

Abstract
Dopa decarboxylase (DDC), a pyridoxal 5′-phosphate (PLP) enzyme responsible for the biosynthesis of dopamine and serotonin, is involved in Parkinson's disease (PD). PD is a neurodegenerative disease mainly due to a progressive loss of dopamine-producing cells in the midbrain. Co-administration of L-Dopa with peripheral DDC inhibitors (carbidopa or benserazide) is the most effective symptomatic treatment for PD. Although carbidopa and trihydroxybenzylhydrazine (the in vivo hydrolysis product of benserazide) are both powerful irreversible DDC inhibitors, they are not selective because they irreversibly bind to free PLP and PLP-enzymes, thus inducing diverse side effects. Therefore, the main goals of this study were (a) to use virtual screening to identify potential human DDC inhibitors and (b) to evaluate the reliability of our virtual-screening (VS) protocol by experimentally testing the “in vitro” activity of selected molecules. Starting from the crystal structure of the DDC-carbidopa complex, a new VS protocol, integrating pharmacophore searches and molecular docking, was developed. Analysis of 15 selected compounds, obtained by filtering the public ZINC database, yielded two molecules that bind to the active site of human DDC and behave as competitive inhibitors with Ki values ≥10 µM. By performing in silico similarity search on the latter compounds followed by a substructure search using the core of the most active compound we identified several competitive inhibitors of human DDC with Ki values in the low micromolar range, unable to bind free PLP, and predicted to not cross the blood-brain barrier. The most potent inhibitor with a Ki value of 500 nM represents a new lead compound, targeting human DDC, that may be the basis for lead optimization in the development of new DDC inhibitors. To our knowledge, a similar approach has not been reported yet in the field of DDC inhibitors discovery.
Introduction
Parkinson's disease (PD) is the most extensively studied pathology within a group of syndromes called “motor system disorders”, whose etiology can be traced back to the loss of dopaminergic neurons of the substantia nigra in the midbrain [1]. Main symptoms of PD include tremors, rigidity, bradykinesia and postural instability; other frequently observed symptoms include depression and other psychiatric disorders, difficulty in swallowing, chewing, and speaking. Early symptoms of PD are usually subtle and occur gradually after 50 years of age. As the symptoms become more severe, patients progressively encounter difficulties in walking, talking, or even completing the simplest tasks; usually, this condition interferes strongly with most daily activities.
At present there is no cure for PD, but a variety of palliatives reducing the severity of disease symptoms exists [2]. In order to replenish dopamine levels at the central nervous system (CNS), L-Dopa is usually administered. The latter is converted to dopamine by Dopa decarboxylase (DDC, E.C. 4.1.1.28), a pyridoxal-5′-phosphate (PLP)-dependent enzyme, which is abundant in the CNS and in the kidney [3]. DDC from pig kidney has been widely characterized with respect to reaction and substrate specificity [4], [5], spectroscopic features of the internal aldimine and of enzyme-intermediate complexes [6], [7], [8], and the role played by residues at or near the active site in the catalysis [9], [10], [11], [12]. Moreover, the crystal structures of DDC, both ligand-free and in complex with the antiParkinson drug carbidopa, have been solved [13].
Although administration of exogenous L-Dopa to PD patients compensates, at least transitorily, for deficiency of dopamine synthesis and often provides dramatic relief from the main symptoms, only 1–5% of L-Dopa reaches the dopaminergic neurons of the brain, being the major part metabolized by the peripheral DDC. Therefore, in order to increase the amount of L-Dopa in the CNS, DDC inhibitors unable to cross the blood-brain barrier (BBB) are usually co-administered with L-Dopa. In this way, not only greater amounts of L-Dopa can reach the brain, thereby substantially increasing its level, but also side effects, either dopamine-related or due to a high concentration of L-Dopa in the blood stream, are diminished [1]. The most commonly used DDC inhibitors in the treatment of PD are carbidopa (L-α-methyl-α-hydrazino-3,4-dihydroxyphenylpropionic acid, MK 485) and benserazide (trihydroxybenzylhydrazine seryl derivative, Ro-4-4602). Pharmacokinetic and metabolic studies in animals and humans have shown that benserazide is completely metabolized before it reaches the arterial blood and that the main metabolic pathway consists of the scission of the molecule between serine and trihydroxybenzylhydrazine (Ro 4-5127) [14]. Thus, it is likely that trihydroxybenzylhydrazine represents the actual DDC inhibitor. Indeed, while benserazide is not a powerful DDC inhibitor [15], carbidopa and trihydroxybenzylhydrazine, both substrate analogs endowed with a substituted hydrazine function, have been found to bind to pig kidney DDC by forming a hydrazone linkage with PLP and work as powerful irreversible DDC inhibitors [16], [17]. Nevertheless, because hydrazine derivatives can react with free PLP and PLP-enzymes, these inhibitors are not entirely selective for DDC, thus resulting in adverse side effects [18], [19]. Although the crystal structure of DDC has been solved ten years ago, no structure-based design studies have been reported to date. Thus, in order to identify competitive and highly selective DDC inhibitors, we decided to undertake a virtual screening (VS) approach combined with in vitro binding experiments.
As a starting point, the structure of pig kidney DDC (∼90% sequence identity with human DDC) in complex with the inhibitor carbidopa (PDB code: 1JS3 [13]) was used to identify the essential features required for DDC binding. Then, a pharmacophore model was generated and validated using an in-house built database of known active and inactive DDC inhibitors, derived from Hartman et al. [20]. The pharmacophore model was first used to filter the lead-like and the drug-like subsets of the public ZINC database (∼8 million compounds [21]) which are tailored to an extended Lipinski's rule of five [22]. Compounds satisfying the pharmacophoric requirements were then instrumental to run docking studies. Hence, compounds showing the highest binding scores were selected, and tested in vitro for their ability to bind and inhibit purified recombinant human DDC. The compounds with the highest inhibitory activity were used to perform a second similarity-based filtering of the public ZINC database to retrieve analogs in order to expand the new classes of DDC inhibitors. The in vitro testing revealed that 9 hits sorted out from the second screening inhibit human DDC in a competitive mode with Ki values in the range 2–15 µM. Subsequently, from a substructure search using the core of the most active compound, a molecule with a Ki value of 500 nM emerged as a promising candidate for further lead optimization.
Results
Virtual Screening
On the basis of the crystal structure of pig kidney DDC (PDB code: 1JS3) in complex with the anti-Parkinson drug carbidopa, the key chemical features responsible for the critical binding interactions were initially derived. A pharmacophore query was used to build a 8-points pharmacophore hypothesis (PH) containing: (1) an aromatic centroid (F1, Aro) located at the geometric center of the catechol ring and its normal projection (F2, PiN), which points at Lys303, (2) a hydrogen bond donor feature (F3, Don) located on the para-hydroxyl substituent of the catechol moiety and its projection (F4, Don2), which points at the side chain of Thr82, (3) a hydrogen bond donor feature (F5, Don) on the meta-hydroxyl substituent of the catechol moiety and its projection (F6, Don2), which points at the phosphate moiety of the PLP cofactor, and (4) a hydrogen bond acceptor feature (F7, Acc) located on the oxygen of the carboxylate group and its projection (F8, Acc2), which points at the side chain of His192. Finally, in order to take into account the shape of the active site of DDC, a steric constraint (excluded volume) was derived from the DDC-carbidopa complex and included in the PH. Such features are summarized in Table 1.
Table 1. Chemical features in the PHs.
| Atoms | Feature | pig DDC - carbidopa | Feature | Final PH |
| Catechol ring | F1 | Aro (1)E | C1 | Aro (1.57)E |
| F2 | PiN (1.4) | C2 | PiN (1.58) | |
| Meta-OH | F3 | Don (1) | C3 | Don (1.28)E |
| F4 | Don2 (1.4) | C4 | Don2 (1.49) | |
| Ortho-OH | F5 | Don (1) | C5 | Don (1) |
| F6 | Don2 (1.4) | C6 | Don2 (1.2) | |
| Carboxylate | F7 | Acc (1) | C7 | Acc (1.49) |
| F8 | Acc2 (1.4) | C8 | Acc2 (1.37) | |
| Active compounds | C9 | Acc2 (1.58)E | ||
| C10 | Acc2 (1.29) |
The table summarizes the type of chemical feature, its location in the initial PH, derived from the pig kidney DDC – carbidopa complex, and in the final PH. The features which were regarded as essential are shown. Numbers in parenthesis indicate the sphere radii of each feature (Å).
Essential.
