

Abstract
Certain chromosome rearrangements display a significant delay in chromosome replication timing (DRT) that is associated with a subsequent delay in mitotic chromosome condensation (DMC). DRT/DMC chromosomes are common in tumor cells in vitro and in vivo and occur frequently in cells exposed to ionizing radiation. A hallmark for these chromosomes is the delayed phosphorylation of serine 10 of histone H3 during mitosis. The chromosome passenger complex, consisting of multiple proteins including Aurora B kinase and INCENP is thought to be responsible for H3 phosphorylation, chromosome condensation and the subsequent segregation of chromosomes. In this report, we show that chromosomes with DRT/DMC contain phosphorylated Chk1, consistent with activation of the S–M phase checkpoint. Furthermore, we show that INCENP is recruited to the DRT/DMC chromosomes during all phases of mitosis. In contrast, Aurora B kinase is absent on DRT/DMC chromosomes when these chromosomes lack serine 10 phosphorylation of H3. We also show that mitotic arrest deficient 2 (Mad2), a member of the spindle assembly checkpoint, is present on DRT/DMC chromosomes at a time when the normally condensed chromosomes show no Mad2 staining, indicating that DRT/DMC activates the spindle assembly checkpoint. Finally, cells with DRT/DMC chromosomes have centrosome amplification, abnormal spindle assembly, endoreduplication and significant chromosome instability.
Keywords: replication checkpoint, chromosome passenger complex, genomic instability, spindle assembly checkpoint, endoreduplication, chromosome instability
Introduction
The mechanisms responsible for chromosome instability (CIN), the most common form of genomic instability that results in gains and losses of whole chromosomes, remains elusive (Lengauer et al., 1997). Two models for the development of CIN involve mutations in cell cycle regulatory pathways or gene dosage imbalances caused by aneuploidy and/or centrosome dysfunction leading to altered DNA replication and repair (Rajagopalan and Lengauer, 2004). Accurate replication and division of DNA is a tightly regulated process with cell cycle checkpoints that monitor the genome and prevent mitosis until DNA synthesis has been completed or DNA aberrations have been resolved, thereby maintaining the integrity of the genome (Kastan and Bartek, 2004). Checkpoints are also important for cancer susceptibility, for toxicities and responses to many cancer therapies (Kastan and Bartek, 2004). There are three distinct, but partially redundant S-phase checkpoints: the replication checkpoint, responds to stalled replication forks and functions to prevent initiation of DNA replication from unfired origins; the intra-S checkpoint, activated by DNA damage and prevents initiation from unfired origins; and the S–M checkpoint, which ensures that the cells do not attempt to divide before the genome has been completely duplicated (Kolodner et al., 2002; Bartek et al., 2004).
The chromosome passenger complex has been shown to play roles in proper chromosome localization, condensation, segregation and cytokenesis (reviewed by Adams et al., 2001). Three members of the passenger complex are: INCENP, Aurora B kinase and survivin. INCENP, a microtubule-binding protein is responsible for binding Aurora B (Honda et al., 2003). Survivin has an essential role in chromosome alignment (Lens et al., 2003) and is important during mitosis for proper localization of INCENP and Aurora B (reviewed by Carmena and Earnshaw, 2003). Aurora B, which phosphorylates histone H3 on serine 10, is important for chromosome condensation, and monitors chromosome biorientation during congression by sensing tension across the centromere established once sister kinetochores are attached to the opposite spindle poles (reviewed by Meraldi et al., 2004). Aurora B has also emerged as an important regulator of the spindle assembly checkpoint. mitotic arrest deficient 2 (Mad2) and budding uninhibited benzimidazole receptor 1 (BubR1) require Aurora B activity to maintain localization once recruited to the kinetochore (Lens et al., 2003).
We have characterized an abnormal chromosomal phenotype that is associated with certain tumor-derived chromosome alterations (Smith et al., 2001). This phenotype is characterized by a significant delay in mitotic chromosome condensation (DMC), a delay in the mitosis-specific phosphorylation of histone H3, and a 2–3 h delay in the replication timing (DRT) of the entire chromosome. We have also developed a chromosome engineering strategy, using Cre/loxP, to generate inter-chromosomal translocations (ICTs) that display this DRT/DMC phenotype (Breger et al., 2005).
In this report, we show that chromosomes with DRT/DMC contain phosphorylated Chk1, consistent with activation of the S-phase checkpoint. However, we found that DNA synthesis could be detected on these delayed chromosomes during mitosis, suggesting that these cells either escaped or adapted from the DNA replication checkpoint. We also show that chromosomes with DRT/DMC have delayed Aurora B recruitment and activate the mitotic spindle assembly checkpoint. Finally, we show that cells containing chromosomes with DRT/DMC have abnormal centrosome amplification, experience endoreduplication at an increased frequency and display dramatic CIN.
