

Summary
C. perfringens possesses at least two functional quorum sensing (QS) systems, i.e., an Agr-like system and a LuxS-dependent AI-2 system. Both of those QS systems can reportedly control in vitro toxin production by C. perfringens but their importance for virulence has not been evaluated. Therefore, the current study assessed whether these QS systems might regulate the pathogenicity of CN3685, a C. perfringens type C strain. Since type C isolates cause both hemorrhagic necrotic enteritis and fatal enterotoxemias (where toxins produced in the intestines are absorbed into the circulation to target other internal organs), the ability of isogenic agrB or luxS mutants to cause necrotizing enteritis in rabbit small intestinal loops or enterotoxemic lethality in mice was evaluated. Results obtained strongly suggest that the Agr-like QS system, but not the LuxS-dependent AI-2 QS system, is required for CN3685 to cause hemorrhagic necrotizing enteritis, apparently because the Agr-like system regulates the production of beta toxin, which is essential for causing this pathology. The Agr-like system, but not the LuxS-mediated AI-2 system, was also important for CN3685 to cause fatal enterotoxemia. These results provide the first direct evidence supporting a role for any QS system in clostridial infections.
Introduction
Clostridium perfringens type C causes disease in livestock by growing in the small intestine and then producing toxins that induce hemorrhagic necrotic enteritis and enterotoxemia, i.e., absorption of intestinally-produced toxins into the circulation so they can damage internal organs outside of the gastrointestinal tract (Uzal and McClane, 2011; McClane et al., 2004). Type C diseases of livestock, which are often rapidly fatal due to severe enteritis or lethal enterotoxemic effects on internal organs, impact agricultural economics so significantly that livestock routinely receive vaccinations with toxoided type C culture supernatants. In humans, type C strains cause similar disease, known as enteritis necroticans (Johnson and Gerding, 1997; Lawrence, 1997), that occurs sporadically throughout the developing world (Johnson and Gerding, 1997); however, type C human disease is historically most associated with the Papua New Guinea highlands, where it is known as Pigbel (Johnson and Gerding, 1997; Lawrence, 1997). Human type C infections also occasionally occur in developed countries, mainly in people with pancreatic disease (Petrillo et al., 2000).
By definition, type C isolates must produce beta toxin (CPB) and alpha toxin (CPA). CPB, a lethal 35 kDa pore-forming protein, has some sequence similarity to Staphylococcus aureus alpha toxin (Smedley et al., 2004). CPA is a lethal 42.4 kDa protein with phospholipase C and sphingomyelinase activities (Titball et al., 1999). Many type C strains produce additional toxins (Fisher et al., 2006) such as perfringolysin O (PFO), a hemolytic pore-forming toxin of the cholesterol-dependent cytolysin family (Heuck et al., 2010). Recent studies using isogenic toxin mutants of CN3685 established that production of CPB, but not CPA or PFO, is essential for this type C strain to cause hemorrhagic necrotic enteritis in a rabbit small intestinal loop model (Sayeed et al., 2008). Studies using purified CPB later showed that all rabbit small intestinal segments, but particularly the jejunum, are sensitive to this toxin (Vidal et al., 2008). Isogenic toxin CN3685 mutants were also used to establish the importance of CPB when this type C isolate causes fatal enterotoxemia in a mouse ID-challenge model (Uzal et al., 2009).
Most bacterial pathogens regulate their virulence factor production in response to environmental cues (Parker and Sperandio, 2009; Calva and Oropeza, 2006). Two-component regulatory systems are a particularly important mechanism for environmental control of bacterial pathogenicity (Beier and Gross, 2006; Calva and Oropeza, 2006). Although understanding of pathogenicity regulation remains rudimentary when C. perfringens vegetative cells cause disease originating in the intestines, recent studies demonstrated that the VirS/VirR two-component regulatory system is very important or essential, respectively, for type C isolate CN3685 to cause fatal enterotoxemia in mice or necro-hemorrhagic enteritis in rabbits (Ma et al., 2011). For rabbit necro-hemorrhagic enteritis, the critical role of VirS/VirR in controlling CN3685 pathogenicity involves regulating CPB production in the intestine (Ma et al., 2011).
A second major family of environmentally-responsive bacterial pathogenicity regulators are quorum sensing (QS) systems, which control gene expression by population density-induced autoinducer signaling molecules (Antunes et al., 2010). One well-studied QS system, named Agr, regulates toxin production in S. aureus (Novick and Geisinger, 2008). The C. perfringens genome encodes portions of the S. aureus Agr system (Myers et al., 2006; Shimizu et al., 2002), including the AgrD peptide and the AgrB membrane endoprotease that processes the AgrD peptide to an active signal-capable form. Recent studies (Ohtani et al., 2009; Vidal et al., 2009a) demonstrated that agrB or agrBD null mutants of C. perfringens type A strain 13 are impaired for producing CPA and PFO, both chromosomally-encoded. Similarly, an agrB null mutant of C. perfringens type A strain F5603 made reduced amounts of CPA and PFO, as well as plasmid-encoded beta2 toxin and enterotoxin (Li et al., 2011). Involvement of the Agr-like QS system in this toxin regulation received support when C. perfringens culture supernatants were shown to contain a signaling molecule capable of restoring toxin production to agrB or agrBD mutants of strain 13 (Ohtani et al., 2009; Vidal et al., 2009a). While the C. perfringens agr locus is a four gene operon including two hypothetical proteins, AgrB and AgrD are necessary and sufficient for obtaining QS regulatory function in this bacterium (Ohtani et al., 2009).
C. perfringens also encodes a second functional QS system, named the AI-2 system, that involves the LuxS enzyme. Previous studies using a luxS mutant reported that AI-2 regulates CPA and PFO production by strain 13 (Ohtani et al., 2002). However, a luxS null mutant of type C strain CN3685 could still upregulate expression of CPB, CPA and PFO upon contact with Caco-2 human enterocyte-like cells under aerobic conditions (Vidal et al., 2009b).
Currently no information is available regarding whether the agr operon, which encodes the essential AgrB and AgrD components of the Agr-like QS system, regulates toxin production by C. perfringens type C isolates in vitro. Equally important, whether the Agr-like QS system or AI-2 QS system regulates C. perfringens virulence has not yet been evaluated. Therefore, in the current study we constructed an isogenic agrB null mutant in type C isolate CN3685 and used that mutant to assess possible Agr-like QS system-mediated control of type C isolate toxin production. That mutant, and a previously constructed luxS null mutant of CN3685, were also employed to evaluate the importance of these two QS systems for the pathogenicity of this type C isolate when it causes either hemorrhagic necrotic enteritis or fatal enterotoxemia.
Results
Analysis of the agr locus in CN3685
The current study first determined whether type C strain CN3685 carries an agr locus similar to that present in strain 13, a type A strain (Ohtani et al., 2009; Vidal et al., 2009a). A PCR assay using primers that amplify the ~2.7 kb agr locus of strain 13 also produced a similar size product using CN3685 DNA (data not shown). When sequenced, this CN3685 PCR product showed a sequence with >98% identity, at the nucleotide level, with the agr locus sequence of strain 13. Translation of this sequence confirmed that the CN3685 agr locus encodes the hypothetical proteins CPE1562 and CPE1563, as well as AgrB and AgrD. RT-PCR transcriptional analysis demonstrated (data not shown) that the CPE1562 ORF, the CPE1563 ORF, agrB and agrD are co-transcribed as an operon, as previously reported for the agr locus in strain 13 (Ohtani et al., 2009; Vidal et al., 2009a).
Construction and characterization of a CN3685 agrB null mutant and complementing strain
To evaluate whether the agr operon found in CN3685 is important for toxin regulation, the agrB gene in this strain was insertionally-inactivated using a Clostridium-modified Targetron™ gene knockout system (Li and McClane, 2008; Sayeed et al., 2008; Chen et al., 2005) to produce a mutant named BMJV10. The presence of an intron in the agrB ORF of BMJV10 was demonstrated by PCR (Fig. 1A) and Southern blot analyses confirmed that only this single intron insertion was present in this strain (Fig. 1B). A complementing strain named BMJV13 was then prepared by electroporating BMJV10 with P3, a pJIR750-derivative (Bannam and Rood, 1993) encoding the wild-type C. perfringens agr operon (Vidal et al., 2009a). As shown in Fig. 1A lane 4, PCR detected the presence of a trans-encoded, wild-type agrB gene in BMJV13 and Southern blot analysis (Fig. 1B) confirmed retention of the single intron insertion in the complementing strain.
Fig. 1. Inactivation of the agrB gene using the Targetron™ gene knockout system to prepare BMJV10 and complementing strain BMJV13.

Panel A, PCR confirmation of the isogenic agrB null mutant and complementing strain. Using DNA isolated from wild-type CN3685, a PCR assay amplified an ~600 bp product using primers that target the agrB gene. Using DNA purified from the agrB null mutant (BMJV10), which has an ~900 bp intron insertion, this same PCR assay amplified an ~1.5 kb PCR product. Purified DNA from the complementing strain BMJV13 yielded the same ~600 bp PCR product as amplified from the wild-type strain. Lane 1, 100 bp molecular weight marker. Panel B, Southern blot hybridization of a labeled intron-specific probe with DNA from wild-type CN3685, the agrB null mutant (BMJV10) or complementing strain (BMJV13). DNA from each strain was digested with EcoRI and electrophoresed on a 1% agarose gel prior to blotting and hybridization with those above mentioned probes. Panel C, RT-PCR analyses for agrB expression by CN3685, BMJV10 and complementing strain BMJV13 grown overnight in TGY broth. Lane 1 shows size markers. Lanes labeled “+” were from samples receiving reverse transcriptase (RT), while lanes labeled “−“ lacked reverse transcriptase to show the absence of DNA contamination.
