 | 0.39 | 0.02 | −0.14 | 0.56 | 1.41 | 0.16 |
| rs12195701 (A/G) | Intronic | 0.21 | 0.21 | 0.03 | 0.26 | 0.07 | 2.85 | 0.004 |
| rs1421368 (G/A) | Intronic | 0.10 | −0.27 | 0.04 | −0.17 | 0.37 | −2.16 | 0.03 |
| rs9472830 (A/G) | Intronic | 0.20 | −0.19 | 0.05 | −0.16 | 0.23 | −2.26 | 0.02 |
| rs1362931 (A/C) | Intronic | 0.20 | −0.19 | 0.05 | −0.16 | 0.23 | −2.25 | 0.02 |
| rs1421372 (A/G) | Intronic | 0.20 | −0.19 | 0.05 | −0.16 | 0.23 | −2.25 | 0.02 |
| rs3799861 (A/G) | Intronic | 0.20 | −0.19 | 0.05 | −0.16 | 0.23 | −2.25 | 0.02 |
| rs7756935 (C/A) | Intronic | 0.21 | −0.16 | 0.11 | −0.15 | 0.25 | −1.98 | 0.05 |
| rs1051931 (A/G) | Non-synonymous Coding | 0.21 | −0.16 | 0.11 | −0.16 | 0.23 | −2.01 | 0.04 |
| rs16874962 (A/G) | Intronic | 0.07 | 0.12 | 0.44 | 0.15 | 0.46 | 1.06 | 0.29 |
| rs17288905 (G/A) | Intronic | 0.084 | −0.04 | 0.76 | −0.02 | 0.94 | −0.29 | 0.77 |
| (B) | * SIRCA&PAMSyN (n=1279) | * PDHS (n=782) | Meta-analysis (n=2061) | |||||
|---|---|---|---|---|---|---|---|---|
| †CRP SNP (Allele: Minor/Major) | Type of SNP | MAF | Beta | P | Beta | P | Z | P |
| rs3093068 (G/C) | 3′ Downstream | 0.06 | −0.32 | 0.04 | −0.09 | 0.70 | −1.82 | 0.07 |
| rs3093059 (G/A) | 5′ Upstream | 0.07 | −0.31 | 0.06 | −0.11 | 0.65 | −1.78 | 0.07 |
| rs12068753 (A/T) | 5′ Upstream | 0.07 | −0.31 | 0.06 | −0.08 | 0.75 | −1.70 | 0.09 |
| rs12084589 (A/C) | 5′ Upstream | 0.07 | −0.31 | 0.06 | −0.05 | 0.83 | −1.63 | 0.10 |
| rs2808631 (G/A) | 3′ Downstream | 0.002 | −1.52 | 0.07 | 0.52 | 0.64 | −1.16 | 0.25 |
| rs1800947 (G/C) | Synonymous coding | 0.07 | 0.19 | 0.23 | 0.29 | 0.18 | 1.77 | 0.08 |
| rs2794521 (G/A) | 5′ Upstream | 0.25 | −0.11 | 0.23 | 0.01 | 0.93 | −0.90 | 0.37 |
| rs1130864 (A/G) | 3′UTR | 0.30 | 0.10 | 0.27 | −0.15 | 0.21 | 0.10 | 0.92 |
| rs2808634 (A/G) | 5′ Upstream | 0.27 | −0.09 | 0.30 | 0.00 | 0.98 | −0.84 | 0.40 |
| rs3093071 (A/C) | 3′ Downstream | 0.01 | −0.23 | 0.54 | −0.21 | 0.67 | −0.75 | 0.45 |
| rs3093066 (A/C) | 3′UTR | 0.001 | −0.57 | 0.57 | −1.52 | 0.33 | −1.04 | 0.30 |
| rs3093069 (C/A) | 3′UTR | 0.001 | −0.57 | 0.57 | 1.54 | 0.49 | −0.01 | 0.99 |
| rs1205 (A/G) | 3′ Downstream | 0.34 | 0.04 | 0.65 | 0.16 | 0.17 | 1.20 | 0.23 |
| rs3093070 (C/A) | 5′ Upstream | 0.02 | −0.13 | 0.69 | −0.67 | 0.56 | −0.22 | 0.50 |
| rs3091244 (A/G) | 3′ Downstream | 0.37 | −0.89 | 0.99 | 0.00 | 0.15 | −0.17 | 0.37 |
PAMSyN=the Philadelphia Area Metabolic Syndrome Network; SIRCA=the Study of Inherited Risk of Coronary Atherosclerosis; PDHS=the Penn Diabetes Heart Study. MAF=minor allele frequency.