Results
DNA synthesis during mitosis
Individual cells can contain multiple chromosomes with the DMC phenotype, and the DMC chromosomes within a given cell can display a wide range in the extent of mitotic chromosome condensation (Figure 1c–f) (Smith et al., 1998). Furthermore, chromosomes with the more extreme undercondensed appearance lack the mitosis-specific phosphorylation of histone H3 on serine 10 (Figure 1a and b and see Smith et al., 2001). Because H3 phosphorylation is typically initiated in late G2 and completed just before nuclear envelope breakdown (Hendzel et al., 1997), these observations suggest that the more extreme undercondensed chromosomes retain an ‘interphase state’ of condensation in cells that contain fully phosphorylated and condensed metaphase chromosomes. This observation, and the 2–3 h DRT (Figure 2a and see Smith et al., 2001), suggested the possibility of active DNA synthesis occurring on these chromosomes during mitosis. Therefore, we tested mitotic CRL5845 cells, previously shown to contain two different DRT/DMC chromosomes, (der(3q) and t(3;9) (Smith et al., 2001)), for DNA synthesis in mitosis by combining phospho-H3 staining with in situ DNA replication (Mills et al., 2000). This analysis detected active DNA synthesis in ~0.1% of the mitotic phospho-H3-positive cells (Figure 2b–j). Nucleotide incorporation in this cell occurred in a small section of the DNA, suggesting that a single chromosome was still synthesizing DNA during mitosis. Furthermore, this section of DNA with nucleotide incorporation retains very little phospho-H3 staining, consistent with previous observations that DMC chromosomes show reduced levels of phospho-H3 staining (Figure 2c, e and g). Fluorescent in situ hybridization (FISH) with a chromosome 3 whole chromosome paint (WCP) was used to test whether the section of this mitotic cell with active DNA synthesis contains chromosome 3 DNA. Figure 2h–j shows that the WCP hybridized to the section of DNA that contained active incorporation, indicating DNA synthesis on a chromosome 3 derivative. Therefore, the delayed DNA synthesis that occurs on chromosomes with DRT/DMC can persist into mitosis.
Figure 1.

Variability of condensation on chromosomes with DRT/DMC from C2C12 microcell hybrids containing i(3q) (Smith et al., 2001). Multiple DMC chromosomes in a single metaphase spread showing variability in the extent of condensation (a). Arrows mark the DMC chromosomes. The same metaphase spread stained with an antibody against serine phospho-H3 showing lack of signal from DMC chromosomes (b). Metaphase spread and magnifications with an extremely undercondensed chromosome probed with a chromosome 3-specific α-satellite probe (red) (c–f). Arrow marks the DMC chromosomes. Box shows area of magnification. Other examples of extremely undercondensed chromosomes. Arrows mark the chromosome 3 probe (i(3q)) (g–i). Box shows area of magnification. The DNA was visualized by staining with DAPI.
Figure 2.

DNA synthesis during mitosis. Schematic representation of the replication timing of chromosomes with DRT/DMC (red) with respect to cell cycle phases (see Smith et al., 2001) (a). In situ DNA replication of mitotic CRL-5845 cells (b–g). Active DNA synthesis was detected using digoxygenin-labeled dUTP followed by FITC-labeled anti-digoxygenin antibody staining. Detection of the mitosis-specific phosphorylation of histone H3 using a phosph-H3-specific antibody, detection was with a Cy3-labeled secondary antibody (d and e). Higher magnification of the mitotic cell shown in panel b (c). Higher magnification of the mitotic cell shown in panel d (e). Merged images from c and e (g). FISH analysis using a chromosome 3-specific paint probe on the same mitotic cell (h). Merged images from e and h (i). Merged image showing in situ DNA synthesis, phospho-H3 staining, chromosome 3 paint and DNA (j). The DNA was visualized using DAPI (b, c, f, g, h, i and j).