When RT-PCR was performed to evaluate agrB expression in these strains, RNA from wild-type CN3685 or the complementing strain BMJV13 supported amplification of an agrB mRNA product (Fig. 1C). In contrast, no RT-PCR product was detected using template RNA isolated from BMJV10. Together these results confirmed intron disruption of the agrB gene in strain BMJV10 and trans-complementation of agrB expression in BMJV13.
Production of CPB, CPA and PFO in TGY broth cultures
Whether the agr operon regulates CPA, CPB and PFO production by wild-type CN3685 under anaerobic conditions, as found in the intestines during disease, was first investigated using cultures grown at 37°C in TGY broth containing thioglycolate. Under these growth conditions, Western blotting detected the presence of CPA, CPB and PFO in CN3685 culture supernatants starting within ~3 h of growth (not shown) and then continuing overnight (Fig. 2A). However, compared against culture supernatants of wild-type CN3685, culture supernatants of the BMJV10 isogenic agrB mutant contained much less of all three toxins after an ~3 h growth (data not shown) and those differences remained detectable even after overnight culture (Fig. 2A). The attenuated CPA, CPB and PFO production by the mutant was specifically attributable to disruption of the agr operon since normal toxin production was regained when the mutant was complemented with a plasmid encoding the wild-type agr operon (Fig. 2A).
Figure 2. Toxin production by CN3685 and its isogenic derivatives grown overnight in TGY broth.

Wild-type CN3685, the agrB null mutant BMJV10, the complementing strain BMJV13 carrying the wild-type agr operon on plasmid P3, and the luxS mutant CPJV19 were grown overnight in TGY medium at 37°C. For CN3685, BMJV10 and BMJV13, equal volumes of each culture supernatant sample were subjected to SDS-PAGE, followed by either, Panel A (top), Western blotting for CPA, CPB, or PFO production or Panel B (left), Silver staining. Since luxS was previously reported to regulate production of CPA and PFO during C. perfringens growth in broth culture (Ohtani et al., 2002), duplicate samples of the overnight culture supernatant of CPJV19 were subjected to SDS-PAGE, followed by either, Panel A (bottom), Western-blotting for CPB production or Panel B (right) Silver staining. Shown in the Figure are representative results that were reproducible over three repetitions. The molecular weight of each toxin is indicated on the right side of the blots in (A) and migration of molecular weight markers is shown in M of the top panel shown in (B).
The reduced production of CPA, CPB and PFO by BMJV10 was not due to slower vegetative growth of this agrB knockout mutant. Comparative studies of wild-type CN3685, the BMJV10 agrB knockout mutant, and the BMJV13 complementing strain detected similar growth of all three strains in TGY broth (data not shown). Silver staining of SDS-containing gels after electrophoresis with culture supernatants samples revealed (Fig. 2B) that the reduced toxin production by the agrB mutant was also not due to a generalized decrease in supernatant proteins as more total protein was present in the culture supernatants of BMJV10 vs. CN3685 or BMJV13. Finally, the reduced presence of toxins in culture supernatants of BMJV10 was attributable to agrB involvement in toxin production, rather than secretion, since the toxins were also not detected using lysed BMJV10 cells (not shown).
In contrast to the reduced CPB production observed for the BMJV10 agrB mutant, culture supernatants of the previously-prepared, isogenic CPJV19 luxS null mutant contained similar CPB levels as did culture supernatants of wild-type CN3685, whether measured at 3,4,5 h post inoculation or overnight (Fig. 2A). CPJV19 also grew similarly as CN3685 in TGY broth (data not shown). Silver staining detected similar patterns and amounts of supernatant proteins for CN3685 and CPJV19 (Fig. 2B).
Evidence that a QS-like signaling effect is involved in agr operon-dependent regulation of CPB production by CN3685
If the agr operon-mediated regulation of toxin production by CN3685 vegetative cells shown in Fig. 2 was mediated by an Agr-like QS mechanism, this effect should involve a diffusible signaling molecule secreted into the supernatant. To test this hypothesis, bacteria were inoculated into the top and bottom chambers of a TGY broth-containing Transwell dish where those two chambers are separated by a membrane filter that is impermeable to C. perfringens vegetative cells but contains pores allowing passage of small molecules. The supernatant samples from the wells were then removed and subjected to CPB Western blotting.
Using this model system, strong CPB production was noted when washed wild-type CN3685 cells were inoculated into both the top and bottom chambers of the dish, which was then incubated under anaerobic conditions for 5 h (Fig. 3). In contrast, no CPB production was observed when washed cells of the cpb null mutant BMC100 (which had been previously constructed and characterized (Sayeed et al., 2008)) were inoculated into both chambers and the dish was then similarly incubated for 5 h. Also consistent with the Fig. 2 results, only very weak CPB production was detected when washed cells of the BMJV10 agrB null mutant were inoculated into both chambers and the dish was then anaerobically cultured for 5 h at 37°C.
Figure 3. Transwell assay to determine if agr operon-mediated regulation of CPB production involves a diffusible signaling molecule.

In a single Transwell plate, two wells (top and bottom) are separated by a membrane filter impermeable to bacteria but having pores that allow diffusion of small molecules. Shown in the top panel is the inoculation strategy used in the experiment. The bottom panels show, respectively, results after the top well of each specified culture supernatant from a 5 h culture was subjected to CPB Western blotting. Shown in the Figure are representative results that were reproducible over three repetitions. The migration of purified CPB is indicated on the Western blot.
However, substantial CPB production was detected after a 5 h anaerobic co-culture at 37°C of washed BMJV10 cells (which retains a functional cpb gene) and BMC100 in the same dish, but separated by a pore-containing support membrane. This effect was independent of which chamber (top or bottom) was inoculated with BMC100 or BMJV10, although CPB was predominantly localized in the chamber containing the agrB mutant (data not shown). Collectively, these Fig. 3 results are consistent with BMC100 producing, via an Agr-like QS system encoded at least in part by the agr operon, a diffusible signal that causes BMJV10 to express increased amounts of CPB.
An AgrD sequence-based synthetic peptide can restore CPB production to the agrB mutant
In S. aureus, the active quorum sensing peptide (named AIP) of the Agr QS system, which is processed from the AgrD polypeptide by the action of AgrB (Novick and Geisinger, 2008), is most commonly an eight amino acid peptide with a centrally-located cysteine residue that comprises part of a thiolactone ring (Fig. 4A). Analysis of the CN3685 AgrD sequence, as determined earlier in this study, revealed the presence of a single cysteine residue. Therefore, we tested whether agr locus involvement in CPB production by CN3685 (as shown in Fig. 2) resembles the Agr QS system of S. aureus by involving an AgrD sequence-derived peptide signal. For this purpose, an 8-mer peptide with a centrally-located cysteine comprising part of a thiolactone ring was synthesized based upon the CN3685 AgrD sequence.
Figure 4. An AgrD sequence-derived peptide can physically complement the agrB null mutant.

Aliquots (25-100 μM final concentrations, as indicated) of CPAIP, an octameric AgrD sequence-derived peptide with a cyclic thiolactone ring (shown in panel A), were added to a TGY tube freshly inoculated with the BMJV10 agrB mutant. After those cultures were grown for 5 h, the culture supernatants were processed for CPB Western blotting (panel B). A 25 mM final concentration of a negative control peptide with a non-AgrD-derived sequence was similarly added to the agrB mutant and then processed as for the CPAIP-treated cultures. For comparison, CPB production by CN3685 and BMJV10 in the absence of peptide is also shown. The Figure shows representative results that were reproducible over three repetitions.
When this peptide, named C. perfringens autoinducing peptide (CPAIP), was added to TGY cultures of the agrB null mutant, it stimulated CPB production. In contrast, equivalent concentrations of a similar-sized, cysteine-containing negative control peptide did not increase CPB production levels. These CPAIP results strongly support the agr locus-mediated regulation of CPB production by the agr locus involving an Agr-like QS effect mediated by an AgrD-derived peptide.
Comparison of Caco-2 cell cytotoxicity induced by anaerobic infection with CN3685, BMJV10, BMJV13 or CPJV19
Our previous data showed that anaerobic infection with type C isolate CN3685 causes the development of cytotoxicity in Caco-2 cells, at least in part, by upregulating toxin production (Ma et al., 2011). In the current study, we investigated whether the Agr-like or the AI-2 QS systems might help to mediate this CN3685-induced development of Caco-2 cell cytotoxicity.
As shown in Fig. 5A, a 1 h anaerobic infection with washed wild-type CN3685 vegetative cells caused significant Caco-2 cell rounding and detachment from confluent monolayers. While cell rounding was also observed under the same infection conditions using the agrB null mutant BMJV10, this morphologic cell damage was comparatively weaker and no cell detachment was observed. Complementation of this mutant to restore agr operon expression reversed this attenuation of morphologic damage at 1 h (Fig. 5A), indicating that the reduced cell damage caused by BMJV10 vs. wild-type CN3685 was attributable to the specific inactivation of the agr operon in the BMJV10 mutant. The attenuation of Caco-2 cell morphologic damage observed during BMJV10 infection did not result from reduced bacterial survival since similar numbers of viable bacterial cells were recovered from cell cultures after a 1-3 h anaerobic infection with CN3685, BMJV10 or BMJV13 (data not shown).