Then, within a set of already experimentally tested compounds [20], those showing no chirality center were included in a training set. On the basis of assays performed on a pig kidney extract [20], they were clustered in inactive (31 compounds causing a loss of DDC activity <10% at 2.2 mM concentration), poorly active (33 compounds causing a loss of DDC activity ≥10% at 2.2 mM concentration), moderately active (29 compounds causing a loss of DDC activity ≥10% at a concentration in the range 220–440 µM), and highly active (9 compounds causing a loss of DDC activity ≥10% at a concentration <110 µM). All active molecules (i.e., poorly, moderately and highly active compounds) were analyzed with the previously developed PH and the conformations of the matching molecules were processed in order to retrieve consensus chemical features (coordinates, radius and feature type). The latter, in turn, were included in the PH (as detailed in the “Materials and Methods section”). For this purpose, the annotation points of the active compounds matching the PH were clustered in ten chemical features (C1–C10, Table 1). C1–C8 coincided with F1–F8 in terms of position (root mean square deviation (RMSD): 0.13 Å) and type of feature, even if the radii of the spheres were slightly different (Table 1). Moreover, two new chemical features were identified: a hydrogen bond acceptor projection feature (C9) roughly pointing at His302 and a hydrogen bond acceptor projection feature (C10) pointing at two structural water molecules (hydrogen bonded to the carboxylate moiety of carbidopa in the DDC-carbidopa complex). The two new projection features, C9 and C10, were then added to the initial PH, and the radii of all the chemical features, F1 to F8, were subsequently adapted according to the values calculated by the consensus function for the C1–C8 features. Therefore, in order to assess which features could be regarded as essential for the subsequent pharmacophore searches (PSs), we generated several alternative PHs, each containing all but one feature, and compared their performance by carrying out PSs of the final training set (initial training set plus 1950 decoys; see “Materials and Methods section” for further details). The final obtained PH (Fig. 1), in which F1, F3 and C9 were set as essential, retrieved 48% active (30% poorly active, 55% moderately active and 88% highly active) and 6% inactive compounds, thus clearly showing a marked improvement as compared to the initial PH, with 27% active (12% poorly actives, 34% moderately active and 55% highly active) and 3% inactive compounds.
Figure 1. Pharmacophore hypothesis of a competive inhibitor of DDC, superimposed on the 1JS6 – carbidopa complex.

The chemical features of the final pharmacophore essential for the binding of a competitive inhibitor to DDC are shown as spheres. Carbidopa is represented as balls and sticks. PLP, water molecules and residues interacting with carbidopa are also shown and labeled.
The final PH was then used to filter the drug-like and lead-like subsets of the ZINC database [21]. This PS retrieved ∼280000 drug-like and ∼180000 lead-like molecules, matching at least 6 chemical features. These virtual hits underwent a second docking-based filtering by means of the Dovis docking tool [23], [24]. All the potential hits (∼460000) were docked into the active site of DDC, and the normal distribution of their scores (reported as predicted pKi values) was derived. The distributions of scores for lead-like and drug-like molecules showed a mean pKi of 5.30±0.62 and 5.57±0.81, respectively (Fig. S1 and S2). 2391 drug-like and 2994 lead-like molecules with significant scores (≥2 standard deviations (SDs from mean) were selected and resubmitted to a further docking step by means of a second docking tool, Autodock Vina [25]. 1496 and 1763 drug-like and lead-like compounds, respectively, showed poses comparable when docked with both Vina and Dovis (RMSD value ≤2 Å). Of these, 17 lead-like and 44 drug-like molecules (whose ZINC code and final ranking are reported in Supplementary Tables S1 and S2) fulfilled also the restraints imposed by the final PH. Then, the similar compounds were clustered, and, on the basis of the predicted pKi and the commercial availability, the most potent of each group was chosen. This protocol gave us 7 drug-like and 8 lead-like molecules that were subjected to in vitro testing. For each compound the chemical structure is reported in Fig. 2 and the number of matched features of the final PH in Table 2.
Figure 2. Chemical structures of selected compounds from the first VS screening.

Table 2. Lead-like (compounds 1, 2, 4, 6, 8, 10, 14, 15) and drug-like (compounds 3, 5, 7, 9, 11, 12, 13) molecules selected by the VS.
| ZINC code | Ambinter | MW(Da) | Matched features | Ki aKi′ b(µM) | BE | Inhibition profile | |
| 1 | 02654612 | T5246255 | 333 | 5 | 106±7a | 11.9 | Competitive |
| 2 | 03324540 | T0516-9932 | 282 | 5 | 390±30a | 12.1 | Competitive |
| 3 | 05936951 | T5347527 | 381 | 4 | n.d. | - | n.d. |
| 4 | 00408890 | STOCK1N-01831 | 295 | 4 | n.d. | - | n.d. |
| 5 | 05252718 | T5607516 | 412 | 4 | 59.8±5.8a | 10.2 | Competitive |
| 6 | 00111962 | STOCK1N-14396 | 326 | 7 | n.d. | - | n.d. |
| 7 | 12603254 | T5999596 | 351 | 6 | n.d. | - | n.d. |
| 8 | 03243721 | T0511-4207 | 332 | 5 | n.d. | - | n.d. |
| 9 | 05185571 | T6014881 | 407 | 5 | n.d. | - | n.d. |
| 10 | 00492694 | STOCK1S-02871 | 266 | 7 | 6.03±0.55a23.1±1.1b | - | Mixed |
| 11 | 08042801 | PB-05725216 | 381 | 6 | 10.7±2.4a | 13.0 | Competitive |
| 12 | 01874906 | STOCK3S-99912 | 459 | 9 | 19.4±1.4b | - | Noncompetitive |
| 13 | 07986167 | T5522252 | 375 | 5 | n.d. | - | n.d. |
| 14 | 00342069 | STK059041 | 322 | 5 | n.d. | - | n.d. |
| 15 | 00134865 | F0433-0503 | 316 | 5 | n.d. | - | n.d. |
The table summarizes, for each compound, the ZINC-code, the Ambinter-code, the molecular weight, the number of matched features of the final PH, the experimental Ki and/or Ki′ values, the BE, and the inhibition profile. n.d., not determined.
Inhibition and binding analysis
The 15 compounds resulting from the virtual screening were tested for their ability to bind to and inhibit human recombinant DDC. At a concentration in which their solubility was maximum, compounds 6, 8, 13, and 15 do not inhibit DDC activity, compounds 3, 4, 7, 9 and 14 exhibit weak DDC inhibition activity (8–25%), while the compound 12 exerts an inhibitory effect of DDC activity of ∼55%. On the other hand, the remaining compounds 1, 2, 5, 10 and 11 inhibit DDC by 70% or more. An IC50 analysis performed on the latter molecules identified five inhibitors of human DDC with well-behaved IC50 curves giving IC50 values equal to 570±66 µM, 1500±220 µM, 124±97 µM, 12.8±0.4 µM, and 27.3±1.6 µM for compounds 1, 2, 5, 10 and 11, respectively. The IC50 curve of compound 11 is shown in Fig. S3A. These molecules and also compound 12 were further analyzed in order to identify the type of inhibition. Compounds 1, 2, 5, 11 behave as typical reversible inhibitors with competitive inhibition, compound 10 with mixed inhibition, and compound 12 with noncompetitive inhibition. The inhibition constant values (Ki and/or Ki′) for each of these inhibitors are reported in Table 2. None of the competitive inhibitors changed the absorbance spectrum of free PLP. On the other hand, the addition of any of the competitive inhibitors to human DDC induced a modification of the CD spectrum which, like in the pig kidney enzyme [26], is characterized by positive dichroic bands at 420 and 335 nm. The modification consists of a decrease of the 420 nm positive signal and a concomitant increase of the 335 nm positive band. The fractional changes in the magnitude of the 420 nm or the 335 nm dichroic band as a function of the compound 1, 2, 5 and 11 concentrations were used to calculate the KD for the inhibitor-DDC complexes formation. The KD values were found to be 240±68 µM, 516±126 µM, 54.9±9.8 µM and 16.1±2.1 µM, for compounds 1, 2, 5, and 11, respectively, in good agreement with the corresponding Ki values. Taken together, these data strongly suggest that all the competitive inhibitors bind to the human DDC active site.
As shown in Fig. 3, the computational docking for compounds 1, 2, 5, and 11 gives the following clues regarding the possible binding mode:
Figure 3. Predicted binding mode of compounds selected after the first VS.

The figure shows the binding mode of (A) compound 1, (B) compound 2, (C) compound 5, and (D) compound 11. Residues and water molecules interacting with the inhibitors are also shown. Figure was rendered with Pymol (http://www.pymol.org/).
Compound 1 (Fig. 3A)
The 1-hydroxybenzotriazole ring of this compound shows a cation - π interaction with Lys303. The nitrogen atom and the hydroxylic group of the 1-hydroxybenzotriazole ring are hydrogen bonded to Thr82 and to the phosphate of the PLP cofactor, respectively. The sulfonamidic nitrogen atom interacts with one of the water molecules bridging the phosphate group of PLP and His192. Moreover, the 3-carboxy-phenyl ring displaces the second water molecule, and interacts with Phe103′ (showing a parallel-displaced stacking arrangement) and Leu353′ (accent indicates residues of the active site provided by the adjacent monomer). Finally, the carboxylic group of this ring is hydrogen bonded to Ser193.