Activation of the replication checkpoint by chromosomes with DRT/DMC
Two possibilities may explain DNA synthesis during mitosis. First, ongoing DNA synthesis on these chromosomes ‘escaped’ detection by the S–M phase checkpoint, allowing DNA synthesis to occur during mitosis. Second, the cells with mitotic DNA synthesis activated the S–M phase checkpoint and subsequently experienced checkpoint adaptation. Checkpoint adaptation occurs in cells that are arrested at a DNA checkpoint but eventually over-ride this arrest and re-enter the cell cycle without repair of the damage (Sandell and Zakian, 1993). Therefore, we assayed cells containing chromosomes with DRT/DMC for activation of the replication checkpoint. When DNA replication is stalled, ataxia-telangiectasia and rad3-related kinase (ATR) phosphorylates serine 345 of Chk1 and activates the checkpoint (Liu et al., 2000). Hence, we assayed for phospho-S-345 of Chk1 in hybrid cells containing human DRT/DMC chromosomes. Figure 3a shows that parental C2C12 cells do not contain any detectable S-345 staining. In contrast, cells containing the i(3q) contain one or two chromosome-sized domains of phospho-S-345 staining (Figure 3b). To determine if these phospho-S-345-positive domains corresponded to the introduced i(3q)s, we processed the same slide for FISH using total human DNA in the mouse C2C12 background (Figure 3c–f). The phospho-S-345-positive domains hybridized to the human DNA probe, indicating that the introduced i(3q)s stained positively with this phospho-Chk1 antibody. A similar analysis of an unrearranged human chromosome 3 introduced into C2C12 cells did not show any detectable phospho-S-345 staining (with over 1000 cells analysed) (Figure 3g and h). These observations suggest that the delayed replication on these chromosomes activates the replication checkpoint leading to phosphorylation of Chk1.
Figure 3.
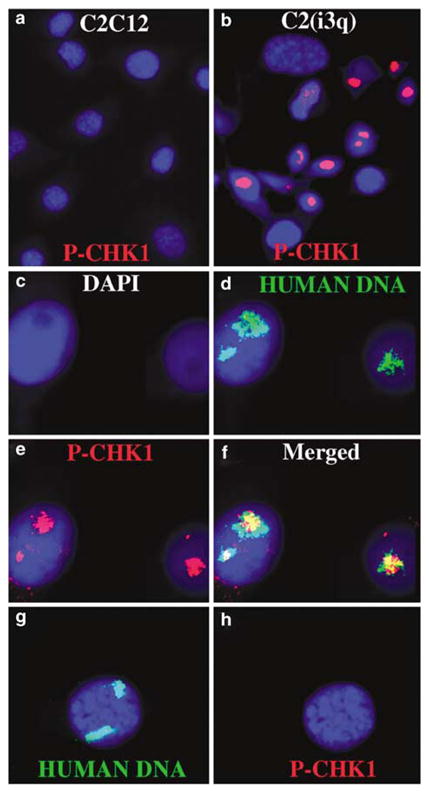
Chromosomes with DRT/DMC have phosphorylated Chk1. Chk1 is phosphorylated on chromosomes with DRT/DMC (a–b). C2C12 and a hybrid with the i(3q) were stained with an antibody against phosphorylated serine 345 of CHK1 (P-CHK1). P-CHK1 was detected using a Cy3-labeled secondary antibody, and nuclei were identified using DAPI. Cells were stained for P-CHK1, images captured and then the slides were processed for FISH using total human DNA as probe (c–h). The human DNA was detected using FITC and the nuclei identified using DAPI.
DMC chromosomes show a delay in the recruitment of Aurora B kinase
Aurora B has been shown to phosphorylate histone H3 on serine 10. To test whether the chromosome passenger complex is present on DMC chromosomes, mitotic spreads were stained for INCENP, phospho-H3 (serine 10) and Aurora B in R268, with an engineered ICT that displays DRT/DMC. R268 cells contain a t(15;16)(q24; q12.1) that displays DRT/DMC only on the chromosome 15 derivative (see Figure 4a and e) (Breger et al., 2005). R268 cells were stained for Aurora B and INCENP and assayed by FISH using chromosomes 15 and 16 centromeric probes. We observed colocalization of Aurora B, INCENP and phosphohistone H3 in interphase nuclei (i.e. late G2), prophase, prometaphase and metaphase (Supplementary Figures 1 and 2). INCENP was detected on the fully condensed metaphase chromosomes along the arms with an increased signal on the centromeres as well as on DMC chromosomes (Figure 4c, d, f and h). In contrast, Aurora B was detected on the fully condensed chromosomes but not on the chromosome 15 derivative with DMC (Figure 4c, d, g and h). However, Aurora B was detected on the chromosome 15 derivative when it was fully condensed, indicating that Aurora B is eventually recruited to the DRT/DMC chromosomes (data not shown). Pretreatment of the cells with colcemid showed much stronger staining on the centromeres by INCENP and Aurora B (see Supplementary Figure 3). Also, the addition of phosphatase inhibitors during mitotic spread preparations had no effect on phospho-H3 staining (data not shown). We conclude that INCENP was present on DMC chromosomes, whereas Aurora B was not. Therefore, the delayed phosphorylation of histone H3 on DMC chromosomes correlates with delayed Aurora B recruitment.
Figure 4.