Figure 5. Anaerobic infection of Caco-2 cells by CN3685 and its isogenic derivatives.

Panel A, Morphological damage to Caco-2 cells. Confluent Caco-2 cells were anaerobically infected at an MOI of 20:1 for 1 hour at 37°C. Inocula used for these infections included fresh MEM containing washed cells of CN3685, BMJV10, BMJV13, or CPJV19. Fresh MEM without bacteria was used as a negative control. Panel B, Lactate dehydrogenase (LDH) release by Caco-2 cells. LDH release was measured as an indicator of Caco-2 cytotoxicity caused by a 1 h anaerobic infection at 37°C using washed vegetative cells of CN3685, BMJV10 and BMJV13. The experiment was repeated three times, and shown are mean results from those three independent repetitions. Error bars depict standard deviation (SD); * depicts a statistically significant (P<0.01) decrease in LDH cytotoxicity compared against infection with washed cells of wild type CN3685. Triton-X 100 (1%) was used as positive control, which gave 100% cell death. After a 1-3 h Caco-2 cell infection with washed vegetative cells of CN3685, BMJV10, or BMJV13 each culture supernatant sample was subjected to Western blotting for Panel C, CPB production or Panel D, CPA production. Also shown, CPB Western blot analyses (panel E) of 1-3 hour culture supernatants removed after a 1-3 h Caco-2 cell infection with washed vegetative cells of CN3685 and the luxS null mutant CPJV19. Western blot results shown in the Figure are representative results that were reproducible over three independent experiments.
Differences between Caco-2 cell cytotoxicity caused by anaerobic infection with washed cells of CN3685, BMJV10 or BMJV13 were then quantitatively compared using a lactate dehydrogenase (LDH) release cytotoxicity assay. Measuring LDH activity in supernatants removed from Caco-2 cell cultures after a 1 h anaerobic infection revealed that infection with washed wild-type CN3685 cells caused significantly more Caco-2 cell death than a similar infection with the isogenic agrB null mutant BMJV10 (Fig. 5B). This cytotoxicity difference was due to inactivation of the agr operon in the mutant since a 1 h anaerobic infection with washed cells of the complementing strain BMJV13 caused similar levels of LDH release from Caco-2 cells as observed after infection using washed cells of the CN3685 parent strain.
In contrast to the results using the agrB mutant, similar cytotoxicity levels were observed in Caco-2 cultures challenged with either wild-type CN3685 or the CPJV19 luxS mutant (data not shown).
Comparison of toxin production after anaerobic infection of Caco-2 cells with CN3685, BMJV10, BMJV13 or CPJV19
Western Blot analysis was performed to evaluate CPB and CPA levels in the supernatants from Caco-2 cultures that had been anaerobically-infected with washed cells of CN3685, BMJV10 or BMJV13. Neither CPB (Fig. 5C) nor CPA (Fig. 5D) production was detected in supernatants removed from Caco-2 cultures after a 1 h anaerobic infection using any of those three strains. However, CPB (Fig. 5C) and CPA (Fig. 5D) production became detectable in Caco-2 culture supernatants after a 2 h anaerobic infection with CN3685 or the BMJV13 complementing strain. In contrast, no CPB or CPA was detected in supernatants of infected Caco-2 cell cultures even after a 3 h anaerobic infection with the agrB null mutant BMJV10 and despite the fact that more total protein was present in culture supernatants after infection with BMJV10 vs. CN3685 (data not shown). Thus, production of CPB and CPA by CN3685 in this in vitro infection model is apparently regulated by the Agr-like QS system encoded, at least in part, by the agr locus. In contrast, similar levels of CPB were made by wild-type CN3685 and CPJV19 in this in vitro infection model (Fig. 5E).
Attempts to detect, by Western blot analysis, the presence of PFO in supernatants removed from Caco-2 cell cultures after a 1-3 h anaerobic infection with washed cells of CN3685, BMJV10, BMJV13 or CPJV19 were unsuccessful (data not shown), suggesting limited early-stage PFO production in this in vitro infection model.
Pathogenicity of wild-type CN3685, BMJV10, BMJV13 and CPJV19 in rabbit small intestinal loops
The Fig. 5 results indicating that the agr operon regulates CPB production in an in vitro Caco-2 cell infection model could suggest that the Agr-like QS system is important for CN3685-induced intestinal disease. This possibility was assessed in rabbit small intestinal loops challenged with washed cells of CN3685, BMJV10, BMJV13 or CPJV19 resuspended in sterile TGY-C, which eliminated effects from preformed toxins made during in vitro growth of bacterial cultures. Trypsin inhibitor was added to all samples to prevent degradation of CPB produced in the intestine by type C isolates (Sayeed et al., 2008; Vidal et al., 2008).
A) Gross pathology of rabbit small intestinal loops infected with CN3685, BMJV10, BMJV13 or CPJV19
After a 6 h treatment with washed cells of wild-type CN3685 or the BMJV13 complementing strain, rabbit loops were distended, with severe mucosal hemorrhage and an apparent presence of increased luminal fluid (Fig. 6A). These effects were visible from both the mucosal and the serosal surfaces. The intestinal wall was thin and lacked natural tone. In contrast, loops challenged with washed cells of the isogenic agrB null mutant BMJV10 or sterile non-toxic TGY-C (negative control) exhibited no gross abnormalities (Fig. 6A). The CPJV19 luxS mutant caused similar gross pathology as observed during a wild-type CN3685 infection (data not shown).
Fig. 6. Gross pathology, fluid accumulation, and histopathology in rabbit intestinal loops inoculated with type C strain CN3685 and its derivatives.

Washed cells of CN3685, the isogenic agrB null mutant (BMJV10), the agr operon complementing strain (BMJV13) or the luxS mutant CPJV19 were inoculated (~108 CFU) into rabbit small intestinal loops and incubated for 6 h. As a negative control, sterile non-toxic TGY-C was also injected into rabbit loops. Panel A, Gross pathology of inoculated loops. Similar gross pathology was caused by CN3685 and CPJV19 (not shown). Panel B, Fluid accumulation in inoculated small intestinal loops. * indicates statistically-significant (P<0.05) differences from the wild-type and BMJV13. Every experiment was independently performed six times. Panel C, Histologic damage in inoculated small intestinal loops tissues were processed by histology and stained using hematoxylin and eosin stain. Sections of treated or control tissues were then photomicrographed at 200X final magnification. Shown are representative photomicrographs of six repetitions for each inoculum.
B) Fluid accumulation in rabbit loops after infection with CN3685, BMJV10, BMJV13 or CPJV19
When luminal fluid levels were quantified, small intestinal loops challenged for 6 h with washed wild-type CN3685 cells had accumulated relatively large amounts of hemorrhagic fluid, which is indicative of mucosal damage (Fig. 6B), matching previous reports (Ma et al., 2011; Sayeed et al., 2008). This effect was statistically different (p<0.05) from fluid levels accumulating in loops treated similarly with washed cells of the agrB mutant BMJV10 or sterile TGY-C (Fig. 6B). In addition, the fluid present in loops treated with either washed cells of BMJV10 or sterile TGY-C was not hemorrhagic.
Confirming a role for the C. perfringens agr operon in this in vivo accumulation of luminal fluids, small intestinal loops challenged for 6 h with washed cells of the BMJV13 complementing strain accumulated more fluid, which was hemorrhagic, than loops challenged with BMJV10. Furthermore, the fluid levels in the BMJV13-treated and CN3685-treated loops did not significantly differ (Fig. 6B). Lastly, no significant difference was detected between luminal fluid levels in loops infected with the CPJV19 luxS mutant vs. CN3685 (Fig. 6B).
C) Histological damage in rabbit small intestinal loops treated with CN3685, BMJV10, BMJV13 or CPJV19
When histological damage was assessed, loops challenged for 6 h with washed CN3685 cells showed (Fig. 6C and Table 1) results similar to previous reports (Sayeed et al., 2008), i.e., their intestinal villi had almost completely disappeared and were replaced by low-denudated bumps, which were occasionally lined by a pseudomembrane comprised of red blood cells, neutrophils, necrotic epithelial cells and cell debris (Fig. 6C). In addition, the lamina propria was diffusely hemorrhagic. In contrast, small intestinal loops similarly treated with washed cells of the BMJV10 isogenic agrB null mutant or sterile TGY-C showed no significant histological abnormalities, except for mild submucosal edema and dilation of submucosal lymphatic vessels. A role for the agr operon in regulating these pathological effects was confirmed by demonstrating that loops treated with washed cells of the complementing strain BMJV13 showed nearly similar lesions as loops challenged with washed CN3685 cells.
Table 1.