For all analyses, the effect allele corresponds to the minor allele. MAF=minor allele frequency.
As an expected negative control (14,32,33), we examined CRP variant associations with CAC in the same sample and found minimal signal, with one SNP having nominal association in SIRCA&PAMSYN (rs3093068, P=0.04); however there were no associations in PDHS nor in combined meta-analysis.
DISCUSSION
We provide novel insight into the pathophysiology of Lp-PLA2 in human. First, we show that unlike TNFα and CRP, circulating Lp-PLA2 does not increase during experimental endotoxemia and therefore does not contribute to human acute phase response. Second, we found that inflammatory macrophages and foam cells, but not circulating monocytes or cultured primary monocytes, generate significant Lp-PLA2. This is consistent with the concept that the majority of Lp-PLA2 in atherosclerotic plaque is derived from local biosynthesis by inflammatory macrophage and foam cells rather than from circulating leukocytes. Third, we found that common variants in PLA2G7 are associated with CAC but had limited relation to circulating Lp-PLA2 mass or activity. This supports an atherogenic role for PLA2G7-Lp-PLA2 in human that may be independent of circulating LpPLA2 mass or activity.
Lp-PLA2 does not contribute to human acute phasee response
We demonstrate that Lp-PLA2 is not an acute phase protein in humans. This is in contrast to rodent models where LPS challenge was shown to induce a rapid increase in plasma and tissue levels of Lp-PLA2 (34). This provides further evidence of fundamental differences between humans and rodents in the physiology and action of Lp-PLA2 (35). Lack of induction in blood and circulating monocytes by endotoxemia in vivo also suggests limited, if any, role for circulating leukocyte production of Lp-PLA2 in atherosclerosis. In contrast, marked in vitro up-regulation in macrophages and foam cells is consistent with a specific role for local vascular production of Lp-PLA2 in atherosclerosis. While it is possible that local macrophage Lp-PLA2 production in plaque may contribute to a portion of circulating Lp-PLA2, it is unlikely to render circulating levels useful as independent biomarkers of Lp-PLA2 actions in atherosclerosis because published data show that circulating Lp-PLA2 mass and activity do not correlate with plaque Lp-PLA2 in patients undergoing elective carotid endarterectomy (36) and because there is substantial confounding of plasma Lp-PLA2 by circulating lipoproteins regardless of tissue source. Overall, these data suggest that levels of Lp-PLA2 mRNA and protein in blood may be poor surrogates of PLA2G7 actions in arterial plaque.
Pro-inflammatory macrophages and foam cells, but not monocytes, generate significant Lp-PLA2
We found that Lp-PLA2 expression was markedly increased during the differentiation of monocytes to macrophages, and further induced in vitro in “foam cell”-like macrophages. This is consistent with constitutive expression and activity in inflammatory macrophages (37) and foam cells in atherosclerosis. Indeed, Lp-PLA2 expression is increased in atherosclerotic lesions in humans (10). In this environment, secreted Lp-PLA2 can hydrolyze oxidized phospholipids and fatty acids on atherogenic lipoproteins, generating reactive lipid mediators thought to promote plaque instability. Inhibition of Lp-PLA2 suppressed oxidized-LDL-induced macrophage apoptosis (38), a feature of inflammatory plaque. Further, in a porcine model of complex atherosclerosis, suppression of Lp-PLA2 retarded atherosclerosis progression and decreased plaque inflammation, necrosis and fibrous cap erosion (7). Compared to placebo, short-term Lp-PLA2 inhibition in human also reduced several markers of plaque inflammation in carotid lesions examined ex vivo (5,6). Overall, these data provide indirect evidence for atherogenic actions of Lp-PLA2 in vascular lesions. Indeed, Lp-PLA2 inhibition is currently being tested in large phase-III clinical trials of CHD in high risk patients (NCT0100072, clinicaltrials.gov).