DMC chromosomes recruit INCENP but do not recruit Aurora B kinase. Metaphase spreads of R268 cells with DMC chromosomes. Metaphase spreads were probed with 15 Cen spectrum green and 16 Cen spectrum red by FISH (a, e). The same metaphase spreads initially stained with an antibody to INCENP and identified with a secondary rabbit antibody linked to Cy3 (b, f). The spreads were also stained with an antibody to Aurora B kinase and identified with a secondary mouse antibody linked to Alexa 488 (c, g). Merged views (d, h). DNA was stained with DAPI. Arrows mark the DMC chromosomes.
Chromosomes with DRT/DMC are delayed in spindle attachment
The spindle assembly checkpoint delays entry into anaphase until the kinetochores of every chromatid pair have attached correctly to spindle microtubules. We reasoned that DRT/DMC coupled with delayed Aurora B recruitment could potentially interfere with kinetochore function. Chromosomes with the DMC phenotype retained CREST staining, suggesting that chromosomes with DRT/DMC have intact kinetochores (Figure 5a and b). Interestingly, DMC chromosomes show a larger region of CREST staining than the fully condensed chromosomes, presumably owing to the undercondensed state of the centromeric region. In addition, the chromosomes with DMC were often located on the periphery, whereas the other fully condensed chromosomes were arranged in a circular pattern with their kinetochores aligned toward the center (see Figure 5b). This analysis occurred without colcemid, suggesting that the fully condensed chromosomes were attached to the mitotic spindle and aligned on the metaphase plate, whereas DMC chromosomes were not.
Figure 5.

Chromosomes with DMC activate the spindle assembly checkpoint. Kinetochore staining on chromosomes with DMC. Mitotic C2C12 cells containing the i(3q) were stained with an antibody against kinetochores (CREST), detection was with a Cy3-labeled secondary antibody (a, b). DNA was detected using DAPI. Cells were harvested for mitotic spreads in the absence of colcemid pretreatment. The arrows mark chromosomes with DMC. Chromosomes with DMC retain Mad2 staining at their kinetochores. CRL5845 cells were grown on slides, fixed and stained with rabbit anti-Mad2 and goat anti-CENP-A antibodies (c–f). The Mad2 antibody was detected with a Cy3-labeled secondary antibody (d, e and g) and the CENP-A was detected using FITC secondary antibody (f and g). The chromosomes were visualized with DAPI (c, e, g and h). Two Mad2-positive and CENP-A-positive kinetochores are present in this cell (arrows). One Mad2-positive chromosome in this cell could not be evaluated for condensation, because it was surrounded by other chromosomes (*). The slide was washed and re-stained for phosphorylation of serine 10 of H3 (h).
One critical component of the spindle assembly checkpoint is Mad2. Mad2 binds to unattached kinetochores and participates in a signal cascade that prevents the metaphase to anaphase transition. We assayed for the presence of Mad2 on the kinetochores of chromosomes with DMC. For this analysis, we stained cells grown on slides for Mad2 and the constitutive kinetochore protein CENP-A. Figure 5c–h shows an undercondensed chromosome that retained Mad2 staining, whereas the other fully condensed chromosomes did not. DMC chromosomes retained a larger region of CENP-A staining and are located on the periphery, suggesting that it was not attached to the spindle. In contrast, all of the fully condensed chromosomes were aligned on the metaphase plate with their kinetochores arranged in a circular pattern. To confirm that the Mad2-positive chromosome displays the DMC phenotype, we stained this cell for H3 phosphorylation on serine 10. The undercondensed chromosome lacks H3 phosphorylation. Note that this cell contains a second Mad2-positive kinetochore. Unfortunately, the condensation state of this chromosome could not be determined, because it was surrounded by condensed chromosomes. Taken together, these observations indicate that the kinetochores of chromosomes with DMC are delayed in their mitotic spindle attachment and retain Mad2 staining, presumably activating the mitotic spindle assembly checkpoint.
Centrosome amplification, abnormal spindle assembly, endoreduplication and CIN
DRT/DMC chromosomes often contained abnormal mitotic figures and hyperdiploid karyotypes (Smith et al., 2001; Breger et al., 2004). Therefore, we analysed centrosome copy number and mitotic spindles using antibodies against γ- and β-tubulin, respectively. C2C12 cells contain one or two centrosomes in ~98% (196/200) of interphase cells (Figure 6a and b). In contrast, hybrid cells that contain a DRT/DMC chromosome contain ≥3 centrosomes in 61.5% (123/200) of inter-phase cells (Figure 6c and d). In addition, C2C12 cells display a typical bipolar array of antiparallel microtubules organized at the poles of mitotic cells (Figure 6e and f). Hybrid cells containing the DRT/DMC chromosome contain more than one spindle (~10%; 21/200) organized by multiple spindle poles (Figure 6g and h), indicating that cells with DRT/DMC display centrosome amplification and abnormal mitotic spindles. C2C12 hybrid cells containing a non-rearranged human chromosome 3 showed normal centrosome copy number (98%; 196/200) and normal spindles (200/200).