Rabbit small intestinal loop histological changes.
| Pathology score for : | ||||||
|---|---|---|---|---|---|---|
| Sample | Desquamation | Necrosis of epithelium |
Necrosis of lamina propria |
Inflammation | Villous blunting |
Overall |
| CN3685 | 4.0 ± 0.7 | 4.3 ± 0.6 | 4.3 ± 0.6 | 3.2 ± 1.37 | 4.2 ± 0.5 | 4.3 ± 0.5 |
| BMJV10 | 0.2 ± 0.0*, a | 0.1 ± 0.0*, a | 0.0 ± 0.0*, a | 0.4 ± 0.4*, a | 0.4 ± 0.4*, a | 0.2 ± 0.1*, a |
| BMJV13 | 2.0 ± 0.7* | 2.1 ± 1.1* | 2.1 ± 1.1* | 1.0 ± 0.5* | 1.8 ± 1.3 | 2.3 ± 1.0* |
| CPJV19 | 3.4 ± 0.8a | 3.5 ± 0.9*, a | 3.4 ± 0.8*, a | 2.7 ± 1.0a | 3.5 ± 0.8a | 3.5 ± 0.7a |
| TGY-C | 0.2 ± 0.4*, a | 0.0 ± 0.0*, a | 0.0 ± 0.0*, a | 0.6 ± 0.5* | 0.0 ± 0.0*, a | 0.3 ± 0.3*, a |
Indicates a statistically significant (P<0.05) difference in comparison with CN3685 infection.
Indicates a statistically significant (P<0.05) difference in comparison with BMJV13 infection.
The degree of damage was scored using a scale of 0 to 5, with 0 indicating no histologic damage and 1, 2, 3, 4 and 5 values indicating increasingly severe damage.
Results shown are mean ± S.D.
Washed cells of the CPJV19 luxS mutant caused small intestinal damage similar to that produced by wild-type CN3685 infection (Fig. 6C and Table 1).
D) Regulation of CPB production in rabbit loops infected with CN3685, BMJV10, BMJV13 or CPJV19
Since CPB is necessary and sufficient to induce hemorrhagic fluid accumulation and intestinal damage in the rabbit small intestinal loop model of C. perfringens necrotizing enteritis (Sayeed et al., 2008; Vidal et al., 2008), an experiment investigated whether the virulence attenuation of BMJV10 shown in Figure 6 could involve in vivo regulation of CPB production by the Agr-like QS system encoded, at least in part, by the agr operon. In this experiment, luminal fluids were removed from rabbit small intestinal loops after a 6 h infection with washed cells of CN3685, the agrB mutant BMJV10 or complementing strain BMJV13.
When those fluids were probed by Western blot utilizing monoclonal anti-CPB antibodies, the presence of CPB was detected in luminal fluids removed from loops challenged for 6 h with washed cells of wild-type CN3685 (Fig. 7, upper panel). In contrast, no CPB was detected in loops challenged for 6 h with washed cells of BMJV10, the agrB mutant, despite the presence of substantial amount of protein in luminal fluid of these loops (Fig. 7, lower panel). The absence of in vivo CPB production by washed BMJV10 cells was reversible by agr operon complementation (Fig. 7, upper panel).
Figure 7. Western blot analysis of in vivo CPB production in intestinal luminal fluids recovered from experimental rabbit small intestinal loops following infection with washed vegetative cells of CN3685, BMJV10, BMJV13, or CPJV19.
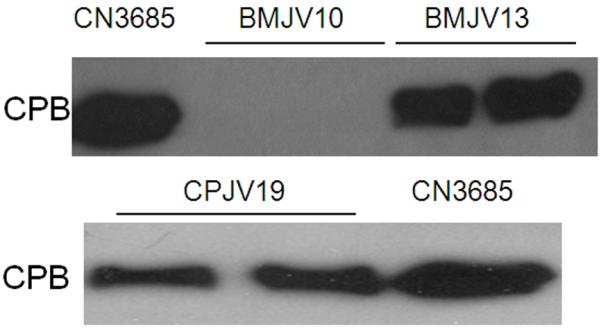
After a 6 h infection, luminal fluids from each loop were removed and then analyzed by SDS-PAGE, followed by Western blotting using a CPB-specific monoclonal antibody. Shown for each strain are loop samples taken from two different rabbits. The migration of purified CPB is indicated to the left of the blots. Shown in the Figure are representative Western blot results that were reproducible over three repetitions.
For comparison, in vivo production of CPB was clearly detectable by Western blotting of intestinal luminal fluids collected from rabbit loops treated for 6 h with washed CPJV19 cells (Fig. 7, lower panel).
Lethality in mice after ID-challenge with wild-type CN3685, BMJV10, BMJV13 or CPJV19
Besides causing intestinal damage in humans and livestock, type C infection can result in fatal enterotoxemias, where toxins produced in the intestines are absorbed into the circulation to damage internal organs beyond the intestines, thus leading to lethality (McClane et al., 2004). Therefore, our recently developed mouse intraduodenal (ID) challenge model (Uzal et al., 2009) was utilized to compare the lethal enterotoxemic effects of ID injections of TGY-C containing washed cells of wild-type CN3685, the isogenic agrB null mutant BMJV10, the complementing strain BMJV13 or the luxS null mutant CPJV19. In this animal model, trypsin inhibitor is also added to all samples to mimic natural disease caused by type C isolates (Ma et al., 2011; Sayeed et al., 2008).
As previously reported by our group (Ma et al., 2011; Uzal et al., 2009), an ID inoculation of washed CN3685 cells produced lethality in most challenged mice (Fig. 8A). Severe clinical signs requiring euthanasia or spontaneous death typically developed between 8 and 24 h post-challenge. Overall clinical signs consisted of a swollen abdomen, depression, inappetence, tachypnea with superficial abdominal breathing, and neurological signs such as circling and rolling.
Fig. 8. Mouse lethality or intestinal colonization of wild-type CN3685 and its derivatives.

Panel A, The percentage of lethality in mice is shown after an intraduodenal inoculation (~108 CFU ml−1) with TGY-C containing washed cells of CN3685, the isogenic agrB mutant (BMJV10), the complementing strain (BMJV13) or the isogenic luxS mutant (CPJV19). As a negative control, sterile non-toxic TGY-C was also injected ID in groups of mice. Lethality was recorded over a 48 h period post-inoculation. Mouse lethality produced by BMJV10 and TGY-C was statistically different (*, P<0.05) from lethality produced by CN3685, BMJV13 or CPJV19. Each experiment was performed independently four times, using groups of four mice (n=16), except the CPJV19 experiment was performed twice, with groups of four mice (n=8). Panel B, Colonization of CN3685 and derivatives in various intestinal sections. After challenge, dilutions of the intestinal contents were plated on blood agar plates and, after anaerobic incubation overnight at 37°C, colonies developing on those plates that had double zones of hemolysis were counted. Results shown are from 5 mice for each C. perfringens strain. No significant differences (P>0.05) in colonization were noted for any strain in any intestinal section.
In contrast, mice receiving an ID injection of washed cells of the BMJV10 agrB mutant developed significantly lower lethality (Fig. 8A). This effect was due to inactivation of the agr operon in the mutant since the complementing strain BMJV13 produced similar lethality levels in challenged mice as did the wild-type strain. No clinical disease or lethality was observed in control mice receiving a similar inoculation of sterile TGY-C (Fig. 8A). Collectively, these results indicate that, in vivo, a functional agr operon is important when CN3685 causes enterotoxemia.
When the ability of the luxS null mutant to cause fatal enterotoxemia in mice was evaluated (Fig. 8A), washed cells of the CPJV19 luxS mutant caused death in most mice challenged by the ID route. This lethal effect was quantitatively similar to that caused by washed cells of wild-type CN3685 (Fig. 8A).
Survival of CN3685, BMJV10, BMJV13 and CPJV19 in rabbit small intestinal loops and the mouse intestinal tract
Luminal fluid recovered from rabbit ileal loops after a 6 h challenge with washed cells of CN3685, BMJV10, BMJV13 or CPJV19 was found to contain ~108 viable C. perfringens cells/ml. In contrast, only ~105 viable C. perfringens cells/ml were recovered from luminal fluid recovered from rabbit small intestinal loops inoculated with sterile TGY-C, verifying that >99% of the C. perfringens present in the luminal fluids were attributable to challenge with CN3685, BMJV10, BMJV13 or CPJV19. Furthermore, these data indicated that the pathogenicity differences observed in Fig. 6 were not due to reduced in vivo survival of BMJV10.
A study was performed in mice to compare regional intestinal colonization by washed cells of CN3685, BMJV10, BMJV13 and CPJV19 after ID inoculation to evaluate whether disruption of the Agr-like QS system affected the site of intestinal colonization or could account for the lethality differences shown in Fig. 8A. When the presence of C. perfringens was quantified in the small intestine, colon and cecum 6 h after challenge with these strains, there was >50-fold more C. perfringens colonization in the small intestine for all strains except CPJV19, which colonized all organs more equally. More importantly, no significant differences were noted in C. perfringens colonization of different organs after challenge with CN3685 or BMJV10, indicating that disruption of Agr QS signaling had not affected colonization patterns and that reduced in vivo survival or colonization differences did not explain the attenuation of BMJV10 shown in Fig. 8A. These Fig. 8B experiments also recovered virtually no viable C. perfringens from the small intestine, colon or cecum of mice challenged with sterile TGY-C, verifying that the colonizing C. perfringens cells recovered from these mice were attributable to challenge with CN3685 or BMJV10.