Genetic variation in PLA2G7 may relate to CHD independent of circulating Lp-PLA2
Several epidemiological studies revealed an association of higher plasma Lp-PLA2 mass and activity levels with risk of CHD (9,39–41). Meta-analyses support a modest CHD relationship independent of traditional risk factors and plasma CRP (39,42,43). Published studies, however, may underestimate the degree of confounding because of incomplete measurement and control for all atherogenic lipoproteins (9). In circulation, Lp-PLA2 associates with both apoB lipoproteins and HDL with the majority found on LDL particles. Since Lp-PLA2 protein and activity are closely linked to circulating apoB lipoproteins (35,44), it is not surprising that genetic factors (e.g. APOC1, PSRC1, ZNF259) that regulate plasma apoB lipoproteins are also associated with plasma Lp-PLA2 (45). Parenthetically, we found modest association of lipid-related genes (e.g., LRP2, LPL, APOA2) with plasma Lp-PLA2 likely reflecting this indirect post-translational influence (Supplemental Table S1A and B). Interpretation of studies of plasma Lp-PLA2 in CHD is challenging partly because circulating lipoproteins may grossly confound the association of plasma Lp-PLA2 with CHD (8) and further because lesion macrophage production may be more relevant to the disease than circulating protein.
While we failed to detect significant association between plasma Lp-PLA2 and common SNPs in PLA2G7, the same PLA2G7 variants were associated with CAC within our study samples. Our preliminary exploration also revealed only nominal associations of PLA2G7 SNPs with Lp-PLA2 mRNA levels in multiple cells and tissues. These eQTL findings should be interpreted cautiously because of limited power, relatively low levels of Lp-PLA2 expression in tested cells, and (unlike CRP) well-characterized cis-acting SNPs for PLA2G7 are lacking. Further, appropriately powered studies are needed to determine whether PLA2G7 SNPs are related to expression of Lp-PLA2 in inflammatory macrophages and foam cells, sources that may be most relevant to atherosclerosis. However, our data suggest caution in using circulating leukocyte Lp-PLA2 mRNA levels as surrogates for effects of PLA2G7 variation on arterial pathology. Overall, our findings support the concept that PLA2G7 may relate to atherosclerosis independent of circulating Lp-PLA2 mRNA and protein.
Published studies of PLA2G7 in CHD are conflicting. In a meta-analysis of individuals of European ancestry, PLA2G7 SNPs did not associate with risk of CHD (N~5,000) (41), although there was a relationship between Lp-PLA2 activity and CHD, and between PLA2G7 SNPs and Lp-PLA2 activity. However in a meta-analysis of over 13,000 Asians, a common non-synonymous PLA2G7 SNP showed evidence of association with CHD (30). Additional non-synonymous SNPs have been associated with carotid plaque in Japanese (31) and recently a loss-of-function variant in PLA2G7 was shown to protect against CHD in Koreans (29). Due to the absence in Caucasian samples of the functional PLA2G7 SNP found in Asians (rs76863441 or V279F), we were not able to evaluate the effect of this functional variant in our samples. However, common variation in PLA2G7 is well covered on the IBC array platform (tag SNP-coverage r2>0.8 for alleles with MAF≥2% in the gene±5KB). (21). Therefore, we are confident that we achieved excellent coverage of common variation in this gene region in Caucasians. While ethnic difference in the presence of allelic variation may exist,, most published data suggest a relationship of PLA2G7 with clinical CHD supporting our CAC findings.
Findings for CRP in our samples are consistent with published data and contrast with that observed for PLA2G7-Lp-PLA2. Thus, while a number of SNPs in CRP had strong associations with circulating CRP levels, there was no relationship between these same SNPs and CAC. These data are in line with hallmark Mendelian randomization studies of clinical CHD outcomes (32,33) and support a model of confounding or reverse causation for CRP associations with CAC and CHD.