Figure 6.
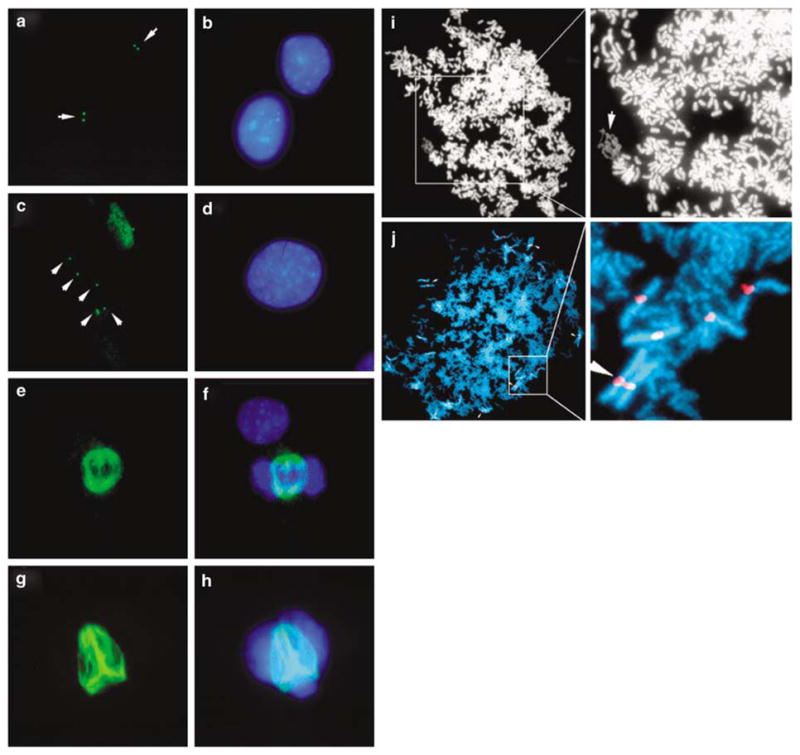
Centrosome amplification, endoreduplication and abnormal mitotic spindles. Immunostaining of γ-tubulin in interphase C2C12 (a and b) and C2C12 cells containing the der(1q) chromosome (c and d). The γ-tubulin antibody was detected with an FITC-labeled secondary antibody. Immunostaining of β-tubulin in mitotic C2C12 (e and f) and C2C12 cells containing the der(1q) chromosome (g and h). Cells were also stained with DAPI for visualization of DNA (b, d, f and h). Two representative metaphase spreads of hyperdiploid karyotypes showing endoreduplication (i–j). Magnification shows the DMC chromosome (arrow) (i). Magnification showing staining for chromosome 3 centromere with the arrow marking duplication of the centromere (j).
When two successive DNA syntheses occur without an intervening mitosis, endoreduplication occurs resulting in tetraploid karyotypes. We noticed that cell populations with DRT/DMC chromosomes often contained mitotic spreads with paired chromosomes (Figure 6i and j). Quantification of the frequency of endoreduplicated cells indicated that hybrid cells with DRT/DMC contained 2% (20/1000) and parental C2C12 cells contained 0.1% (1/1000), indicating an increased frequency of endoreduplication in cells with DRT/DMC.
We next determined if hybrids with DRT/DMC chromosomes also displayed CIN. Figure 7a shows that C2C12 cells displayed a relatively stable tetraploid karyotype with the majority of cells containing 80 chromosomes (Figure 7d). Figure 7b shows a representative metaphase spread harvested from C2C12 cells. In contrast, hybrid cells containing DMC chromosomes displayed karyotypes with every spread containing different numbers of chromosomes (majority containing greater than 156 chromosomes; Figure 7a). Figure 7c shows a representative spread harvested from cells containing the der(1q). These observations indicate that cells containing DRT/DMC chromosomes display CIN.
Figure 7.

CIN in cells with DRT/DMC. Mitotic chromosome spreads were stained with DAPI and scored for the number of chromosomes/spread, a minimum of 20 metaphase spreads were scored for each sample. Chromosome counts on control C2C12 cells and on C2C12 microcell hybrids containing the i(3q) [C2i(3q)-1], the der(3q) [C2(der3q)-1], the t(3;9) [C2(t3:9)-1] or the der(1q) [C2der(1q)-1] (a). A representative mitotic spread from C2C12 cells containing 80 chromosomes (b). A representative mitotic spread from a microcell hybrid with the t(3;9) chromosome containing >200 chromosomes (c). Chromosome counts on control C2C12 cells and on C2C12 microcell hybrids containing a non-rearranged chromosome 3 (C2(3n)-1 and C2(3n)-2) (d).