Discussion
To help cope with its presence in such varied environmental niches as soil, water, feces, the intestines and muscle, C. perfringens possesses at least two QS systems (Ohtani et al., 2009; Vidal et al., 2009a; Ohtani et al., 2002). The first of those QS systems partially resembles the Agr system of S. aureus, where an agr locus is transcribed as an operon consisting of four genes encoding the AgrA, B, C and D proteins (Novick and Geisinger, 2008). Similar agr loci are also present in the genomes of other clostridial pathogens, including C. difficile, C. botulinum and C. sporogenes (Cooksley et al., 2010). C. botulinum possesses two agr-like operons, one apparently controlling sporulation and the other regulating production of botulinum neurotoxin (Cooksley et al., 2010). Recent studies (Ohtani et al., 2009; Vidal et al., 2009a; Li et al., 2011) have established that the agr operon, presumably acting via an Agr-like QS system, also regulates sporulation and toxin production by C. perfringens type A strains (see discussion below).
The current study further evaluated agr operon control of in vitro C. perfringens toxin production. It first demonstrated that, as for C. perfringens type A strain 13 (Ohtani et al., 2009), the agr operon regulates production of PFO and CPA during growth of type C strain CN3685 in TGY broth. More significantly, this study presents the first evidence that the agr operon also controls CPB expression under similar in vitro growth conditions (note that strain 13 does not carry a cpb gene). These findings indicate that the agr operon is also important for toxin regulation in non-type A strains of C. perfringens. To date, the agr operon has been shown to regulate production of chromosomally-encoded (CPA and PFO) and plasmid-encoded (CPB and CPB2) toxins by C. perfringens vegetative cells (this study; Ohtani et al., 2009; Vidal et al., 2009a), as well as enterotoxin production by sporulating cells of this bacterium (Li et al., 2011).
CPB regulation by the agr operon was shown to involve a diffusible signal, consistent with a QS effect. Furthermore, a CN3685 AgrD sequence-derived synthetic peptide was specifically able to stimulate CPB production. These results support agr operon control of CPB production acting via a QS system and the Agr-like QS system as an emerging global regulator of in vitro C. perfringens toxin production.
As mentioned in the Introduction, there are similarities and differences between the Agr QS systems of S. aureus and C. perfringens. Both bacteria possess AgrD orthologues; however, except for the presence of a serine at residue 2 and a cysteine at residue 4, no primary sequence homology exists between the AgrD-derived AIP-I of S. aureus (Lyon et al., 2002) and the AgrD-derived CPAIP of C. perfringens tested in this study. Furthermore, since the C. perfringens agr operon apparently does not encode AgrA and AgrC orthologues, it has been suggested that the VirS/VirR two-component system assumes the role of AgrA/AgrC for the C. perfringens Agr-like QS system (Ohtani et al., 2009). Consistent with that hypothesis, several studies (including the current one) have established that production of CPA, PFO, beta2 toxin and CPB is under at least partial control of both the VirS/VirR two-component system and the Agr-like QS system (Li et al., 2011; Ma et al., 2011; Ohtani et al., 2009; Lyristis et al., 1994) However, future studies should definitively test the relationship between the VirS/VirR two-component system and the Agr-like QS system in regulating expression of these toxin genes.
While several pathogenic clostridial spp. possess an agr locus that likely regulates sporulation and toxin production in vitro by an Agr-like QS mechanism (Li et al., 2011; Cooksley et al., 2010), it had not yet been evaluated whether the agr operon is involved in regulating the virulence of any pathogenic clostridial spp during infection. The current study first addressed this issue indirectly by showing that the agr operon, presumably acting via a QS effect, contributes to cytotoxicity induced by a CN3685 anaerobic infection of Caco-2 cells. We then directly established the importance of the agr operon when CN3685 causes either hemorrhagic necrotic enteritis in rabbits or fatal enterotoxemias in mice. The mechanism of agr operon-mediated CN3685 virulence regulation likely involves an Agr-like QS system since, i) the agrB and agrD genes are necessary and sufficient for agr operon regulation of C. perfringens toxin production in vitro (Ohtani et al., 2009), ii) the current study showed that a functional agr locus is required for CN3685 to produce a diffusible signal that can regulate CPB production in vitro, iii) the CPAIP peptide, which is derived from a portion of the CN3685 AgrD sequence, was able to specifically induce in vitro CPB production by the agrB null mutant and iv) CPB production was previously shown to be required or important, respectively, for CN3685 to cause necro-hemorrhagic enteritis or fatal enterotoxemias (Uzal et al., 2009; Sayeed et al., 2008).
The current study further demonstrated that, in the absence of a functional wild-type agr operon, CN3685 fails to produce detectable levels of CPB in rabbit small intestinal loops. Since CPB production is essential for this type C strain to cause pathology in rabbit small intestinal loops (Sayeed et al., 2008), Agr-like control of CPB production provides a mechanism to explain the virulence attenuation of the agrB null mutant in this animal model of necro-hemorrhagic enteritis. No attempt was made in the current study to evaluate in vivo CPB production in the mouse ID challenge enterotoxemia model since our previous study using the same CN3685 wild-type strain could not detect CPB production in this model (Uzal et al., 2009), probably because CPB levels in the lumen of CN3685-infeced mice are reduced by the open gastrointestinal tract in the mouse ID model, which allows toxin excretion and continuous protease influx into the intestines to facilitate CPB degradation. However, since, i) CPB is very important for this type C strain to cause lethality in the mouse model (Uzal et al., 2009) and ii) the agr operon regulates production of CPB by CN3685 in the rabbit small intestinal loop model (this study), the agr operon may also control enterotoxemic lethality in the mouse ID challenge model via an Agr-like QS system that regulates in vivo CPB production.
Since washed CN3685 cells were used for infection in the current pathogenicity studies, our in vivo results implicating Agr-like QS regulation of CPB production in rabbit small intestinal loops support the functioning of this QS system in the intestinal environment. An Agr QS system was previously shown to be important for regulating the virulence of Listeria monocytogenes (Riedel et al., 2009), where this QS system is important for intestinal invasion. In contrast, when C. perfringens apparently uses its Agr-like QS system to cause hemorrhagic necrotic enteritis or enterotoxemias, this bacterium does not typically invade the intestinal epithelium during infection; instead, C. perfringens apparently uses its Agr-like QS system to regulate toxin production in the intestines.
Establishing the importance of the Agr-like QS system for CN3685 pathogenicity did not eliminate the possible involvement of other QS systems in the virulence of this strain. The importance of the AI-2 QS system for C. perfringens toxin regulation and virulence has been unclear. A previous study (Ohtani et al., 2002) using luxS null mutants reported that the LuxS-dependent AI-2 QS system controls production of CPA and PFO in vitro, although this regulation was relatively modest. However, since a luxS null mutant of CN3685 remained able to rapidly upregulate production of CPA, PFO, and CPB when used to infect Caco-2 cells under aerobic conditions (Vidal et al., 2009b), the current study explored whether CN3685 requires the LuxS-mediated AI-2 QS system for in vitro production of CPB under anaerobic conditions or for in vivo virulence. These analyses determined that LuxS production is not necessary for CN3685 to express CPB in vitro or during either hemorrhagic necrotic enteritis in rabbit small intestinal loops or fatal enterotoxemias in mice. Collectively, the pathogenicity results obtained in the current study using the agrB and luxS mutants indicated that, i) not all QS systems are necessary for CN3685 to cause disease and ii) the Agr-like and AI-2 systems do not play redundant roles in the pathogenicity of this type C strain, at least in our animal models.
The dispensability of the AI-2 QS system for CN3685 intestinal virulence, as detected in the current study, is interesting since this QS system is important for the virulence of several Gram-negative enteric pathogens, including enteropathogenic E. coli (EPEC), C. jejuni and V. cholerae (Kaper and Sperandio, 2005; Sperandio et al., 1999). Instead, CN3685 apparently relies primarily upon its Agr-like QS system for enteric virulence. Since the Agr QS system is limited to only certain Gram-positive bacteria, it might provide C. perfringens with greater signaling specificity than would reliance upon the AI-2 QS system shared by many other intestinal bacteria. However, the AI-2 QS system still could make contributions to C. perfringens intestinal virulence that are not picked up by the animal pathogenicity models used in these studies. Alternatively, the AI-2 QS system could be important when other C. perfringens strains cause extraintestinal diseases, such as gas gangrene, or it could be important for C. perfringens survival in non-disease settings.
In summary, the current study provides the first evidence for the importance of a QS system in any clostridial infection. Specifically, our results support the importance of the agr operon, by encoding components of an Agr-like QS system, for the virulence of C. perfringens strain CN3685. When this type C strain causes intestinal disease and fatal enterotoxemia originating in the small intestine, the Agr-like QS system apparently regulates virulence, at least in part, by controlling CPB production in vivo. Our results further indicate that not all QS systems need be functional for this C. perfringens strain to be pathogenic since inactivating the LuxS-mediated AI-2 QS system did not reduce the virulence of CN3685 when causing intestinal disease or enterotoxemia in our animal models. Further studies are needed to identify all molecular signaling events involved in the increasingly-important clostridial Agr-like QS virulence regulatory system.
Experimental procedures
Strains and bacterial culture media
C. perfringens strains used in this study are listed in Supplementary Table 1. The bacterial culture media used throughout this work included FTG (fluid thioglycolate medium; Difco Laboratories), TGY (3% tryptic soy broth [Becton-Dickinson]; 2% glucose [Sigma Aldrich], 1% yeast extract [Becton-Dickinson], 0.1% sodium thioglycolate [Sigma Aldrich]), and brain heart infusion (BHI) agar (Becton-Dickinson). For in vivo studies, the sodium thioglycolate in TGY was replaced with 0.1% cysteine (creating TGY-C). Strain BMJV13 was always grown with chloramphenicol (15 μg ml−1). The luxS null mutant (CPJV19) of CN3685 had been previously constructed and characterized, both genotypically and phenotypically (Vidal et al., 2009b).