Limitations of the present study, and future outlook
Our study has several limitations. First, our studies are correlative and do not define causality. We have not studied loss-of-function or gain-of-function variants in PLA2G7 for their relation to CAC or CHD and therefore cannot infer Lp-PLA2 directional actions in atherosclerosis. However, expression data in inflammatory macrophages and foam cells coupled to preliminary studies of Lp-PLA2 inhibition in human atherosclerosis support an atherogenic role for human PLA2G7. Second, recent studies have shown stronger associations of PLA2G7 with circulating Lp-PLA2 measures than in our sample. This may relate to our smaller sample size, heterogeneity in the SIRCA and PDHS study samples, or differences in Lp-PLA2 assays used across studies. The PLA2G7-Lp-PLA2 system, however, may be a poor target for Mendelian randomization studies for several reasons including heterogeneous environmental and genetic influences on circulating levels, PLA2G7 actions in atherosclerosis are likely independent of circulating Lp-PLA2, and well-characterized cis-acting SNPs to use as instrumental variables for PLA2G7 are lacking. Finally, although not a direct measure of coronary atherosclerosis, studies show that CAC provides a quantitative estimate of coronary atherosclerosis (46) and is a useful predictor of CHD events (47).
In conclusion, we have demonstrated that Lp-PLA2, in contrast to CRP, is not an acute phase protein in humans. Lp-PLA2 has limited expression in circulating leukocytes or unstimulated monocytes ex vivo but is induced during differentiation to macrophages and in foam cells. Thus, robust biomarkers of Lp-PLA2 action in atherosclerosis and of its pharmacological modulation in vascular tissues are lacking. Common variation in PLA2G7, but not in CRP, is related to the burden of CAC, suggesting that PLA2G7 may indeed modulate human atherosclerosis. Our data provide support for the atherogenicity of Lp-PLA2 in human while highlighting the challenges in using plasma Lp-PLA2 as a biomarker of CHD and in determining drug-dosing and therapeutic efficacy in atherosclerosis.
Supplementary Material
Acknowledgments
Funding: This work was supported by a Clinical and Translational Science Award (UL1RR024134), a Diabetes and Endocrine Research Center (P20-DK 019525) award (both from the National Institute of Health to the University of Pennsylvania), and a P50 HL-083799-SCCOR Project award from the National Institute of Health (to M.P.R.). GlaxoSmithKline provided research grant support for measurement of LpPLA2 mass and activity.
Abbreviations
- SNP
single nucleotide polymorphism
- CAC
coronary artery calcification
- CHD
coronary heart disease
- LPS
lipopolysaccharide
- CRP
C-reactive protein
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
Footnotes
Relationship with Industry: M.P.R. and D.J.R. received research grant support from GlaxoSmithKline. Employees of GlaxoSmithKline did not contribute to study design, data interpretation, and editing of the report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol. 2005;25:923–31. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 2.Wilensky RL, Macphee CH. Lipoprotein-associated phospholipase A(2) and atherosclerosis. Curr Opin Lipidol. 2009;20:415–20. doi: 10.1097/MOL.0b013e3283307c16. [DOI] [PubMed] [Google Scholar]
- 3.Kim JY, Hyun YJ, Jang Y, et al. Lipoprotein-associated phospholipase A2 activity is associated with coronary artery disease and markers of oxidative stress: a case-control study. Am J Clin Nutr. 2008;88:630–7. doi: 10.1093/ajcn/88.3.630. [DOI] [PubMed] [Google Scholar]
- 4.Daniels LB, Laughlin GA, Sarno MJ, Bettencourt R, Wolfert RL, Barrett-Connor E. Lipoprotein-associated phospholipase A2 is an independent predictor of incident coronary heart disease in an apparently healthy older population: the Rancho Bernardo Study. J Am Coll Cardiol. 2008;51:913–9. doi: 10.1016/j.jacc.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohler ER, 3rd, Ballantyne CM, Davidson MH, et al. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2008;51:1632–41. doi: 10.1016/j.jacc.2007.11.079. [DOI] [PubMed] [Google Scholar]
- 6.Serruys PW, Garcia-Garcia HM, Buszman P, et al. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–82. doi: 10.1161/CIRCULATIONAHA.108.771899. [DOI] [PubMed] [Google Scholar]