Compound 2 (Fig. 3B)
The 1-hydroxybenzotriazole ring interacts similarly to the one of compound 1. One of the oxygen atoms of the sulfonamidic group is hydrogen bonded with the two water molecules interacting with the carboxylic group of carbidopa in the carbidopa-DDC-complex. The piperidinic ring interacts hydrophobically with the residues Tyr79, Phe80 and Thr246.
Compound 5 (Fig. 3C)
The 2-carbonyl-2,3-dihydro-benzimidazole ring of this compound shows a cation- π interaction with Lys303, and its nitrogen and carbonyl atoms interact with the amide carbonyl of Pro81 and with Thr82, respectively. Furthermore, the oxygen atoms of the sulfonamidic group interact with several water molecules, which interact with the carboxylic group of carbidopa in the carbidopa-DDC complex. In particular, one of the oxygen atoms is hydrogen bonded to the water molecules bridging the phosphate group of PLP and His192, and the other sulfonamidic oxygen atom is hydrogen bonded to one of the two solvent exposed water molecules. The amidic carbonyl moiety interacts with His192 and a water molecule. Finally, the two aromatic rings, benzene and furan, show hydrophobic interactions with Phe103′, Leu353′, Tyr79, Phe80 and Thr246.
Compound 11 (Fig. 3D)
The aromatic catechol ring of this compound approximately overlaps with the catechol ring of carbidopa (and, possibly with the one of L-Dopa) and, similarly to the latter, it shows a cation- π interaction with Lys303. The ortho and meta hydroxyl groups of the catechol ring are hydrogen bonded to the phosphate moiety of the PLP cofactor and to Thr82, respectively. The catechol is connected to a 3-mercapto-1,2,4-triazole heterocycle through a rigid methylidene-amino moiety. The N1 atom of the heterocycle is hydrogen bonded to His192, and the thiol group to the phosphate moiety of the PLP cofactor. The heterocycle is well positioned to displace the water molecules (which were found to interact with carbidopa in the crystal structure of the carbidopa-DDC complex; PDB code: 1JS3) bridging the phosphate group of PLP and His192. The triazole ring and the 2,4-dichloro-phenyl ring show hydrophobic interactions with several residues located at the opposite sides of the active site entrance, namely Phe103′ and Leu353′, as well as Tyr79, Phe80 and Thr246. Moreover, the triazole ring and Phe103′ show a parallel-displaced stacking arrangement.
Second Screening (Similarity Search)
Following the initial identification of active compounds, a similarity search was carried out by using the most potent hits (compounds 5 and 11) as query. First, starting from the ZINC database of small molecules, a fingerprinted database was generated, and then the two identified compounds were used as a query to find similar compounds. To this end, the MACCS structural keys [27] and the Tanimoto coefficient (TC) [28] were used as fingerprint system and similarity metric, respectively. Then, each compound was ranked according to the TC, calculated with respect to the query, and those showing a TC>0.80 were selected and further filtered on the basis of the commercial availability. The candidate molecules related to compound 5 showed variations limited to the 2-(((furan-2-yl)-methylamino)-carbonyl) moiety (namely ZINC03886086 and ZINC22621067). On the other hand, a series of compounds showing variations on substitution pattern of either the hydroxyl groups on the catechol ring or the halogen atoms on the second aromatic ring has been selected among the molecules related to compound 11. This procedure was adopted in an attempt to obtain information on the structure/activity relationship of the above mentioned positions. To this end, two molecules from the Ambinter database, which did not figure in the ZINC database, were also selected. This procedure gave two molecules related to compound 5 and twenty molecules related to compound 11 (see Table S3 and S4 for the ranking of the selected compounds). All of them, whose structures are shown in Fig. 4, were subjected to in vitro testing.
Figure 4. Chemical structures of selected compounds from the secondVS screening.

Inhibition and binding analysis
To probe the reliability of this second screening we tested the effect of the 22 compounds on human DDC activity using the “in vitro” assay. The decarboxylase activity was determined in the absence and in presence of each compound, and IC50 values were determined for compounds 17, 21, 22, 23, 24, 25, 28, 30, 31, 34, 35, and 37 (Table 3). Dose-response curves of compounds 17, 21, 23, 25, 34, and 37 are reported in Fig. S3A. It was not possible to define the IC50 values for the remaining compounds since at a concentration in which their solubility was maximum they inhibit DDC activity less than 55%. The analysis of the inhibition profile reveals that compounds 16, 17, 18, 20, 21, 23, 24, 25, 28, 30, 31, 34, and 37 behave as competitive inhibitors of human DDC, while compounds 22, 27, 29, 32, and 33 inhibit decarboxylase activity in a mixed mode, and compounds 26 and 35 are noncompetitive inhibitors. The Ki and/or Ki′ values of these inhibitors are listed in Table 3. Changes in the CD bands of the visible spectrum of DDC induced by the addition of each of the competitive inhibitors are indicative of their binding to the active site of the enzyme. Compounds 21 and 34 cause a decrease of the 420 nm dichroic signal and a concomitant appearance of a positive dichroic signal at 388 nm as well as an increase of the 335 nm dichroic band, while the remaining competitive inhibitors induce a decrease of the 420 nm band and an increase of the 335 nm band. Fig. 5 shows the dichroic spectral changes induced by compound 37 on human DDC. Analysis of the data at 420 nm or at 335 nm as a function of inhibitor concentration yields the following KD values: 17.5±5.1 µM, 28.2±3.2 µM, 2.5±0.7 µM, 4.7±0.5 µM, 2.4±0.1 µM, 21.1±3.5 µM, 25.1±3.5 µM, 31.7±3.4 µM, 7.1±0.9 µM and 1.5±0.2 µM (inset of Fig. 5) for compounds 17, 20, 21, 23, 25, 28, 30, 31, 34, and 37, respectively. These values are in good agreement with the corresponding Ki values. Unfortunately, the KD values for the compounds 16, 18, and 24 cannot be measured because of their low solubility. Like the competitive inhibitors of the first series, none of these competitive inhibitors alters the absorbance spectrum of free PLP. In addition, when human DDC complexed with each of the competitive inhibitors was subjected to extensive dialysis against 100 mM potassium phosphate buffer, pH 7.4, the CD spectra of the dialyzed enzymes were identical to that of free DDC. This clearly indicates a reversible binding mode of these molecules.
Table 3. Structural analogs of the most potent hit compounds (compounds 5 and 11) obtained after the first screening.
| ZINC-code | Ambinter | MW(Da) | IC50(µM) | Ki aKi′ b(µM) | BE | Inhibition profile | |
| 16 | 03886086 | PB-90199554 | 335 | n.d. | 59.8±0.3a. | 12.7 | Competitive |
| 17 | 22621067 | PB-251389058 | 408 | 16.8±1.7 | 4.4±1.4a | 13.1 | Competitive |
| 18 | 01183789 | PB-90040583 | 365 | n.d. | 63.2±3.1a | 11.5 | Competitive |
| 19 | 01183732 | PB-90046346 | 365 | n.d. | n.d. | - | n.d. |
| 20 | 01184616 | PB-90047186 | 365 | n.d. | 36.8±4.2a | 12.1 | Competitive |
| 21 | 00550832 | STK103726 | 347 | 6.3±0.9 | 2.3±0.4a | 16.3 | Competitive |
| 22 | 03044871 | STK508886 | 328 | 6.7±0.9 | 15.1±0.1a46.7±0.5b | - | Mixed |
| 23 | - | PB-05713794 | 312 | 16.6±.3 | 5.7±0.1a | 16.8 | Competitive |
| 24 | 00549544 | STOCK3S-13636 | 331 | 77.1±2.4 | 120±14a | 11.9 | Competitive |
| 25 | 00520318 | STOCK3S-69837 | 347 | 15.2±1.3 | 3.7±0.2a | 15.7 | Competitive |
| 26 | 00548914 | STOCK3S-09410 | 347 | n.d. | 65.2±−4.1b | 12.1 | Nonconmpetitive |
| 27 | 00344575 | STOCK3S-13173 | 312 | n.d. | 24.8±1.2a43.1±1.9b | - | Mixed |
| 28 | 00554457 | STOCK3S-64376 | 331 | 17±1 | 14±1a | 14.7 | Competitive |
| 29 | 00553626 | STOCK3S-56595 | 331 | n.d. | 44.4±2.3a208±3b | - | Mixed |
| 30 | 01478099 | STOCK3S-62307 | 381 | 29.8±4.8 | 12.2±0.4a | 12.9 | Competitive |
| 31 | 00199179 | STOCK3S-19327 | 296 | 83±12 | 33.4±5a | 15.1 | Competitive |
| 32 | - | STOCK3S-20068 | 523 | n.d. | 4.5±0.3a17±0.2b | - | Mixed |
| 33 | 00532465 | STOCK4S-26311 | 314 | n.d. | 16.5±0.8a325±39b | - | Mixed |
| 34 | 00530774 | STOCK4S-17596 | 330 | 11±0.8 | 10.4±3.3a | 15.8 | Competitive |
| 35 | 01473651 | STOCK2S-87581 | 381 | 29±2 | 127±16b | - | Noncompetitive |
| 36 | 04898088 | STOCK2S-83947 | 331 | n.d. | n.d. | - | n.d. |
| 37 | 00344577 | STOCK2S-84925 | 312 | 2.4±0.3 | 1.8±1.1a | 18.4 | Competitive |
MACCS structural keys system (BIT_packed) and the Tanimoto coefficient were used as fingerprint system and similarity metric, respectively. The table summarizes, for each compound, the ZINC-code, the Ambinter-code, the molecular weight, the experimental IC50, Ki and/or Ki′values, the BE, and the inhibition profile. n.d., not determined.