Cell culture
Human-derived enterocyte-like Caco-2 cells were routinely cultured in Eagle’s Minimum Essential Medium (MEM) (Sigma) supplemented with heat-inactivated 10% fetal calf serum (FCS) (Mediatech Incorporated), 1% non-essential amino acids (Sigma), 1% glutamine (Sigma), penicillin (100 U ml−1), and streptomycin (100 μg ml−1). Caco-2 cell cultures were normally maintained by harvesting with 0.25% trypsin (Gibco), seeding harvested cells in culture flasks containing fresh cell culture medium, and incubating those flasks at 37°C in a 5% CO2 humidified atmosphere.
Purification of CPB
An isolated colony of CN3685 was cultured at 37°C for 8 h in TGY broth (3 l) to produce CPB. The toxin was purified from supernatants of those cultures by anion exchange chromatography using a DEAE-CL6B Sepharose column (Sigma) and an ÄKTA prime system (Amersham Bioscience), as previously described (Sayeed et al., 2008; Vidal et al., 2008). The toxin preparation was evaluated by densitometric scanning of Coomassie Blue-stained gels after SDS-PAGE of the purified CPB and by Western blotting using a monoclonal anti-CPB antibody. The final preparation was found to be ~95% homogeneous for CPB and showed no apparent contamination with CPA or PFO.
Sequencing of the agr locus in CN3685
DNA was purified from CN3685 using the MasterPure Gram-positive DNA purification kit (Epicentre, Wisconsin). PCR was then performed on this DNA using primers that were designed based upon strain 13 agr locus sequences (Vidal et al., 2009a). The ~2.9 kb PCR product amplified from CN3685 DNA with these primers was then sequenced at Beckman Coulter Genomics (Danver, MA) and the obtained sequence were deposited in GenBank under accession number JN648378.
RT-PCR transcriptional analysis of the agr operon in CN3685
RT-PCR analysis of the CN3685 agr locus to demonstrate co-transcription of the CPE 1563 ORF, the CPE 1562 ORF, agrB and agrD as an operon was performed as described previously during similar analysis of the strain 13 agr locus (Vidal et al., 2009a).
Construction of a CN3685 agrB null mutant (BMJV10) using the TargeTron™ gene knockout system and preparation of a complementing strain (BMJV13)
The agrB gene was inactivated using pJIR750agri, which carries an agrB-targeted intron (Li et al., 2011). That plasmid was electroporated into the parent type C isolate CN3685, as described previously (Sayeed et al., 2008). Transformants were then plated onto BHI agar plates containing 15 μg ml−1 of chloramphenicol. After colonies were PCR-screened (see Figure 1 legend) a putative agrB mutant was further characterized by Southern blotting and PCR analyses (see Results). All PCR-screening reactions were performed using Taq 2X Master Mix (New England Biolabs) and primers (final concentration of 1 μM) in a Techne (Burkhardtsdorf, Germany) thermocycler. To prepare a complementing strain, the agrB mutant was subcultured daily for ten days in FTG to cure the intron plasmid, creating the cured agrB mutant named BMJV10. This strain was then transformed by electroporation with plasmid P3, which encodes the C. perfringens agr operon (Vidal et al., 2009a), to create the complementing strain BMJV13.
Southern blot analyses
C. perfringens DNA was purified using the MasterPure Gram-positive DNA purification kit (Epicentre, Wisconsin). Each purified DNA sample (2.5 μg) was then digested overnight with EcoRI at 37°C according to the manufacturer’s (New England Biolabs) instructions. The digested DNA samples were electrophoresed on a conventional 1% agarose gel and the separated DNA digestion products were then transferred onto nylon membranes (Roche) for hybridization with an intron-specific probe. The probe was prepared by PCR using the PCR DIG Probe Synthesis Kit (Roche) and primers listed in Supplementary Table 1 (AgrB-IBS and AgrB-EBS2). After hybridization of the probe, the Southern blot was developed using reagents from the DIG DNA labeling and detection kit (Roche), according to the manufacturer’s instructions.
RT-PCR analyses for agrB expression
Total RNA was extracted from overnight TGY cultures of C. perfringens CN3685 or its isogenic derivatives as previously described (Vidal et al., 2009a). Briefly, the pellet was resuspended in 200 μl of acetate solution (20 mM sodium acetate [pH 5], 1 mM EDTA, 0.5% sodium dodecyl sulfate [SDS, Bio-Rad]). That suspension received 200 μl of saturated phenol (Fisher scientific) and was then thoroughly resuspended before incubation at 60°C in a water bath with vigorous shaking for 5 min. After centrifugation (17,000 x g at 4°C for 5 min), the nucleic acid-containing supernatant received cold ethanol and centrifuged (17,000 x g at 4°C) for 5 min to obtain the RNA pellet. This pellet was washed twice with cold 70% ethanol and finally resuspended in 100 μl of DNase-free, RNase-free water. All RNA samples were additionally treated with 2 U of DNase I (Promega) at 37°C for 30 min. To stop DNase I activity, a DNase I inhibitor (Promega) was added to the reaction tube. RNA was quantified by absorbance at 260 nm and stored in 20 μl aliquots at −80°C. The quality of RNA preparations was further verified by running 1 μg of the sample on an agarose gel and staining by ethidium bromide.
RT-PCR reactions were performed on those DNase-treated RNA samples using the AccesQuick RT-PCR system (Promega). Briefly, 50 ng of each RNA sample were reverse-transcribed to cDNA at 45°C for 45 min and then used as template for PCR reactions (denaturing at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1 min) with primers agrFwd and agrRev (flanking the agrB ORF). Control RT-PCR reactions were similarly performed, except for the omission of reverse transcriptase.
Analysis of CPB, CPA and PFO production in TGY cultures
CN3685, BMJV10, BMJV13, and CPJV19 were grown overnight in FTG at 37°C. An aliquot (100 μl) of each culture was inoculated into 10 ml of TGY and then incubated for 2, 3, 4, or 6 h or overnight at 37°C. TGY cultures were centrifuged at 8,000 x g for 10 min and equal volumes of each culture supernatant were mixed with SDS-PAGE loading buffer and boiled for 5 min. Duplicate lanes of each sample were electrophoresed on a 12% polyacrylamide gel containing SDS; half of that gel was silver-stained and proteins on the other half of the gel were transferred onto nitrocellulose membranes for Western blotting. Those membranes were blocked with TBS-Tween 20 (0.05% v/v) and non-fat dry milk (5% w/v) for 1 h and then probed with either rabbit polyclonal anti-PFO antiserum (Li et al., 2011), mouse monoclonal anti-CPB antibody (Sayeed et al., 2008), or monoclonal anti-CPA antibody (Vidal et al., 2009a). Bound antibody was then detected after incubation with a horseradish peroxidase-conjugated secondary anti-species specific antibody and addition of SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Evaluation of whether an agr operon-dependent secreted factor controls CPB production in vitro
As described previously (Vidal et al., 2009a), a physical barrier impermeable to bacteria, but allowing passage of small molecules, was used to study whether agr operon-dependent effects on gene regulation involve a QS mechanism, which would include a diffusible signaling molecule. This barrier, which separated bacteria inoculated into a bottom chamber from those inoculated into a top chamber, was created by installing a Transwell filter (0.4 μm pore size, Corning) in a 6-well culture plate (Corning). A 3 ml aliquot of sterile TGY was added to each well and different combinations of overnight TGY cultures of wild-type CN3685, the CN3685 cpb null mutant (BMC100) or the CN3685 agrB null mutant (BMJV10) were inoculated (30 μl) into either the top or bottom chamber of each well. These combinations were: 1) CN3685/CN3685, 2) BMC100/BMC100, 3) BMJV10/BMJV10 or, 4) BMJV10/BMC100. The cultures were incubated anaerobically at 37°C for 5 h and the supernatants obtained by centrifugation were then Western-blotted with a mouse monoclonal anti-CPB antibody, as described.