- 7.Wilensky RL, Shi Y, Mohler ER, 3rd, et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med. 2008;14:1059–66. doi: 10.1038/nm.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tellis CC, Tselepis AD. The role of lipoprotein-associated phospholipase A2 in atherosclerosis may depend on its lipoprotein carrier in plasma. Biochim Biophys Acta. 2009;1791:327–38. doi: 10.1016/j.bbalip.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Thompson A, Gao P, Orfei L, et al. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 375:1536–44. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hakkinen T, Luoma JS, Hiltunen MO, et al. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1999;19:2909–17. doi: 10.1161/01.atv.19.12.2909. [DOI] [PubMed] [Google Scholar]
- 11.Kolodgie FD, Burke AP, Skorija KS, et al. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2523–9. doi: 10.1161/01.ATV.0000244681.72738.bc. [DOI] [PubMed] [Google Scholar]
- 12.Lehrke M, Reilly MP, Millington SC, Iqbal N, Rader DJ, Lazar MA. An inflammatory cascade leading to hyperresistinemia in humans. PLoS Medicine. 2004;1:e45. doi: 10.1371/journal.pmed.0010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson PD, Mehta NN, Wolfe ML, et al. Innate immunity modulates adipokines in humans. J Clin Endocrinol Metab. 2007;92:2272–9. doi: 10.1210/jc.2006-2545. [DOI] [PubMed] [Google Scholar]
- 14.Reilly MP, Wolfe ML, Localio AR, Rader DJ. C-reactive protein and coronary artery calcification: The Study of Inherited Risk of Coronary Atherosclerosis (SIRCA) Arterioscler Thromb Vasc Biol. 2003;23:1851–6. doi: 10.1161/01.ATV.0000092327.60858.4A. [DOI] [PubMed] [Google Scholar]
- 15.Reilly MP, Iqbal N, Schutta M, et al. Plasma leptin levels are associated with coronary atherosclerosis in type 2 diabetes. J Clin Endocrinol Metab. 2004;89:3872–8. doi: 10.1210/jc.2003-031676. [DOI] [PubMed] [Google Scholar]
- 16.Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M, Jr, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. 1990;15:827–32. doi: 10.1016/0735-1097(90)90282-t. [DOI] [PubMed] [Google Scholar]
- 17.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 18.Bagheri R, Qasim AN, Mehta NN, et al. Relation of plasma fatty acid binding proteins 4 and 5 with the metabolic syndrome, inflammation and coronary calcium in patients with type-2 diabetes mellitus. Am J Cardiol. 106:1118–23. doi: 10.1016/j.amjcard.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 20.Shen H, Bielak LF, Ferguson JF, et al. Association of the vitamin D metabolism gene CYP24A1 with coronary artery calcification. Arterioscler Thromb Vasc Biol. 30:2648–54. doi: 10.1161/ATVBAHA.110.211805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keating BJ, Tischfield S, Murray SS, et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reilly MP, Li M, He J, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 377:383–92. doi: 10.1016/S0140-6736(10)61996-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74:765–9. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta NN, McGillicuddy FC, Anderson PD, et al. Experimental Endotoxemia Induces Adipose Inflammation and Insulin Resistance in Humans. Diabetes. 2009 doi: 10.2337/db09-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fowler S. Characterization of foam cells in experimental atherosclerosis. Acta Med Scand Suppl. 1980;642:151–8. doi: 10.1111/j.0954-6820.1980.tb10947.x. [DOI] [PubMed] [Google Scholar]
- 27.Heinzen EL, Ge D, Cronin KD, et al. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol. 2008;6:e1. doi: 10.1371/journal.pbio.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nica AC, Parts L, Glass D, et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang Y, Waterworth D, Lee JE, et al. Carriage of the V279F null allele within the gene encoding Lp-PLA is protective from coronary artery disease in South Korean males. PLoS ONE. 6:e18208. doi: 10.1371/journal.pone.0018208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Hao Y, Mo X, et al. PLA2G7 gene polymorphisms and coronary heart disease risk: a meta-analysis. Thromb Res. 126:498–503. doi: 10.1016/j.thromres.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 31.Miwa Y, Kamide K, Takiuchi S, et al. Association of PLA2G7 polymorphisms with carotid atherosclerosis in hypertensive Japanese. Hypertens Res. 2009 doi: 10.1038/hr.2009.151. [DOI] [PubMed] [Google Scholar]