Figure 5. Visible CD spectral changes induced by compound 37 on human DDC.

CD spectra of human holoDDC (—) and in the presence of the compound 37 at the indicated concentration in 0.1 M potassium phosphate buffer, pH 7.4. Inset: ellipticity change of DDC plus compound 37 at 420 nm and 335 nm as a function of inhibitor concentration. The enzyme concentration was 6 µM.
The binding mode of the compounds resulting from the second screening was also investigated by molecular docking means (applying the procedure described in “Materials and Methods Section”). This analysis provided us with the following information:
Inhibitors (16 and 17) related to compound 5
Compounds 16 and 17 (Fig. 6A) are predicted to have binding properties very similar to the ones of compound 5. This is not unexpected given their structural resemblance. However, it should be noted that (i) the carboxyl group of compound 16 could form a salt bridge with Arg447, (ii) the benzene ring of compound 17, which replaces the furan ring of compound 5, is predicted to assume a partial parallel-displaced stacking arrangement with Phe103′, and (iii) the nitrogen atoms of the 2-carbonyl-2,3-dihydro-benzimidazole ring of both compounds, which are slightly translated with respect to compound 5, are hydrogen bonded to Thr82 and the phosphate group of the PLP cofactor.
Figure 6. Predicted binding mode of compounds selected after the second VS (similarity search).

The figure shows the binding mode of (A) compound 37 and (B) compound 17. Residues and water molecules interacting with the inhibitors are also shown. Figure was rendered with Pymol (http://www.pymol.org/).
Inhibitors (18–37) related to compound 11
All these compounds are predicted to have binding features very similar to the ones of compound 11, even if some differences in the interactions of their hydroxyl groups with the protein and/or the coenzyme can be noted. In Fig. 6B the predicted binding mode of compound 37 is shown.
Third Screening
In order to identify molecules with increased activity and obtain insights into structure-activity relationships, we further expanded the class of active compounds resulting from the second VS by a substructure search carried out using the core of the most active compound 37 (Fig. 7). The OpenBabel substructure search tool (Open Babel v. 2.3.1, 2011) was used to filter the Ambinter database of small molecules, using as query the following SMILES notation (Weininger, 1988): [OH]c2[cH][cH]c(C = Nn1cnnc1)[cH]c2[OH]. Since the most potent compounds obtained in the second VS (see compounds 21, 37, 21) have a catechol ring with meta- and para- hydroxylic groups, the explicit hydrogens of the unsubstituted benzene carbon atoms and of the hydroxylic oxygen atoms were included in the SMILES notation in order to avoid the selection of compounds with substitution patterns involving these positions.
Figure 7. Core of compound 37.

The core of compound 37 was used as a query (the Smiles annotation string used is shown in the text), using OpenBabel, to filter the Ambinter database. Positions where no substitutions were allowed during the search are coloured in red.
The substructure search retrieved 73 hits from the Ambinter database. Among these hits, a series of compounds were selected based on several criteria including i) commercial availability; ii) predicted pose in the active site of DDC, according to docking and modeling analysis; iii) clustering, based on similarity and subsequent removal of redundancy. At the end of this step, 10 compounds, whose chemical structures are reported in Fig. 8, were selected: a compound (38) representing the core of compound 37 and a closely related derivative (39) showing methyl and thiol substituents at position 3 and 5, respectively, of the 1,2,4-triazole ring; one compound showing a substitution of the benzene ring for a smaller furane ring (compound 40); six compounds with different substitution patterns of the benzene ring (compounds 41, 42, 43, 44, 45, 47); one compound with a methoxy linker between the 1,2,4-triazole and the benzene ring of compound 37 (compound 46). All of them were subjected to in vitro testing.
Figure 8. Chemical structures of selected compounds from the thirdVS screening.

Inhibition and binding analysis
The inhibition activity of these compounds was evaluated by human DDC “in vitro” assay. Compound 47 at 200 µM (the concentration of its maximum solubility) does not inhibit DDC activity. On the other hand, the IC50 and the Ki values for competitive inhibition of the remaining compounds have been determined and listed in Table 4. Dose-response curves of compounds 39, 40, 41, 42, 43, 44, 45, and 46 are shown in Fig. S3B. None of these active compounds binds free PLP but, when added to human DDC, they cause CD spectral changes identical to those of compound 37. The KD values of compounds 38, 39, 41, 42, 43, and 45, determined as above reported, were found to be 24.5±4.4 µM, 9.1±0.3 µM, 6.1±0.5 µM, 2.7±0.5 µM, 2.8±0.5 µM and 1.5±0.3 µM, respectively. These values are very close to the corresponding Ki. In the case of compounds 40, 44, and 46, the enzyme concentration required for CD measurements far exceeds their Ki values, which precludes the determination of the KD values of these molecules.
Table 4. Structural analogs of the most potent hit compound (37) obtained after the second screening.
| Ambinter | MW(Da) | IC50(µM) | Ki(µM) | BE | |
| 38 | Amb1808242 | 204 | 76.0±5.8 | 24.5±1.4 | 22,6 |
| 39 | Amb8754110 | 250 | 27.5±3.3 | 10.8±0.8 | 19,9 |
| 40 | Amb666646 | 302 | 3.5±0.2 | 1.4±0.1 | 19,4 |
| 41 | Amb5967895 | 326 | 30.1±2.1 | 9.8±0.5 | 15,4 |
| 42 | Amb655238 | 326 | 5.2±0.4 | 2.0±0.2 | 17,5 |
| 43 | Amb685531 | 326 | 3.8±0.6 | 1.9±0.1 | 17,5 |
| 44 | Amb2470350 | 342 | 1.7±0.2 | 0.5±0.1a | 18,4 |
| 45 | Amb2472735 | 342 | 6.2±0.3 | 2.0±0.1a | 16,7 |
| 46 | Amb666103 | 342 | 3.5±0.3 | 1.1±0.1 | 17,4 |
| 47 | Amb782700 | 342 | >200 | n.d. | n.d. |
The table summarizes, for each compound, the Ambinter-code, the molecular weight, the experimental IC50, Ki′ values, and the BE. n.d., not determined.
Ability to cross the Blood-Brain Barrier (BBB) of identified compounds
All identified inhibitors were analyzed to estimate their ability to cross the blood-brain barrier (BBB), according to a simple prediction model developed by Clark et al. [29]. According to this model, compounds with predicted values of logBB less than −0.3 are not considered capable of crossing the BBB, while values greater than zero are predictive of concentration in the brain higher than in the blood. All of the identified inhibitors have logBB less than −0.3, and ranging from −0.57 to −1.65, highlighting the poor BBB penetration propensity of these compounds. Moreover, the most active among the compounds related to 5 and 11, namely compounds 17 and 44, showed logBB values of −1.24 and −1.30, respectively, comparable with the one of carbidopa that is equal to −1.50.
Discussion
PD is one of the most common neurodegenerative disorders. It is characterized clinically by parkinsonism (resting tremor, bradykinesia, rigidity, and postural instability) and pathologically by the loss of dopaminergic neurons in the substantia nigra. Aromatic hydrazine derivatives (e.g., carbidopa and benserazide) are used in combination with L-Dopa to treat the symptoms of PD or Parkinson-like symptoms. Being unable to cross the BBB, the former drugs work by inhibiting only DDC at the peripheral level. Several side effects (from nausea, hypotension, arrhythmias, and gastrointestinal bleeding to more serious psychiatric symptoms, including auditory and/or visual hallucinations, confusion, psychosis and suicidal thoughts) ensuing from co-administration of L-Dopa with carbidopa or benserazide have been reported [30]. These adverse effects can be ascribed, at least partially, to the mode of action of carbidopa and trihydroxybenzylhidrazine (the in vivo hydrolysis product of benserazide) that, by reacting non-enzymatically with free PLP, would cause PLP depletion [18], [19], [31]. It has also been found that the condensation products formed between PLP and hydrazine derivatives are the most potent inhibitors of pyridoxine kinase, an enzyme involved in the biosynthesis of PLP [32], [33]. In addition, it must be pointed out that PD patients treated with L-Dopa and DDC peripheral inhibitors show high levels of plasmatic homocysteine [34], [35] found to be inversely correlated with PLP concentrations in the treated patients [36]. Based on these reports, it is likely that admnistration of L-Dopa in combination with carbidopa or benserazide could be responsible for an alteration of the PLP-dependent metabolism. Thus, there is currently a great interest in the identification of highly potent, reversible and selective DDC inhibitors.