Discussion
DRT/DMC chromosomes are common in tumor cells and occur frequently in cells exposed to ionizing radiation (IR) (Smith et al., 2001; Breger et al., 2004). In this report, we found that DNA synthesis could be detected on DRT/DMC chromosomes during mitosis, and that DRT/DMC chromosomes contain phosphorylated Chk1. Furthermore, DRT/DMC chromosomes recruit INCENP, but are delayed in recruitment of Aurora B. In addition, DRT/DMC chromosomes retain Mad2 at a time when the fully condensed chromosomes have lost Mad2 staining. Finally, cells with DRT/DMC chromosomes show abnormal centrosome amplification, endoreduplication and significant CIN.
Chk1, important in the S phase and the DNA damage-induced G2/M checkpoints, appears to have a basal level of activity during normal S-phase progression (reviewed by Bartek et al., 2004). Our results suggest that Chk1 is activated by the delayed replication associated with DRT/DMC chromosomes. In addition, Chk1 phosphorylation remains in cis on the DRT/DMC chromosomes. Mutations in Grapes, the Drosophila Chk1 homolog, also show defects in chromosome condensation during metaphase (Yu et al., 2000). Our results are consistent with this observation and suggest that activated Chk1 participates in the delayed condensation phenotype observed on these chromosomes.
Our observations also suggest that chromosomes with DRT/DMC activate the S–M phase checkpoint. However, cells with DRT/DMC chromosomes are able to enter mitosis with active DNA synthesis. Analysis of the hydroxyurea (HU)-induced S–M phase checkpoint indicated that cells containing DRT/DMC chromosomes retain a functional checkpoint (data not shown). Therefore, because DNA synthesis could be detected on DRT/DMC chromosomes during mitosis at a low frequency (~0.1%) compared to the frequency of DMC (~50%) (Smith et al., 2001), these observations suggest that the S–M phase checkpoint is activated by DRT but that adaptation of the replication checkpoint had occurred in a fraction of cells.
Important distinctions between DRT and HU treatment are: first, DRT is characterized by continuing DNA replication, whereas HU treatment is characterized by stalled DNA replication; second, DRT occurs only on a subset of the chromosomes within a cell, whereas HU treatment affects every active replication fork on every chromosome. Adaptation to the DNA damage checkpoint in yeast was observed only when a single double-strand break was present (Sandell and Zakian, 1993). Thus, adaptation to the replication checkpoint may occur only in the presence of active DNA synthesis and not in the presence of stalled replication, or perhaps only when a fraction of the replication forks are affected. In addition, our observations suggest that one aspect of the replication checkpoint functions on each chromosome independently delaying mitotic chromosome condensation only on the affected chromosome (i.e. DMC). This aspect of the S–M phase checkpoint would not be apparent in cells treated with HU, because stalled replication would affect every chromosome and consequently prevent mitotic condensation on all of the chromosomes simultaneously.
Aurora B is important for the proper microtubule–kinetochore interaction and activation of the spindle assembly checkpoint (reviewed by Meraldi et al., 2004). However, Mad2 and BubR1 are recruited normally to kinetochores with impaired Aurora B function (Lens et al., 2003). Mad2 localizes to DMC chromosomes, suggesting that the spindle assembly checkpoint has sensed an unattached kinetochore. Cells that display the DRT/DMC phenotype show a significant amount of abnormal spindle assembly and endoreduplication, very reminiscent of G2 and spindle assembly checkpoint adaptation (Andreassen et al., 2003). This adaptation could lead to the inheritance of more subtle genetic changes or damaged chromosomes, propagating further instability. Our data suggest that DRT/DMC chromosomes activate both Chk1- and Mad2-dependent checkpoints without causing a permanent arrest. Furthermore, our observations identify Aurora B kinase as a potential link between activated Chk1, chromosome condensation and retention of Mad2.
Most cancers display CIN (Lengauer et al., 1997), which is a dominant trait and is independent of p53 mutations. Another common form of genomic instability is characterized by the generation of frequent chromosome rearrangements, including the formation of marker chromosomes and gene amplification. We refer to this process as translocation instability (TIN) and believe that these two forms of genetic instability are distinct. Recent studies in yeast indicate that suppression of spontaneous gross chromosomal rearrangements (TIN) involves redundant S-phase checkpoint pathways, suggesting that spontaneous ‘DNA replication errors’ can lead to chromosome rearrangements (Kolodner et al., 2002). In addition, checkpoint adaptation in yeast has been shown to precede both spontaneous and damage-induced genomic instability (Galgoczy and Toczyski, 2001). Checkpoint adaptation is required to achieve the highest possible viability of yeast cells exposed to DNA-damaging agents (Galgoczy and Toczyski, 2001). Thus, checkpoint adaptation serves to increase resistance to DNA damage, but in doing so allows cells to undergo mutagenic events. Our results suggest that the TIN associated with the DRT/DMC phenotype is a consequence of adaptation of the DNA replication checkpoint allowing cells to enter mitosis with unreplicated DNA.