- 32.Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359:1897–908. doi: 10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- 33.Elliott P, Chambers JC, Zhang W, et al. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. Jama. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Memon RA, Fuller J, Moser AH, Feingold KR, Grunfeld C. In vivo regulation of plasma platelet-activating factor acetylhydrolase during the acute phase response. Am J Physiol. 1999;277:R94–103. doi: 10.1152/ajpregu.1999.277.1.R94. [DOI] [PubMed] [Google Scholar]
- 35.Stafforini DM, Tjoelker LW, McCormick SP, et al. Molecular basis of the interaction between plasma platelet-activating factor acetylhydrolase and low density lipoprotein. J Biol Chem. 1999;274:7018–24. doi: 10.1074/jbc.274.11.7018. [DOI] [PubMed] [Google Scholar]
- 36.Atik B, Johnston SC, Dean D. Association of carotid plaque Lp-PLA(2) with macrophages and Chlamydia pneumoniae infection among patients at risk for stroke. PLoS ONE. 5:e11026. doi: 10.1371/journal.pone.0011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X, McIntyre TM, Zimmerman GA, Prescott SM, Stafforini DM. Molecular characterization of the constitutive expression of the plasma platelet-activating factor acetylhydrolase gene in macrophages. Biochem J. 2003;375:351–63. doi: 10.1042/BJ20030636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carpenter KL, Dennis IF, Challis IR, et al. Inhibition of lipoprotein-associated phospholipase A2 diminishes the death-inducing effects of oxidised LDL on human monocyte-macrophages. FEBS Lett. 2001;505:357–63. doi: 10.1016/s0014-5793(01)02840-x. [DOI] [PubMed] [Google Scholar]
- 39.Ballantyne CM, Hoogeveen RC, Bang H, et al. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2004;109:837–42. doi: 10.1161/01.CIR.0000116763.91992.F1. [DOI] [PubMed] [Google Scholar]
- 40.Packard CJ, O’Reilly DS, Caslake MJ, et al. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. West of Scotland Coronary Prevention Study Group. N Engl J Med. 2000;343:1148–55. doi: 10.1056/NEJM200010193431603. [DOI] [PubMed] [Google Scholar]
- 41.Casas JP, Ninio E, Panayiotou A, et al. PLA2G7 genotype, lipoprotein-associated phospholipase A2 activity, and coronary heart disease risk in 10 494 cases and 15 624 controls of European Ancestry. Circulation. 121:2284–93. doi: 10.1161/CIRCULATIONAHA.109.923383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corson MA, Jones PH, Davidson MH. Review of the evidence for the clinical utility of lipoprotein-associated phospholipase A2 as a cardiovascular risk marker. Am J Cardiol. 2008;101:41F–50F. doi: 10.1016/j.amjcard.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 43.Koenig W, Khuseyinova N. Lipoprotein-associated and secretory phospholipase A2 in cardiovascular disease: the epidemiological evidence. Cardiovasc Drugs Ther. 2009;23:85–92. doi: 10.1007/s10557-008-6135-6. [DOI] [PubMed] [Google Scholar]
- 44.Persson M, Nilsson JA, Nelson JJ, Hedblad B, Berglund G. The epidemiology of Lp-PLA(2): distribution and correlation with cardiovascular risk factors in a population-based cohort. Atherosclerosis. 2007;190:388–96. doi: 10.1016/j.atherosclerosis.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 45.Suchindran S, Rivedal D, Guyton JR, et al. Genome-wide association study of Lp-PLA(2) activity and mass in the Framingham Heart Study. PLoS Genet. 6:e1000928. doi: 10.1371/journal.pgen.1000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rumberger JA, Schwartz RS, Simons DB, Sheedy PF, 3rd, Edwards WD, Fitzpatrick LA. Relation of coronary calcium determined by electron beam computed tomography and lumen narrowing determined by autopsy. Am J Cardiol. 1994;73:1169–73. doi: 10.1016/0002-9149(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 47.Kondos GT, Hoff JA, Sevrukov A, et al. Electron-beam tomography coronary artery calcium and cardiac events: a 37-month follow-up of 5635 initially asymptomatic low- to intermediate-risk adults. Circulation. 2003;107:2571–6. doi: 10.1161/01.CIR.0000068341.61180.55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.