A hierarchical filtering approach was used in this work to identify novel inhibitors of DDC. The hits identified in the initial VS were tested in vitro on human DDC. This initial information was used to guide a second round and a third round of VS, in order to retrieve potentially more potent substances and expand the new classes of inhibitors, thereby gaining insights into their structure-activity relationships. In the last years, a lot of effort has been made to improve the rank order in VS approaches. Often, compounds are ranked according to their estimated free energy of binding, and molecular docking is commonly used to this purpose. However, its high computational cost and required time set an upper limit to the amount of compounds that can be processed. One way to solve this issue is to carry out the docking analysis on a subset of the initial database, filtering the latter by other means. Thus, to improve the rank order in VS approaches, pharmacophoric modelling was used in this work, as previously proposed by others [37], [38]. In this way, we were able to select only those compounds (∼105), which show the essential chemical features for DDC binding. During the whole VS process, we didn't make use of additional scoring functions as a consensus [39], [40], [41], other than the ones implemented in Dovis 2.0 and Autodock Vina. However we exploited, in addition to docking score criteria, a pose dependent selection criterium (see “Materials and Methods section”). In this way, final compounds were selected if they 1) showed a comparable docking pose as obtained by two independent search algorithms, 2) were top ranking hits, as assessed by two independent scoring functions, and 3) satysfied the restraints imposed by the pharmacophore model To our knowledge, the VS protocol that we used, in which Pharmacophore-Based Virtual Screening is integrated as a pre-processing and post-processing step with the main docking-based virtual screening step, has never been applied before. Notably, it resulted in a high success rate (5 active compounds out of 15) during the initial in vitro testing on human DDC. Such complex VS approach outperformed simpler approaches such as 2D similarity or substructure searching. In fact, when the latter were used with carbidopa as query (by the MACCS structural keys and the TC as fingerprint systems and similarity metric, respectively) on the same drug-like and lead-like subsets, compounds 1, 2, 5 and 11 were ranked, according to TC, in positions ≥7.5×105. Therefore, they would have not been identified.
By means of our VS approach we identify three new classes of compounds acting as competitive human DDC inhibitors: (i) compounds 1 and 2, (ii) compound 5 and its analogs, and (iii) compound 11 and its analogs. These classes show scaffolds composed by two differentially substituted aromatic cycles, connected by a rigid hydrophobic linker with an electron-rich group. The first aromatic ring is represented by a catechol ring (compound 11 and its analogs), or a 2-carbonyl-2,3-dihydro-benzimidazole (compound 5 and its analogs) or a hydroxybenzotriazole (compounds 1 and 2) bicyclic ring. The linker is represented by a 4-N-[3-mercapto-1,2,4-triazol-4-yl]-carboximidoyl moiety (compound 11 and analogs) or a sulfonamidic group (compounds 1, 2, 5 and its analogs). The second ring is represented by a substituted chloro-benzene ring (compound 11 and analogs), or a piperidine ring (compound 2), a carboxy-benzene ring (compounds 1 and 16), a 2-(((furan-2-yl)-methylamino)-carbonyl)-benzene ring (compound 5) or a 2-(((phenyl)-amino)-carbonyl)-benzene ring (compound 17).
When taking into account the chemical structure as well as the inhibition and binding data of the compounds, simple structure-activity observations can be derived. For molecules related to compound 5, the substitution of the furan-2-yl-methyl moiety with a benzene ring results into an about 13-fold increase of affinity (compare compounds 5 and 17), while the substitution of the furan-2-yl-methylamino-carbonyl moiety with a carboxylic group does not improve affinity (compare 5 and 16). For the molecules related to compound 11, it can be observed that (i) the shift of the two hydroxyl substituents from the ortho-meta to the meta-para positions results in compounds with comparable binding affinity (compare compounds 11 and 30), (ii) the removal of chlorine subtituents from the ortho-meta) hydroxy-substituted compound (compound 11) results in a decrease of Ki (compare compounds 11 and 23), (iii) the removal or substitution of one or two chlorine subtituents from the meta-para hydroxy-substituted compound (compound 30) results in a decrease in Ki values in the following order: compounds 30 (ortho-para-Cl)≈34 (para-F)>25 (ortho-Cl)>21 (para-Cl)≈37 (unsubstituted), (iv) the removal of a hydroxylic group from the ortho-meta (compound 11) or meta-para (compound 30) hydroxy-substituted compounds results in a increase of Ki in the following order: compound 11≈ compound 30<compound 20 (meta-hydroxy)<compound 18 (para-hydroxy)<compound 19 (ortho-hydroxy, poorly active), (v) the removal or substitution of chlorine from the mono-ortho-hydroxy (compound 19) and mono-meta-hydroxy (compound 20) compounds results in compounds either of different affinity (compounds 24, 28, 31, 36) or different inhibition profile (compounds 29, 32, 33). Among those showing a competitive inhibition profile, compound 31 (unsubstituted) and compound 28 (ortho-Cl) are the most potent, and (vi) the presence of a hydroxylic-substitution pattern different from that of the catechol moiety, ortho-meta or meta-para, or of a mono hydroxylic-substituted ring, results in a deviation from the competitive inhibition profile, i.e., mixed (compounds 22, 27, 32) or noncompetitive (compounds 26 and 35) inhibition.
According to their predicted binding mode (see results) the three classes of active compounds share some or all of the following key interactions: (i) cation - π interaction with Lys303 and one or two hydrogen bonds with the phosphate of PLP, Thr82 or the carbonyl of Pro81; (ii) hydrogen bond interactions with the active-site water network and/or with His192; (iii) stacking interaction with Phe103′; (iv) hydrophobic interactions with Tyr79, Phe80, Thr246 and Leu353′. The hydrophobic interactions with Tyr79, Phe80, Thr246 and Phe103′ represent two new pharmacophoric features of our developed structure/ligand-based pharmacophore model, and it is noteworthy that the compounds 17, 21, 23, 25, and 37 showing the lowest Ki (<10 µM) fulfill all these binding requirements, while, compound 2, missing several key interactions, exhibits the highest Ki value (Ki = 390 µM). It should be noted that in the spatial structure of ligand free DDC and its complex with carbidopa a flexible loop between residues 328 and and 339 is invisible in the electron density map [13]. Limited proteolysis and site-directed mutagenesis data have indicated that substrate-induced conformational changes occur at this loop and that its closure is an essential step in the decarboxylation mechanism [4], [42]. On the basis of our docked pose it is predicted that this conformationally dynamic loop forms a cleft that accommodates the benzene ring of compound 37 and its derivatives. Although the lack of structural information on this loop does not allow the rational design of this part of the molecule to improve the binding efficiency, the results of out third VS allow to draw some conclusions about the influence of the cleft on inhibitor binding and its ability to accommodate different chemical groups. In particular, we noticed that: (ii) the presence at the 5 position of the thiol substituent, which is predicted to interact via hydrogen-bond with the phosphate tail of PLP, improves binding efficiency as can be appreciated by comparing compound 38 and 39, (ii) the substitution of the benzene ring for a smaller furane has only minimal influence on activity (compare compounds 37 and 40), (iii) the analysis of compounds in which the benzene ring is decorated with a methyl (compounds 41, 42, 43) or a methoxy substituent (compounds 44, 45 and 47) in ortho-, metha- and para- positions, respectively, allows to infer that, although the cleft is able to accommodate compounds bulkier than 37, a net preference of metha- and para- over ortho- substitutions could be observed.
In conclusion, this research has allowed the identification of compounds which are predicted do not cross the BBB, and, unlike carbidopa and thrydroxybenzylhydrazine (the in vitro hydrolysis product of benserazide) behave as reversible competitive inhibitors with Ki values in the low micromolar range. The most potent inhibitor binds human DDC with a Ki of 500 nM. Moreover, the finding that they do not interact with free PLP allows us to envisage that these inhibitors could have an intrinsic chemical reactivity lower than that of carbidopa and the metabolic product of benserazide. Thus, the compounds identified and tested here may provide precious guidelines for the drug development process for the PD therapy.
Materials and Methods
Materials
L-Dopa, PLP, 2,4,6-trinitrobenzene-1-sulfonic acid, isopropyl-β-D-thiogalactopyranoside, protease inhibitor cocktail (P8849) were Sigma products. DDC inhibitors were purchased from Ambinter (Paris, France). Purity (90% or higher) of these compounds was confirmed on the basis of NMR and/or LCMS data provided by vendors. All other chemicals were of the highest purity available.