Synthesis of an AgrD-sequence based synthetic peptide
Synthesis of CPAIP was based on analogy with AIP-1 of the S. aureus Agr QS system. The CPAIP sequence is TSACLWFT, with a thiolactone ring connecting the C and T residues. Synthesis of this peptide was carried out at the University of Pittsburgh Genomics and Proteomics Core Facility using standard FMOC chemistry and TBTU/Hobt activation using a Liberty CEM microwave synthesizer. FMOC-Tyr(tBu), FMOC-Ser(tBu), FMOC-Ala, FMOC-Cys(mmt), FMOC-Leu, FMOC-Trp(Boc) and FMOC-Phe were purchased from Peptides International and used for the stepwise assembly of the linear sequence on a H-Thr(tBu)-2-ClTrt resin (1% DVB, 200-400 mesh, 0.550 mmole g−1, EMD Chemicals, Inc). After completion of the chain assembly, the N-terminal amino group was reversibly protected using Boc Anhydride/DIPEA/DMF for 2 h at room temperature. Cleavage of the partially protected peptide fragment from the resin was accomplished using 1% TFA in DCM and filtered into a solution of 10% pyridine in methanol followed by evaporation to dryness under high vacuum on a Buchi Rotavapor system at 37°C. Thiolactone formation was initiated by adding 10.2 mg of 1-hydroxy-7-azabenzotriazole (HoAt), 43.5 mg of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDAC-HCL), 13.05 ml N,N-diisopropylethylamine (DIPEA) and 4-(N,N-dimethylamino) pyridine (DMAP) to 98 mg of the partially protected peptide fragment in Dichloromethane (DCM). The mixture was layered with Nitrogen and stirred overnight at room temperature. The mixture was then evaporated to dryness under high vacuum on a Buchi Rotavapor system at 37°C. The thiolactone-fragment was then cleaved with TFA:H20:EtSMe (Sigma-Aldrich) (90:5:3:2) for 2 h at room temperature and then precipitated with Diethyl Ether (Fisher Scientific). Following 3 ether washes, the resultant product was dried and then purified by C-18 RP-HPLC on a Waters Alliance chromatography system using standard Acetonitrile/0.1% TFA gradient conditions. Analytical C-18 RP-HPLC characterization followed by MALDI-TOF analysis on an Applied Biosystems Voyager workstation confirmed the expected mass of the target protein product. As a negative control, the 8-mer peptide IFQGCQFR was used.
CPAIP signaling of the agrB mutant to produce CPB in vitro
Aliquots (25 μM, 50 μM or 100 μM, final concentrations) of the CPAIP peptide were added to a TGY tube that had been freshly inoculated with agrB knockout mutant BMJV10 and those cultures were then grown for 5 h at 37°C under anaerobic conditions. Wild type CN3685 or the agrB null mutant BMJV10 (without CPAIP) were similarly cultured for comparison of CPB production. As a control, 25 μM of a negative control peptide was added to another TGY tube freshly inoculated with BMJV10 and then incubated at 37°C under anaerobic conditions. Supernatants of each sample were collected and a 25 μl sample was used for Western Blot detection for CPB production with a mouse monoclonal anti-CPB antibody, as described earlier.
Analysis of CPB production in the presence of Caco-2 cell cultures
Wild-type CN3685, the BMJV10 agrB null mutant, the BMJV13 agr operon complementing strain or the luxS null mutant CPJV19 were grown overnight in TGY at 37°C. Bacterial cells were obtained by centrifugation at 8,000 x g for 10 min and then washed three times with cold PBS (pH 7.4). Pellets were resuspended in the appropriate volume of cold MEM without FBS or antibiotics and used to infect a dish containing confluent polarized Caco-2 cell cultures (moi 1:20) or a dish containing MEM alone as a control. Infected cultures were incubated at 37°C under anaerobic conditions. After 1, 2 or 3 h post-infection, culture supernatants were collected and centrifuged. Similar amounts (20 μl) of each sample were electrophoresed, on an SDS-containing 12% polyacrylamide gel, transferred to nitrocellulose membranes, and Western blotted using either a mouse monoclonal anti-CPB antibody or a mouse monoclonal anti-CPA antibody.
Evaluation of agr operon-mediated cytotoxicity of Caco-2 cell cultures
Cell-free supernatants from Caco-2 cell cultures that had been infected for 1 h with CN3685, BMJV10 or BMJV13 were assayed for released lactate dehydrogenase (LDH) using a LDH cytotoxicity detection kit (Roche). Each sample was read at 490 nm using a multi-well plate reader (Bio-Rad). Supernatants from Caco-2 cell cultures treated for 1 h, under the same conditions, with 1% Triton X-100 were used as a control to establish the 100% cytotoxicity level (Ma et al., 2011). The development of morphological changes, indicating cytotoxicity, was also followed every hour using a Zeiss Axiovert 25 inverted microscope. Pictures of infected-Caco-2 cells were taken using a Canon Powershot G5 fitted to the Zeiss Axiovert 25 microscope.
Virulence testing of CN3685, CPJV19, BMJV10 and BMJV13 in rabbit small intestinal loops
Fasted young adult, male or female, New Zealand White rabbits (Charles River, CA) were pre-medicated with acepromazine, xylazine and burprenorphine. Anaesthesia was then induced with ketamine and maintained with inhalatory isofluorane. A laparotomy was performed via the mid line and the small intestine was exposed. Lengths (~2 cm) of jejunum were isolated by ligation, leaving an empty segment of gut between the loops. Care was taken to avoid over-distension of bowel loops and interference with the blood supply. During surgery, the serosal surface of the loops was kept wet by frequent soaking with normal saline solution. Loops received an inoculum containing, i) purified CPB (20 μg) plus trypsin inhibitor (TI, 150 μg ml−1), as a positive control, ii) sterile TGY-C plus TI, as a negative control or iii) washed vegetative cells (~108 CFU) of CN3685, BMJV10, BMJV13 or BMJV19 resuspended in fresh TGY-C plus TI. The abdominal incision was closed by separate muscle and skin sutures and the animals were kept deeply anaesthetized throughout the experiment.
After 6 h, the rabbits were euthanized by an overdose of sodium barbiturate (Beuthanasia, Schering-Plough Animal Health, Kenilworth, NJ). The abdominal cavity was then re-opened, and the small intestinal loops were excised in the same order that they had been inoculated. Loops were cut out and weighed, before and after the fluid was removed, examined grossly and length was measured. Fluid secretion was expressed as the loop weight-to-length ratio (g cm−1). For histological analysis, all tissues were fixed by immersion in 10% buffered, pH 7.4 formalin for 24-48 h, followed by dehydration through graded alcohols to xylene before being embedded in paraffin wax. Four μm thick sections were cut and stained with hematoxylin and eosin according to standard procedures. Tissue sections were examined by a pathologist in a blinded fashion, using a quantitative scoring system as described previously (Sayeed et al., 2008). The degree of damage was scored using a scale of 0 to 5, with 0 indicating no histologic damage and 1, 2, 3, 4 and 5 values indicating increasingly severe damage. Histologic parameters considered in this evaluation included mucosal necrosis, desquamation of the epithelium, inflammation, villous blunting, edema and hemorrhage.
Evaluation of in vivo CPB production in rabbit small intestinal loops
Intestinal fluids collected from rabbit small intestinal loops were centrifuged at 20,000 x g for 10 min at 4°C to remove cell debris and intestinal content. The cleared supernatant was then assayed for the presence of CPB by Western blotting. Briefly, a 10 μl aliquot of fluid, or purified CPB (0.5 μg) used as a control, was subjected to electrophoresis on a SDS-containing, 12% polyacrylamide gel. After electrotransfer of separated proteins onto a nitrocellulose membrane, the membrane was blocked with TBS-Tween 20 (0.05% v/v) and non-fat dry milk (5% w/v) for 1 h at room temperature and then probed with a mouse monoclonal anti-CPB antibody overnight at 4°C. After incubation with an anti-mouse secondary antibody for 1 h at room temperature, the presence of CPB on the blot was detected by the addition of SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Analysis of mouse lethality induced by CN3685, BMJV10, BMJV13 or CPJV19
Washed vegetative cells of wild-type CN3685, the CPJV19 luxS null mutant, the BMJV10 agrB null mutant, and the BMJV13 agr operon complementing strain were each tested in our previously-described mice ID inoculation model for type C enterotoxemia (Uzal et al., 2009). Each inoculum preparation contained ~108 CFU ml−1 washed vegetative cells of the indicated strain suspended in fresh TGY-C along with TI (150 μg ml−1); those samples were then inoculated intraduodenally into mice. Mice were fasted 24 h before inoculation but allowed access to water up to 1 h before the start of the experiments. Their abdomens were disinfected with iodine solution (Betadine, Purdue Pharma LP, Connecticut) immediately before surgery. The animals were anesthetized by intraperitoneal administration of Avertine (Winthrop Laboratories, New York). A midline laparotomy was performed and 1 ml of each washed vegetative cell preparation was injected into the mouse duodenum immediately distal to the stomach. The injections were performed by insertion of a 0.5-inch, 27-gauge needle into the duodenum, oblique to the intestinal lumen in a direction away from the stomach. After inoculation, the incision in the peritoneum, abdominal muscles and skin was sutured in one plane using super glue (Henkel Corporation, Avon, Ohio). The surgical procedure lasted approximately 2 min per animal and all mice were awake within 30 min after surgery. The assay endpoint included spontaneous death, development of severe clinical signs necessitating euthanasia, or survival without clinical alterations after 48 h.
Determination of C. perfringens cell numbers inoculated into rabbit small intestinal loops or present in those loops after a 6 h incubation
A standard dilution-plating technique was used to determine the colony-forming units (CFU) in each ml of inoculum. Serial dilutions of each inoculum were prepared in TGY-C and 100 μl of those dilutions were plated onto blood agar plates. After a 24 h anaerobic incubation at 37°C, the number of colonies present on each plate was counted and a CFU ml−1 calculation was made based upon that number and the dilution factor used. To determine the survival and colonization of strains in rabbit small intestinal loops, the loops were opened after 6 h and the fluid content was collected using aseptic technique. The number of C. perfringens cells present per ml of the collected intestinal fluids was then determined by plating serial dilution of those fluids onto blood agar plates and then counting colonies with the characteristic double zone of hemolysis for C. perfringens.