Cells containing chromosomes with DRT/DMC induced by exposure to IR often contain hyperdiploid karyotypes (Breger et al., 2004). In this report, we show that introduction of a DRT/DMC chromosome into a karyotypically stable cell line results in centrosome amplification, abnormal mitotic spindles, endoreduplication and abnormal numbers of chromosomes. A recent study found that chromosome non-disjunction events lead to failed cytokinesis and the generation of tetraploid cells with subsequent multipolar spindles and ultimately aneuploid karyotypes (Shi and King, 2005). Our results suggest that DRT/DMC chromosomes activate the spindle assembly checkpoint. However, we often observe cells with paired chromosomes and hyperdiploid karyotypes. Taken together, our results suggest that DRT/DMC chromosomes experience non-disjunction events at an increased frequency, and have failed cytokinesis resulting in centrosome amplification, endoreduplication and ultimately in CIN and TIN.
Materials and methods
Cell culture
C2C12, CRL5845 and HeLa were from the American Type Culture Collection (ATCC, Manassas, VA, USA). All were grown in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (Hyclone Laboratories, Logan, UT, USA). The microcell hybrid C2i(3q)-1 was derived from RH30 cells (from Dr P Houghton) (Smith et al., 1998) and the C2(3n)-1 are C2C12 microcell hybrids containing an intact chromosome 3, derived from GM11713 (ATCC). R268 cells, derived from HTD114 fibrosarcoma cell line contain a t(15;16)(q24; q12.1) generated using Cre/lox (Breger et al., 2005).
Mitotic preparations, immunofluorescence and FISH
Chromosome preparations were harvested either with colcemid pretreatment (0.1 μg/ml) for 3 h or in the absence of colcemid treatment as described (Smith et al., 2001). Briefly, cells were grown, harvested with trypsin and incubated in 0.075 M KCl with or without 1 × phosphatase inhibitors (Sigma, St Louis, MO, USA). Cells were then fixed with 75% methanol, 25% acetic acid and spread on glass slides. Chromosomes were stained with antibodies to phosphoserine 10 histone H3 (Upstate, Chicago, IL, USA), INCENP (a generous gift from the Wellcome Trust, Dr William Earnshaw) and Aurora B (AIM 1, BD Biosciences; San Jose, CA, USA). Cultured cells were stained with antibodies to Mad2 (Covance, Berkeley, CA, USA), CENP-A (Upstate), β-tubulin (Sigma), γ-tubulin (Sigma) and CREST (a generous gift from Dr David Ward). Signal was identified with secondary antibodies with Cy3-tags (Jackson Immunochemicals, West Grove, PA, USA) or alexa fluor 488 tags (Molecular Probes (Invitrogen, Carlsbad, CA, USA)).
DNA probes (total human DNA or chromosome 3 α-satellite) were nick-translated using standard protocols to incorporate biotin-11-dUTP (deoxyuridinetriphosphate) or digoxygenin-dUTP. Hybridizations were carried out on slides at 37°C for 16 h. Final probe concentrations varied from 40–60 ng/μl. Signal detection was carried out as described (Trask and Pinkel, 1990). Amplification of the biotinylated probe signal utilized alternating incubations of slides with anti-avidin (Vector, Burlingame, CA, USA) and fluorescein isothiocyanate (FITC)-Extravidin (Sigma). Chromosome-specific painting probes were used according to the manufacturer’s recommendations (Vysis, Des Plaines, IL, USA). Centromeric probes for chromosomes 15 and 16 were also obtained for use in FISH according to the manufacturer’s recommendation (Vysis). Amplification of digoxygenated probes utilized alternating incubations of slides with FITC-tagged sheep antibodies made in rabbit and FITC-tagged rabbit antibodies made in sheep Boehringer Mannheim (Roche Applied Sciences, Indianapolis, IN, USA). Slides were stained with propidium iodide or 4′,6-diamidino-2-phenylindole (DAPI), coverslipped and viewed under ultraviolet fluorescence with appropriate filters using epifluorescence microscopy (Zeiss, Thornwood, NY, USA).