Databases of chemicals
The drug-like and lead-like subsets of the ZINC database [21] were downloaded and used to generate low-energy conformations. The “Conformation import” function of MOE (The Molecular Operating Environment; Chemical Computing Group®, Montreal, Canada) was applied to this purpose. For each compound, the following settings were applied here and thereafter, if not otherwise stated: maximum 250 conformers for each compound; no input filters used; constraints options kept at their default values; only the conformers with strain energy less than 4 kcal/mol retained.
All of the compounds of the training set [20] were built with MOE, energy minimized using the mmff94x force field, and used to build a series of energetically plausible conformations. A set of 1950 decoy molecules, provided by Schrodinger (Schrodinger®, L.L.C., New York), was imported in MOE and series of energetically reasonable conformations were generated for each compound, using the “conformation import” function of MOE. Therefore, the final training set is composed by the union of the initial training set plus decoys.
Target preparation
Since a potential competitive inhibitor is expected to bind to the internal aldimine of DDC, we made use of PDB structure 1JS6 for pharmacophoric modeling purposes. To this end, 1JS6 was structurally aligned to 1JS3 using the “MOE-ProSuperpose function”; then, we manually inserted carbidopa into the active site of 1JS6, on the basis of the 1JS3 structure. The obtained complex was visually inspected in order to verify the absence of steric clashes between carbidopa and the residues at the active site.
Pharmacophore Model generation
Pharmacophore modeling calculations were performed using MOE. The MOE PQ function was used in order to build the PH starting from the 1JS6 – carbidopa complex. The “Unified Annotation Scheme” was used to assign the pharmacophoric annotation points (such as H-bond donor, H-bond acceptor, hydrophobic). Chemical features (coordinates, type of chemical feature and sphere radius) corresponding to atoms or groups of atoms of carbidopa were inserted in the PH. MOE's default values for the radii of atom-based chemical features (1 Å) and projections (1.4 Å) were used. An excluded volume, comprising residues at the active site of DDC located within a radius of 15 Å from any atom of carbidopa, was inserted in the PH.
The active compounds of the training set were analyzed by PS using the PH. During each PS the “partial match” option was activated and the number of chemical features to be matched was always set to “at least three” (default setting, if not otherwise stated). The aromatic centroid feature of the catechol ring of carbidopa was set as “essential”. For each compound, all of the matching conformations were selected. For each of the selected conformation, the number of matched features and the RMSD between the centers and the corresponding annotation points were derived. The conformations for each selected compound were sorted by decreasing number of matched features and increasing RMSD; for each compound only the highest ranked conformation was selected. This procedure was applied also in the following PSs.
Of the 71 active compounds analyzed, all satisfied the restraints imposed by the PH, and they were subsequently subjected to the “pharmacophore consensus function” of MOE. The “pharmacophore consensus function” suggests, from a set of structurally aligned molecules, common chemical features (coordinates, radius and feature type) that can be in turn inserted in a PH. When the “pharmacophore consensus function” was used, the following settings were applied: tolerance radius set to 1, the consensus score set to “weighted conformations” and the threshold set to 30%. Thus, the PH was modified according to the new data obtained from the pharmacophore consensus analysis: new identified features were added to the PH and the radii of the spheres of all the chemical features were increased/decreased accordingly.
In order to assess which feature of the PH should be regarded as essential for the subsequent PSs, we generated several alternative PHs, by discharging each time one feature. Then, we evaluated each PH for its ability to rank active and inactive compounds. To this end, we first derived a new dataset of compounds, merging the training set and the decoys. Then, for each PH, we measured the fraction of active and inactive compounds that were present amongst the best 3.46% ranking hits (corresponding to the ideal situation, in which all 71 active compounds are ranked in the first 71 positions of the ranked hits), according to the PS. When the PH with all the chemical features was used, 48% of the active molecules and 10% of the inactive molecules of the training set were retrieved in 3.46% of the top ranking hits. Among the generated alternative PHs, the one showing the highest decrease in terms of retrieved active compounds lacked the C9 feature. Moreover, the PHs showing the highest increase in terms of retrieved inactive compounds were those lacking F3 (16%) and C10 (16%).
Finally, to verify the effects of the changes brought to the PH in the previous steps, we evaluated the ability of the PH to rank active and inactive compounds when used with its initial settings (derived from the 1JS6 – carbiDOPA complex) and with the final settings (see Table 1). To this end, the same database was used as before and we measured the fraction of active (poorly active, moderately active and highly active) and inactive compounds, which were present amongst the best 3.46% ranking hits, according to the PS.
Docking settings
The drug-like and lead-like compounds retrieved from the PS were docked into the active site of DDC. To this end Dovis 2.0 [23], [24] was applied. Initially, the complex PLP-carbidopa was re-docked in the active site of DDC (PDB code: 1JS3), in order to optimize the parameters of the docking program to be used during the high-throughput docking. To this end, 1JS6 was structurally aligned to 1JS3 using the “MOE-ProSuperpose function”; then, we manually inserted carbidopa into the active site of 1JS6, on the basis of the 1JS3 structure. The obtained complex was visually inspected in order to verify the absence of steric clashes between carbidopa and the residues at the active site. To this end, an energy grid with 40×40×40 (numbers refer to the number of grid points in xyz), centered on the ε-amino group of the active site Lys303 of pig kidney DDC, and a 0.375-point spacing was used. Atom-specific affinity maps were then computed for each atom observed in the ligand, and a distance-dependent dielectric constant was adopted. Several docking simulations were initially run. During each simulation, grid dimension and center values were systematically varied, while other parameters were maintained at their default values, in order to find which combination of values was able to reproduce the binding pose of carbidopa bound to PLP present in the three dimensional structure. The selected values for grid dimensions and center were respectively 36×36×36 and x = 43.573 y = 37.127 z = 68.616 (coordinates are referred to the 1JS3). During the high-throughput docking, the Lamarckian Genetic Algorithm was chosen as search algorithm, using the following values: number of individuals in population, 150; maximum number of energy evaluation, 250000; maximum number of generation, 27×103; rate of gene mutation, 0.02. Water molecules were excluded from the docking calculations. All other parameters were kept at their default values.
After initial parameter optimization, the active compounds of the training set plus decoys were docked in the active site of DDC by means of Dovis 2.0, and ranked according to their predicted energy of binding. Only 11% of highly active compounds were retrieved within approximately the first 15% of the screened database. Then, in an attempt to improve the docking step, we applied a procedure involving two independent docking tools (Dovis 2.0 and AutoDock Vina), and we considered only those compounds showing a similar pose (RMSD<2 Å). Moreover, compounds were ranked according to the mean of their predicted energy of binding. By applying this second approach, 44% of highly active and only 3% of inactive compounds were retrieved within approximately the top-ranking 15% of the screened dataset. Therefore, the latter approach was adopted for the complete VS protocol.
When Autodock Vina [25] was used, the same grid parameters described previously were adapted, the exhaustiveness option was set to 8, and all other options were kept at their default values. Since a potential competitive inhibitor is expected to bind to the internal aldimine of DDC, we made use of PDB structure 1JS6 for subsequent docking purposes.
Post-filtering phase
A post-filtering step, based on the initial PH hypothesis, was coupled to the main docking-step. The latter, introducing additional structural knowledge on the “hot spots” of the active site, increased the performance of the VS protocol. Indeed, when the training set plus decoys was analyzed by the complete VS-protocol (docking plus pharmacophore search), 44% of highly active, 10% of moderately active, 3% of poorly active and 6% of inactive compounds were ranked in the top 0.6% of the screened database.
During the post-filtering phase, the poses of the docked drug-like molecules obtained in the previous docking-based virtual screening step were ranked according to their score, obtained with the scoring function [24] implemented in Dovis 2.0. The mean and standard deviation of the scores were then calculated, and those compounds scoring at >2 standard deviation from mean were selected and re-docked with Autodock Vina. Only those compounds showing a similar (RMSD<2 Å) docked pose, as assessed by Dovis 2.0 and AutoDock Vina, were kept. Finally, the selected poses were filtered with the previously developed PH with the “absolute position” option activated. In this way, when analyzed during PS, the poses were not allowed to be translated or rotated, and only those in agreement with the PH were kept. Lead-like molecules were processed similarly.
Assessment of the binding efficiency (BE) and BBB of selected compounds
When assessing the druggability of newly identified molecules, it is useful to monitor their molecular weight. This parameter relates to a contrasting trend: (1) an important increase in molecular weight is often required to reach the appropriate level of potency, (2) key properties that determine the druggability of a molecule tend to worsen when the molecular weight increases over a certain point [43]. Therefore, when establishing the viability of a molecule both potency and molecular weight need to be monitored and well balanced. The concept of BE represents a suitable means of assessing this relationship; it is, for a given compound, a measure of the binding energy per unit of mass relative to its target. Several definitions have been proposed, but throughout this work the following has been be used:
| (1) |
being Ki expressed in molarity (micromoles/L), and the molecular weight in kDa. It has been observed that 90% of lead molecules and drugs have efficiencies above 12.4 and 14.7, respectively [43].