Determination of C. perfringens cell numbers present in the mouse intestines 6 h after ID inoculation
The survival and colonization of strains in the mouse small intestine, colon and cecum was determined by challenging groups of 5 mice, ID, with each of the strains described above. The mice were euthanized after 6 h and the whole contents of the small intestine, colon and cecum were collected separately, diluted in TGY, and plated as described above to calculate CFU g−1 of content.
Statistical analyses
For statistical analyses, each experiment in rabbits was performed with at least 2 repetitions per animal in each of 8 different rabbits. In mice, each experiment was repeated three times in groups of 4 mice. All statistical analyses were done using the SAS 9.2 software. The fluid accumulation data were analyzed using the two-way ANOVA analysis of variance with post hoc test and the histology data by the Friedman test. Mouse lethality was analyzed by the Fisher’s exact test and the LDH cytotoxicity data were analyzed using the two-tailed, unpaired Student’s t-test.
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Animal Care and Use Committee of the University of California, Davis (Permit numbers: 13222 and 16383).
Supplementary Material
Acknowledgements
This research was generously supported by grant R01 AI-056177-08 from the National Institute of Allergy and Infectious Disease. The authors wish to thank Dr. P. Hauer for providing the CPB and CPA monoclonal antibodies used in the study and Drs. Kazi Islam and Raymond Yurko for peptide synthesis at the University of Pittsburgh Genomics and Proteomics Core Facility. We thank Dr. Jianming Chen for help with graphics.
References
- Antunes LC, Ferreira RB, Buckner MM, Finlay BB. Quorum sensing in bacterial virulence. Microbiology. 2010;156:2271–2282. doi: 10.1099/mic.0.038794-0. [DOI] [PubMed] [Google Scholar]
- Bannam TL, Rood JI. Clostridium perfringens-Escherichia coli shuttle vectors that carry single antibiotic resistance determinants. Plasmid. 1993;29:233–235. doi: 10.1006/plas.1993.1025. [DOI] [PubMed] [Google Scholar]
- Beier D, Gross R. Regulation of bacterial virulence by two-component systems. Curr Opin Microbiol. 2006;9:143–152. doi: 10.1016/j.mib.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Calva E, Oropeza R. Two-component signal transduction systems, environmental signals, and virulence. Microb Ecol. 2006;51:166–176. doi: 10.1007/s00248-005-0087-1. [DOI] [PubMed] [Google Scholar]
- Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl Environ Microbiol. 2005;71:7542–7547. doi: 10.1128/AEM.71.11.7542-7547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooksley CM, Davis IJ, Winzer K, Chan WC, Peck MW, Minton NP. Regulation of neurotoxin production and sporulation by a putative agrBD signaling system in proteolytic Clostridium botulinum. Appl Environ Microbiol. 2010;76:4448–4460. doi: 10.1128/AEM.03038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher DJ, Fernandez-Miyakawa ME, Sayeed S, Poon R, Adams V, Rood JI, et al. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect Immun. 2006;74:5200–5210. doi: 10.1128/IAI.00534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuck AP, Moe PC, Johnson BB. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell Biochem. 2010;51:551–577. doi: 10.1007/978-90-481-8622-8_20. [DOI] [PubMed] [Google Scholar]
- Johnson S, Gerding DN. Enterotoxemic infections. In: Rood JI, McClane BA, Songer JG, Titball RW, editors. The Clostridia: Molecular Biology and Pathogenesis. Academic Press; London, United Kingdom: 1997. pp. 117–140. [Google Scholar]
- Kaper JB, Sperandio V. Bacterial cell-to-cell signaling in the gastrointestinal tract. Infect Immun. 2005;73:3197–3209. doi: 10.1128/IAI.73.6.3197-3209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence GW. The pathogenesis of enteritis necroticans. In: Rood JI, McClane BA, Songer JG, Titball RW, editors. The Clostridia: Molecular Biology and Pathogenesis. Academic Press; London, United Kingdom: 1997. pp. 198–207. [Google Scholar]
- Li J, McClane BA. A novel small acid soluble protein variant is important for spore resistance of most Clostridium perfringens food poisoning isolates. PLoS Pathog. 2008;4:e1000056. doi: 10.1371/journal.ppat.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen J, Vidal JE, McClane BA. The Agr-like quorum-sensing system regulates sporulation and production of enterotoxin and beta2 toxin by Clostridium perfringens type A non-food-borne human gastrointestinal disease strain F5603. Infect Immun. 2011;79:2451–2459. doi: 10.1128/IAI.00169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon GJ, Wright JS, Muir TW, Novick RP. Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus. Biochemistry. 2002;41:10095–10104. doi: 10.1021/bi026049u. [DOI] [PubMed] [Google Scholar]
- Lyristis M, Bryant AE, Sloan J, Awad MM, Nisbet IT, Stevens DL, Rood JI. Identification and molecular analysis of a locus that regulates extracellular toxin production in Clostridium perfringens. Mol Microbiol. 1994;12:761–777. doi: 10.1111/j.1365-2958.1994.tb01063.x. [DOI] [PubMed] [Google Scholar]
- Ma M, Vidal J, Saputo J, McClane BA, Uzal F. The VirS/VirR two-component system regulates the anaerobic cytotoxicity, intestinal pathogenicity, and enterotoxemic lethality of Clostridium perfringens Type C isolate CN3685. MBio. 2011;2:e00338–00310. doi: 10.1128/mBio.00338-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClane BA, Uzal FA, Fernandez-Miyakawa M, Lyerly D, Wilkins TD. The Enterotoxigenic Clostridia. In: Dworkin M, F. S, Rosenburg E, Schleifer KF, Stackebrandt E, editors. The Prokaryotes. Springer-Verlag; New York, NY: 2004. pp. 698–752. [Google Scholar]
- Myers GS, Rasko DA, Cheung JK, Ravel J, Seshadri R, DeBoy RT, et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 2006;16:1031–1040. doi: 10.1101/gr.5238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet. 2008;42:541–564. doi: 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- Ohtani K, Hayashi H, Shimizu T. The luxS gene is involved in cell-cell signalling for toxin production in Clostridium perfringens. Mol Microbiol. 2002;44:171–179. doi: 10.1046/j.1365-2958.2002.02863.x. [DOI] [PubMed] [Google Scholar]
- Ohtani K, Yuan Y, Hassan S, Wang R, Wang Y, Shimizu T. Virulence gene regulation by the agr system in Clostridium perfringens. J Bacteriol. 2009;191:3919–3927. doi: 10.1128/JB.01455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CT, Sperandio V. Cell-to-cell signalling during pathogenesis. Cell Microbiol. 2009;11:363–369. doi: 10.1111/j.1462-5822.2008.01272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrillo TM, Beck-Sague CM, Songer JG, Abramowsky C, Fortenberry JD, Meacham L, et al. Enteritis necroticans (pigbel) in a diabetic child. N Engl J Med. 2000;342:1250–1253. doi: 10.1056/NEJM200004273421704. [DOI] [PubMed] [Google Scholar]
- Riedel CU, Monk IR, Casey PG, Waidmann MS, Gahan CG, Hill C. AgrD-dependent quorum sensing affects biofilm formation, invasion, virulence and global gene expression profiles in Listeria monocytogenes. Mol Microbiol. 2009;71:1177–1189. doi: 10.1111/j.1365-2958.2008.06589.x. [DOI] [PubMed] [Google Scholar]
- Sayeed S, Uzal FA, Fisher DJ, Saputo J, Vidal JE, Chen Y, et al. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol. 2008;67:15–30. doi: 10.1111/j.1365-2958.2007.06007.x. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, et al. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci U S A. 2002;99:996–1001. doi: 10.1073/pnas.022493799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley JG, 3rd, Fisher DJ, Sayeed S, Chakrabarti G, McClane BA. The enteric toxins of Clostridium perfringens. Rev Physiol Biochem Pharmacol. 2004;152:183–204. doi: 10.1007/s10254-004-0036-2. [DOI] [PubMed] [Google Scholar]
- Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB. Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 1999;96:15196–15201. doi: 10.1073/pnas.96.26.15196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titball RW, Naylor CE, Basak AK. The Clostridium perfringens alpha-toxin. Anaerobe. 1999;5:51–64. doi: 10.1006/anae.1999.0191. [DOI] [PubMed] [Google Scholar]
- Uzal FA, McClane BA. Recent progress in understanding the pathogenesis of Clostridium perfringens type C infections. Vet Microbiol. 2011;153:1–2. doi: 10.1016/j.vetmic.2011.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzal FA, Saputo J, Sayeed S, Vidal JE, Fisher DJ, Poon R, et al. Development and application of new mouse models to study the pathogenesis of Clostridium perfringens type C Enterotoxemias. Infect Immun. 2009;77:5291–5299. doi: 10.1128/IAI.00825-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal JE, Chen J, Li J, McClane BA. Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS One. 2009a;4:e6232. doi: 10.1371/journal.pone.0006232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal JE, Ohtani K, Shimizu T, McClane BA. Contact with enterocyte-like Caco-2 cells induces rapid upregulation of toxin production by Clostridium perfringens type C isolates. Cell Microbiol. 2009b;11(9):1306–1328. doi: 10.1111/j.1462-5822.2009.01332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal JE, McClane BA, Saputo J, Parker J, Uzal FA. Effects of Clostridium perfringens beta-toxin on the rabbit small intestine and colon. Infect Immun. 2008;76:4396–4404. doi: 10.1128/IAI.00547-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.