Replication studies
In situ DNA replication was carried out essentially as described (Mills et al., 2000), except that cells were plated on conconavalin A (Sigma)-treated slides to enhance attachment of mitotic cells. Cells were permeabilized with 25 μm digitonin Fluka (Sigma-Aldrich Chemie GmbH, Buchs SG, Switzerland), incubated with nucleotide mix containing 50 μm Dig-dUTP (Boehringer Mannheim) for 30 min at 37°C, fixed in 4% paraformaldehyde and stained with an antibody against serine 10 phosphorylated histone H3. The slides were then processed for FISH using a chromosome 3 painting probe (Vysis), as described above.
Replication checkpoint and immunofluorescence
Cells were grown on glass slides, fixed in 4% paraformaldehyde and stained with an antibody against phosphorylated serine 345 of Chk1 Cell Signaling (Danvers, MA, USA). A total of ≥2000 cells per experiment were scored by microscopy (Zeiss) in three independent experiments. Phosphorylation of Chk1 on chromosomes with DRT was assayed using and then processed for FISH using total human DNA as probe, as described above.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health NIH/NIGMS F32-GM071176-01A1 (BHC), The Laura and Greg Norman National Childhood Cancer Foundation (BHC), NCI CA104693 (MT) and NCI CA97021 (MT).
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Adams R, Carmena M, Earnshaw W. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001;11:49–54. doi: 10.1016/s0962-8924(00)01880-8. [DOI] [PubMed] [Google Scholar]
- Andreassen P, Lohez O, Margolis R. G2 and spindle assembly checkpoint adaptation, and tetraploidy arrest: implications for intrinsic and chemically induced genomic instability. Mutat Res. 2003;532:245–253. doi: 10.1016/j.mrfmmm.2003.08.020. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- Breger K, Smith L, Thayer M. Engineering translocations with delayed replication: evidence for cis control of chromosome replication timing. Hum Mol Genet. 2005;14:2813–2827. doi: 10.1093/hmg/ddi314. [DOI] [PubMed] [Google Scholar]
- Breger K, Smith L, Turker M, Thayer M. Ionizing radiation induces frequent translocations with delayed replication and condensation. Cancer Res. 2004;64:8231–8238. doi: 10.1158/0008-5472.CAN-04-0879. [DOI] [PubMed] [Google Scholar]
- Carmena M, Earnshaw W. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- Galgoczy D, Toczyski D. Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol Cell Biol. 2001;21:1710–1718. doi: 10.1128/MCB.21.5.1710-1718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendzel M, Wei Y, Mancini M, Van Hooser A, Ranalli T, Brinkley B, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Honda R, Korner R, Nigg E. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003;14:3325–3341. doi: 10.1091/mbc.E02-11-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan M, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kolodner R, Putnam C, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler K, Vogelstein B. Genetic instabilities in human cancers. Nature. 1997;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- Lens S, Wolthius R, Klompmaker R, Kauw J, Agami R, Brummelkamp T, et al. Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J. 2003;22:2934–2947. doi: 10.1093/emboj/cdg307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui X, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg E. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Gen Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Mills A, Coleman N, Morris L, Laskey R. Detection of S-phase cells in tissue sections by in situ DNA replication. Nat Cell Biol. 2000;2:244–245. doi: 10.1038/35008670. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Lengauer C. CIN-ful cancers. Cancer Chem Pharmacol. 2004;53:S65–S68. doi: 10.1007/s00280-004-0889-8. [DOI] [PubMed] [Google Scholar]
- Sandell L, Zakian V. Loss of yeast telomere: arrest, recovery, and chromosome loss. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- Shi Q, King R. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 2005;437:1038–1042. doi: 10.1038/nature03958. [DOI] [PubMed] [Google Scholar]
- Smith L, Liu S, Goodrich L, Jacobson D, Degnin C, Bentley N, et al. Duplication of ATR inhibits MyoD, induces aneuploidy and eliminates radiation induced G1 arrest. Nat Genet. 1998;19:39–47. doi: 10.1038/ng0598-39. [DOI] [PubMed] [Google Scholar]
- Smith L, Plug A, Thayer M. Delayed replication timing leads to delayed chromosome condensation and chromosomal instability of chromosome translocations. Proc Natl Acad Sci USA. 2001;98:13300–13305. doi: 10.1073/pnas.241355098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask B, Pinkel D. Fluorescence in situ hybridization with DNA probes. Methods Cell Biol. 1990;33:383–400. doi: 10.1016/s0091-679x(08)60542-7. [DOI] [PubMed] [Google Scholar]
- Yu K, Saint R, Sullivan W. The Grapes checkpoint coordinates nuclear envelope breakdown and chromosome condensation. Nat Cell Biol. 2000;2:609–615. doi: 10.1038/35023555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.