All identified inhibitors were analyzed to estimate their ability to pass the BBB according to a simple prediction model developed by Clark [29], which takes into account the octanol/water partition coefficient (logP), calculated according to Moriguchi (MlogP; [44]), and the topological polar surface area. Moreover, values for MlogP topological polar surface area were calculated using the AlogPS online software [45] and MOE, respectively. Predicted log BB was calculated using the formula:
| (2) |
Compounds with predicted values of logBB less than −0.3 are not considered capable of crossing the BBB, while values greater than zero are predictive of concentration in the brain higher than in the blood.
Expression and Purification of Human DDC
Human DDC was expressed and purified by mean of the procedure previously described [46]. The enzyme concentration was determined by using an εM of 1.3×105 M−1 cm−1 at 280 nm [26]. PLP content of wild-type was determined by releasing the coenzyme in 0.1 M NaOH and by using εM = 6600 M−1 cm−1 at 388 nm.
Assay of the inhibitory activity
DDC activity was determined by measuring the production of dopamine with a spectrophotometric assay outlined by Sherald et al. [47] and modified by Charteris and John [48]. The reaction was carried out in 0.1 M potassium phosphate buffer pH 7.4 at 25°C for 1 min and stopped by inactivation of the enzyme at 100°C for 90 sec. The inhibitory assay was performed by measuring the decarboxylase activity of DDC with 50 nM enzyme, 0.1 mM L-Dopa, and 10 µM PLP in the presence and in the absence of a fixed amount of each compound. Various amounts of dimethyl sulphoxyde (DMSO) were necessary to solubilize the compounds in 100 mM potassium phosphate buffer, pH 7.4. In any case, the final DMSO concentration in the assay mixture was kept lower than 1% (v/v), i.e., under experimental conditions in which decarboxylase activity is not affected. Dose-response curves were generated to determine the concentration required to inhibit 50% of decarboxylase activity (IC50). Compounds were evaluated at 9 concentrations, and each in duplicate. IC50 values were derived by nolinear regression analysis. The related inhibition type of the compounds on human DDC was identified from Lineweaver–Burk plots, and Ki and/or Ki′ values for the inhibitors tested were calculated by the software Origin 7.0.
Enzyme-ligand equilibrium dissociation constant (KD)
The equilibrium dissociation constant for each tested compound from DDC was determined by monitoring the decrease in the 420 nm CD signal of DDC (7 µM) in the presence of various ligand concentrations in 100 mM potassium phosphate buffer, pH 7.4. In any case, the maximun DMSO concentration in the mixture was kept lower than 1% (v/v). The KD values were obtained from the fitting of the data to the equation
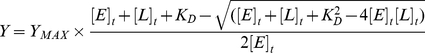 |
where [E]t and [L]t represent the total concentrations of DDC dimer and ligand, respectively, Y refers to the visible CD signal change at 420 nm at the ligand concentration [L], and Ymax refers to the CD signal when all enzyme molecules are complexed with the ligand. CD measurements were performed with a Jasco J-710 spectropolarimeter using quartz cuvettes with 1 cm path length.
Supporting Information
Normal distribution plot of predicted pKi values for drug-like molecules. 280000 drug-like molecules were docked in the active site of DDC with the Dovis 2.0 docking tool [23], [24] and the distribution of their predicted pKi was derived. The normal distribution showed a mean pKi of 5.57±0.81.
(TIF)
Normal distribution plot of predicted pKi values for lead-like molecules. 180000 lead-like molecules were docked in the active site of DDC with the Dovis 2.0 docking tool [23], [24] and the distribution of their predicted pKi was derived. The normal distribution showed a mean pKi of 5.30±0.62.
(TIF)
Dose-response curve of the most potent compounds. (A) Compounds obtained from the first and the second screening indicated with the following colors: black, magenta, light green, blue, orange, dark green and red, for compound 11, 17, 21, 23, 25, 34 and 37, respectively. (B) Compounds obtained from the third screening indicated with the following colors: dark green, purple, orange, magenta, blue, black, red and light green, for compound 39, 40, 41, 42, 43, 44, 45 and 46, respectively. Data were fitted using the standard IC50 equation with Origin 7.0 software (OriginLab).
(TIF)
Ranking of the drug-like compounds, as assessed by VS protocol.
(DOC)
Ranking of lead-like candidates, as assessed by VS protocol.
(DOC)
Ranking of selected compounds, obtained by applying the similarity search of the entire ZINC database (∼9.0×106 compounds), using compound 11 as query.
(DOC)
Ranking of selected compounds, obtained by applying the similarity search of the entire ZINC database (∼9.0×106 compounds), using compound 5 as query.
(DOC)
Acknowledgments
This paper is dedicated to Prof. Donatella Barra on the occasion of her 70th birthday. The technical assistance of Dr. Giulia Chiavegato is kindly acknowledged.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants from the Italian Ministero dell'Università e della Ricerca, from the “Consorzio Interuniversitario per le Applicazioni di Supercalcolo per Università e Ricerca” (STD10-281; CASPUR, Roma, Italy), and by the Consorzio Interuniversitario per le Biotecnologie (CIB). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hauser RA. Levodopa: past, present, and future. Eur Neurol. 2009;62:1–8. doi: 10.1159/000215875. [DOI] [PubMed] [Google Scholar]
- 2.Gardian G, Vecsei L. Medical treatment of Parkinson's disease: today and the future. Int J Clin Pharmacol Ther. 2010;48:633–642. doi: 10.5414/cpp48633. [DOI] [PubMed] [Google Scholar]
- 3.Opacka-Juffry J, Brooks DJ. L-dihydroxyphenylalanine and its decarboxylase: new ideas on their neuroregulatory roles. Mov Disord. 1995;10:241–249. doi: 10.1002/mds.870100302. [DOI] [PubMed] [Google Scholar]
- 4.Bertoldi M, Frigeri P, Paci M, Voltattorni CB. Reaction specificity of native and nicked 3,4-dihydroxyphenylalanine decarboxylase. J Biol Chem. 1999;274:5514–5521. doi: 10.1074/jbc.274.9.5514. [DOI] [PubMed] [Google Scholar]
- 5.Bertoldi M, Moore PS, Maras B, Dominici P, Voltattorni CB. Mechanism-based inactivation of dopa decarboxylase by serotonin. J Biol Chem. 1996;271:23954–23959. doi: 10.1074/jbc.271.39.23954. [DOI] [PubMed] [Google Scholar]
- 6.Moore PS, Bertoldi M, Dominici P, Borri Voltattorni C. Aromatic amino acid methyl ester analogs form quinonoidal species with Dopa decarboxylase. FEBS Lett. 1997;412:245–248. doi: 10.1016/s0014-5793(97)00788-6. [DOI] [PubMed] [Google Scholar]
- 7.Voltattorni CB, Minelli A, Dominici P. Interaction of aromatic amino acids in D and L forms with 3,4-dihydroxyphenylalanine decarboxylase from pig kidney. Biochemistry. 1983;22:2249–2254. doi: 10.1021/bi00278a030. [DOI] [PubMed] [Google Scholar]
- 8.Voltattorni CB, Minelli A, Turano C. Spectral properties of the coenzyme bound to DOPA decarboxylase from pig kidney. FEBS Lett. 1971;17:231–235. doi: 10.1016/0014-5793(71)80153-9. [DOI] [PubMed] [Google Scholar]
- 9.Bertoldi M, Castellani S, Bori Voltattorni C. Mutation of residues in the coenzyme binding pocket of Dopa decarboxylase. Effects on catalytic properties. Eur J Biochem. 2001;268:2975–2981. doi: 10.1046/j.1432-1327.2001.02187.x. [DOI] [PubMed] [Google Scholar]
- 10.Dominici P, Moore PS, Castellani S, Bertoldi M, Voltattorni CB. Mutation of cysteine 111 in Dopa decarboxylase leads to active site perturbation. Protein Sci. 1997;6:2007–2015. doi: 10.1002/pro.5560060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominici P, Tancini B, Borri Voltattorni C. Chemical modification of pig kidney 3,4-dihydroxyphenylalanine decarboxylase with diethyl pyrocarbonate. Evidence for an essential histidyl residue. J Biol Chem. 1985;260:10583–10589. [PubMed] [Google Scholar]
- 12.Tancini B, Dominici P, Barra D, Voltattorni CB. An essential arginine residue at the binding site of pig kidney 3,4-dihydroxyphenylalanine decarboxylase. Arch Biochem Biophys. 1985;238:565–573. doi: 10.1016/0003-9861(85)90201-2. [DOI] [PubMed] [Google Scholar]
- 13.Burkhard P, Dominici P, Borri-Voltattorni C, Jansonius JN, Malashkevich VN. Structural insight into Parkinson's disease treatment from drug-inhibited DOPA decarboxylase. Nat Struct Biol. 2001;8:963–967. doi: 10.1038/nsb1101-963. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz DE, Brandt R. Pharmacokinetic and metabolic studies of the decarboxylase inhibitor benserazide in animals and man. Arzneimittelforschung. 1978;28:302–307. [PubMed] [Google Scholar]
- 15.Borri-Voltattorni C, Minelli A, Borri P. Interaction of N-(DL-seryl)N′-(2,3,4-trihydroxybenzyl)-hydrazine with L-dopa decarboxylase from pig kidney. Experientia. 1977;33:158–160. doi: 10.1007/BF02124039. [DOI] [PubMed] [Google Scholar]
- 16.Borri Voltattorni C, Minelli A, Borri P. The interaction of 2,3,4-trihydroxybenzylhydrazine with DOPA decarboxylase from pig kidney. Life Sci. 1981;28:103–108. doi: 10.1016/0024-3205(81)90372-6. [DOI] [PubMed] [Google Scholar]
- 17.Borri-Voltattorni C, Minelli A, Borri P. Interaction of L-alpha-methyl-alpha-hydrazino-3,4 dihydroxyphenylpropionic acid with dopa-decarboxylase from pig kidney. FEBS Lett. 1977;75:277–280. doi: 10.1016/0014-5793(77)80102-6. [DOI] [PubMed] [Google Scholar]
- 18.Bender DA. Effects of benserazide, carbidopa and isoniazid administration on tryptophan-nicotinamide nucleotide metabolism in the rat. Biochem Pharmacol. 1980;29:2099–2104. doi: 10.1016/0006-2952(80)90179-3. [DOI] [PubMed] [Google Scholar]
- 19.Bender DA, Earl CJ, Lees AJ. Niacin depletion in Parkinsonian patients treated with L-dopa, benserazide and carbidopa. Clin Sci (Lond) 1979;56:89–93. doi: 10.1042/cs0560089. [DOI] [PubMed] [Google Scholar]
- 20.Hartman WJ, Akawie RI, Clark WG. Competitive inhibition of 3, 4-dihydroxyphenylalanine (DOPA) decarboxylase in vitro. J Biol Chem. 1955;216:507–529. [PubMed] [Google Scholar]
- 21.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 23.Jiang X, Kumar K, Hu X, Wallqvist A, Reifman J. DOVIS 2.0: an efficient and easy to use parallel virtual screening tool based on AutoDock 4.0. Chem Cent J. 2008;2:18. doi: 10.1186/1752-153X-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics Chapter. 2008;8:Unit 8 14. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 25.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dominici P, Moore PS, Borri Voltattorni C. Dissociation, unfolding and refolding trials of pig kidney 3,4-dihydroxyphenylalanine (dopa) decarboxylase. Biochem J. 1993;295(Pt 2):493–500. doi: 10.1042/bj2950493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MACCS structural keys. 14600 Catalina Street, San leandro, CA 94577: MDL Information Systems Inc; [Google Scholar]
- 28.Tanimoto TT. 1957. IBM Internal Report.
- 29.Clark DE. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood-brain barrier penetration. J Pharm Sci. 1999;88:815–821. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 30.Standaert DG, Young AB, Schleifer LS, editors. Treatment of central nervous system degenerative disorders. pp. 533–538. 11 ed: McGraw-Hill Medical Publishing Division, New York.
- 31.Bender DA. Inhibition in vitro of the enzymes of the oxidative pathway of tryptophan metabolism and of nicotinamide nucleotide synthesis by benserazide, carbidopa and isoniazid. Biochem Pharmacol. 1980;29:707–712. doi: 10.1016/0006-2952(80)90544-4. [DOI] [PubMed] [Google Scholar]
- 32.Ebadi MS, Russell RL, McCoy EE. The inverse relationship between the activity of pyridoxal kinase and the level of biogenic amines in rabbit brain. J Neurochem. 1968;15:659–665. doi: 10.1111/j.1471-4159.1968.tb08964.x. [DOI] [PubMed] [Google Scholar]
- 33.McCormick DB, Snell EE. Pyridoxal phosphokinases. II. Effects of inhibitors. J Biol Chem. 1961;236:2085–2088. [PubMed] [Google Scholar]
- 34.Allain P, Le Bouil A, Cordillet E, Le Quay L, Bagheri H, et al. Sulfate and cysteine levels in the plasma of patients with Parkinson's disease. Neurotoxicology. 1995;16:527–529. [PubMed] [Google Scholar]
- 35.Kuhn W, Roebroek R, Blom H, van Oppenraaij D, Przuntek H, et al. Elevated plasma levels of homocysteine in Parkinson's disease. Eur Neurol. 1998;40:225–227. doi: 10.1159/000007984. [DOI] [PubMed] [Google Scholar]
- 36.Miller JW, Selhub J, Nadeau MR, Thomas CA, Feldman RG, et al. Effect of L-dopa on plasma homocysteine in PD patients: relationship to B-vitamin status. Neurology. 2003;60:1125–1129. doi: 10.1212/01.wnl.0000055899.24594.8e. [DOI] [PubMed] [Google Scholar]
- 37.Chen Z, Li HL, Zhang QJ, Bao XG, Yu KQ, et al. Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets. Acta Pharmacol Sin. 2009;30:1694–1708. doi: 10.1038/aps.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McInnes C. Virtual screening strategies in drug discovery. Curr Opin Chem Biol. 2007;11:494–502. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 39.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 40.Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- 41.Wang R, Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J Chem Inf Comput Sci. 2001;41:1422–1426. doi: 10.1021/ci010025x. [DOI] [PubMed] [Google Scholar]
- 42.Bertoldi M, Gonsalvi M, Contestabile R, Voltattorni CB. Mutation of tyrosine 332 to phenylalanine converts dopa decarboxylase into a decarboxylation-dependent oxidative deaminase. J Biol Chem. 2002;277:36357–36362. doi: 10.1074/jbc.M204867200. [DOI] [PubMed] [Google Scholar]
- 43.Perola E. An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J Med Chem. 2010;53:2986–2997. doi: 10.1021/jm100118x. [DOI] [PubMed] [Google Scholar]
- 44.Moriguchi IHS, Liu Q, Nakagome I, Matsushita Y. Simple method of calculating Octanol/Water Partition Coefficient. Chem Pharm Bull. 1992;40:4. [Google Scholar]
- 45.Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, et al. Virtual computational chemistry laboratory–design and description. J Comput Aided Mol Des. 2005;19:453–463. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- 46.Montioli R, Cellini B, Borri Voltattorni C. Molecular insights into the pathogenicity of variants associated with the aromatic amino acid decarboxylase deficiency. J Inherit Metab Dis. 2011 doi: 10.1007/s10545-011-9340-6. [DOI] [PubMed] [Google Scholar]
- 47.Sherald AF, Sparrow JC, Wright TR. A spectrophotometric assay for Drosophila dopa decarboxylase. Anal Biochem. 1973;56:300–305. doi: 10.1016/0003-2697(73)90194-2. [DOI] [PubMed] [Google Scholar]
- 48.Charteris A, John R. An investigation of the assay of dopamine using trinitrobenzensulphonic acid. Anal Biochem. 1975;66:365–371. doi: 10.1016/0003-2697(75)90604-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Normal distribution plot of predicted pKi values for drug-like molecules. 280000 drug-like molecules were docked in the active site of DDC with the Dovis 2.0 docking tool [23], [24] and the distribution of their predicted pKi was derived. The normal distribution showed a mean pKi of 5.57±0.81.
(TIF)
Normal distribution plot of predicted pKi values for lead-like molecules. 180000 lead-like molecules were docked in the active site of DDC with the Dovis 2.0 docking tool [23], [24] and the distribution of their predicted pKi was derived. The normal distribution showed a mean pKi of 5.30±0.62.
(TIF)
Dose-response curve of the most potent compounds. (A) Compounds obtained from the first and the second screening indicated with the following colors: black, magenta, light green, blue, orange, dark green and red, for compound 11, 17, 21, 23, 25, 34 and 37, respectively. (B) Compounds obtained from the third screening indicated with the following colors: dark green, purple, orange, magenta, blue, black, red and light green, for compound 39, 40, 41, 42, 43, 44, 45 and 46, respectively. Data were fitted using the standard IC50 equation with Origin 7.0 software (OriginLab).
(TIF)
Ranking of the drug-like compounds, as assessed by VS protocol.
(DOC)
Ranking of lead-like candidates, as assessed by VS protocol.
(DOC)
Ranking of selected compounds, obtained by applying the similarity search of the entire ZINC database (∼9.0×106 compounds), using compound 11 as query.
(DOC)
Ranking of selected compounds, obtained by applying the similarity search of the entire ZINC database (∼9.0×106 compounds), using compound 5 as query.
(DOC)